

Abstract
Serum amyloid A action in immune response and deposition in inflammation-linked amyloidosis involve SAA-lipid interactions. We show that SAA sequesters neutral and anionic phospholipids and their hydrolytic products to form nanoparticles, suggesting a synergy with phospholipase A2. The lipid charge and shape affect SAA protection from proteolysis, aggregation and fibrillogenesis.
Graphical abstract
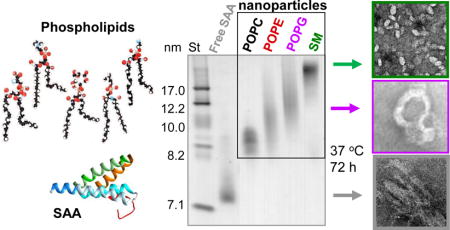
Serum amyloid A can solubilize diverse phospholipids and their hydrolytic products to form lipoprotein nanoparticles, which hampers amyloid fibril formation
Serum amyloid A (SAA, 12 kDa) is a plasma protein whose N-terminal 6-9 kDa fragments, termed amyloid A (AA), deposit systemically in AA amyloidosis, a potentially life-threatening complication of chronic inflammation.1,2 After acute injury or inflammation, plasma levels of SAA increase in 1-2 days up to 1000-fold and then swiftly drop. The physiologic advantage of this dramatic but transient increase is unknown and the precise role of SAA in immune response is unclear. Normal functions of SAA and its pathologic deposition as amyloid involve SAA-lipid interactions.1–4 Most human SAA circulates bound to plasma high-density lipoproteins (HDL). HDL are soluble nanoparticles (8-12 nm), each containing several hundred lipids stabilized by surface-bound apolipoproteins.5 In acute inflammation, SAA binds to HDL and diverts HDL cholesterol transport for cell repair.1 Since SAA is a Cambrian protein that pre-dates HDL,3 its primordial function must pre-date HDL binding. We posit that this function is to clear membrane debris from injured cells.6 This idea stems from several findings: i) SAA spontaneously clears a model phospholipid palmitoyl oleoyl (PO) phosphatidyl-choline (PC) to form HDL-size particles;6–8 ii) SAA crystal structures9,10 reveal a concave apolar surface formed by the amphipathic α-helices h1 and h3 (residues 1-27 and 51-69, ESI Fig. S1), which likely binds HDL11 and enables SAA to sequester lipids in nanoparticles.6 Similar to other apolipoproteins,5 SAA binding to HDL at pH7 stabilizes α -helices and inhibits amyloid formation,8,12 probably by blocking the apolar amyloidogenic segments in h1 and h3 that bind lipids13. However, SAA1 aggregation upon internalization by cells disturbs cell membranes, causing lysosomal leakage and apoptosis12; these effects likely stem from the lipid-dependent structural changes in SAA oligomers at lysosomal pH14. The effects of specific lipids on SAA fibril formation are just beginning to be elucidated.13
Here, in addition to the zwitterionic POPC that mimics the major lipids in the plasma membrane and in HDL, we explored SAA interactions with i) other neutral phospholipids, sphingomyelin (SM) and PO phosphatidyl ethanolamine (PE), which are important constituents of the plasma membrane and HDL; ii) anionic lipids such as PO phosphatidyl glycerol (PG), which are enriched in the inner leaflet of the plasma membrane but become exposed upon cell injury, and free fatty acids such as oleic acid (OA), which are produced upon lipolysis that is upregulated in inflammation (ESI Table S1).
We used recombinant full-length murine SAA1, a major amyloidogenic isoform that binds HDL (see ESI Methods, Fig. S1). First, SAA was incubated at 24 °C with multilamellar vesicles (MLVs) of POPC, POPE, POPG or SM at a protein:lipid molar ratio of 1:1 or 1:100 (“low” or “high lipid”) as described in Methods. SAA rapidly cleared these MLVs to form lipoproteins (Fig. 1A), which were isolated by gel filtration and observed by the non-denaturing PAGE and transmission electron microscopy (TEM). Lipoprotein stoichiometry was determined using biochemical assays (ESI Table S2). The lipoprotein size ranged from about 7.5 to 20 nm depending on the nature of the lipid (POPC < POPG < POPE < SM) and the protein:lipid ratio; “high-lipid” preparations yielded larger lipid-loaded particles (Fig. 1B, C).
