

Abstract
Although anti-estrogen therapies are successful in many patients with estrogen receptor alpha-positive (ERα+) breast cancer, 25-40% fail to respond. Although multiple mechanisms underlie evasion of these treatments, including tumor heterogeneity and drug-resistant cancer stem cells (CSCs), further investigations have been limited by the paucity of preclinical ERα+ tumor models. Here we examined a mouse model of prolactin-induced aggressive ERα+ breast cancer, which mimics the epidemiologic link between prolactin exposure and increased risk for metastatic ERα+ tumors. Like a subset of ERα+ patient cancers, the prolactin-induced adenocarcinomas contained two major tumor subpopulations that expressed markers of normal luminal and basal epithelial cells. CSC activity was distributed equally across these two tumor subpopulations. Treatment with the selective estrogen receptor downregulator (SERD), ICI 182,780 (ICI), did not slow tumor growth, but induced adaptive responses in CSC activity, increased markers of plasticity including target gene reporters of Wnt/Notch signaling and epithelial-mesenchymal transition, and increased double positive (K8/K5) cells. In primary tumorsphere cultures, ICI stimulated CSC self-renewal, and was able to overcome the dependence of self-renewal upon Wnt or Notch signaling individually, but not together. Our findings demonstrate that treatment of aggressive mixed lineage ERα+ breast cancers with a SERD does not inhibit growth, but rather evokes tumor cell plasticity and regenerative CSC activity, predicting likely negative impacts on patient tumors with these characteristics.
Keywords: breast cancer, luminal B subtype, anti-estrogen therapy, cancer stem cells, lineage heterogeneity
Introduction
The majority of clinical breast cancers express estrogen receptor alpha (ERα). Estrogen is the major mitogenic driver of most of these cancers, and adjuvant anti-estrogen therapies are successful for many patients. However, a substantial subset of these women initially exhibit or acquire resistance to these treatments and may succumb to metastatic disease (1,2). While extensive research is illuminating the causes of this estrogen insensitive growth, little is known about the consequences of inhibiting estrogen activity on other aspects of tumor phenotype which may contribute to tumor behavior and outcomes, such as tumor heterogeneity and the activity of cancer cells with stem cell-like characteristics.
Intratumoral heterogeneity is increasingly recognized as an important player in tumor progression and treatment resistance (3–5). One component of diversity in breast cancers is epithelial lineage. Many clinical cancers contain tumor subpopulations with features of normal mammary luminal and basal cells, distinguished by patterns of cytokeratin and surface protein expression. While tumor cells in many ERα+ cancers display features of normal luminal cells and express primarily cytokeratin 8 (K8), cells expressing cytokeratins associated with basal cells (e.g., cytokeratin 5, K5) are found in some of these cancers and have been associated with poorer outcomes and a higher frequency of cells displaying stem-like characteristics (6–8). The relationships between these tumor subpopulations are not fully understood. In the normal mammary gland, the hierarchical model posits that stem cells, which reside in the basal compartment, can differentiate into both the luminal and basal epithelial cell lineages (9). In a similar fashion, cancer stem cells (CSCs), also referred to as tumor-initiating cells or cancer stem-like cells, have been defined as capable of regenerating tumors which exhibit the heterogeneity of the original tumor (10,11). A growing literature suggests that CSCs resist many current therapeutic approaches, thereby contributing to cancer recurrence (5). Thus understanding how therapies directed at the estrogen receptor affect cancer cell heterogeneity and CSCs in ERα+ breast cancers is of considerable interest.
Ovarian hormones are critical regulators of mammary epithelial subpopulations. Although estrogen itself does not increase progenitor/stem cell activity, it increases expression of progesterone receptors, which mediate expansion of the stem cell population (12,13). The effect of estrogenic signals on CSC activity in cancers is not as clear; both 17β-estradiol (E2) and clinical ER antagonists have been reported to increase CSC activity in vitro (14,15). However, these studies are confounded by differences in the source of the tumor cells, method of treatment, and assays for this activity (14,15). The paucity of ERα+ preclinical models has restricted the scope of in vivo studies. Xenografts of a few breast cancer cell lines have been the mainstay of this work, which is now being extended to a small number of ERα+ patient derived xenografts (PDXs). Examination of the effects of estrogens/anti-estrogens on CSC activity in many of these systems is further complicated by effects of these agents on proliferation/survival of estrogen responsive tumors.
Critical study of the dynamic relationships among cancer epithelia and CSC activity, and their responses to anti-estrogen therapy requires in vivo models. Robust in vivo models of ERα+ disease, particularly those with an intact immune system, are rare (16,17). High levels of circulating prolactin (PRL) are associated with increased risk of developing metastatic ERα+ breast cancer in postmenopausal women (18,19), and smaller studies have associated higher PRL activity with therapeutic resistance (20). The NRL-PRL transgenic mouse mimics the expression of PRL within the mammary glands of women (21). Nulliparous NRL-PRL females spontaneously develop aggressive mammary carcinomas with high incidence. Many of these tumors express ERα and share features with clinical ERα+ breast tumors of the Luminal B subtype, including low progesterone receptor (PR) expression and high rates of estrogen-independent proliferation (1,22,23). These characteristics permit examination of the effects of estrogen activity, independent of altered tumor growth.
In this study, we utilized the NRL-PRL mouse model to probe the effects of the selective estrogen receptor downregulator (SERD), ICI 182,780 (ICI) on lineage heterogeneity and cancer stem cells in ERα+ tumors. Like a subset of patient ERα+ tumors, PRL-induced ERα+ adenocarcinomas were composed of two major epithelial subpopulations, which expressed markers of normal luminal and basal cells. These subpopulations contained CSC activity at similar frequencies. Treatment with ICI did not slow tumor growth, but transiently disrupted lineage diversity by reducing the proportion of epithelia with basal characteristics, and lowered CSC activity. However, with ongoing treatment, heterogeneity was restored, associated with an increased frequency of double positive K8+/K5+ cells. This was accompanied by increased CSC activity, and transcripts for targets of canonical Wnt and Notch signals and markers of stem cell activity and the epithelial–mesenchymal transition (EMT). Analysis of CSC activity using tumorsphere assays in vitro showed that ICI stimulated CSC self-renewal, and was able to override inhibition of Wnt and Notch signals individually, but not together, indicating these pathways can compensate for one another. Our studies demonstrate that mixed lineage ERα+ breast cancers contain highly plastic tumor cells, and show that estrogenic signals are one component of a paracrine network that maintains lineage balance, independent of effects on cell growth. Our findings suggest that use of anti-estrogen therapies on the subset of clinical tumors modeled here may not only fail to slow cancer growth, but also may lead to more aggressive growth of plastic breast CSCs.
Materials and methods
Experimental mice
All mice were bred, housed, and handled in accordance with the Guide for Care and Use of Laboratory Animals in AAALAC-accredited facilities. All procedures were approved by the University of Wisconsin-Madison Institutional Animal Care and Use Committee. NRL-PRL mice were generated and maintained on the FVB/N strain background as described (24). Primary tumors from nulliparous NRL-PRL and MMTV-neu females (25),and those that developed from p53−/− mammary transplants in syngeneic FVB/N recipients (26), were resected, cut into 1mm3 fragments, and serially transplanted bilaterally into the caudal mammary glands of 8-week old FVB/N females. At the time of collection for passage, tumors were divided for preparation of single cell suspensions and histological examination (described below).
Histology and immunohistochemistry
Tumor portions, adjacent mammary tissue, and uteri were fixed overnight in 10% neutral buffered formalin, embedded in paraffin, and sectioned (8 μm). After quenching endogenous peroxidase activity, sections were exposed to citric acid for antigen retrieval, blocked using goat serum, and incubated with the appropriate primary antibody overnight as described (22). For immunohistochemistry (IHC), sections were incubated with secondary antibody for one hour, followed by peroxidase streptavidin (Vector Laboratories, PK-4005) and 3,3′ diaminobenzidine (ImmPACT DAB, Vector Laboratories, SK-4105). Sections were counterstained with hematoxylin. For immunofluorescence, sections were incubated with secondary antibodies and stained with DAPI. For some studies, positively stained tumor cells were quantified by counting 100 cells in each of five randomly distributed 20X fields-of-view from three different tumors. All images were captured using an E600 Eclipse fluorescence microscope utilizing Nikon NIS-Elements imaging software. The primary and secondary antibodies used are listed in Supplementary Table 1.
