

Abstract
Aberrant epidermal growth factor receptor (EGFR) signaling is a common driver of glioblastoma (GBM) pathogenesis, however, the downstream effectors that sustain this oncogenic pathway remain unclarified. Here we demonstrate that tripartite motif-containing protein 59 (TRIM59) acts as a new downstream effector of EGFR signaling by regulating STAT3 activation in GBM. EGFR signaling led to TRIM59 upregulation through SOX9 and enhanced the interaction between TRIM59 and nuclear STAT3, which prevents STAT3 dephosphorylation by the nuclear form of T cell protein tyrosine phosphatase (TC45), thereby maintaining transcriptional activation and promoting tumorigenesis. Silencing TRIM59 suppresses cell proliferation, migration, and orthotopic xenograft brain tumor formation of GBM cells and glioma stem cells (GSCs). Evaluation of GBM patient samples revealed an association between EGFR activation, TRIM59 expression, STAT3 phosphorylation, and poor prognoses. Our study identifies TRIM59 as a new regulator of oncogenic EGFR/STAT3 signaling and as a potential therapeutic target for GBM patients with EGFR activation.
Keywords: TRIM59, glioma, STAT3, EGFR, TC45
Introduction
A hallmark of human cancers is aberrantly active oncogenic signaling stimulated by amplified and overexpressed genes (1). Epidermal growth factor receptor (EGFR) is frequent amplified and overexpressed, and plays a prominent role in glioblastoma (GBM) (2–4). Amplification and overexpression of EGFR or EGFRvIII (a constitutively active EGFR mutant) confer a worse prognosis in glioma patients (2, 5, 6). EGFR/EGFRvIII drives tumorigenesis by multiple down-stream pathways, including through activation of signal transducer and activator of transcription 3 (STAT3) signaling, thereby stimulating cancer cell proliferation, survival, and chemoresistance (7–9). STAT3 signaling can be activated through amplification and mutation of EGFR, phosphorylation of the enhancer of zeste homolog 2 (EZH2), and activation of the janus kinase 2 (JAK2) (7–9). In other cancer and immune cells, STAT3 activity can be inhibited through either dephosphorylating JAK in the cytoplasm by SHP1, SHP2 or PTP1B (10), or directly dephosphorylation in the nucleus by the nuclear form of T cell protein tyrosine phosphatase (TC45) (11–13). In addition to these proteins identified in the EGFR/STAT3 signaling axis, additional components remain uncharacterized for their roles in promoting tumorigenesis.
TRIM59 is a member of the tripartite motif-containing (TRIM) protein superfamily, and has a TRIM or RBCC motif consisting of a RING-finger domain (R), a B‐box domain (B), and a coiled-coil domain (CC)(14). TRIM59 was initially identified as an early signal transducer in SV40 Tag and Ras oncogenic pathways in murine prostate cancer models (15). Subsequently, TRIM59 was found to be upregulated in human gastric tumors, promoting tumorigenesis by enhancing ubiquitination and degradation of p53 (16). TRIM59 was also upregulated in human lung cancer, osteosarcoma, and cervical cancer (17–19). Moreover, TRIM59 interacts with ECSIT as an adaptor protein required for the Toll-like receptor-mediated transduction pathway in HeLa cells (20). TRIM59 has been implicated in mediating tumor progression, but the mechanisms regarding how it facilitates tumorigenesis have not been elucidated.
In this study, we reveal TRIM59 as a new effector for EGFR/EGFRvIII-driven tumorigenesis. Our data shows that TRIM59 expression is upregulated by EGFR/EGFRvIII in GBM through SOX9. EGFR signaling promotes TRIM59-STAT3 interaction in the nucleus. Specifically, Y218/Q221 sites of TRIM59 are required for STAT3 activation and TRIM59-STAT3 interaction. TRIM59 promotes STAT3 activity by inhibiting TC45-mediated dephosphorylation of STAT3, leading to enhanced EGFR/EGFRvIII-driven tumorigenesis. The importance of this novel pathway is highlighted by the co-expression of p-EGFRY1173, TRIM59, and p-STAT3Y705 in a large number of glioma clinical samples. Co-expression of p-EGFRY1173 and TRIM59 also correlates with poor survival of GBM patients.
Materials and Methods
Cell lines
LN229 and U87 (21) GBM cells were from ATCC (Manassas, VA, USA). Patient-derived glioma stem cell (GSC) lines, GSC 1123 and GSC 83 were from Dr. Ichiro Nakano (22). Glioma cells were cultured in 10% FBS/DMEM, and GSC cells were maintained in DMEM/F12 supplemented with B27 (1:50), heparin (5 mg/ml), basic FGF (20 ng/ml), and EGF (20 ng/ml) as we previously described (23). All cell lines in this study were authenticated using STR DNA fingerprinting in March 2017 by Shanghai Biowing Applied Biotechnology Co., Ltd (Shanghai, China), and mycoplasma infection was detected using LookOut Mycoplasma PCR Detection kit (Sigma-Aldrich). Only lower-passage cell lines were used for the study.
