

Abstract
Background
Patients with follicular lymphoma (FL) have heterogeneous outcomes. Predictor models able to distinguish, at diagnosis, patients at high versus low risk of progression are still needed.
Methods
The primary objective of this study was to use gene-expression profiling data to build a signature predictive of outcome in patients treated in the rituximab era. In a retrospectively assembled training cohort of 134 pretreatment FL patients from the prospective randomized PRIMA trial, we developed an expression-based predictor of progression-free survival (PFS) that was further evaluated in FFPE samples obtained from three independent international cohorts, using NanoString technology. The validation cohorts comprised a distinct set of patients from the PRIMA trial (n=178), a cohort from the University of Iowa/Mayo Clinic Lymphoma SPORE (n=201) and the Hospital Clinic University of Barcelona (n=109). All tissue samples consisted of pretreatment diagnostic biopsies and were confirmed as FL grade 1-3a. The patients were all treated with regimens containing rituximab and chemotherapy, possibly followed by either rituximab maintenance or ibritumomab-tiuxetan consolidation.
Findings
The expression levels of 395 genes were associated with a risk of progression. Twenty-three genes reflecting both B-cell biology and tumor microenvironment were retained to build a predictive model, which identified a population at an increased risk of progression (p<0.0001). In a multivariate Cox model for PFS adjusted on rituximab maintenance treatment and FLIPI-1, this predictor was found to independently predict progression (adjusted hazard ratio (HR) of the high-risk compared to the low-risk group: 3.68; 95%CI: 2.19-6.17). The digital gene expression data met quality criteria for 460/488 (94%) FFPE samples of the validation cohorts. The predictor performances were confirmed in each of the individual validation cohorts (adjusted HR [95%CI] comparing high risk to low risk groups were respectively 2.57 [1.65-4.01], 2.12 [1.32-3.39] and 2.11 [1.01-4.41]). In the combined validation cohort, the median PFS values were 3.1 (95%CI: 2.4-2.8) and 10.8 (95%CI: 10.1-NR) years in the high- and low-risk groups, respectively. The risk of lymphoma progression at 2 years was twice as high in the high-risk group (38% (95%CI: 29-46) versus 19% (95%CI: 15-24)). In a multivariate analysis, the score predicted PFS independently of anti-CD20 maintenance treatment and of the FLIPI score (hazard ratio for the combined cohort, 2.30; 95%CI, 1.72-3.07).
Interpretation
We developed a robust 23-gene expression-based predictor of PFS, applicable to routinely available FFPE biopsies from FL patients at diagnosis. This score may allow individualizing therapy for patients with FL according to the patient risk category.
Funding
Roche Company, SIRIC Lyric, LYSARC, NIH and the Henry J. Predolin Foundation, Spanish Plan Nacional de Investigacion SAF2015-64885-R.
Introduction
Follicular lymphoma (FL) is the most common indolent lymphoma and is characterized by prolonged median survival, usually exceeding 10 years.1 Treatment options range from watchful waiting to CD20-directed immunotherapy alone or in combination with chemotherapy.2 New non-cytotoxic combinations are also currently being evaluated.3 Patient outcomes are, however, highly heterogeneous, and a significant proportion of patients are at risk of early progression and/or transformation into high-grade lymphoma.4 Currently, Follicular Lymphoma International Prognostic Index (FLIPI-1 and FLIPI-2) scores are the best clinical pre-treatment predictors of outcome, although they are unable to accurately capture the group of patients who progress within 2 years.5–7 There have also been proposals to combine the mutation status of several genes with FLIPI to improve the identification of FL patients at high risk of progression.8,9 Hence, new predictor models able to distinguish, at diagnosis, patients with markedly distinct outcomes are still needed to personalize treatment approaches.
Gene-expression signatures previously reported in FL in the pre-rituximab era have highlighted the influence of non-malignant tumor-infiltrating cells.10 However, the prognostic impact of immune cells in the microenvironment, assessed using immunohistochemistry, has remained controversial, potentially due to heterogeneity of patient cohorts, methodologies, and statistical endpoints across the studies.11–14 Overall, there is still a lack of robust confirmatory studies with which to translate these original findings for routine patient management.
We hypothesized that a gene-expression profiling (GEP)-based study, reflecting both the tumor biology and microenvironment, would allow building a composite signature predictive of outcome in patients treated in the rituximab era. We thus investigated the biological features of FL tumors in a retrospective analysis using samples of patients from a large phase III trial.15 We developed a gene-expression-based predictor of progression-free survival (PFS) applicable to formalin-fixed paraffin-embedded (FFPE) tissues, a resource generated during routine diagnostic work-up. We validated this predictor model in three distinct, independent cohorts of patients, all receiving immunochemotherapy as first-line treatment. Beyond the predictor model, we used these data to evaluate gene-expression signatures reflecting different aspects of tumor biology for their association with outcome.
Methods
Study design and patients
A gene-expression-based predictor model was produced using a training cohort of FL patients and then tested for clinical validation in three independent cohorts (Figure 1). All tissue samples (training and all three validation cohorts) consisted of pretreatment diagnostic biopsies (obtained within 12 months before treatment initiation) and were confirmed as FL grade 1-3a16 by expert hematopathologists. This study was conducted in accordance with the Declaration of Helsinki. All patients signed a consent form for participation in specific biological studies.
Figure 1. Outline of the overall study design.
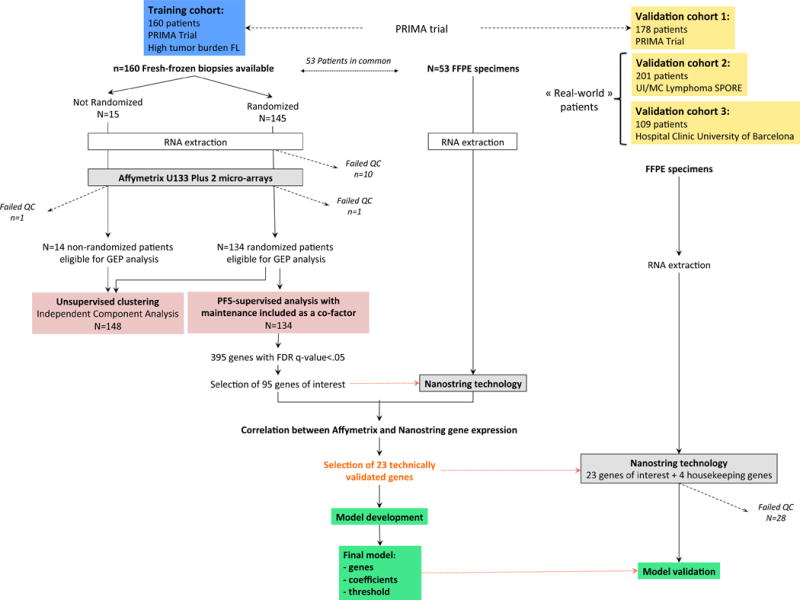
Fresh-frozen tissue (FFT) tumor biopsies were prospectively obtained from 160 untreated patients enrolled in the international PRIMA trial. RNA with sufficient quality (RIN>6.5) was obtained for 149/160 cases and gene-expression profiling was performed using Affymetrix U133 Plus 2.0 micro-arrays. A multivariate Cox regression analysis identified genes whose expression was associated with PFS independent of maintenance treatment in the subgroup of randomized patients. Expression levels from 95 curated genes were then determined by means of digital expression profiling (NanoString technology) in 53 FFPET samples of the training set, allowing assessment of the technical replication of expression levels for each gene between technologies. Genes with high correlation (>0.75) were included in a L2-penalized Cox model adjusted on rituximab maintenance to build a PFS-predictive score. The model was further evaluated using NanoString technology in 488 FFPET samples from 3 independent international cohorts of patients: a distinct validation set from the PRIMA trial (n=178), and two others obtained in large centers (respectively the Mayo Clinic/Iowa SPORE project, n=201 and the Barcelona Hospital Clinic, n=109). An unsupervised analysis of the gene-expression data generated in the training cohort was also performed independently.