Tumor isolation and dissociation
Single cell suspensions were prepared from tumors by mincing into 1mm3 fragments and dissociating in Epicult-B Mouse Medium (Stem Cell Technologies, #05610) supplemented with 5% fetal bovine serum (FBS), 10 ng/mL epidermal growth factor (EGF), 300 U/mL collagenase, and 100 U/mL hyaluronidase (Stem Cell Technologies, #07912) at 37°C for one hour. Red blood cells were lysed using ammonium chloride, and then cells were incubated for one minute in trypsin-EDTA (Stem Cell Technologies, #07901) containing 5 mg/mL dispase (Stem Cell Technologies, #07913) and 0.1 mg/mL DNase I (Stem Cell Technologies, #07900), and filtered through 40 μm strainers.
Flow cytometry
Single cell suspensions were stained with DAPI for live/dead analysis, and conjugated antibodies for CD31-APC (BD Pharmingen, #551262), CD45-APC (BD Pharmingen, #559864), EPCAM-PE (BD Pharmingen, #563477), and CD49f-FITC (BD Pharmingen, #555735) for 30 minutes on ice. Cells were washed with Hanks’ Balanced Salt Solution supplemented with 2% FBS before fluorescence activated cell sorting (FACS) using a BD FACSAria II with BD FACSDiva software. Sorted cells were collected into either 50% FBS in phosphate-buffered saline (PBS) for in vivo transplantation or TRIzol LS (Life Technologies, #10296-028) for RNA extraction. Flow cytometry after treatment with ICI 182,780 was performed with a BD LSRFortessa. The gating strategy for examination of tumor cell subpopulations is shown in Supplementary Fig. 1. Gates were set using fluorescence minus one (FMO) controls.
In vivo limiting dilution transplantation
Sorted viable tumor cells were suspended in 50% Matrigel/PBS at appropriate dilutions (100, 1,000, 5,000, and 10,000 cells). Left caudal mammary fat pads of recipient 3-week old FVB/N females were cleared of endogenous epithelial cells. Cell suspensions were pipetted into a pocket created in the cleared fat pad using fine scissors. Recipient fat pads were collected when tumors reached 1.5cm in diameter or 8 months after transplantation. The estimated frequencies of cancer stem cells were calculated using extreme limiting dilution analysis (ELDA) software (27).
In vivo treatment with ICI 182,780
In order to examine the effect of treatment on individual tumors, 100,000 dissociated viable tumor cells suspended in 50% Matrigel/PBS were transplanted into cleared left caudal mammary fat pads of 3-week old FVB/N females. Subsequent tumor growth was measured daily using digital calipers. Once tumors reached a diameter of 7 mm, the mice were either left untreated or administered ICI 182,780 (ICI, Fulvestrant, 250 mg/kg sc, AstraZeneca Pharmaceuticals). One cohort was euthanized two days after treatment; another cohort was given a second injection of ICI after seven days to maintain anti-estrogen activity, and euthanized 14 days after initiation of treatment. At this time, tumor size approached end stage, as defined by our IACUC. Continued anti-estrogenic activity was confirmed by measurement of uterine wet-weights.
Quantitative real-time PCR
RNA from unsorted cells was isolated using the RNeasy mini-kit (Qiagen, #74104). RNA from FACS-sorted cells was extracted with TRIzol-LS and amplified using the MessageBOOSTER cDNA synthesis kit for qPCR (Epicentre, #MB060124) or the Ovation PicoSL WTA System V2 (NuGEN Technologies, #3312) according to the manufacturer’s instructions. Synthesized cDNA was quantified using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and quality was assessed using an Agilent 2100 Bioanalyzer. qRT-PCR was performed as previously described (22) except that transcript levels of Esr1, Krt5, and Krt8 from FACS-sorted cells were normalized to Hprt mRNA. The following primer sequences were used: 18S: F: 5′-CGC CGC TAG AGG TGA AAT TCT-3′, R: 5′-CGA ACC TCC GAC TTT CGT TCT-3′; Sox2: F: 5′CCA CCA ATC CCA TCC AAA TT-3′, R: 5′-CAA AAA GAA GTC CCA AGA TCT CTC A-3′; Twist1: F: 5′-CAC GCA GTC GCT GAA CGA-3′, R: 5′-ATG TAC CTG GCC GCC AGT T-3′; Hes1: F: 5′-TAC CCC AGC CAG TGT CAA CA-3′, R: 5′-CCT TCG CCT CTT CTC CAT GA-3′; Axin2: F: 5′-CCA GGC TGG AGA AAC TGA AAC T-3′, R: 5′-CCT GCT CAG ACC CCT CCT TT-3′; Krt5: F: 5′-CCT GGT GGA GGA CTA CAA GAA CA-3′, R: 5′-CAC ATC CTT CTT CAA CAT CAC AAA C-3′; Krt8: F: 5′-TGA ACA ACA AGT TCG CCT CCT T-3′, R: 5′-TCC ACT TGG TCT CCA GCA TCT-3′; Bmi1: F: 5′-AGT TCG GCC AAC TTG CAA AA-3′, R: 5′-GCC TTG TCA CTC CCA GAG TC-3′; Wnt4: F: 5′-AGC CGG GCA CTC ATG AAT C-3′, R: 5′-CCC GCA TGT GTG TCA AGA TG-3′; Esr1: F: 5′-GCC AGA ATG GCC GAG AGA-3′, R: 5′-TCA TTG CAC ACG GCA CAG T-3′; and Hprt: F: 5′-TGC TGA CCT GCT GGA TTA CA-3′, R: 5′-TTT ATG TCC CCC GTT GAC TGA-3′.
Tumorsphere formation assay
In vitro tumorsphere assay parameters as previously described (28,29) were modified for robust sphere formation using cells prepared from NRL-PRL tumors. Cell density was validated based on a linear range of tumorsphere forming efficiency. Single cell suspensions prepared from harvested tumors were seeded at 25,000 cells/well into ultra-low attachment 96-well plates (Corning, #3474) in mammosphere medium consisting of DMEM-F12 with 20 ng/mL EGF, 20n g/mL bFGF, 1X B27 supplement (Gibco, #17504044), 5 μg/mL insulin, and 100 μg/mL gentamicin for six replicates. Cells were cultured at 37°C for 10 days and tumorspheres greater than 50 μm in diameter were counted. In order to examine treatment responsiveness in vitro, freshly seeded cells from Tumor 1 were subjected to various treatments during development of the primary tumorspheres. These included vehicle (0.1% ethanol, 0.1% DMSO, or both), 1 μM ICI 182,780, 10 nM 17β-estradiol, 100 μM iCRT14 (Sigma-Aldrich, SML-0203), 50 μM γ-Secretase Inhibitor (GSI) (Sigma-Aldrich, S2188), and 100 nM trichostatin A (TSA) (Sigma-Aldrich, T8552). For some experiments, established untreated primary tumorspheres were treated with the inhibitors above for 6 hours, and then harvested for RNA analyses. To assess CSC self-renewal capacity, replicates of primary tumorspheres were pooled, dispersed with trypsin, resuspended in mammosphere medium without treatments, and reseeded at 25,000 cells/well to ascertain ability to form secondary tumorspheres. For some experiments, K5+ and K8+ cells in primary tumorspheres were visualized by immunofluorescence, and stained cells in ten individual tumorspheres from each treatment condition were quantified.
Tumor microarray imaging and analysis
K5 and K8 expression were examined in a human breast cancer tissue microarray (TMA) containing patient tumors and normal breast samples (University of Wisconsin Carbone Cancer Center BioBank) (30). In brief, slides were deparaffinized, hydrated and permeabilized, then incubated with anti-K8 (Developmental Studies Hybridoma Bank, University of Iowa, TROMA-1), 1:50 in antibody diluent, (Da Vinci Green-BioCare Medical), followed by goat anti-rat IgG biotinylated secondary antibody (BioCare Medical), Streptavidin-HRP (BioCare Medical), and Deep Space Black chromogen (BioCare Medical). This was followed by anti-K5 (Covance Antibody Products, cat#PRB-160P), 1:4000, Mach 2 rabbit AP polymer (BioCare Medical) and Warp Red chromogen (BioCare Medical). Slides were counterstained with CAT hematoxylin (BioCare Medical), scanned using the Vectra imaging system (PerkinElmer Life Sciences), and analyzed using InForm 1.4 software (PerkinElmer Life Sciences), as described (30). Cytokeratin and hematoxylin signals were resolved using Nuance 3.0.2 (PerkinElmer Life Sciences), pseudocolored and merged to generate the images shown.