Figure 1.
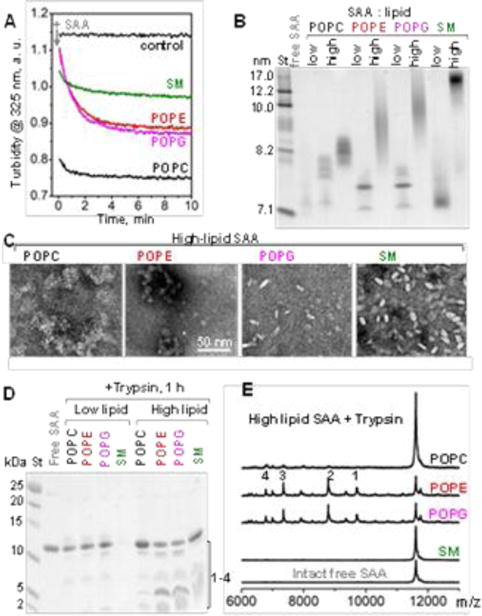
Formation and characterization of SAA:phospholipid complexes. (A) Clearance time course of MLVs by SAA at 24 °C monitored by turbidity. Arrow shows t=0 when SAA was added to MLVs at a 1:2 protein:lipid weight ratio. POPC MLV alone provided a control. (B) Non-denaturing PAGE of free SAA and its “low-lipid” and “high-lipid” complexes (see ESI Table S2). Lipid-fee SAA, which forms oligomers, is shown for comparison. (C) TEM of negatively-stained ”high-lipid” complexes. (D) SDS PAGE shows limited proteolysis of SAA in lipid complexes after 1 h of tryptic digestion. (E). MALDI-TOF mass spectrometry of the tryptic fragments generated using “high-lipid” SAA complexes. Intact free SAA is shown for comparison. The molecular mass of SAA fragments 1-4 observed in POPE and POPG complexes and the corresponding residue fragments were: 1) 9,714 Da (1-94), 2) 8,791 Da (1-76), 3) 7,348 Da (1-70), and 4) 6,336 Da (1-56).
Lipoprotein formation involved SAA folding at 25 °C, from a largely unordered to an α -helical conformation, observed by far-ultraviolet (UV) circular dichroism (CD) (ESI Fig. S2). The helical content depended on the amount and the nature of the lipid; higher helicity was seen in “high-lipid” complexes wherein POPC, POPE and POPG induced up to 55% helix, with less helical structure induced by SM (ESI Fig. S2 and Table S2). Similarly, SAA bound to HDL showed up to 50% helix.8 In stark contrast to free SAA that showed a reversible thermal unfolding below 25 °C, the unfolding of SAA-lipid complexes was partially irreversible with the midpoint Tm in the 45-55 °C range (ESI Fig. S2). Together, these results revealed that SAA spontaneously clears neutral and anionic lipids and forms binary complexes 7-20 nm in size, with increased helical content (up to 55%) and increased thermal stability (Tm~45-55 °C) as compared to the intrinsically disordered lipid-free SAA (Tm~15°C) (ESI Fig. S2).
Tryptic digestion of lipoproteins was monitored by SDS PAGE and MALDI TOF. Unlike free SAA that was largely fragmented by trypsin after 5 min (ESI Fig. S3), lipid-bound SAA was less fragmented after 1 hour (Fig. 1D). Surprisingly, although SAA in “high-lipid” complexes showed higher helical content (ESI Table S2, Fig. S2), “low-lipid” complexes were better protected from proteolysis (Fig. 1D). Perhaps the compact structure of the “low-lipid” complexes, suggested by their sharp gel bands (Fig. 1B), conferred their proteolytic protection. In contrast, “high-lipid” SAA complexes with POPG and POPE were especially labile to proteolysis (Fig. 1D, E). MALDI-TOF revealed N-terminal proteolytic fragments, e.g. residues 1-76, which constitute AA deposits in vivo (Fig. 1E). Hence, these fragments may be produced in vivo either from the major lipid-bound form of SAA or from its transient lipid-free form.