Plasmids
TRIM59 cDNA was amplified from U87 cells, sequenced, and then subcloned into the pLVX-Puro and pcDNA3 vectors (Clontech) with an HA tag. SOX9 cDNA was also amplified from U87 cells and then subcloned into the pcDNA3 vector. HA-TRIM59 deletion constructs were made as previously described (20). pcDNA3-Flag-STAT3, pcDNA3-Flag-STAT3-Y705F, and pcDNA3-Myc-STAT3 were derived from pLEGFP-WT-STAT3, which was a gift from George Stark (Lerner Research Institute, Cleveland Clinic, Addgene plasmid #71450) (24). pcDNA3-Flag-TC45 was derived from pEFneo-HA-TC45 (12). HA-TRIM59Y218F/Q221A point mutation was generated using a site-directed mutagenesis kit (Invitrogen) following the manufacturer’s protocol. TRIM59 shRNAs were purchased from Genechem (Shanghai, China).
Immunoprecipitation and Western blotting assays
Immunoprecipitation and Western blotting analyses were performed as we previously described (23). Briefly, cells were lysed, centrifuged, and then protein concentrations were determined. Equal amounts of cell lysates were immunoprecipitated with specific antibodies and protein G-agarose beads (Invitrogen). Standard Western blotting was done with antibody against β-actin (I-19), STAT1 (C-111), STAT5 (G-2), and STAT3 (H-190) (Santa Cruz Biotechnology); Flag (M2, Sigma-Aldrich); HA (#66006-1-Ig), Lamin B1 (Proteintech Group); TRIM59 (ab69639, Abcam); EGFR (D38B1), phospho-STAT3 (Y705) (D3A7), phospho-EGFR (Y1173) (53A5), STAT3 (124H6), TCPTP (TC45)(D7T7D), Myc (9B11), Tubulin (#2148), and HA (C29F4) (Cell Signaling Technology).
Cell proliferation and colony formation assays
Cell proliferation analysis was performed using a WST-1 assay kit (Roche) and colony formation analysis was performed as previously described (25). Briefly, for cell proliferation analysis, cells were split, seeded, and proliferation measured using the WST-1 assay kit. For colony formation assay, 10,000 cells were seeded in a 0.4% Noble Agar top layer with a bottom layer of 0.8% Noble Agar in each of the triplicate wells of a 12-well plate. Cell culture media was changed every 3 days thereafter. Colonies were scored after 2-3 weeks and data were analyzed.
shRNA knockdown and transfection assays
shRNA knockdown and transfection assays were performed as previously described (23). Lentiviruses were produced by co-transfecting specified DNA and packaging plasmids (26) into 293T cells using Lipofectamine 2000 reagent according to manufacturer’s instruction (#52758, Invitrogen). Viruses were concentrated by ultracentrifugation and added into the culture media supplemented with 8 μg/ml polybrene. Transduced human GBM cells were harvested, and then expressions of exogenous proteins or inhibition of target genes by knockdown vectors were validated by Western blotting assay.
RNA isolation and qRT-PCR
Total RNA was isolated from cells using the Trizol Plus RNA Purification Kit (Thermo Fisher Scientific) and was reversely transcribed into cDNA using the Reverse Transcription Kit (Takara) according to the manufacturer’s instructions. qRT–PCR was performed using the Power SYBR Green Master Mix (Life Technologies) in the Applied Biosystems StepOne Plus Real-Time Thermal Cycling Block. Primers are listed in Table S1. Results were analyzed using the 2−(ΔΔCt) method.
RNA-Seq and Gene Set Enrichment Analysis (GSEA)
Total RNA was extracted and purified using the Qiagen RNeasy Mini kit (Valencia, CA, USA) according to the manufacturer’s instructions. The quality of RNA was assessed by a bioanalyzer before sequencing. Libraries for poly(A)+ RNA were prepared according to the Illumina protocol. Libraries were sequenced on Illumina HiSeqX Ten platforms. The criteria of differentially expressed genes were genes that showed a > 2 fold gene expression changes with a false discovery rate (FDR) < 0.05. GSEA was performed using the GSEA software (27). RNA-Seq data reported in this study have been deposited with the Gene Expression Omnibus under the accession GEO ID: GSE95386 and GSE106557.