Abbreviations: FFT: fresh-frozen tissues; FFPE: formalin-fixed paraffin-embedded tissues; PFS: progression-free survival.
Training cohort
Fresh-frozen tissue (FFT) tumor biopsies were prospectively obtained from 160 untreated patients participating in the phase III randomized PRIMA trial, which evaluated rituximab maintenance after rituximab-chemotherapy induction in high-tumor-burden FL patients (NCT00140582). In this trial, patients were first treated by one of the three rituximab-chemotherapy regimens (R-CHOP: rituximab, cyclophosphamide, doxorubicine, vincristine, prednisone; R-CVP: rituximab, cyclophosphamide, vincristine, prednisone; R-FCM: rituximab, fludarabine, cyclophosphamide, mitoxantrone). After this induction phase, patients who obtained a complete or partial response were randomly assigned to observation or rituximab maintenance for 2 years (see appendix p 2 for inclusion criteria and design of the PRIMA trial).15 Response and progression were defined with international standard criteria.17 Patients of the training cohort were French and Belgian patients, exclusively selected based on the availability of FFT samples. The clinical characteristics and median follow-up (6.6 years) of this training cohort were similar to those of patients from the whole PRIMA study (appendix p 24). RNA of sufficient quality (RIN>6.5) was obtained for 149/160 cases and further processed. FFPE biopsies were also available for 53 of these patients for technical validation of gene expression quantification on fixed tissue.
Validation cohorts
The model generated from the training cohort was further evaluated in three independent cohorts of patients for whom FFPE biopsies obtained at diagnosis were available, including a distinct validation set from the PRIMA trial (limited to FFPE samples from French and Belgian patients of the PRIMA trial who were not part of the training cohort: cohort 1, n=178) and two other sets of newly diagnosed patients, followed in large clinical centers, the University of Iowa/Mayo Clinic Lymphoma SPORE18 (UIMC, cohort 2, n=201) and the Hospital Clinic University of Barcelona (BCN, cohort 3, n=109). Cohorts 2 and 3 consisted of newly diagnosed patients who were prospectively recruited in observational studies. They were all treated with regimens containing rituximab and chemotherapy, possibly followed by either rituximab maintenance or ibritumomab-tiuxetan consolidation and with a median follow-up exceeding five years (Table 1). Progression, as identified during routine follow-up practices, was defined with similar criteria as in the training cohort. Given that patients having received R-chemotherapy in these validation cohorts did not have to fulfill PRIMA trial inclusion criteria, their characteristics (including distribution in the different FLIPI categories) may differ.
Table 1. Demographics and clinical characteristics of patients in the training cohort and the three validation cohorts.
The results obtained from the training set were further validated in three independent cohorts of patients drawn from a distinct validation set in the PRIMA study (cohort 1), the University of Iowa/Mayo Clinic Lymphoma SPORE (cohort 2) and the Hospital Clinic University of Barcelona (cohort 3).
| Cohort Number of patients |
Training cohort N=134 no./total no. (%)* |
Validation cohort 1 N=172 no./total no. (%)* |
Validation cohort 2 N=186 no./total no. (%)* |
Validation cohort 3 N=102 no./total no. (%)* |
|---|---|---|---|---|
|
| ||||
| Follow-up duration | ||||
| median, IQR (yrs) | 6.6 (6.0-7.0) | 6.8 (6.5-7.1) | 5.5 (3.9-7.8) | 5.8 (4.1-8.7) |
|
| ||||
| Time from biopsy to initial treatment | ||||
| median, range (months) | 1 (0-5) | 1 (0-7) | 0.8 (0-7.2) | 0.7 (0-4) |
|
| ||||
| Baseline characteristics | ||||
| Age > 60 years | 47/134 (35) | 51/172 (30) | 76/186 (41) | 38/102 (37) |
| Male sex | 68/134 (51) | 84/172 (49) | 109/186 (59) | 44/102 (43) |
| Ann Arbor stage III/IV | 123/134 (92) | 158/172 (92) | 149/184 (81) | 77/102 (75) |
| ECOG PS ≥ 1 | 43/134 (32) | 45/172 (26) | 71/185 (38) | 15/101 (15) |
| B symptoms present | 30/134 (22) | 54/172 (31) | 38/184 (21) | 16/101 (16) |
| Bone Marrow involvement | 83/129 (64) | 100/168 (60) | 87/169 (51) | 59/102 (58) |
| Elevated LDH | 43/134 (32) | 57/171 (33) | 59/160 (37) | 25/95 (26) |
| Hemoglobin level < 12 g/dL | 29/134 (22) | 32/172 (19) | 33/169 (20) | 18/94 (19) |
| Elevated β2-microglobulin | 81/128 (63) | 93/159 (58) | 31/43 (72) | 49/92 (53) |
|
| ||||
| FLIPI score | ||||
| 0-1 risk factors | 23/134 (17) | 41/171 (24) | 51/186 (27) | 38/96 (40) |
| 2 risk factors | 53/134 (40) | 56/171 (33) | 62/186 (33) | 32/96 (33) |
| 3-5 risk factors | 58/134 (43) | 74/171 (43) | 73/186 (39) | 26/96 (27) |
|
| ||||
| Histological grade | ||||
| 1-2 | 113/134 (84) | 132/172 (77) | 123/186 (66) | 64/102 (63) |
| 3A | 12/134 (9) | 22/172 (13) | 63/186 (34) | 28/102 (27) |
| Undetermined/other** | 9/134 (7) | 18/172 (10) | 0 | 10/102 (10) |
|
| ||||
| Induction regimen | ||||
| R-CHOP | 128/134 (96) | 157/172 (91) | 96/186 (52) | 102/102 (100) |
| R-CVP | 6/134 (4) | 15/172 (9) | 59/186 (32) | 0 |
| R-Bendamustine | 0 | 0 | 29/186 (16) | 0 |
| other | 0 | 0 | 2/186 (1) | 0 |
|
| ||||
| Maintenance regimen | ||||
| No | 76/134 (57) | 96/172 (56) | 152/186 (82) | 59/102 (58) |
| Yes | 58/134 (43) | 76/172 (55) | 34/186 (18) | 43/102 (42) |
|
| ||||
| Rituximab | 58/58 (100) | 76/76 (100) | 34/34 (100) | 41/43 (95) |
| Ibritumomab-tiotexan | 0 | 0 | 0 | 2/43 (5) |
Because of rounding, percentages may not total 100.
Undetermined/other included FL of undetermined grade (except 3b), FL with a very small component of DLBCL<10% and FL with diffuse area.
Abbreviations: BM, bone marrow; LDH, lactate dehydrogenase; R, rituximab; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; CVP, cyclophosphamide, vincristine, prednisone; FCM, fludarabine, cyclophosphamide, mitoxantrone; NR, not reached.