Statistical analysis
Statistical analyses were performed using Prism v.5 (GraphPad Software, Inc., San Diego, CA). Independent experiments were analyzed by one-way ANOVA, followed by Tukey’s multiple comparison tests. Differences were considered significant at p<0.05.
Results
NRL-PRL mice spontaneously develop histologically diverse ERα+ adenocarcinomas, which contain distinct luminal and basal epithelial subpopulations
To assess tumor lineage heterogeneity and responses to anti-estrogen therapy in aggressive ERα+ breast cancers, we utilized the diverse ERα+ mammary adenocarcinomas that develop spontaneously in response to high local PRL exposure in NRL-PRL nulliparous females (21,22). These carcinomas can be serially orthotopically passaged to syngeneic recipients without alteration of histotype or ERα expression (Supplementary Fig. 2A), permitting extensive analyses of a single tumor. We examined two independent adenocarcinomas, with differences in morphology (Tumor 1, Fig. 1Ai; Tumor 2, Fig. 1Aii), and time required for regeneration after transplantation (mean time to palpable tumors: Tumor 1, 47 days; Tumor 2, 88 days). Like the majority of PRL-induced carcinomas, many epithelia in both tumors contained detectable nuclear ERα+ (54±6.3%, 53±6.2%, respectively) (Fig. 1Aiii,iv) with low progesterone receptor (PR) expression (Fig. 1Av,vi).
Figure 1. NRL-PRL mice develop ERα+ adenocarcinomas composed of luminal and basal epithelial subpopulations.
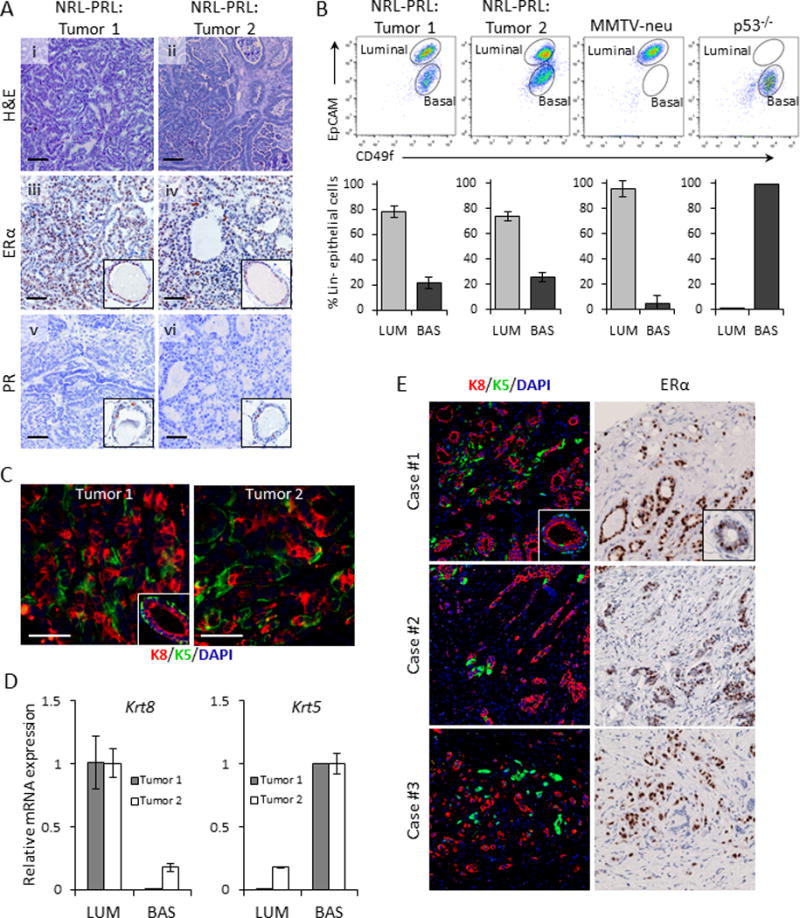
(A, i,ii) Adenocarcinomas of varying histotypes (“Tumor 1” (i) and “Tumor 2” (ii)) develop spontaneously in nulliparous female NRL-PRL mice (hematoxylin and eosin stain). (A, iii,iv) ERα expression in Tumor 1 (iii) and Tumor 2 (iv). (A, v,vi) Undetectable progesterone receptor (PR) expression in both Tumor 1 (v) and Tumor 2 (vi). Insets, ERα and PR staining in luminal epithelium; cross sections of mammary ducts. Scale bars, 50 μm. (B) Representative flow cytometric plots showing differences in tumor subpopulations among NRL-PRL, MMTV-neu, and p53−/− tumors. Bar graphs represent percentages of luminal (EPCAMhiCD49f+) and basal (EPCAMmedCD49f+) cells relative to total Lin-tumor cells (N=3; mean±S.D.). (C) Immunofluorescent staining of Tumors 1 and 2 using lineage-specific markers, cytokeratin-8 (K8) (red) and cytokeratin-5 (K5) (green). Inset shows a cross section from a normal mammary duct. Scale bars, 50 μm. (D) Relative Krt8 and Krt5 mRNA levels in FACS sorted luminal and basal subpopulations from the two tumors. (E) ERα+ patient tumor cores from a clinical breast tumor microarray showing pseudocolored K8 (red) and K5 (green) immunostaining (left panel) and ERα immunostaining with hematoxylin counterstain of a nearby section from the same core (right panel). Insets show staining of a normal mammary duct.
Intratumoral heterogeneity has been implicated in breast cancer aggression and therapy resistance (3–5). Relative expression levels of the cell-surface markers, EpCAM (epithelial cell adhesion molecule) and CD49f (integrin-α6), are frequently used to identify normal mammary luminal and basal cell subpopulations (9), as well as tumor cells sharing characteristics of these lineages. Analysis of tumor cells from the NRL-PRL tumors by flow cytometry revealed distinct luminal (EpCAMhiCD49f+) and basal (EpCAMmedCD49f+) subpopulations (Fig. 1B). Interestingly, the ratios of these subpopulations were strikingly consistent among passages of the same tumor, between these two tumors, and indeed, other independent tumors examined (Supplementary Fig. 2B). In contrast, tumors generated from luminal, but ERα- (MMTV-neu) and claudin-low (p53−/−) murine models of breast cancer exhibited a single epithelial population (luminal or basal, respectively). The heterogeneity of both PRL-induced tumors was also reflected in expression of the mammary lineage-specific cytokeratins, K8 (luminal) and K5 (basal) (Fig. 1C). Fractionation of the luminal and basal subpopulations by FACS in combination with qRT-PCR confirmed that these subpopulations were highly enriched for the expected cytokeratin mRNA (Fig. 1D). To identify the subset of clinical ERα+ tumors that may be modeled by PRL-induced murine tumors, we examined a tissue microarray (TMA) of primary breast cancers for K5 and K8 expression. Although many samples from ERα+ carcinomas displayed only K8+ staining, other high grade tumors exhibited a mixture of K8+ and K5+ cells, resembling the bilinearity of the PRL-induced carcinomas (Fig. 1E). Together, these findings suggest that these PRL-induced carcinomas model a subset of patient ERα+ cancers which display marked epithelial lineage heterogeneity.
Cancer stem cell frequency is similar between luminal and basal subpopulations
To examine functional CSC activity in these distinct tumor subpopulations, we performed limiting-dilution transplantation analysis of sorted luminal and basal tumor cells. Although the frequency of CSCs differed between the two tumors (interestingly related to the difference in time required to regenerate these tumors after passage), the luminal or basal subpopulations isolated from the same adenocarcinoma exhibited similar CSC frequencies (Table 1). Immunohistochemical and flow cytometric analyses confirmed that the secondary tumors derived from either the luminal or basal subpopulations displayed the morphology (Fig. 2A), ERα expression (Fig. 2B) and subpopulation heterogeneity (Fig. 2C) of the parent tumor, defining properties of CSCs.