Luciferase promoter assay
For the luciferase promoter assay, TRIM59 promoter was amplified from U87 cells using the primers: 5′-acacacatgagccaccacg-3′ and 5′-ggatgctgagagccgccccga-3′ and subcloned into the pGL3 vector (Promega). pGL3-TRIM59 promoter was co-transfected with or withtout a SOX9 expression plasmid into U87 cells using the Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s recommendation. pRL-TK (Renilla luciferase, Promega) was used as a control. Luciferase and Renilla signals were measured using the Dual-Luciferase Reporter Assay kit (Promega) according to a protocol provided by the manufacturer (Berthold technologies).
Tumorigenesis studies
Athymic nu/nu female mice aged 6-8 weeks (SLAC, Shanghai, China) were used. Mice were randomly divided into 5 per group. In total, 5 × 105 U87 GBM cells or 6,000 GSC83 GSCs transduced with a luciferase reporter to measure tumor growth were stereotactically implanted into the mouse brain as previously described (25). Mice were euthanized when neuropathological symptoms developed. Tumor volumes were measured using the largest tumor cross-section for each samples and estimated as (W2 × L)/2, W < L (28) using H&E stained sections or measured in vivo using luciferase activity after injection of D-luciferin. Bioluminescence imaging was performed using the IVIS Lumina imaging station (Caliper Life Sciences). All animal experiments were approved by Shanghai Jiao Tong University Institutional Animal Care and Use Committee (IACUC).
IHC of human glioma specimens
In accordance to a protocol approved by Shanghai Jiao Tong University Institutional Clinical Care and Use Committee, according to the Declaration of Helsinki, clinical brain tissue specimens were collected at Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China. The investigators obtained informed written consent from the subjects. These specimens were examined and diagnosed by pathologists at Ren Ji Hospital. The tissue sections from paraffin-embedded de-identified human GBM specimens were stained with against TRIM59 (1:50), p-EGFRY1173 (1:50), and p-STAT3Y705 (1:50) antibodies. Non-specific IgGs were used as negative controls. IHC staining was scored as 0-7 according to the percentage of positive cells, as previously described (25). Tumors with 0 or 2 staining scores were considered as low expressing and those with 3 to 7 scores were considered high expressing. Two separate individuals who were blinded to the slides examined and scored each sample.
Statistics
GraphPad Prism version 5.0 for Windows (GraphPad Software Inc., San Diego, CA, USA) was used to perform one-way analysis of variance (ANOVA) with Newman-Keuls post hoc test or an unpaired, two-tailed Student’s t-test. Spearman’s rank correlation analysis was used to investigate the correlation of protein expression levels in human clinical GBM specimens, and Kaplan-Meier survival analysis was carried out by log-rank test using SPSS version 20.0 for Windows (International Business Machines Corp.). A p-value of less than 0.05 was considered statistically significant.
Results
TRIM59 expression level is associated with a poor prognosis of glioma patients
To identify the roles of TRIM59 in glioma progression, we first assessed expression of TRIM59 in clinical specimens of glioma patients. We performed IHC staining assays in a total 152 clinical brain tissue specimens, including 6 normal brain tissues, 17 WHO grade II, 22 WHO grade III and 107 GBM. As shown in Fig. 1A, TRIM59 staining was negative or weak in normal brain tissues and low in WHO grade II tumors. TRIM59 was expressed in the majority of high-grade gliomas (WHO grade III and GBM tumors), and was particularly high in GBM (Fig. 1A and 1B). These observations suggest that TRIM59 expression is correlated with glioma grade.
Figure 1. TRIM59 expression is correlated with a poor prognosis of GBM patients.