Gene-expression profiling experiments
Gene-expression profiling was first performed in the training cohort using the Affymetrix GeneChip® Human Genome U133 Plus 2.0 array (data available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE93261). Digital gene expression quantification of selected genes was then conducted on RNA from FFPE samples by means of NanoString technology, both in some biopsies of the training set (n=53) and in the validation cohorts (appendix pp 2,3 for detailed experimental methods).
Statistical analysis
Predictive model building
All genes (14,356 probesets representing unique genes and considered expressed above noise level, appendix p 4) were tested for association with PFS in the training cohort. As rituximab maintenance significantly impacted PFS in the PRIMA trial (appendix p 9), a multivariate Cox regression analysis including maintenance treatment as a cofactor was applied to the 134 randomized patients. Initial clinical features (such as FLIPI score) were not included in the model, in order to design a fully biologically based model. P-values were adjusted for multiple testing, and only genes significant at the False Discovery Rate (FDR) threshold of 5% were retained. Of the 395 genes whose expression was significantly associated with PFS, we selected an initial set of 95 genes (see appendix pp 4, 10 for selection criteria). The expression levels of these 95 manually curated genes and four housekeeping genes were then measured using NanoString in a subset of 53 FFPE samples from the training cohort to assess the technical replication between the two technologies. Genes with a correlation coefficient >0.75 were retained in an L2-penalized (Ridge) Cox model adjusted for rituximab maintenance to build a multigene score predicting PFS independently of the effect of the maintenance treatment. The choice of Ridge penalization was guided by the multicollinearity between the predictors. The global performance of the model was determined by means of the concordance statistic (C-statistic).19 An optimal threshold on the score to separate patients into low- and high-risk groups was determined using the MaxStat package20 to select the cutoff value producing the maximal log-rank score in the training cohort.
The locked model, including the multigene score and the threshold, was first evaluated in the three validation groups separately. The three groups were subsequently pooled for additional analyses. Sensitivity and specificity of the score for the prediction of the risk of lymphoma progression at two years were assessed on the combined validation cohorts (appendix p 6). All statistical analyses, model training and testing were conducted using R. Detailed descriptions on statistical methods are provided in the appendix p 4.
Unsupervised clustering and functional enrichment analyses
Gene clustering was performed on microarray expression data from the training cohort by means of an independent component analysis (ICA).21 In contrast with PFS-supervised analysis, this unsupervised analysis was performed on all patients from the training cohort (n=148 passing QC). Briefly, ICA identifies expression signatures (“components”) capturing major underlying sources of variation in the data. Permutations were applied to determine the maximal number of non-random components. For each component, the most influential genes, i.e. those with highest weights (“leading genes”) were retrieved using a standard deviation threshold of 3. To infer the potential biological relevance of ICA components, we compared their leading genes with gene sets of the Molecular Signature Database (MSigDB) v5.0 and Leukemia/Lymphoma Molecular Profiling Project (LLMPP) collections (SignatureDB)22 (appendix p 5).
Role of the funding source
The funding sources had no involvement in study design, collection, analysis, and interpretation of the data, and in writing of the report or decision to submit this paper for publication. SHu, BT, JPJ, ET, MC, SB, AV, and GS had access to the raw Affymetrix microarray expression data. SHu, BT, JPJ, BA, CR, DG, and GS had access to the raw Nanostring data. The LYSARC (Lymphoma Study Academic Research organisation) provided access to raw clinical data of PRIMA patients to SHu, BT, JPJ, CH, PF, HT, PB, FJ, and GS. LM, ALG and EC had access to raw clinical data of BCN patients. ALF, SMA, BKL, FO, and JRC had access to raw clinical data of UIMC patients. BT and JPJ conducted the final bioinformatics and statistical analyses. The corresponding author had full access to all the data in the study and the final responsibility for the decision to submit for publication.
Results
Predictive model of progression-free survival
In the PFS-supervised analysis, differential expression of 395 genes (list in appendix p 24) was significantly associated with outcome in the training cohort. Higher expression levels were associated with longer PFS for 228 genes and with shorter PFS for 167 genes (appendix p 11). Of those 395 genes, 95 were selected by manually curating for each gene to integrate technical, statistical and biological aspects. To build a PFS predictive model applicable to FFPE samples, the expression of those 95 curated genes was measured using NanoString technology on 53 FFPE tissues derived from the same biopsies as in the training set. Twenty-three genes with correlation coefficients >0.75 between the two technologies and sample types (microarray on FFT versus NanoString on FFPE samples) were retained (appendix p 12). This panel included genes previously described to be involved in B-cell development (VPREB1, FOXO1, FCRL2, AFF3, TCF4), apoptosis, and DNA damage response (RASSF6, GADD45A), cell cycle (E2F5, USP44), cell migration (CXCR4, SEMA4B, EML6, DCAF12, VCL, RGS10), immune regulation (CXCR4, KIAA0040, TAGAP, ORAI2, KIAA0040, METRNL), and other processes (PRDM15, ABCB1, ALDH2, SHISA8) although some of these genes are involved in multiple additional pathways.
The 23-gene expression signature scores ranged from 0.621 to 1.504 in the training cohort. The C-statistic was 0.709 (95% confidence interval [CI]: 0.644-0.773), outperforming FLIPI-1 (0.578, 95%CI: 0.501-0.655). A score of 1.075 was determined as the optimal threshold (appendix p 13) to separate patients into high- (n=47, 35%) and low-risk (n=87, 65%) groups (p<0.0001, log-rank test) (Figure 2). In a multivariate Cox model for PFS adjusted on rituximab maintenance treatment and FLIPI-1, this predictor was found to independently predict progression (adjusted hazard ratio (HR) of the high-risk compared to the low-risk group: 3.68; 95%CI: 2.19-6.17; p<0.0001). The 5-year PFS rates were 26% (95%CI: 16-43) and 73% (95%CI: 64-83) for the high- and low-risk groups, respectively. Gene coefficients and score thresholds allowing score calculation from either Affymetrix or Nanostring values are given in appendix p 44.
Figure 2. Progression-free survival of patients from the training cohort, according to the predictor score.
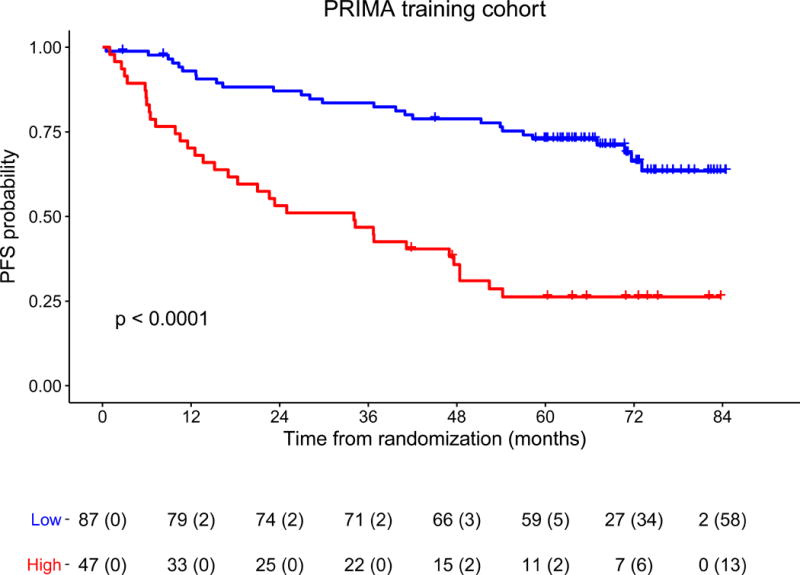
Kaplan-Meier estimates of progression-free survival in randomized patients of the training cohort, since the time of randomization in PRIMA trial. An optimal threshold was set to separate patients into high- (n=47, 35% of the patients, red curve) and low-risk (n=87, 65% of the patients, blue curve) groups with significantly different outcomes (p<0.0001, log rank test). The 5-year PFS rates were 26% (95%CI: 16-43) and 73% (95%CI: 64-83) for the high-and low-risk groups, respectively. For each time point the number of patients at risk and (number of patients censored) are indicated.