Table 1. Frequency of cancer stem cell activity in luminal and basal tumor subpopulations.
CSC frequencies were estimated by limiting-dilution transplantation of sorted tumor cells into cleared fat-pads of syngeneic mice. Tabulation of tumor takes was input into extreme limiting dilution analysis software (see Methods). We assume significance if goodness-of-fit (GOF) is >0.05, allowing calculation of CSC frequencies.
| Tumor designation | Cell subpopulation | # of cells injected | Take rate | Frequency of CSCs (95% CI) (pGOF) |
|---|---|---|---|---|
| Tumor 1 | Luminal | 10000 | 4/4 | 1/3174 |
| (EPCAMhiCD49f+) | 5000 | 3/4 | (1/1383 - 1/7285) (0.62) | |
| 1000 | 1/4 | |||
| 100 | 0/4 | |||
| Basal | 10000 | 4/4 | 1/4430 | |
| (EPCAMmedCD49f+) | 5000 | 2/4 | (1/1951 - 1/10058) (0.70) | |
| 1000 | 1/4 | |||
| 100 | 0/4 | |||
| Tumor 2 | Luminal | 10000 | 2/4 | 1/16933 |
| (EPCAMhiCD49f+) | 5000 | 1/4 | (1/5462 - 1/52490) (0.53) | |
| 1000 | 0/4 | |||
| 100 | 0/4 | |||
| Basal | 10000 | 2/4 | 1/12477 | |
| (EPCAMmedCD49f+) | 5000 | 1/4 | (1/4525 - 1/34402) (0.46) | |
| 1000 | 1/4 | |||
| 100 | 0/4 |
Figure 2. Transplantation of limiting numbers of cells from the luminal and basal tumor subpopulations regenerates carcinomas with characteristics of the parental tumor.
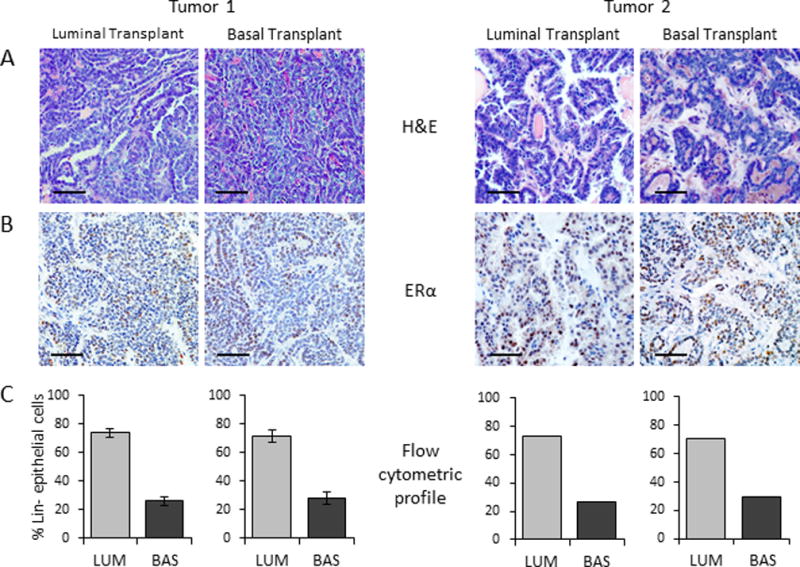
Representative photomicrographs of secondary tumors derived from sorted luminal and basal subpopulations demonstrating similar morphology, ERα expression and heterogeneity of the parent tumor. (A)H&E, (B) ERα IHC, and (C) flow cytometric analysis (Tumor 1: N=3, mean±S.D.; Tumor 2: N=1; others were too small for analysis). Scale bars, 50 μm.
ICI 182,780 treatment in vivo does not alter tumor growth, but transiently depletes basal cells and CSCs, and triggers compensatory pathways that restore heterogeneity and increase CSC activity
The underlying causes of ERα+ breast cancer relapse in the face of anti-estrogen treatment are not well understood. We therefore examined the effects of treatment with the SERD, ICI 182,780 (ICI) on the heterogeneity and CSC activity of the PRL-induced ERα+ adenocarcinomas. We orthotopically transplanted unsorted tumor cells to cleared mammary fat pads of FVB/N cycling females, and treated mice bearing established tumors with ICI for 2 or 14 days (Fig. 3A). Anti-estrogenic bioactivity of ICI was confirmed by measurement of uterine weight at the time of tumor collection (Fig. 3B). ICI did not affect tumor histotype (Supplementary Fig. 3A), volumetric growth (Fig. 3C, Supplementary Fig. 3B), proliferation indices or apoptosis determined by Ki67 expression (Fig. 3D) and cleaved caspase 3 labeling (Supplementary Fig. 3C, D), respectively.
Figure 3. Treatment with the estrogen receptor antagonist, ICI 182,780, does not alter growth of passaged tumors, but transiently depletes the tumor subpopulation with basal characteristics.
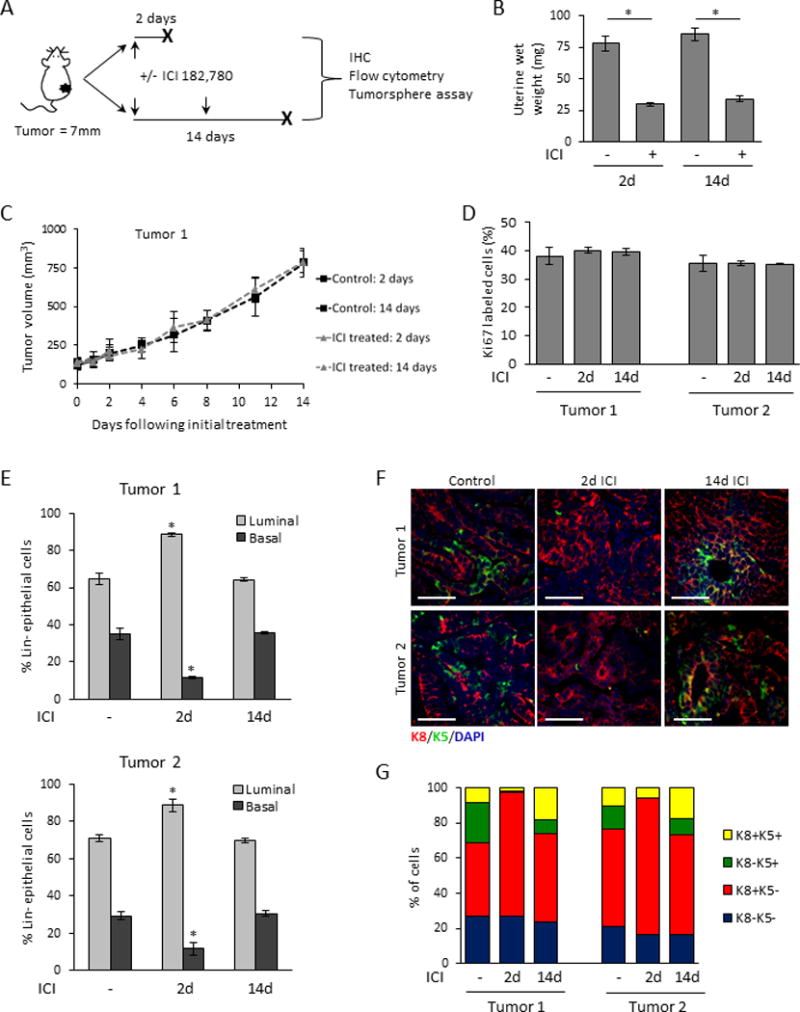
(A) Schema of the in vivo treatment methodology. When transplanted tumor cells developed into tumors of 7mm in diameter, mice were treated with or without ICI 182,780 (ICI) as described in the Methods, and tumors were harvested after 2 or 14 days of treatment. (B) Uterine wet-weights of treated and untreated mice confirm the systemic anti-estrogenic activity of ICI 182,780 (N=3; mean±S.D.). (C) ICI did not alter the rate of growth of established tumors (Tumor 1 shown, Tumor 2 in Supplementary Fig. 2A). (N=3; mean±S.D.). (D) ICI did not alter the rate of proliferation in established tumors measured by Ki-67 staining. (N=3; mean±S.D.). (E) Flow cytometric analysis in response to in vivo ICI 182,780, 2 or 14 days after initiation of treatment (N=3; mean±S.D.). (F) Representative K5 and K8 immunofluorescence at 2 and 14 days after initiation of treatment. (G) Quantification of K8/K5 staining was performed as described in the Methods. N= 3 independent tumors. (Means±S.D. and statistical evaluations are shown in Supplementary Table 2).