The time course of amyloid formation at 37 °C was monitored by binding of a fluorescent dye thioflavin T (ThT) (Fig. 2A). In free SAA, a sigmoidal increase in the ThT emission suggested formation of amyloid-like structure via the nucleation-propagation mechanism.15 In contrast, “low-lipid” SAA complexes as well as the “high-lipid” complexes with POPC or SM showed no changes in emission. Interestingly, “high-lipid” complexes with POPG or POPE showed a gradual increase in emission, with no lag (nucleation) phase and an exponential decay (rather than a sigmoidal) propagation phase that had lower amplitude as compared to free SAA (Fig. 2A). TEM of samples taken after 72 hours of incubation at 37 °C confirmed fibril formation in free SAA and in “high-lipid” complexes with POPE or POPG; in contrast, “high-lipid” SAA complexes with POPC and SM remained morphologically intact and no fibrils were observed (Fig. 2B). Since “low-lipid” complexes, which had a more inert structure, did not form fibrils, “high-lipid” complexes were used for further studies.
Figure 2.
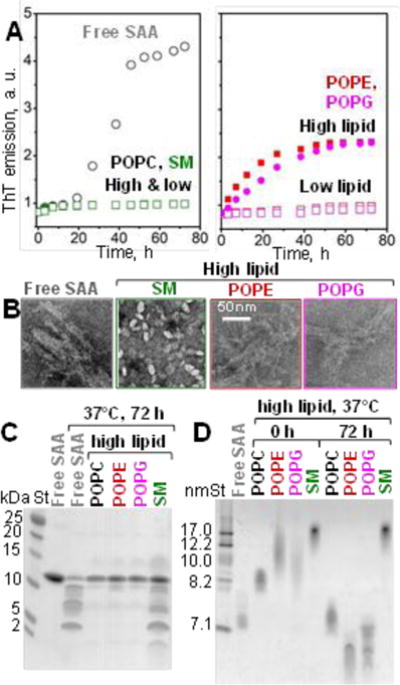
Effects of phospholipids on SAA amyloid formation at 37 °C in “high-lipid” complexes. (A) Time course of amyloid formation monitored by ThT fluorescence. Each data point is a mean of four independent measurements. (B) TEM of negatively stained samples after 72 h incubation at 37 °C; each sample contained 1 mg/ml SAA, either lipid-free or in high-lipid complexes. (C) SDS PAGE of SAA samples after 72 h incubation at 37 °C. (D) Non-denaturing PAGE of SAA samples before (0 h) and after (72 h) incubation at 37 °C.
Far-UV CD spectra of incubated “high-lipid” complexes of SAA with POPC or SM (which did not form fibrils) showed a highly helical structure, particularly with POPC; in contrast, free SAA and SAA in “high-lipid” complexes with POPG and POPE (which formed fibrils) showed little ordered secondary structure at 25 °C (ESI Fig. S4). Together, these results showed that SAA binding to POPC and SM stabilizes α -helices during incubation at 37 °C and inhibits amyloid formation, while binding to POPE and POPG does not inhibit amyloid formation but alters its kinetics.
To understand differential effects of lipids on amyloid formation upon SAA incubation at 37 °C, we probed protein and lipoprotein integrity during incubation. SDS PAGE showed that free SAA (which formed fibrils) was largely fragmented after 72 h at 37 °C; SAA was also fragmented in “high-lipid” complexes with SM (when no fibrils were observed), consistent with relatively low helical content of SAA with SM (ESI Fig. S2, “high lipid”); no protein fragments formed in complexes with POPC (no fibrils), POPE and POPG (fibrils) (Fig. 2C) wherein the protein showed high helical content (ESI Table S2, Fig. S2). Clearly, protein fragmentation correlated inversely with the initial protein helical content, but not with the amyloid formation.
Unexpectedly, non-denaturing PAGE showed that SAA complexes with POPC, POPE and POPG, but not SM, converted into smaller particles upon prolonged incubation at 37 °C (Fig. 2D). Since SDS PAGE detected no protein fragmentation during incubation with PO, such a remodelling could have resulted from PO hydrolysis. In fact, free fatty acid content significantly increased during incubation at 37 °C of “high-lipid” SAA samples with POPE, POPG and POPC, but not with SM (ESI Fig. S5). Consequently, remodelling of SAA complexes with POPC, POPG and POPE, but not SM, upon incubation at 37 °C (Fig. 2D) probably stems from the PO hydrolysis.