Abbreviations: PFS: progression-free survival.
Validation of the model in independent cohorts
The 23-gene expression model was then tested in three independent cohorts (Figure 3). The digital gene expression data met quality criteria for 460/488 FFPE samples, achieving an overall success rate of 94%, with similar performances across the three cohorts (cohort 1: 172/178, 97%; cohort 2:186/201, 93%; cohort 3: 102/109, 94%). When the model was first assessed separately in each cohort, similar performances to those obtained in the training cohort were observed, with C-statistic values of 0.650 (95%CI: 0.587-0.712), 0.619 (95%CI: 0.558-0.681) and 0.614 (95%CI: 0.509-0.720) in cohorts 1, 2, and 3, respectively. The score threshold applied the high-risk group classification to 34% (59/172, cohort 1), 23% (42/186, cohort 2), and 21% (21/102, cohort 3) of patients, and all these groups had a significantly worse outcome than their low-risk counterparts (Figure 4A-C). In a multivariate Cox model adjusted on maintenance treatment and FLIPI-1, the score was an independent predictor of PFS in each validation cohort (adjusted HR [95%CI] comparing high risk to low risk groups: cohort 1, 2.57 [1.65-4.01]; cohort 2, 2.12 [1.32-3.39]; cohort 3, 2.11 [1.01-4.41]).
Figure 3. The gene expression-based predictor for FL patients tested in the validation cohorts.
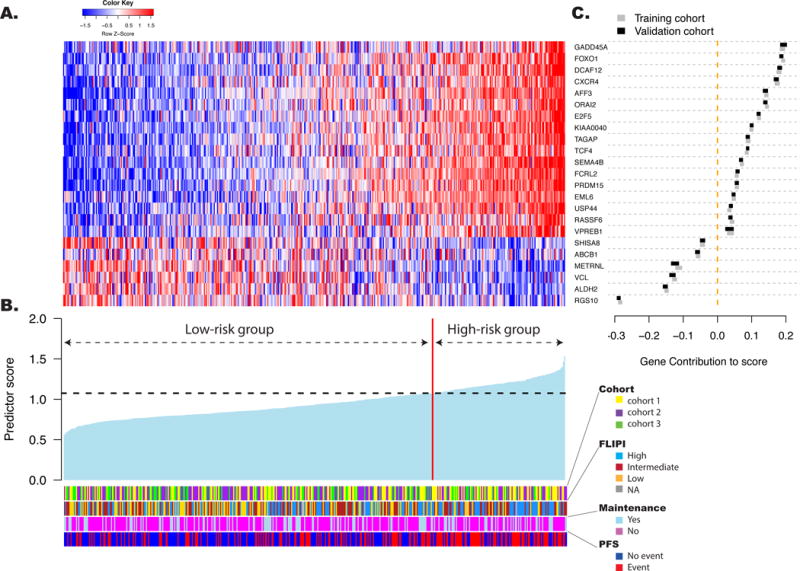
The predictor is a linear combination of the log2-transformed normalized gene expression levels weighted by individual gene coefficients. A: The relative gene expression levels of the 23 genes in the predictive model are presented in the form of a heat map. Each column represents a single patient from the combined validation cohorts, arranged according to the predictor score, with lowest score on the left. Each row represents a gene from the model, ordered by gene contribution to the score. B: The score from the predictor for patients in the validation cohorts. The patients are arranged as in panel A. The vertical red line separates patients into high- (n=122, 27% of the patients) and low-risk (n=338, 73% of the patients) groups according to the threshold (horizontal line) determined in the training cohort. The clinical and treatment characteristics of the patients are depicted. Cohort 1 included patients from the PRIMA trial, cohort 2 from the University of Iowa/Mayo Clinic Lymphoma SPORE and cohort 3 from the Hospital Clinic University of Barcelona. C: The relative contributions of each of the 23 genes to score variation. The X axis position of the boxes represents the absolute average contribution of the genes (calculated as the mean expression in a given cohort, multiplied by the coefficient assigned to the gene in the score). The width of the boxes shows the contribution of each gene to the score variation (calculated as the standard deviation of the gene in the cohort multiplied by its coefficient). Gene contributions are presented in both the training cohort (grey) and the combined validation cohort (black).
Figure 4. Kaplan-Meier estimates of progression-free survival predicted by the 23-gene signature score among patients of the three validation cohorts and according to FLIPI score.
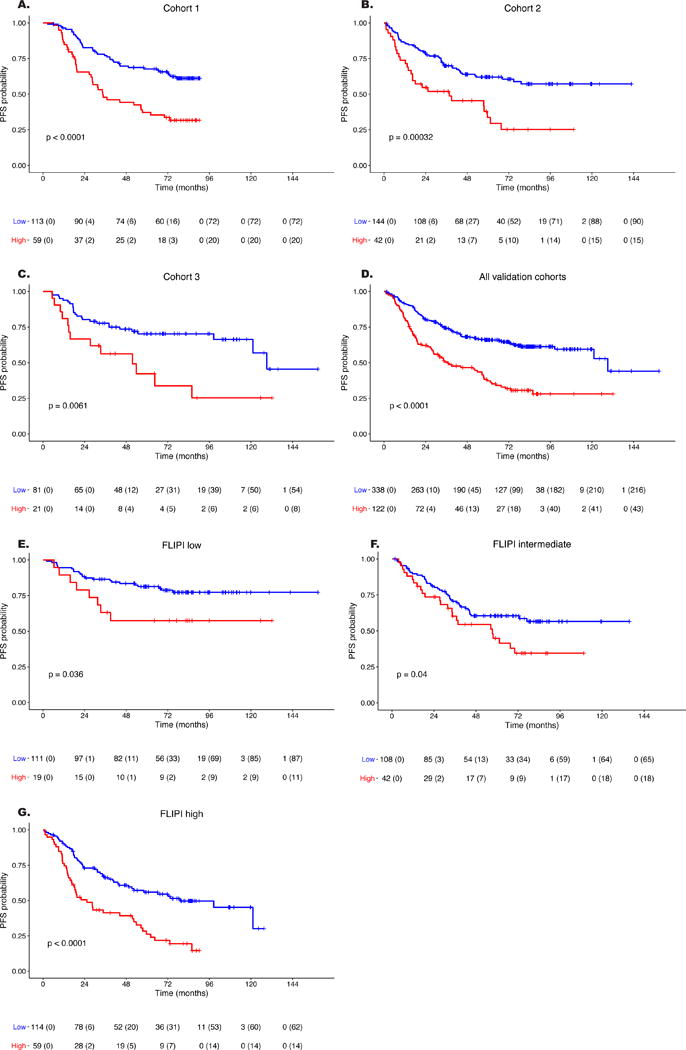
The threshold set in the training cohort separated patients into high- and low-risk groups (red and blue curves, respectively). The 23-gene score significantly predicted PFS in patients from each validation cohort (A-C: Cohort 1, 2, and 3, respectively) as well as in the combined validation cohorts (D) and in each FLIPI subgroup (E-G: low, intermediate and high risk FLIPI scores, respectively). Logrank test p-values for each of the comparisons are reported. FLIPI score was available for 453 patients. Abbreviations: PFS: progression-free survival; 95%CI: 95% confidence interval.