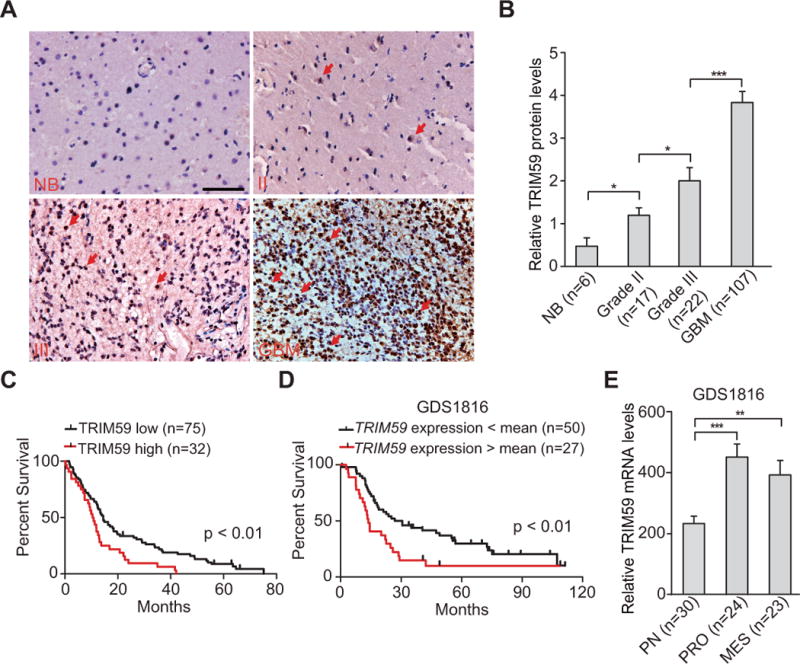
A, IHC staining of TRIM59 expression in normal brain tissues and clinical glioma specimens. Scale bars: 50 μm. Arrows show positive staining. NB, normal brain tissues. II, WHO grade II gliomas. III, WHO grade III gliomas. B, Quantitative analysis of TRIM59 protein expression in A. C, Kaplan–Meier analysis of patients with high TRIM59 protein-expressing GBM versus low TRIM59 protein-expressing tumors in A. Statistical analysis was performed by log-rank test in a SPSS software. Median survival (in months): low, 14.47±1.25; high, 10.43±0.97. Black bars, censored data. D, Kaplan–Meier analysis of patients with high TRIM59 mRNA-expressing gliomas versus low TRIM59 mRNA-expressing tumors. Expression data of TRIM59 mRNA were downloaded from the GDS1816 dataset of Grade III tumors and GBM (29) and analyzed. Median survival (in months): low, 26.83±7.17; high, 13.77±0.91. E, Expression level of TRIM59 mRNA is significantly higher in proliferation (PRO) and mesenchymal (MES) subtype tumors than in proneural (PN) subtype tumors. Error bars ± SD. *, P <0.05. **, P <0.01. ***, P <0.001. Data represent two independent experiments with similar results.
To further assess the relationship of TRIM59 expression and glioma patient survival, we performed Kaplan–Meier survival analysis in our GBM samples and revealed a statistically significant worse prognosis for glioma patients with high TRIM59 protein levels compared with those with low levels (Fig. 1C). The median patient survival times of these patients were 10.43±0.97 and 14.47±1.25 months, respectively (P < 0.01). We also downloaded one microarray dataset of clinical WHO grade III and GBM tumors, GDS1816 (29), and examined the relationship of TRIM59 mRNA expression and glioma patient survival by Kaplan–Meier survival analysis. As shown in Fig. 1D, Kaplan–Meier survival analysis demonstrated that glioma patients with high TRIM59 mRNA levels show a statistically significant worse prognosis compared with those with low levels, with the median patient survival times of 13.77±0.91 and 26.83±7.17 months, respectively (P < 0.01). We also examined TRIM59 mRNA expression in different clinical subtype glioma samples, and found that compared with proneural (PN) subtype tumors, TRIM59 is highly expressed in proliferation (PRO) and mesenchymal (MES) subtype tumors (Fig. 1E). Taken together, these observations strongly indicate that TRIM59 expression is closely associated with progression and poor prognosis in glioma patients.
TRIM59 is transcriptionally upregulated by EGFR/EGFRvIII through SOX9 in GBM
Since TRIM59 is expressed relatively high in clinical proliferation subtype gliomas with EGFR amplification and mutation, we hypothesize that TRIM59 is important for EGFR/EGFRvIII-driven glioma tumorigenesis. To validate this, we first analyzed expression of TRIM59 using Western blotting assay in isogenic LN229 and U87 GBM cells with, or without EGFRvIII. This analysis revealed that TRIM59 was significantly upregulated in EGFRvIII-expressing GBM cells compared with that in the controls (Fig. 2A). In U87 GBM cells with stable overexpression of EGFR, EGF stimulation also markedly increased TRIM59 expression (Fig. 2B). Treatment with the EGFR tyrosine kinase inhibitor, erlotinib significantly inhibited EGF-stimulated TRIM59 expression (Fig. 2B). We further assessed the expression of TRIM59 mRNA in LN229 and U87 GBM cells with, or without EGFRvIII, and found that TRIM59 mRNA expression levels were significantly increased in GBM cells transduced with EGFRvIII compared with the controls (Fig. 2C). This data demonstrates that TRIM59 is upregulated by activated EGFR in GBM cells.
Figure 2. TRIM59 expression is upregulated by activated EGFR in GBM cells.