However, closer examination of the tumors revealed that ICI transiently altered the ratio of luminal (EpCAMhiCD49f+) and basal (EpCAMmedCD49f+) cells as measured by flow cytometry. Two days after initiation of ICI treatment, the proportion of epithelial cells that expressed basal surface proteins in both tumors was reduced by over 50%, but after 14 days of continued treatment, the relative proportions of the original tumor were restored (Fig. 3E). These temporary changes in epithelial subpopulations were mirrored in cytokeratin expression. Tumors harvested two days after initiation of ICI treatment contained only rare K5+ cells. However, after continued treatment (14 days), expression of K5 had recovered. Interestingly, many K5+ cells at this time also co-expressed K8 (Fig. 3F,G; Supplementary Table 2).
To examine the effect of ICI on CSC activity, we employed the in vitro tumorsphere assay (28,29). Two days after initiation of ICI treatment in vivo, epithelia from each tumor exhibited significantly decreased tumorsphere formation. However, after 14-days of in vivo treatment, the ability of epithelia from Tumor 1 to form tumorspheres had recovered to control levels and that of Tumor 2 had also slightly increased (Fig. 4A).
Figure 4. ICI 182,780 antagonizes cancer stem cells and alters markers for EMT, Notch, and Wnt activity.
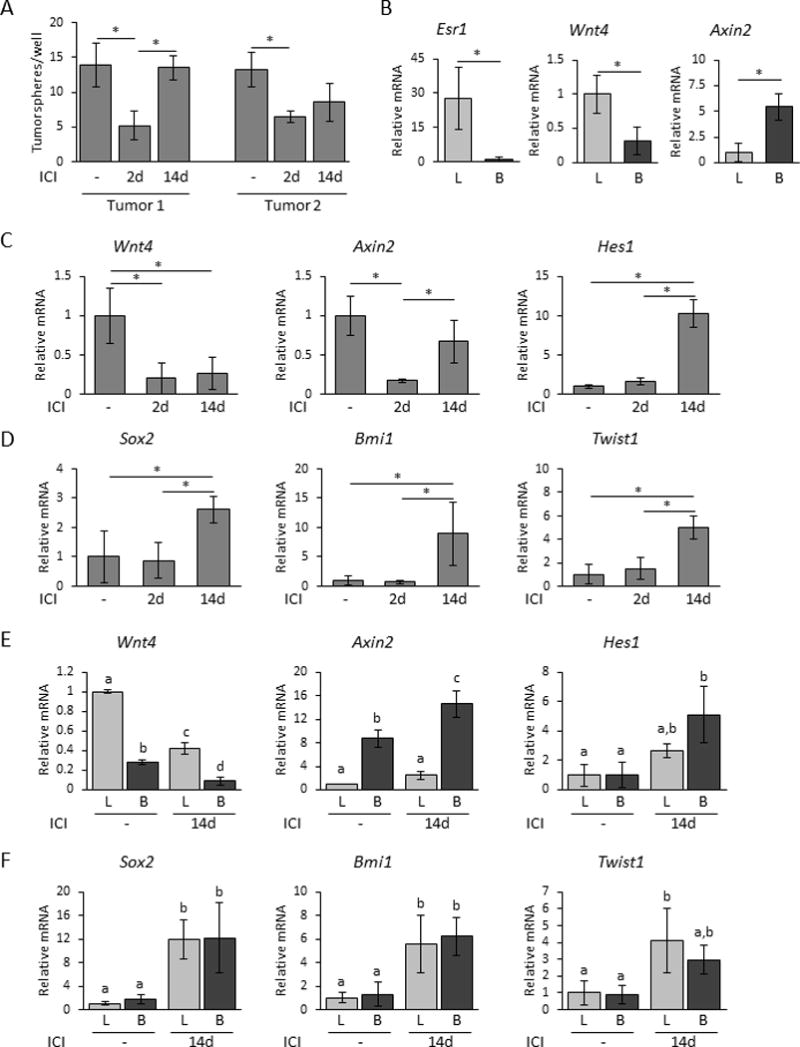
(A) Primary tumorsphere forming ability of tumor cells from control or ICI 182,780 treated mice (see Methods; N= 3 independent tumors; mean±S.D.). (B) Relative mRNA levels of Esr1, Wnt4, and Axin2 from FACS-sorted luminal and basal subpopulations from Tumor 1 without treatment (N=3; mean±S.D.). Light bars, luminal cells (L); dark bars, basal cells (B). Asterisks indicate significant differences determined by Student’s t test, p<0.05. (C,D) Relative mRNA levels of Wnt4, Axin2, Hes1 (C), and Sox2, Bmi1, Twist1 (D) from control, 2 day and 14 day ICI-treated tumor-burdened mice (N=3-5; mean±S.D.). Asterisks indicate significant differences determined by one-way ANOVA followed by Tukey’s multiple comparison tests, p<0.05. (E,F) Relative levels of Wnt4, Axin2, Hes1 mRNAs (E), and Sox2, Bmi1, Twist1 mRNAs (F) in FACS-sorted luminal and basal tumor subpopulations harvested from control or 14 day ICI-treated mice (N=3; mean±S.D.). Light bars, luminal cells (L); dark bars, basal cells (B). Different letters indicate significant differences (one-way ANOVA followed by Tukey’s multiple comparison tests, p<0.05).
To further characterize the ICI response, we examined transcripts in subpopulations of Tumor 1 without treatment for components of the Wnt pathway, which are implicated in regulation of basal cell populations and stem cell activity, and are regulated by steroid hormones in the normal gland. As expected, Esr1 transcripts were highly enriched in cells expressing a luminal surface-marker profile (Fig. 4B). Wnt4 mRNA also was enriched in the luminal compartment, whereas transcripts for Axin2, a target of canonical Wnt signals, were enriched in cells expressing basal surface-markers (Fig. 4B), resembling the pattern of paracrine Wnt signaling components in normal mammary glands (12,31,32). After 2d of ICI treatment, Wnt4 mRNA had fallen about 4-fold, as did transcripts for Axin2 (Fig. 4C). However, after 14 days of ICI activity, Wnt4 mRNA remained reduced, but levels of Axin2 mRNA were restored. At this time, transcripts for the Notch target gene, Hes1, had risen 10-fold over untreated levels, indicating increased Notch activity (Fig. 4C). Transcripts for Sox2, Bmi1, and Twist1 were also significantly elevated after 14 days of ICI treatment (Fig. 4D), suggesting a rise in stem-like and EMT traits. Ezh2, Dmnt1, and Dmnt3b mRNAs were not significantly altered by ICI treatment. In order to identify the cell populations responding to this altered endocrine environment, we compared levels of transcripts in the luminal and basal subpopulations after 14 days of ICI to those without treatment. After 14 days, canonical Wnt signals as reflected in Axin2 transcripts had recovered in the basal subpopulation, Notch activity indicated by Hes1 mRNA had risen in both subpopulations, particularly the basal cells (Fig. 4E), and stem cell markers were elevated in both subpopulations (Fig. 4F).
These results indicate dynamic responses of ERα+ tumors to SERD treatment, independent of effects on tumor growth. The transient changes in tumor subpopulations induced by ICI, in combination with the appearance of double positive K8+/K5+ cells and lack of evidence for altered proliferation or death, suggests plasticity of tumor cells, which is modulated by ICI. Furthermore, our data are consistent with disruption of paracrine Wnt signals from luminal to basal cells as one component of the initial response, followed by compensatory responses in multiple pathways associated with stem-like characteristics.