These results allowed us to pool the three cohorts for a combined analysis (n=460, median follow-up of the combined cohort: 6.6 years, IQR: 4.9-7.2). The C-statistic in the combined cohort was 0.628 (95%CI: 0.587-0.668), showing similar performances to FLIPI (n=453 patients evaluable, C-statistic 0.621, 95%CI: 0.583-0.659; appendix p 14). The median PFS values of the overall validation cohort were 3.1 (95%CI: 2.4-4.8) and 10.8 (95%CI: 10.1-not reached) years in the high- and low-risk groups, respectively (p<0.0001) (Figure 4D). The prognostic impact of the predictor was maintained in each group of induction chemotherapy (appendix p 15).
Early relapse within 2 years after diagnosis (POD24) has recently been shown to define a group of patients at high risk of death.7 We confirmed the impact of POD24 status on overall survival in the combined validation cohort (n=438 patients evaluable, appendix p 16). As the POD24 is not defined at the time of diagnosis, it is of critical importance to identify, as soon as the time of diagnosis, the patients who will experience early progression. In the combined validation cohort, the 2-year progression rate was 19% (95%CI: 15%–24%) in patients with a low predictor score (low-risk group) but rose to 38% (95%CI: 29%-46%) in patients with a high predictor score (high-risk group), showing the model’s ability to predict early relapse. The score predicted POD24 with a sensitivity of 43% and a specificity of 79%, resulting respectively in positive predictive value of 38% and a negative predictive value of 82%. However, this signature score was not associated with either OS from the time of diagnosis (appendix p 17) or lymphoma-specific survival (appendix p 18).
Patients in the high-risk group presented with a significantly higher frequency of B-symptoms, bone marrow involvement, and Ann Arbor stages III-IV as well as higher FLIPI scores but were also significantly younger and had lower histological grades (Table 2). The score predicted PFS in the combined cohort independently of maintenance treatment and FLIPI-1 score (adjusted HR of the high-risk compared to the low-risk group: 2.30 [1.72-3.07]). When the patients were grouped according to their FLIPI-1 risk category, the score significantly predicted PFS in all subgroups (Figure 4E-G and Table 3). In particular, the model stratified patients with high-risk FLIPI into groups differing by more than four years in median PFS (2.1 versus 6.6 years) and 50% of those with high-risk FLIPI and high-risk score experienced progression within two years, thus meeting criteria of high risk of early death by POD24.
Table 2. Patient characteristics according to the prognostic group defined by the gene-expression predictor.
Clinical and treatment characteristics were compared using Fisher’s exact test.
| high-risk N=122 no./total no. (%)* |
low-risk N=338 no./total no. (%)* |
P-value | |
|---|---|---|---|
|
| |||
| Cohort | |||
| 1 = PRIMA trial | 59/122 (48) | 113/338 (33) | |
| 2 = UI/MC - SPORE cohort | 42/122 (34) | 144/338 (43) | |
| 3 = BCN | 21/122 (17) | 81/338 (24) | |
|
| |||
| Baseline characteristics | |||
| Age > 60 years | 31/122 (25) | 134/338 (40) | 0.0050 |
| Male sex | 69/122 (57) | 168/338 (50) | 0.194 |
| Ann Arbor stage III/IV | 111/120 (93) | 273/338 (81) | 0.0027 |
| ECOG PS ≥ 1 | 45/121 (37) | 86/337 (26) | 0.015 |
| B symptoms present | 41/121 (34) | 67/336 (20) | 0.0080 |
| Bone Marrow involvement | 95/116 (82) | 152/324 (47) | <0.0001 |
| Elevated LDH | 42/111 (38) | 99/315 (31) | 0.217 |
| Hemoglobin level < 12 g/dL | 28/114 (25) | 55/321 (17) | 0.083 |
| Elevated β2-microglobulin | 53/80 (66) | 120/214 (56) | 0.115 |
|
| |||
| FLIPI score | |||
| 0-1 risk factors | 19/120 (16) | 111/333 (33) | |
| 2 risk factors | 42/120 (35) | 108/333 (32) | 0.0006 |
| 3-5 risk factors | 59/120 (49) | 114/333 (34) | |
|
| |||
| Histological grade | |||
| 1-2 | 102/122 (84) | 217/338 (64) | |
| 3A | 16/122 (13) | 97/338 (29) | 0.0004 |
| Undetermined/other** | 4/122 (3) | 24/338 (7) | |
|
| |||
| Induction regimen | |||
| R-CHOP | 90/122 (74) | 265/338 (78) | |
| R-CVP | 21/122 (17) | 53/338 (16) | 0.596 |
| R-Bendamustine | 10/122 (8) | 19/338 (6) | |
| other | 1/122 (1) | 1/338 (0) | |
|
| |||
| Maintenance regimen | |||
| No | 79/122 (65) | 228/338 (67) | 0.587 |
| Yes | 43/122 (35) | 110/338 (33) | |
Because of rounding, percentages may not total 100.
Undetermined/other included FL of undetermined grade (except 3B, n=16), FL with component DLBCL<10% (n=10) and FL with diffuse area (n=2).
Table 3. Progression-free survival according to the predictor score, in each validation cohort and in FLIPI risk categories.
Median PFS, 2-year and 5-year PFS values are indicated for each validation cohort and each FLIPI subgroup. Hazard ratios were calculated in a multivariate Cox model adjusted on rituximab maintenance treatment and FLIPI-1 in each validation cohort. The multivariate Cox model was applied to 171, 186 and 96 patients of cohorts 1, 2 and 3, respectively, because information regarding FLIPI was missing for some patients. In the combined cohort, the model was thus applied to 453 patients with FLIPI scores. In FLIPI subgroups, a univariate Cox model was applied.
| Cohort | Risk group | n (%) | Progression-Free Survival | Multivariate Cox model HR [95%CI] |
||
|---|---|---|---|---|---|---|
| Median (yr) [95%CI] | 2 yr (%) [95%CI] | 5 yr (%) [95%CI] | ||||
| 1 (Prima) n=172 |
high-risk | 59 (34) | 2.9 [2.4-5.8] | 66 [54-79] | 37 [27-52] | 2.57 [1.65-4.01] |
| low-risk | 113 (66) | NR [NR-NR] | 83 [76-90] | 68 [59-77] | ||
| 2 (UI/MC) n=186 |
high-risk | 42 (23) | 3.1 [1.4-5.6] | 55 [41-72] | 34 [21-55] | 2.12 [1.32-3.39] |
| low-risk | 144 (77) | NR [6.6-NR] | 79 [73-86] | 62 [54-71] | ||
| 3 (BCN) n=102 |
high-risk | 21 (21) | 4.3 [1.3-NR] | 67 [49-90] | 42 [24-73] | 2.11 [1.01-4.41] |
| low-risk | 81 (79) | 10.8 [10.1-NR] | 80 [72-89] | 70 [61-81] | ||
| All n=460 |
high-risk | 122 (27) | 3.1 [2.4-4.8] | 62 [54-71] | 37 [29-48] | 2.30 [1.72-3.07] |
| low-risk | 338 (73) | 10.8 [10.1-NR] | 81 [76-85] | 66 [61-71] | ||
| Univariate Cox model | ||||||
| Low n=130 |
high-risk | 19 (15) | NR [2.8-NR] | 79 [63-100] | 57 [39-85] | 2.31 [1.03-5.18] |
| low-risk | 111 (85) | NR [NR-NR] | 88 [83-94] | 81 [74-89] | ||
| Int n=150 |
high-risk | 42 (28) | 4.8 [2.9-NR] | 74 [61-88] | 45 [31-65] | 1.68 [1.02-2.77] |
| low-risk | 108 (72) | NR [6-NR] | 81 [74-89] | 60 [52-71] | ||
| High n=173 |
high-risk | 59 (34) | 2.1 [1.4-4.5] | 50 [39-65] | 26 [17-41] | 2.37 [1.58-3.56] |
| low-risk | 114 (66) | 6.6 [4.3-NR] | 73 [65-82] | 56 [47-67] | ||
Abbreviations: HR: hazard ratio; PFS: progression-free survival; 95%CI: 95% confidence interval.