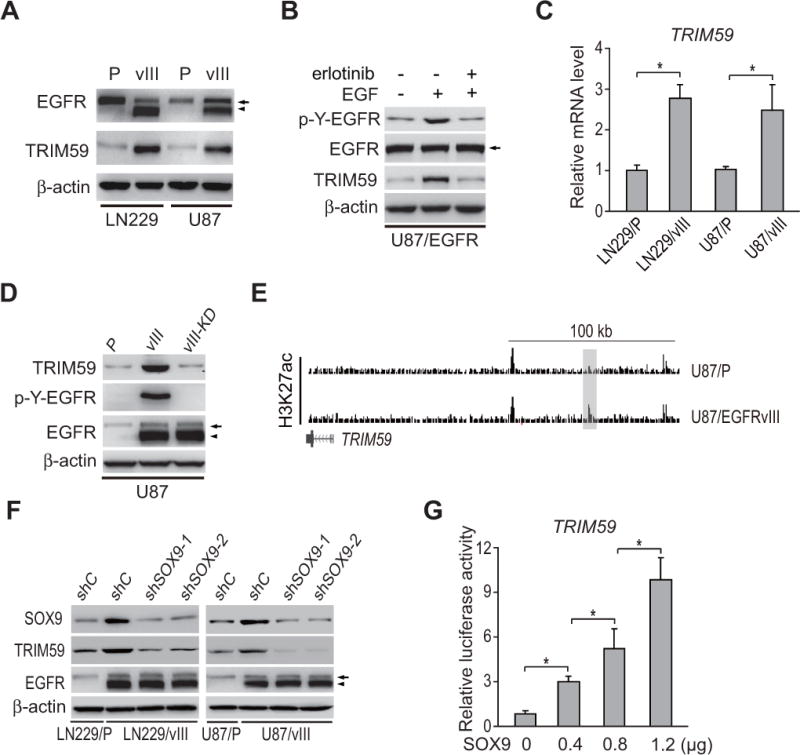
A, Effects of EGFRvIII overexpression on TRIM59 protein expression in U87 and LN229 GBM cells. P, parental cells; vIII, U87 or LN229 cells expressing EGFRvIII. β-actin was used as a loading control. B, Erlotinib treatment inhibited EGF-stimulated EGFR phosphorylation and EGFR-upregulated TRIM59 expression in U87 GBM cells with stable expression of EGFR. EGF (20 ng/ml) stimulated U87 GBM cells with, or without, Erlotinib (10 μM) for 24 h. C, qRT-PCR analysis of effects of EGFRvIII on TRIM59 mRNA expression. D, WB analysis of effect of kinase dead EGFRvIII (EGFRvIII-KD) on TRIM59 expression. E, Snapshots of UCSC genome browser (genome.ucsc.edu) of H3K27ac ChIP-Seq data (GSE72468) at the loci of TRIM59. The shaded H3K27ac peaks indicated putative EGFRvIII-responsive enhancer. F, Effects of SOX9 depletion on EGFRvIII-stimulated TRIM59 protein expression. G, Overexpression of SOX9 promotes TRIM59 transcription in a dose-dependent manner. A luciferase reporter of TRIM59 promoter was transiently transfected into U87 cells with the different amount of SOX9 expression plasmid DNA. Arrows, EGFR. Arrow heads, EGFRvIII. Error bars ± SD. *, P <0.05. Data represent two independent experiments with similar results.
To test whether EGFRvIII’s effect on TRIM59 expression depends on its kinase activity, we used a kinase activity-deficient EGFRvIII construct (EGFRvIII-KD; Fig. 2D) (30). In U87 GBM cells, EGFRvIII significantly elevated TRIM59 expression relative to the parental control line while EGFRvIII-KD did not (Fig. 2D), indicating that the enhanced TRIM59 expression is dependent on EGFRvIII kinase activity.
EGFRvIII was shown to induce transcription and epigenetic remodeling of the enhancer landscape to promote glioma tumorigenesis through SOX9 and FOXG1 transcription factor networks (31). To reveal the regulatory mechanisms of TRIM59 upregulation by EGFR/EGFRvIII, we further analyzed histone H3 lysine 27 acetylation (H3K27ac) ChIP-Seq data (GSE72468) at the loci of TRIM59, and detected a significant increase in H3K27 acetylation at the putative enhancers near TRIM59 in U87/EGFRvIII GBM cells compared with U87 GBM cells (Fig. 2E), suggesting that EGFRvIII upregulates TRIM59 expression by activating its enhancers. Next, we performed SOX9 and FOXG1 knockdown experiments in LN229/EGFRvIII and U87/EGFRvIII cells using two separate shRNAs. Compared with the controls, SOX9 knockdown in EGFRvIII-expressing cells markedly inhibited EGFRvIII-upregulated TRIM59 protein expression (Fig. 2F), whereas FOXG1 knockdown had no effects (Fig. S1). Moreover, overexpression of SOX9 activated TRIM59 expression in U87 GBM cells (Fig. 2G). Taken together, our data support that TRIM59 is transcriptionally upregulated by EGFR/EGFRvIII through SOX9 in GBM cells.