Phospholipid hydrolysis in vivo to produce lyso-phospholipids and free fatty acids is mediated mainly by the secretory phospholipase A2 (PLA2). To test how this lipolysis affects SAA aggregation, “high-lipid” SAA complexes were hydrolysed with PLA2 as described in ESI Methods. A strikingly similar pattern of lipoprotein remodelling into smaller particles was observed upon PLA2 lipolysis (Fig. 3A) and upon prolonged incubation at 37 °C without PLA2 (Fig. 2C), in excellent agreement with the gel filtration (Fig. S6). These results ascertained that lipoprotein remodelling into smaller particles stemmed from PO hydrolysis.
Figure 3.
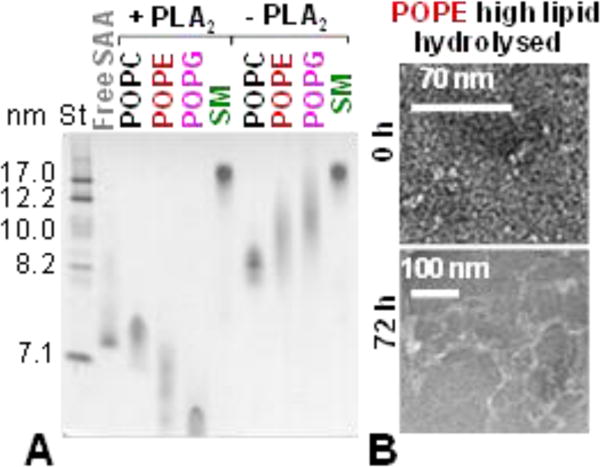
Remodelling of “high-lipid” SAA complexes by PLA2 and its effects on fibrillation. (A) Non-denaturing PAGE of the complexes that have been incubated at 37 °C for 3 h with (+PLA2) or without PLA2 (-PLA2). Intact free SAA is shown for comparison. Gel filtration data (Fig S6) ascertain these results. (B) TEM of “high-lipid” SAA:POPE complexes hydrolysed with PLA2; the images were taken before (0 h) and after 72 h incubation at 37 °C (72 h).
To determine how the products of the PLA2 reaction affect amyloid formation by SAA, “high-lipid” SAA complexes that have been hydrolysed with PLA2 as described in ESI Methods were incubated at 37 °C for 72 h. TEM showed fibril formation upon incubation of SAA complexes with POPE (Fig. 3B) and POPG, but not POPC or SM. ESI Fig. S7 shows additional representative data for SAA complexes with POPE (which formed amyloid) and with POPC (which did not). We conclude that, regardless of the PO hydrolysis (either spontaneous or by PLA2), formation of lipoprotein complexes with POPC and SM, but not with POPE or POPG, protects SAA from fibril formation.
PLA2 produces equimolar amounts of lyso-phospholipids and free fatty acid. The role of lyso-phospholipids alone was explored in recent studies of SAA fragments showing that binding to zwitterionic, but not anionic, lipids prevents amyloid formation.13 However, the effect of free fatty acids on the SAA conformation and aggregation has not been explored.
To directly probe this effect, SAA was incubated with oleic acid (OA) as described in ESI Methods. Non-denaturing PAGE showed similar migration for SAA with and without OA (Fig. 4A). Nevertheless, OA induced SAA folding into an 40% α -helical conformation at 25 °C (Fig. 4B), suggesting direct interactions between SAA and OA. Such interactions were also supported by limited tryptic digestion that was greatly delayed in SAA in the presence of OA (ESI Fig. S8). Next, SAA complexes with OA were incubated at 37 °C and amyloid formation was monitored by ThT fluorescence (Fig. 4C). Similar to POPG and POPE (Fig. 2A), OA did not block amyloid formation but abrogated the lag phase and decreased the maximal emission intensity. TEM confirmed that SAA in the presence of OA formed fibrils upon 72 h incubation at 37 °C; far-UV CD spectra of this fibrillary material were similar to those of other fibrillar SAA, suggesting a partially ordered secondary structure (Fig. 4D). We conclude that OA binds SAA and induces a highly helical conformation, which decelerates the proteolysis and alters the kinetics of SAA misfolding into amyloid but does not block this misfolding.
Figure 4.