The biological basis and prognostic significance of gene-expression signatures
Expression data previously generated from FL tumors and reactive lymph nodes or tonsils23 were used as an appraisal for evaluating which cell types (lymphoma cells and/or tumor microenvironment) expressed the 23 genes included in the model (appendix p 19). Most genes were more highly expressed in B cells compared to the microenvironment (except for GADD45A, ABCB1, RGS10). A differential intensity of expression between tumor and non-tumor B cells was observed for a few genes whose expression was associated with either longer (ABCB1, SHISA8, ALDH2, VCL) or shorter (FOXO1, PRDM15, EML6, SEMA4B) time to progression.
As a previous GEP study reported a negative impact of the macrophage part on outcome in patients treated in the pre-rituximab era,10 we then evaluated the previously described IR1 (T-cells and macrophages signature) and IR2 (macrophages and dendritic cells) signatures in the training cohort (n=134 randomized patients, appendix p 4). When combined into a composite score using the weights specified as originally described, this score did not predict PFS in our series. When evaluated separately, both signatures were associated with a longer PFS (IR1: p=0.0056, IR2: p=0.0018, log-rank test; appendix p 20).
To further investigate the biological basis of expression patterns acting in FL tumors, we performed an exploratory unsupervised analysis of the gene-expression data generated in the training cohort (including randomized and non-randomized patients, n=148) using independent component analysis (ICA). After discarding those potentially associated with experimental batches (appendix p 5), we identified 19 independent gene signatures (appendix p 21). One of these signatures, ICA13, was highly associated with patient outcomes, patients with a high ICA13 score (ie, higher expression of the main genes contributing to ICA13 signature, appendix p 45) having a shorter PFS (p=0.00091) (appendix p 21). It comprised genes expressed by germinal-center (GC) B-cells such as AICDA, BACH2, TCF4, ORAI2, E2F5, CXCR4 or POU2AF1 (appendix p 45). A significant overlap was observed between leading genes of this component (n=133) and the 167 bad-prognosis genes in the initial supervised analysis (24 genes in overlap indicated in appendix p 45, p<0.0001). This ICA13 was also highly correlated with our 23-gene predictor score (p<0.0001; appendix p 21). Enrichment analyses revealed that this signature also overlapped with B-cell progenitor signatures as well as with genes overexpressed in Burkitt lymphoma compared to diffuse large B-cell lymphoma (DLBCL) (appendix p 46). Comparison with other datasets obtained from normal and malignant B-cell subsets (appendix p 7) showed that ICA13 was highly expressed in centroblasts located in the GC dark zone and was particularly enriched in Burkitt lymphomas and, to a lesser extent, in normal pre-B cells (appendix p 22).
Discussion
We have developed a gene-expression-based predictor of PFS in high-tumor-burden FL that identifies, at diagnosis, those patients who have an increased risk of progression when treated initially with rituximab-chemotherapy. The model was tested in three independent cohorts of patients, where it confirmed its ability to accurately identify a high-risk population independently of the FLIPI score and maintenance by rituximab. These results argue that our model relies on biologically relevant features of tumor cells involved in disease progression, and not merely associates with known clinical predictors. The proportion of high-risk patients slightly differed between the validation cohorts (34%, 23%, and 21% of patients in cohorts 1, 2, and 3, respectively), which might be explained by the specific selection for high-tumor burden patients in the PRIMA trial.
The predictor was developed to be used on the NanoString platform,24 which enables accurate and reproducible quantification of RNA obtained from FFPE samples25 (or even on core biopsies). The NanoString platform is now present in many clinical laboratories that can apply the score calculation exactly as described in our manuscript. Despite the variable geographic origin and the age of FFPE blocks used in this study, sufficient quality of gene expression was obtained in 94% of samples, confirming that this technology represents a promising option for a gene-expression-based clinical test.
In our study, the genes of the 23-gene model were selected based on several experimental criteria, but were also manually curated for their biologic significance, which could introduce potential bias. Nevertheless, our process identified a robust model that was externally validated. Limitations of our study include the lack of patients having received bendamustine-rituximab, and the fact the 23-gene model could not predict patients overall survival or the risk of transformation, given the few events observed in those cohorts. Another validation cohort would also have been helpful to confirm the unsupervised clustering analyses, but those were exploratory to investigate potential links between lymphoma biology and the predictive model. Furthermore, while it reflects multiple aspects of the biology of the tumor including lymphoma cells as well as their microenvironment, it would be premature to use its results in order to identify patients with specific biological characteristics that would benefit from particular targeted therapies. Finally, spatial heterogeneity has been shown to greatly influence mutation detection in FL. It is uncertain whether the same limitation would be observed with gene-expression profiling of the tumors.
Other predictors of FL outcome have been recently proposed that combine mutation data with FLIPI,8,9 and our score showed similar performances (sensitivity, specificity, positive and negative predictive values) to predict PFS or POD24.9 Although the GEP-based predictor might be combined together with gene mutations to improve patient stratification, we aimed at using a single and highly reproducible technique that would be easily applied in the clinical setting.
Outcome prediction by gene-expression signatures has been previously reported in FL in the pre-rituximab era by the LLMPP, which emphasized the role of non-malignant tumor-infiltrating cells. When applying the LLMPP algorithm10 in our training cohort, we found that both IR1 and IR2 signatures were associated with longer PFS. The lack of association of the IR2 signature with a poor outcome here is in agreement with immunohistochemistry studies performed in the rituximab era.14,26,27 The correlation of macrophage infiltration with patients prognosis is still controversial, but may vary according to the use of rituximab or different chemotherapeutic regimens.14 In particular, it has been recently suggested that doxorubicin-containing regimen may abrogate the negative impact of CD163+ TAMs.14
We also investigated the biological processes acting in FL tumors at diagnosis using an unsupervised analysis. We identified in an exploratory analysis a gene signature (ICA13) that was strongly associated with poor prognosis. Although the two analyses were performed independently, nine out of the 23 genes retained in the predictor (CXCR4, DCAF12, E2F5, ORAI2, PRDM15, RASSF6, TAGAP, TCF4, USP44) were part of the ICA13 signature whose score was highly correlated with our predictor. We also observed that some of the 23 genes had a distinct expression pattern between FL tumor B-cells and normal B-cells. Altogether, these data support that our 23-gene expression-based model recovers biologically meaningful attributes of lymphoma cells that are truly associated with the risk of disease progression. The ICA13 component revealed characteristics of “Burkitt-like” cells and/or dark zone centroblastes and/or immature pre-B cells. Although some progenitor-like transcriptional programs might be reactivated within GC during normal B-cell development, the adverse prognostic significance of such signature in FL is provocative, as is the strong negative impact of the expression of the surrogate light chain VPREB1 transcript. An Embryonic Stem Cell-like transcriptional program underlying histological transformation was described in FL patients,28 and the expression of the surrogate light chains was observed in some FL cases that transformed into B-lymphoblastic lymphomas.29 Altogether, one might hypothesize that (i) a subset of FL cells could arise from self-renewing ancestral cells and retain a partially active progenitor-like expression program, or may reacquire such program following re-entry cycles in the GC;30 and (ii) such features, also reminiscent of Burkitt cells and/or progenitor B cells, are related to disease aggressiveness in a subset of patients.