TRIM59 knockdown inhibits EGFRvIII-stimulated STAT3 activity
The functional roles of TRIM59 in cancers are not fully characterized. To test whether TRIM59 is critical for EGFR/EGFRvIII-driven glioma tumorigenesis, we knocked down TRIM59 in LN229/EGFRvIII and U87/EGFRvIII cells using two separate shRNAs, and performed gene set enrichment analysis (GSEA) in TRIM59-knockdown LN229/EGFRvIII cells versus control cells (Fig. 3A). EGFR-regulated transcription factor STAT3-targeted gene signature (32) was significantly altered in TRIM59-knockdown GBM cells (Fig. 3A). Then, we performed Western blotting assays and found that compared with the controls, depletion of TRIM59 in EGFRvIII-expressing cells markedly inhibited EGFRvIII-promoted STAT3 phosphorylation (Fig. 3B). Moreover, TRIM59 knockdown significantly inhibited EGFRvIII-induced transcription of STAT3 target genes, SOCS3 and c-FOS (10) (Fig. 3C).
Figure 3. Knockdown of TRIM59 inhibits EGFRvIII-stimulated STAT3 activity.
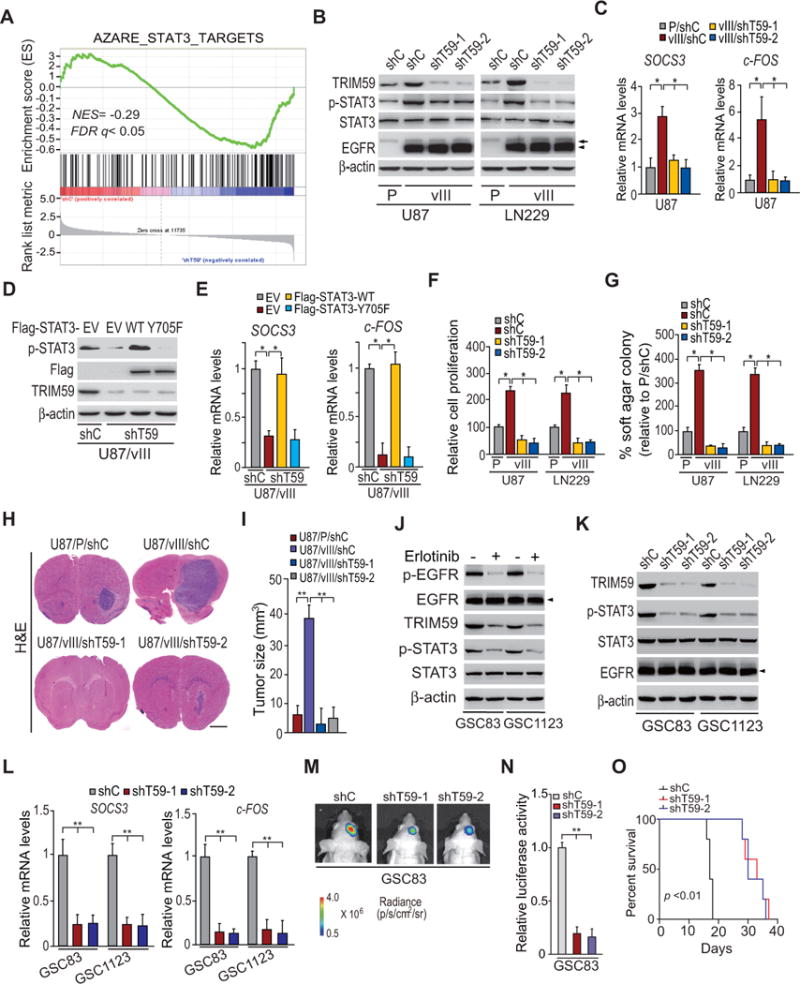
A, Gene set enrichment analysis (GSEA) of STAT3 target genes using ranked gene expression changes in TRIM59-knockdown LN229/EGFRvIII cells compare to control cells. NES, normalized enrichment score. B, WB assays of the effects of TRIM59 knockdown with two different shRNAs (shT59-1 and shT59-2) compared to a control shRNA in LN229 and U87 GBM cells on EGFRvIII-stimulated STAT3 phosphorylation. Arrows, EGFR. Arrow heads, EGFRvIII. C, qRT-PCR analyses of effects of TRIM59 depletion on SOCS3 and c-FOS expression in B. D, WB assays of overexpression of Flag-tagged STAT3 wild type (WT) or an inactive form of STAT3 (Y705F) mutant or an empty vector (EV) in U87/EGFRvIII GBM cells with or without TRIM59 knockdown. E, qRT-PCR analyses of the effects of overexpression of Flag-STAT3 WT or Y705F mutant on TRIM59 depletion-inhibited expression of SOCS3 and c-FOS in D. F and G, Effects of knockdown of TRIM59 on in vitro cell proliferation (F) and cell colony formation (G). H, shRNA knockdown of TRIM59 inhibits EGFRvIII-promoted U87 tumor growth in the brain. I, Quantification of tumor size in H. J, Effects of EGFRvIII on expression of TRIM59 and p-STAT3. GSC83 and GSC11233 GSCs were treated with Erlotinib (10 μM) for 24 h. β-Actin was used as a control. Arrow heads, EGFRvIII. K and L, Knockdown of TRIM59 inhibits p-STAT3 and EGFRvIII-induced transcription of SOCS3 and c-FOS in GSC83 and GSC11233 GSCs. M, Representative bioluminescence images of brains with indicated GSC83 control or TRIM59 knockdown GSCs at 13 days after implantation. Images represent results of five mice per group of two independent experiments. N, Quantification of the bioluminescence activity in M. O, Kaplan–Meier analysis of animals with shT59-expressing GSC83 GSC tumors versus shC-expressing tumors. Statistical analysis was performed by log-rank test. Median survival (in days): shC, 17; shT59-1, 33; shT59-2, 30. Error bars ± SD. *P < 0.05. Data are representative of two or three independent experiments with similar results.