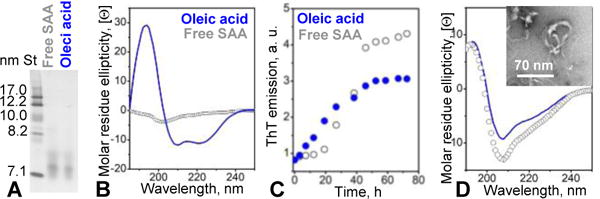
Effect of oleic acid on the SAA secondary structure and amyloid formation. (A) Non-denaturing PAGE of SAA in the absence (gray) and in the presence of OA (blue). SAA (0.5 mg/ml) was treated with 5 mM OA as described in ESI Methods. (B) Far-UV CD spectra show that OA induced helical folding in SAA at 25 °C, from the largely unfolded to 40% α -helical conformation. (C) Time course of amyloid formation by SAA in the absence and in the presence of OA monitored by ThT fluorescence. (D) Far-UV CD spectrum of fibrillar OA-treated or lipid-free SAA after 72 h incubation at 37 °C. Insert: TEM of OA-treated SAA after incubation.
In summary, this study reports several new findings. First, besides POPC, SAA also rapidly clears MLVs of other neutral (SM, POPE) and anionic phospholipids (POPG), which are important membrane constituents, and converts them into 7-20 nm particles (Fig. 1A-C). SAA also binds the hydrolytic products of these lipids, including lyso-phospholipids13 and free fatty acids (Fig. 4). Hence, SAA binds diverse lipids regardless of their charge (negative or neutral) and shape, which defines spontaneous lipid curvature (positive, negative or zero, ESI Table S1). SAA also solubilizes dimyristoyl PC,7,8 cholesterol,16 and a lipophilic vitamin retinol.10 The size of the SAA-lipid particles suggests that lipids are sequestered via the concerted action of multiple protein molecules. We propose that several copies of SAA cooperate to form a lipid-filled hydrophobic cavity lined with the apolar faces of α -helices h1 and h3.6 We postulate that rapid energy-independent solubilization of diverse lipids reflects the primordial function of SAA in immune response - clearance of cell membrane debris from injured sites.
Second, our results reveal differential effects of phospho-lipids on SAA proteolysis and amyloid formation. Our studies clearly show that binding to POPC, which mimics the major lipids in the plasma membrane, protects SAA from proteolysis (Fig. 1D) and amyloid formation (Fig. 2C). In contrast, lipid-rich SAA complexes with POPE and POPG, as well as the complexes with OA are labile to proteolysis and amyloid formation (Fig. 1D; Fig. 2A,B; Fig. 4). These differences must reflect distinct protein and lipid packing in different complexes. Besides, the relatively labile SAA conformation in complexes with POPE (which is reportedly anionic in nanoparticles),17 POPG and OA might be affected by the electrostatic repulsion between the negative lipid charge and the electronegative patch on the SAA surface formed by helices h2 and h4.6
Third, our results suggest that SAA facilitates phospholipid hydrolysis by secretory PLA2 via two mechanisms. First, such a hydrolysis critically depends on the positive (convex) curvature of the lipid surface.18 By forming nanoparticles, SAA confers positive curvature to diverse lipids facilitating their hydrolysis by PLA2 (Figs. 3, S7). This finding is important since in inflammation PLA2 is upregulated up to 3,000-fold concomitantly with SAA, suggesting a concerted action of these proteins. Notably, phospholipid hydrolysis remodels SAA complexes into small ~7 nm particles (Figs. 2D, 3A, S6). Compared to their larger lipid-loaded counterparts, the small particles have a more compact structure that helps protect SAA from proteolysis (Fig. 1D). Furthermore, SAA ability to bind the hydrolytic products of PLA2, including lyso-phospholipids13 and free fatty acids (Fig. 4), suggests another mode of synergy between SAA and PLA2. We propose that SAA not only solubilizes lipids into nanoparticles and thereby facilitates phospholipid hydrolysis by secretory PLA2, but also helps remove the reaction products. Whether or not the resulting SAA:lipid complexes form amyloid depends on their lipid composition and, in particular, on the nature of the lipid head group. We show that SAA complexes with POPG and POPE and their corresponding lyso-lipids13 are more prone to amyloid formation than their POPC-containing counterparts.