In conclusion, we have established a 23-gene predictive score able to identify in routine practice two groups of patients with FL with markedly distinct outcomes when treated with immunochemotherapy. This predictor captures multiple aspects of the biology of the tumor and the heterogeneous composition of its microenvironment. Together with clinical parameters such as the FLIPI index, this score may already allow to better adjust current therapeutic options according to the patient risk category. For patients at low risk of progression, shorter treatments and with a low toxicity profile should be considered. For patients with high-risk FLIPI and a high 23-gene score, having a 50% risk estimate of lymphoma progression at 2 years, new options should be developed. This group represents an ideal target population to investigate innovative combinations aiming to improve their outcome. Further studies are required to determine whether this model is valid in low-tumor-burden patients (currently managed with watchful waiting or single-agent rituximab) and in patients treated with novel agents that may interfere both with tumor B-cells and with their microenvironment.12 This 23-gene predictor may thus represent a promising tool to further develop personalized therapy in patients with FL.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed on August 15, 2017, with no date restrictions, for all original publications (ie, review articles were excluded) with the search terms “follicular lymphoma” in the title, “gene expression OR expression signature” in the title or abstract, and “prognosis OR prognostic OR prediction OR predictive OR progression OR risk OR transformation OR survival” anywhere in the text. This search identified a total of 52 original publications. The relevant publications addressing the impact of gene expression on outcome in FL patients were of two types: some studies used transcriptome-wide profiling to identify signatures predictive of progression, death or histologic transformation, whereas others focused on somatic alterations in single genes, impacting the global transcriptional profile and outcome. The seminal LLMPP study highlighted the role of non-malignant tumor-infiltrating cells on FL prognosis in the pre-rituximab era, enabling to build a molecular predictor of overall survival, based on two expression signatures of immune response (IR1 and IR2). Another study identified a T-cell signature of favorable prognosis and a proliferation signature associated with relapse. Other groups, using RT-PCR and/or immunohistochemistry to assess the expression levels of single genes, confirmed the influence of different immune subsets on the risk of disease progression or death, but sometimes with conflicting results and in heterogeneously treated patient cohorts.
Several other studies focused on molecular alterations driving histologic transformation. One study identified the role of an embryonic stem cell signature probably sustained by MYC activation in tFL. Another one pinpointed 6 signatures related to the NF-kB pathway, with high scores found in biopsies preceding transformation, although not necessarily detected at the time of diagnosis.
Finally, other studies identified the clinical significance of somatic alterations and/or expression pattern in single genes. Although not directly linked to our gene expression-focused search, new clinico-genetic predictors (m7-FLIPI and POD24-PI), combining the mutation status of several genes with the FLIPI score, have been proposed to improve the identification of FL patients at high risk of progression.
Added value of this study
To overcome the limitations of previous studies due to heterogeneous treatment cohorts, small size and/or no validation cohorts, we conducted a progression-free survival (PFS)-supervised analysis on expression data obtained in a large cohort of patients in the setting of a clinical trial. Our study identified a 23-gene score able to predict the risk of progression in FL patients at diagnosis, independently of the FLIPI score and use of anti-CD20 maintenance therapy. Importantly, we developed this predictor to be fully applicable to routinely available formalin-fixed, paraffin-embedded biopsies. Moreover, results were further validated in 3 independent international cohorts of patients homogeneously treated with immunochemotherapy. To the best of our knowledge, this is the largest study to date of gene-expression profiling able to predict the risk of progression of FL patients receiving first-line immunochemotherapy, confirmed in 3 independent cohorts. Furthermore, our study identified a gene signature characteristic of B-cell centroblasts that was prognostic, underlining that beside tumor microenvironment, tumor B-cell biology itself contributed to the clinical aggressiveness of the disease.
Implications of all the available evidence
Despite recent progress in the stratification and management of FL patients, a significant proportion are still underserved by current standard treatment and experience rapid progression of their disease. Our gene-expression predictor could be of valuable interest in the clinical setting to identify patients at high-risk but also low-risk of progression and consequently adjust the therapeutic strategy, including enrollment for innovative treatments.
Acknowledgments
The authors thank Mrs. Nadine Vailhen, the Platform of Biological Resources, Henri Mondor Hospital, Créteil (bio-bank ID number: BB-0033-00021), and the Lymphoma Study Association (LYSA) pathology platform (Henri Mondor Hospital, Créteil) for FFPE sample storage for the PRIMA training and validation cohorts.
This work was supported by a funding from the Roche Company, by the SIRIC LYric Grant INCa-DGOS-4664, by the LYSARC (Lymphoma Study Academic Research organisation) and by the Spanish Plan Nacional de Investigacion SAF2015-64885-R.
The funding of the University of Iowa/Mayo Clinic (UIMC) Lymphoma SPORE was supported by NIH grant P50 CA97274 and the Henry J. Predolin Foundation.
The authors are indebted to Gilles Thomas for his initial input in this project and would like to thank Bertrand Nadel and Sandrine Roulland for helpful discussions, and Stephanie Cox for her assistance.
American Journal Experts, an independent medical writing agency, provided editing assistance for the manuscript.
GS reports grants from Roche Genentech, during the conduct of the study; personal fees from Amgen, personal fees from BMS, personal fees from Celgene, personal fees from Janssen, personal fees from Novartis, personal fees from Merck, personal fees from Roche, personal fees from Servier, personal fees from Morphosys, personal fees from Gilead, personal fees from Kite pharma, outside the submitted work. CH reports personal fees from Roche, during the conduct of the study; personal fees from Janssen, personal fees from Gilead, personal fees from Biogen, personal fees from Sandoz, personal fees from Pfizer, personal fees from Takeda, personal fees from Novartis, personal fees from Amgen, outside the submitted work. PF reports personal fees from Roche, outside the submitted work. BL reports grants and personal fees from Genentech/Roche, personal fees from Celgene, personal fees from Abbvie, personal fees from Gilead, personal fees from Sandoz, outside the submitted work. KT reports grants and personal fees from Celgene, outside the submitted work. HT reports grants and personal fees from Celgene, personal fees and non-financial support from Roche, personal fees from Karyopharm, personal fees from Astra-Zeneca, outside the submitted work. FJ reports personal fees from roche, personal fees from Janssen, and personal fees from Celgene, outside the submitted work. ALG reports grants and other personal fees from Roche, outside the submitted work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributors
SHu and GS designed the study and performed litterature search. SHu, CH, PF, HT, FJ, FO, PB, GS, ALF, SMA, BKL, JRC, LM, ALG, and EC collected the samples and the clinical data. Hemopathologists ATG, LX, ALF, and EC reviewed the cases. ET, BA, MC, LB, CR, SB, DG, and AV performed the experiments and generated the data. SHu, BT, and JPJ analyzed the data. SHu, BT, and GS wrote the manuscript and draw the figures. All authors substantially contributed to data interpretation and revision of the manuscript.