To validate that TRIM59 is important for EGFR-stimulated STAT3 activity, we overexpressed Flag-tagged STAT3 wild type (WT), or an inactive form mutant (Y705F), in U87/EGFRvIII cells transfected with a TRIM59 shRNA. As shown in Fig. 3D and 3E, overexpression of STAT3 WT rescued TRIM59 knockdown-suppressed STAT3 phosphorylation and EGFRvIII-induced transcription of SOCS3 and c-FOS in U87/EGFRvIII/shT59 cells, whereas overexpression of STAT3Y705F did not.
To support that TRIM59 is critical for EGFR/EGFRvIII-driven glioma tumorigenesis, we performed assays to investigate effects of TRIM59 knockdown on EGFR/EGFRvIII-stimulated cell proliferation, colony formation in soft agar in vitro, and tumor growth using orthotopic xenografts. As shown in Fig. 3F and 3G, compared with the controls, depletion of TRIM59 in EGFRvIII-expressing cells markedly inhibited EGFRvIII-promoted cell proliferation and colony formation in soft agar. When engineered U87 cells were implanted into the brains of animals, knockdown of endogenous TRIM59 significantly reduced EGFRvIII-stimulated tumor growth relative to non-silencing control xenografts (Fig. 3H and 3I). These data demonstrate that TRIM59 is important for EGFRvIII-stimulated STAT3 activity and glioma tumor growth.
To further determine whether TRIM59 is critical for glioma tumorigenesis, we analyzed the effects of TRIM59 on patient-derived glioma stem cells (GSCs) using molecular biology, in vitro, and in vivo assays (25, 33). As shown in Fig. 3J, TRIM59 was expressed in both mesenchymal subtype GSCs (GSC83 and GSC1123) previously characterized by high levels of endogenous EGFRvIII (25) and are highly tumorigenic in orthotopic mouse xenografts (22, 25). Treatment of EGFR inhibitor, erlotinib, significantly inhibited EGFR phosphorylation (p-EGFR), STAT3 phosphorylation (p-STAT3), and TRIM59 expression (Fig. 3J). Knockdown of endogenous TRIM59 using two separate shRNAs in both GSC lines markedly suppressed p-STAT3 and EGFRvIII-induced transcription of SOCS3 and c-FOS (Fig. 3K and 3L). Intracranial xenografts demonstrated that TRIM59 knockdown significantly inhibited GSC growth in vivo (Fig. 3M and 3N), and extended animal survival compared with the control (Fig. 3O), validating our observations in EGFRvIII GBM cell lines (Fig. 3B to 3I). These data also support that TRIM59 is important for EGFRvIII tumors. We also found that TRIM59 is co-expressed with SOX9 in classical (CL) and mesenchymal (MES) subtype Grade III and GBM tumors in the Chinese Glioma Genome Atlas (CGGA) datasets (Fig. S2A and S2B). Moreover, SOX9 is co-expressed with EGFR in both tumor subtypes (Fig. S2C and S2D). This further demonstrates that SOX9 enhances TRIM59 expression to regulate CL and MES subtype glioma tumorigenicity.