In vitro anti-estrogen treatment increases tumorsphere self-renewal
To further investigate the effects of ICI on CSC activity, we examined its effects during development of primary tumorspheres from Tumor 1 in vitro, which allowed for investigation of effects directly on the self-renewal capacity of the CSCs apart from the complex in vivo tumor environment. Neither ICI nor 17β-estradiol (E2) altered the number of primary tumorspheres that formed, indicating that ICI did not affect function of existing CSCs (Fig. 5A, left panel). However, dissociation of the spheres and subsequent replating without additional treatment showed that ICI-, but not E2-treated primary tumorspheres, developed significantly more secondary tumorspheres (Fig. 5A, right panel), indicating that ICI increased the self-renewal of CSCs. Moreover, although ICI did not alter the size of primary tumorspheres (Fig. 5B, left panel), consistent with the lack of effects on proliferation observed in vivo, ICI modulated cell lineages, as determined by cytokeratin expression. Primary tumorspheres were composed primarily of K8+ cells. However, a subset expressed K5, generally in conjunction with K8 (Supplementary Fig. 4A, B). ICI significantly increased the proportion of these K8+/K5+ double positive cells (Fig. 5B, right panel, Supplementary Fig. 4B), confirming that it can modulate cell fate in a defined system independent of the in vivo environment. Like ICI, tamoxifen increased formation of secondary tumorspheres, but neither inhibition of progesterone/glucocorticoid activity with RU486 nor prolactin activity with Δ1-9-G129R-hPRL (33) affected primary or secondary tumorsphere formation (Supplementary Fig. 5A). An inhibitor for the G-coupled estrogen receptor, G-36, failed to reduce the ICI-induced increase in self-renewal, indicating that this estrogen responsive receptor did not mediate the ICI response (Supplementary Fig. 5B).
Figure 5. In vitro ICI 182,780 treatment of tumor cells increases CSC self-renewal independent of Wnt and Notch pathways.
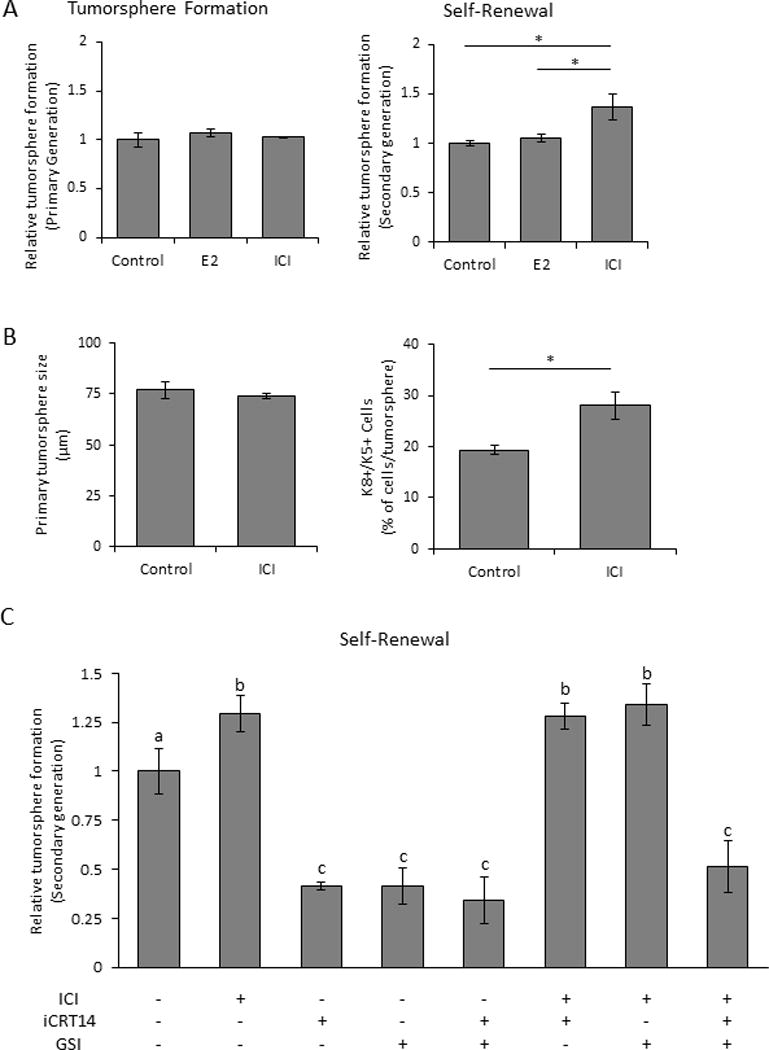
(A) Primary (left panel) and secondary (self-renewal, right panel) tumorsphere formation from Tumor 1 cells treated with 17β-estradiol (E2) or ICI during primary tumorsphere formation, relative to ethanol vehicle (See Methods). (B) Size (left panel) and frequency of double-positive (K8+/K5+) cells (right panel) of primary tumorspheres that developed with ethanol or ICI treatment (see Methods). Asterisks indicate significant differences (unpaired Student’s t-test, p<0.05). (C) Relative secondary tumorsphere generation (self-renewal) from primary tumorspheres treated with inhibitors of the Wnt (iCRT14) or Notch (GSI) pathways, with or without ICI co-treatment. Different letters indicate significant differences (one-way ANOVA followed by Tukey’s multiple comparison tests, p<0.05). A-C, N=3 independent experiments; mean±S.D.
To further understand the effect of ICI on CSC self-renewal, we used inhibitors to block signaling cascades implicated in cell fate decisions during formation of primary tumorspheres (Supplementary Fig. 5C), and examined effects on the ability of ICI to increase formation of secondary tumorspheres. As expected, Wnt and Notch signals were required for optimal self-renewal, but ICI overrode the individual inhibition of both pathways (Fig. 5C). However, when both pathways were inhibited, ICI was unable to increase CSC self-renewal, suggesting that Wnt and Notch signals are able to compensate for one another for this function. In contrast, ICI was unable to overcome the effective reduction in ability to form secondary tumorspheres induced by the HDAC inhibitor, trichostatin-A (Supplementary Fig. 5D). Inhibition of EGFR, SRC family kinases, mTOR, FAK, and JAK2 had no effect on self-renewal with or without ICI. Together, these results indicate that ICI does not alter the activity of existing CSCs to generate tumorspheres, but modulates cell lineage decisions during formation of the primary tumorspheres, enabling increased CSC self-renewal.
Discussion
A deeper understanding of tumor heterogeneity, population dynamics, and their responses to standard therapies, is critical to go beyond treatments which only reduce the bulk of the tumor, and to identify strategies which ultimately prevent tumor relapse. Little is known about how anti-estrogens affect these processes in ERα+ breast cancers, independent of effects on tumor growth. Here we demonstrated that PRL-induced ERα+, but estrogen independent, mammary cancers exhibited epithelial lineage heterogeneity, similar to some clinical ERα+ breast cancers (6–8). CSCs were not limited to a single subpopulation, but were active at similar frequencies in both the luminal and basal compartments. Treatment with the SERD, ICI, failed to affect tumor growth, but rather transiently perturbed this heterogeneity and reduced CSC activity. With longer treatment, tumor heterogeneity and CSC activity were restored, associated with increased canonical Wnt and Notch signaling, and transcript markers linked to stem cell activity and EMT. These results reveal a key role for ER-mediated signals in plasticity of these tumors and CSC activity, and expose potential deleterious effects of endocrine treatments.
The NRL-PRL transgenic mouse models elevated exposure to PRL (21), epidemiologically associated with higher risk for aggressive ERα+ breast cancers in postmenopausal women (18,19). In nulliparous NRL-PRL females, local PRL acts independently and cooperatively with ovarian steroids to alter the dynamic differentiation of mammary epithelial subpopulations (34). It expands progenitor/stem epithelial populations in conjunction with increased canonical Wnt signals, and reduces steroid-driven maturation of luminal cells, associated with altered transcriptional networks predicted to perturb differentiation, actions which are likely to contribute to the development of breast cancer. Although the majority of these tumors are ERα+, PR+ cells are very rare; however, we cannot rule out low levels of expression as a result of high turnover (35). As shown herein, these adenocarcinomas exhibit a mixed lineage, which may reflect PRL actions on the epithelial hierarchy. The substantial K5+ subpopulation resembles that in a subset of ERα+ clinical cancers (6–8), and allowed us to examine population dynamics between tumor cells with luminal and basal features.