Declaration of interests
The other authors declared no conflicts of interest.
References
- 1.Bachy E, Houot R, Morschhauser F, et al. Long-term follow up of the FL2000 study comparing CHVP-interferon to CHVP-interferon plus rituximab in follicular lymphoma. Haematologica. 2013;98:1107–14. doi: 10.3324/haematol.2012.082412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghielmini M, Vitolo U, Kimby E, et al. ESMO Guidelines consensus conference on malignant lymphoma 2011 part 1: diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL) and chronic lymphocytic leukemia (CLL) Ann Oncol Off J Eur Soc Med Oncol ESMO. 2013;24:561–76. doi: 10.1093/annonc/mds517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nowakowski GS, Ansell SM. Therapeutic targeting of microenvironment in follicular lymphoma. Hematol Educ Program Am Soc Hematol Am Soc Hematol Educ Program. 2014;2014:169–73. doi: 10.1182/asheducation-2014.1.169. [DOI] [PubMed] [Google Scholar]
- 4.Relander T, Johnson NA, Farinha P, Connors JM, Sehn LH, Gascoyne RD. Prognostic factors in follicular lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28:2902–13. doi: 10.1200/JCO.2009.26.1693. [DOI] [PubMed] [Google Scholar]
- 5.Solal-Céligny P, Roy P, Colombat P, et al. Follicular lymphoma international prognostic index. Blood. 2004;104:1258–65. doi: 10.1182/blood-2003-12-4434. [DOI] [PubMed] [Google Scholar]
- 6.Federico M, Bellei M, Marcheselli L, et al. Follicular lymphoma international prognostic index 2: a new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J Clin Oncol Off J Am Soc Clin Oncol. 2009;27:4555–62. doi: 10.1200/JCO.2008.21.3991. [DOI] [PubMed] [Google Scholar]
- 7.Casulo C, Byrtek M, Dawson KL, et al. Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33:2516–22. doi: 10.1200/JCO.2014.59.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pastore A, Jurinovic V, Kridel R, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111–22. doi: 10.1016/S1470-2045(15)00169-2. [DOI] [PubMed] [Google Scholar]
- 9.Jurinovic V, Kridel R, Staiger AM, et al. Clinicogenetic risk models predict early progression of follicular lymphoma after first-line immunochemotherapy. Blood. 2016;128:1112–20. doi: 10.1182/blood-2016-05-717355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dave SS, Wright G, Tan B, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351:2159–69. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- 11.de Jong D, Koster A, Hagenbeek A, et al. Impact of the tumor microenvironment on prognosis in follicular lymphoma is dependent on specific treatment protocols. Haematologica. 2009;94:70–7. doi: 10.3324/haematol.13574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Jong D, Fest T. The microenvironment in follicular lymphoma. Best Pract Res Clin Haematol. 2011;24:135–46. doi: 10.1016/j.beha.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Sander B, de Jong D, Rosenwald A, et al. The reliability of immunohistochemical analysis of the tumor microenvironment in follicular lymphoma: a validation study from the Lunenburg Lymphoma Biomarker Consortium. Haematologica. 2014;99:715–25. doi: 10.3324/haematol.2013.095257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kridel R, Xerri L, Gelas-Dore B, et al. The Prognostic Impact of CD163-Positive Macrophages in Follicular Lymphoma: A Study from the BC Cancer Agency and the Lymphoma Study Association. Clin Cancer Res. 2015;21:3428–35. doi: 10.1158/1078-0432.CCR-14-3253. [DOI] [PubMed] [Google Scholar]
- 15.Salles G, Seymour JF, Offner F, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet. 2011;377:42–51. doi: 10.1016/S0140-6736(10)62175-7. [DOI] [PubMed] [Google Scholar]
- 16.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition - WHO - OMS. 4th. Lyon, France: IARC; 2008. [Google Scholar]
- 17.Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non-Hodgkin’s lymphomas. NCI Sponsored International Working Group. J Clin Oncol Off J Am Soc Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 18.Maurer MJ, Bachy E, Ghesquières H, et al. Early event status informs subsequent outcome in newly diagnosed follicular lymphoma. Am J Hematol. 2016;91:1096–101. doi: 10.1002/ajh.24492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uno H, Cai T, Pencina MJ, D’Agostino RB, Wei LJ. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat Med. 2011;30:1105–17. doi: 10.1002/sim.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hothorn T, Lausen B. On maximally selected rank statistics. R News. 2002:3–5. [Google Scholar]
- 21.Rotival M, Zeller T, Wild PS, et al. Integrating genome-wide genetic variations and monocyte expression data reveals trans-regulated gene modules in humans. PLoS Genet. 2011;7:e1002367. doi: 10.1371/journal.pgen.1002367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaffer AL, Wright G, Yang L, et al. A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunol Rev. 2006;210:67–85. doi: 10.1111/j.0105-2896.2006.00373.x. [DOI] [PubMed] [Google Scholar]
- 23.Pangault C, Amé-Thomas P, Ruminy P, et al. Follicular lymphoma cell niche: identification of a preeminent IL-4-dependent T(FH)-B cell axis. Leukemia. 2010;24:2080–9. doi: 10.1038/leu.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–25. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 25.Reis PP, Waldron L, Goswami RS, et al. mRNA transcript quantification in archival samples using multiplexed, color-coded probes. BMC Biotechnol. 2011;11:46. doi: 10.1186/1472-6750-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canioni D, Salles G, Mounier N, et al. High numbers of tumor-associated macrophages have an adverse prognostic value that can be circumvented by rituximab in patients with follicular lymphoma enrolled onto the GELA-GOELAMS FL-2000 trial. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26:440–6. doi: 10.1200/JCO.2007.12.8298. [DOI] [PubMed] [Google Scholar]
- 27.Taskinen M, Karjalainen-Lindsberg M-L, Nyman H, Eerola L-M, Leppä S. A high tumor-associated macrophage content predicts favorable outcome in follicular lymphoma patients treated with rituximab and cyclophosphamide-doxorubicin-vincristine-prednisone. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13:5784–9. doi: 10.1158/1078-0432.CCR-07-0778. [DOI] [PubMed] [Google Scholar]
- 28.Gentles AJ, Alizadeh AA, Lee S-I, et al. A pluripotency signature predicts histologic transformation and influences survival in follicular lymphoma patients. Blood. 2009;114:3158–66. doi: 10.1182/blood-2009-02-202465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slot LM, Hoogeboom R, Smit LA, et al. B-Lymphoblastic Lymphomas Evolving from Follicular Lymphomas Co-Express Surrogate Light Chains and Mutated Gamma Heavy Chains. Am J Pathol. 2016;186:3273–84. doi: 10.1016/j.ajpath.2016.07.027. [DOI] [PubMed] [Google Scholar]
- 30.Sungalee S, Mamessier E, Morgado E, et al. Germinal center reentries of BCL2-overexpressing B cells drive follicular lymphoma progression. J Clin Invest. 2014;124:5337–51. doi: 10.1172/JCI72415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.