In our view, spontaneous formation of HDL-like particles at circa pH7 reflects beneficial action of SAA in immunity and is not linked to cytotoxicity. Like SAA, other amyloid-forming apolipo-proteins also contain amphipathic α -helices5. One example is α -synuclein that forms amyloid in neurodegenerative diseases. Similar to SAA, α -synuclein folds into α -helices upon lipid binding and forms HDL-like particles with anionic phospholipids, which may benefit remodeling of synaptic membranes19. The membrane-disrupting ability of α -syniclein and other amyloid proteins, such as Aβ peptide in Alzheimer’s disease or amylin in diabetes-2, has been linked to cytotoxicity in vivo. To our knowledge, such a link has not been established firmly for SAA.
Supplementary Material
Acknowledgments
This study was supported by the National Institutes of Health grant GM067260, the Stewart Family Amyloid Endowment Fund and the Deutsche Forschungsgemeinschaft grant FA456/15-1.
Footnotes
Electronic Supplementary Information (ESI) available: Materials and Methods, Tables S1 and S2, Figures S1-S8. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
References
- 1.Kisilevsky R, Manley PN. Amyloid. 2012;19:5. doi: 10.3109/13506129.2011.654294. [DOI] [PubMed] [Google Scholar]
- 2.Westermark GT, Fändrich M, Westermark P. Annu Rev Pathol. 2015;10:321. doi: 10.1146/annurev-pathol-020712-163913. [DOI] [PubMed] [Google Scholar]
- 3.Sun L, Ye RD. Gene. 2016;583(1):48. doi: 10.1016/j.gene.2016.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kollmer M, Meinhardt K, Haupt C, Liberta F, Wulff M, Linder J, Handl L, Heinrich L, Loos C, Schmidt M, Syrovets T, Simmet T, Westermark P, Westermark GT, Horn U, Schmidt V, Walther P, Fändrich M. Proc Natl Acad Sci USA. 2016;113(20):5604. doi: 10.1073/pnas.1523496113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das M, Gursky O. Adv Exp Med Biol. 2015;855:175. doi: 10.1007/978-3-319-17344-3_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frame N, Jayaraman S, Gantz D, Gursky O. J Struct Biol. 2017;200:293. doi: 10.1016/j.jsb.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takase H, Furuchi H, Tanaka M, Yamada T, Matoba K, Iwasaki K, Kawakami T, Mukai T. Biochim Biophys Acta. 2014;1842(10):1467. doi: 10.1016/j.bbalip.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Jayaraman S, Haupt C, Gursky O. J Lipid Res. 2015;56(8):1531. doi: 10.1194/jlr.M059162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu J, Yu Y, Zhu I, Cheng Y, Sun PD. Proc Natl Acad Sci USA. 2014;111(14):5189. doi: 10.1073/pnas.1322357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derebe MG, Zlatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, MacMillan JB, Williams NS, Hooper LV. Elife. 2014;3:e03206. doi: 10.7554/eLife.03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frame NM, Gursky O. FEBS Lett. 2016;590:866. doi: 10.1002/1873-3468.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claus S, Meinhardt K, Aumüller T, Puscalau-Girtu I, Linder J, Haupt C, Walther P, Syrovets T, Simmet T, Fändrich M. EMBO Rep. 2017;18(8):1352. doi: 10.15252/embr.201643411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka M, Nishimura A, Takeshita H, Takase H, Yamada T, Mukai T. Chem Phys Lipids. 2017;202:6. doi: 10.1016/j.chemphyslip.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Jayaraman S, Gantz DL, Haupt C, Gursky O. Proc Natl Acad Sci USA. 2017;114(32):E6507. doi: 10.1073/pnas.1707120114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, Roy R, Singh S. J Struct Biol. 2005;151(3):229. doi: 10.1016/j.jsb.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Liang JS, Schreiber BM, Salmona M, Phillip G, Gonnerman WA, de Beer FC, Sipe JD. J Lipid Res. 1996;37:2109. [PubMed] [Google Scholar]
- 17.Her C, Filoti DI, McLean MA, Sligar SG, Alexander Ross JB, Steele H, Laue TM. Biophys J. 2016;111(5):989. doi: 10.1016/j.bpj.2016.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leidy C, Mouritsen OG, Jorgensen K, Peters NH. Biophys J. 2004;87:408. doi: 10.1529/biophysj.103.036103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eichmann C, Campioni S, Kowal J, Maslennikov I, Gerez J, Liu X, Verasdonck J, Nespovitaya N, Choe S, Meier BH, Picotti P, Rizo J, Stahlberg H, Riek R. J Biol Chem. 2016;291(16):8516. doi: 10.1074/jbc.M115.707968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.