Recent studies have reported a relatively rare progesterone-responsive K5-expressing cell population in up to 50% of all clinical luminal tumors, and this lineage heterogeneity is associated with treatment resistance and recurrence (7,8). We found that the relative proportions of these subpopulations in the PRL-induced carcinomas were strikingly consistent, not only among passages of the same tumor, but in tumors arising from limiting dilutions of sorted luminal or basal epithelia from independent parent carcinomas. This suggests that this balance confers benefit to the tumor, and implies juxtacrine or paracrine signals that regulate “quorum”; indeed, this has been suggested as a driver of cancer progression (3–5,36,37). Interestingly, the luminal and basal compartments in the PRL-induced tumors contained similar levels of CSC/tumor-regenerating activity, whereas stem cells of the normal mammary gland are strictly confined to the basal cell community (9). While not extensively examined in clinical cancers, CSC activity in cells with luminal characteristics has also been described in some primary patient cancers (38).
Although ICI failed to slow proliferation of these tumors, it induced a durable change in tumor cell plasticity. After just two days of ICI administration, the growth of tumors was unaffected, but the proportion of basal cells and CSC activity had declined. This was reflected in decreased expression of a major luminal effector of normal mammary stem cell activity, Wnt4, and reduced canonical Wnt target expression (Axin2). Consistent with our findings, 17β-estradiol has been reported to strongly upregulate Wnt4 expression in ERα+ breast cancer cells (39), and canonical Wnt signaling is implicated in CSC activity in other cancers (40–42).
Interestingly, after 14 days of ICI treatment, the original lineage ratio and CSC activity were restored, and the proportion of double positive, K8+/K5+, tumor cells had increased 2-3 fold. K8+/K5+ cells resemble a bipotent epithelial subpopulation observed in the normal gland using lineage-tracing (31,43), and have been reported in mammary cancers induced by constitutive activation of PI3K (44,45). Furthermore, there was a 2-3 fold induction of several markers of plasticity, including Hes1, Twist1, Bmi1 and Sox2. Cell plasticity often coincides with stem cell traits, including asymmetric self-renewal and drug resistance (4,5). Interestingly, at this time after prolonged ICI treatment, Axin2 mRNA had recovered to control levels, although Wnt4 transcripts remained low, suggesting that canonical Wnt signals were now initiated by other members of this complex pathway (46,47). This was accompanied by elevation of Hes1 transcripts, indicating increased Notch signals, which have been shown to be upregulated by anti-estrogens and can also upregulate CSC activity (40,48). Moreover, rising Sox2, Bmi1, and Twist1 mRNAs suggest augmented stem cell self-renewal and EMT. Crosstalk among these genes and the Wnt and Notch pathways is well documented (9,41,49). These results in an ERα+ model of breast cancer resemble the Wnt-mediated cooperation between luminal and basal subclones described for mixed lineage tumors that develop in MMTV-Wnt1 females (37,50). Together, they suggest that ICI-induced reduction of Wnt4 expression observed in the current study underlies the decline in the basal tumor subpopulation and fall in CSC activity, and support the importance of other initiators of Wnt signals which are not dependent on estrogen in the restoration of lineage heterogeneity and CSC activity.
In order to investigate the effect of ER-mediated signals on CSC activity and epithelial lineages apart from the in vivo tumor microenvironment, we utilized the tumorsphere assay. This nonadherent system is widely used to assay stem activity, permitting comparison to other studies (28). ICI treatment during formation of primary tumorspheres from PRL-induced tumors did not affect numbers of primary tumorspheres, consistent with other reports that CSCs do not express ERα (14,15), but did increase CSC self-renewal, evident in higher numbers of secondary tumorspheres. Using a similar assay, Simões et al. reported that anti-estrogen treatment increased CSC self-renewal in ERα+ breast cancer cell lines and PDX models (48). The inability of ICl to inhibit growth in our system, in contrast to the cell lines and PDX models examined by Simões and colleagues, permits us to reject the model that ICI enriches for CSCs by selecting against more differentiated cells. Instead, our data suggest that ICI alters tumor cell differentiation/plasticity, resulting in a net increase in CSC activity. This is supported by the ICI-induced increase in double positive K8+/K5+ cells in primary tumorspheres in our studies independent of effects on tumorsphere size, and the unsurprising ability of the HDAC inhibitor, trichostatin-A, to block the ICI-induced increase in CSC self-renewal. Additionally, CSC self-renewal was sensitive to both Notch and Wnt signals. The importance of Notch signals in CSC self-renewal in PRL-induced tumors resembles the results in ERα+ models reported by Simões and colleagues (48). However, unlike that report, which demonstrated that simultaneous inhibition of Notch and ER-mediated signals cooperatively reduced CSC renewal, ICI treatment of primary tumorspheres herein overrode inhibition of Notch signals. Wnt signals have received less study in ERα+ breast cancer, but have been linked to tamoxifen resistance (48). In our study, ICI also overcame Wnt-induced inhibition of CSC self-renewal, as for Notch signals. However, the inability of ICI to override the simultaneous inhibition of both the Notch and Wnt pathways demonstrates the complementarity of these pathways in these bilineage cancers. The regulation and crosstalk among these pathways on CSC activity in ERα+ cancers, particularly those composed of mixed lineages, deserves additional study.
Together, our data demonstrate that tumor epithelia in PRL-induced mixed lineage ERα+ cancers, which resemble a subset of clinical luminal breast cancers, are plastic with respect to lineage surface marker expression and CSC activity. They indicate that ER-mediated signals contribute to the local network which maintains equilibria of epithelial subpopulations and stem cell niche(s) in these cancers. The rapid compensation for the inhibition of estrogenic signals demonstrates the bidirectional fluidity of these subpopulations and underscores the tight regulation of this balance. In addition to Notch signals, which are associated with CSC activity and anti-estrogen resistance of luminal breast cancers, our studies implicate canonical Wnt signals in maintenance of tumor subpopulation diversity and CSC activity. Clinical ERα+ breast cancers that exhibit lineage heterogeneity are largely understudied. Our studies reveal that the subset of ERα+ cancers which are considered resistant to anti-estrogens as they are not dependent on estrogen for growth, nonetheless may exhibit a dynamic response to these therapies with undesirable consequences.
Supplementary Material
Significance.
This study suggests that treatment of a subset of ERα+ breast cancers with anti-estrogen therapies may not only fail to slow growth but promote aggressive behavior by evoking tumor cell plasticity and regenerative CSC activity.
Acknowledgments
We would like to thank Debra Rugowski, and the staff of the UWCCC Flow Cytometry Laboratory, the Translational Science Biocore Biobank and the UW-Madison Biotron Laboratory for assistance with these studies. We are also grateful to Drs. Lisa Arendt, Joan Jorgensen, Robert Lipinski, Ruth Sullivan, and Chad Vezina for their helpful discussions throughout the project, and Florence Boutillon for production of recombinant PRLR antagonist.
Grant Support
This work was supported in part by NIH R01 CA157675 (L.A. Schuler), NIEHS T32 ES007015 (M.P. Shea), Era of Hope DOD-BCRP award W81XWH-06-1-0491 (C.M. Alexander), NCI T32 CA009135 and Kuwait Foundation for the Advancement of Sciences 2013-6302-03 (S.A. Fakhraldeen), and the University of Wisconsin Carbone Cancer Center (UWCCC) Support Grant NIH P30 CA014520 (all authors). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NCI, NIH.
Abbreviations
- CSC
cancer stem cell
- EMT
epithelial-mesenchymal transition
- ERα
estrogen receptor alpha
- FACS
fluorescence activated cell sorting
- PDX
patient derived xenograft
- PRL
prolactin
- SERD
selective estrogen receptor downregulator
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Ades F, Zardavas D, Bozovic-Spasojevic I, Pugliano L, Fumagalli D, de Azambuja E, et al. Luminal B breast cancer: molecular characterization, clinical management, and future perspectives. J Clin Oncol. 2014;32:2794–803. doi: 10.1200/JCO.2013.54.1870. [DOI] [PubMed] [Google Scholar]
- 2.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) Davies C, Godwin J, Gray R, Clarke M, Cutter D, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378:771–84. doi: 10.1016/S0140-6736(11)60993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tabassum DP, Polyak K. Tumorigenesis: it takes a village. Nat Rev Cancer. 2015;15:473–83. doi: 10.1038/nrc3971. [DOI] [PubMed] [Google Scholar]
- 4.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–37. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Easwaran H, Tsai H-C, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–27. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abd El-Rehim DM, Pinder SE, Paish CE, Bell J, Blamey RW, Robertson JFR, et al. Expression of luminal and basal cytokeratins in human breast carcinoma. J Pathol. 2004;203:661–71. doi: 10.1002/path.1559. [DOI] [PubMed] [Google Scholar]
- 7.Goodman CR, Sato T, Peck AR, Girondo MA, Yang N, Liu C, et al. Steroid induction of therapy-resistant cytokeratin-5-positive cells in estrogen receptor-positive breast cancer through a BCL6-dependent mechanism. Oncogene. 2016;35:1373–85. doi: 10.1038/onc.2015.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kabos P, Haughian JM, Wang X, Dye WW, Finlayson C, Elias A, et al. Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res Treat. 2011;128:45–55. doi: 10.1007/s10549-010-1078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014;28:1143–58. doi: 10.1101/gad.242511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727–38. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 12.Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465:803–7. doi: 10.1038/nature09091. [DOI] [PubMed] [Google Scholar]
- 13.Asselin-Labat M-L, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010;465:798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- 14.Finlay-Schultz J, Sartorius CA. Steroid hormones, steroid receptors, and breast cancer stem cells. J Mammary Gland Biol Neoplasia. 2015;20:39–50. doi: 10.1007/s10911-015-9340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simões BM, Alferez DG, Howell SJ, Clarke RB. The role of steroid hormones in breast cancer stem cells. Endocr Relat Cancer. 2015;22:T177–86. doi: 10.1530/ERC-15-0350. [DOI] [PubMed] [Google Scholar]
- 16.Dabydeen SA, Furth PA. Genetically engineered ERα-positive breast cancer mouse models. Endocr Relat Cancer. 2014;21:R195–208. doi: 10.1530/ERC-13-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohibi S, Mirza S, Band H, Band V. Mouse models of estrogen receptor-positive breast cancer. J Carcinog. 2011;10:35. doi: 10.4103/1477-3163.91116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tworoger SS, Eliassen AH, Zhang X, Qian J, Sluss PM, Rosner BA, et al. A 20-year prospective study of plasma prolactin as a risk marker of breast cancer development. Cancer Res. 2013;73:4810–9. doi: 10.1158/0008-5472.CAN-13-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tikk K, Sookthai D, Johnson T, Rinaldi S, Romieu I, Tjønneland A, et al. Circulating prolactin and breast cancer risk among pre- and postmenopausal women in the EPIC cohort. Ann Oncol. 2014;25:1422–8. doi: 10.1093/annonc/mdu150. [DOI] [PubMed] [Google Scholar]
- 20.Tworoger SS, Hankinson SE. Prolactin and breast cancer etiology: an epidemiologic perspective. J Mammary Gland Biol Neoplasia. 2008;13:41–53. doi: 10.1007/s10911-008-9063-y. [DOI] [PubMed] [Google Scholar]
- 21.O’Leary KA, Shea MP, Schuler LA. Modeling prolactin actions in breast cancer in vivo: insights from the NRL-PRL mouse. Adv Exp Med Biol. 2015;846:201–20. doi: 10.1007/978-3-319-12114-7_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arendt LM, Rugowski DE, Grafwallner-Huseth TA, Garcia-Barchino MJ, Rui H, Schuler LA. Prolactin-induced mouse mammary carcinomas model estrogen resistant luminal breast cancer. Breast Cancer Res. 2011;13:R11. doi: 10.1186/bcr2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Creighton CJ. The molecular profile of luminal B breast cancer. Biologics. 2012;6:289–97. doi: 10.2147/BTT.S29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA. Prolactin induces ERα-positive and ERα-negative mammary cancer in transgenic mice. Oncogene. 2003;22:4664–74. doi: 10.1038/sj.onc.1206619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7:389–97. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- 26.O’Leary KA, Rugowski DE, Sullivan R, Schuler LA. Prolactin cooperates with loss of p53 to promote claudin-low mammary carcinomas. Oncogene. 2013;33:3075–82. doi: 10.1038/onc.2013.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Kim S, Alexander CM. Tumorsphere assay provides more accurate prediction of in vivo responses to chemotherapeutics. Biotechnol Lett. 2014;36:481–8. doi: 10.1007/s10529-013-1393-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw FL, Harrison H, Spence K, Ablett MP, Simões BM, Farnie G, et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia. 2012;17:111–7. doi: 10.1007/s10911-012-9255-3. [DOI] [PubMed] [Google Scholar]
- 30.Fakhraldeen SA, Clark RJ, Roopra A, Chin EN, Huang W, Castorino J, et al. Two isoforms of the RNA binding protein, coding region determinant-binding protein (CRD-BP/IGF2BP1), are expressed in breast epithelium and support clonogenic growth of breast tumor cells. J Biol Chem. 2015;290:13386–400. doi: 10.1074/jbc.M115.655175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Amerongen R, Bowman AN, Nusse R. Developmental stage and time dictate the fate of Wnt/β-catenin-responsive stem cells in the mammary gland. Cell Stem Cell. 2012;11:387–400. doi: 10.1016/j.stem.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 32.Rajaram RD, Buric D, Caikovski M, Ayyanan A, Rougemont J, Shan J, et al. Progesterone and Wnt4 control mammary stem cells via myoepithelial crosstalk. EMBO J. 2015;34:641–52. doi: 10.15252/embj.201490434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernichtein S, Kayser C, Dillner K, Moulin S, Kopchick JJ, Martial JA, et al. Development of pure prolactin receptor antagonists. J Biol Chem. 2003;278:35988–99. doi: 10.1074/jbc.M305687200. [DOI] [PubMed] [Google Scholar]
- 34.O’Leary KA, Shea MP, Salituro S, Blohm CE, Schuler LA. Prolactin alters the mammary epithelial hierarchy, increasing progenitors and facilitating ovarian steroid action. Stem Cell Reports. 2017;9:1167–79. doi: 10.1016/j.stemcr.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diep CH, Daniel AR, Mauro LJ, Knutson TP, Lange CA. Progesterone action in breast, uterine, and ovarian cancers. J Mol Endocrinol. 2015;54:R31–53. doi: 10.1530/JME-14-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–44. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 37.Kim S, Goel S, Alexander CM. Differentiation generates paracrine cell pairs that maintain basaloid mouse mammary tumors: proof of concept. PLoS One. 2011;6:e19310. doi: 10.1371/journal.pone.0019310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Villadsen R, Sørlie T, Fogh L, Grønlund SZ, Fridriksdottir AJ, et al. Tumor initiating but differentiated luminal-like breast cancer cells are highly invasive in the absence of basal-like activity. Proc Natl Acad Sci U S A. 2012;109:6124–9. doi: 10.1073/pnas.1203203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–34. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445–64. doi: 10.1038/nrclinonc.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu W, Kang Y. Cell lineage determinants as regulators of breast cancer metastasis. Cancer Metastasis Rev. 2016;35:631–44. doi: 10.1007/s10555-016-9644-y. [DOI] [PubMed] [Google Scholar]
- 42.Wei W, Lewis MT. Identifying and targeting tumor-initiating cells in the treatment of breast cancer. Endocr Relat Cancer. 2015;22:R135–55. doi: 10.1530/ERC-14-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rios AC, Fu NY, Lindeman GJ, Visvader JE. In situ identification of bipotent stem cells in the mammary gland. Nature. 2014;506:322–7. doi: 10.1038/nature12948. [DOI] [PubMed] [Google Scholar]
- 44.Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature. 2015;525:114–8. doi: 10.1038/nature14669. [DOI] [PubMed] [Google Scholar]
- 45.Van Keymeulen A, Lee MY, Ousset M, Brohée S, Rorive S, Giraddi RR, et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature. 2015;525:119–23. doi: 10.1038/nature14665. [DOI] [PubMed] [Google Scholar]
- 46.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 47.Alexander CM, Goel S, Fakhraldeen SA, Kim S. Wnt signaling in mammary glands: plastic cell fates and combinatorial signaling. Cold Spring Harb Perspect Biol. 2012;4:a008037–a008037. doi: 10.1101/cshperspect.a008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simões BM, O’Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, et al. Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep. 2015;12:1968–77. doi: 10.1016/j.celrep.2015.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Piva M, Domenici G, Iriondo O, Rábano M, Simões BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6:66–79. doi: 10.1002/emmm.201303411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cleary AS, Leonard TL, Gestl SA, Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508:113–7. doi: 10.1038/nature13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.