EGFR promotes TRIM59 interaction with STAT3 in the nucleus
To reveal how TRIM59 mediates EGFRvIII-stimulated STAT3 activity, we first transfected HA-tagged TRIM59 into LN229/EGFRvIII and U87/EGFRvIII GBM cells, and then performed immunoprecipitation and Western blotting analyses using anti-HA and anti-STAT3 antibodies. As shown in Fig. 4A, TRIM59 interacted with STAT3 in both GBM cells. Moreover, their interaction was significantly enhanced by overexpression of EGFRvIII (Fig. 4B). Our analysis of immunoprecipitation and Western blotting using cytoplasmic and nuclear fractions further revealed that the association between TRIM59 and STAT3 occurred in the nucleus (Fig. 4C).
Figure 4. EGFR promotes TRIM59 interaction with STAT3 in the nucleus.
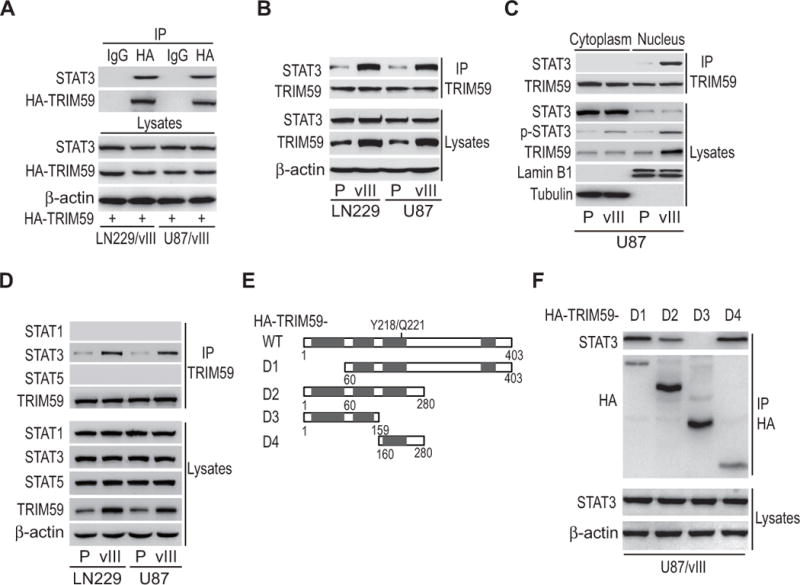
A, IP and WB analyses of the association of STAT3 with ectopic HA-TRIM59 in LN229/EGFRvIII and U87/EGFRvIII GBM cells. IgG was used as a control. B, Effects of EGFRvIII overexpression on the association of STAT3 with TRIM59. C, The interaction of TRIM59 and STAT3 occurs primarily in the nucleus. Cytoplasmic and nuclear proteins from U87 GBM cells, with or without EGFRvIII overexpression, were separated and subjected to IP as indicated. The nuclear marker Lamin B1 and the cytoplasmic marker Tubulin were used to demonstrate the purity of fractions. D, TRIM59 specifically interacts with STAT3. IP/WB analyses of the association of TRIM59 with STAT1, STAT3 and STAT5 in LN229 and U87 GBM cells. E, Schematics of TRIM59WT and various TRIM59 deletion mutants. F, STAT3 interacts with TRIM59 with amino acid residues 160-280. TRIM59WT or the indicated TRIM59 mutants was transfected into U87/EGFRvIII GBM cells. Data are representative of two or three independent experiments with similar results.
Next, to investigate the specificity of TRIM59-STAT3 interaction, we determined the interaction between TRIM59 with STAT1 or STAT5 in U87 and LN229 GBM cells with or without EGFRvIII overexpression. As shown in Fig. 4D, TRIM59 did not bind to STAT1 or STAT5 in control or EGFRvIII cells.
Finally, to further identify which region or domain in TRIM59 mediates its association with STAT3, we generated several deletion mutants lacking various functional binding domains as indicated in Fig. 4E, and then separately transfected them into U87/EGFRvIII GBM cells. Mutants D1, D2, and D4, but not mutant D3, could bind to STAT3 (Fig. 4F), suggesting that the middle region of amino acid residues 160-280 of TRIM59 is required for interacting with STAT3. These data suggest that EGFRvIII specifically promotes TRIM59 interaction with STAT3 in the nucleus.
Y218/Q221 sites of TRIM59 are important for TRIM59-STAT3 interaction
Since the middle region of amino acid residues 160-280 of TRIM59 does not contain any previously defined signaling motif, we performed in silico analysis using The Eukaryotic Linear Motif Resource for Functional Sites of Proteins (http://elm.eu.org) and identified one potential consensus motif, YXXQ (consistent with a STAT3 SH2-binding domain) (34–36), in TRIM59 at amino acid residues 218-221(Fig. 4E). To assess whether this YXXQ motif is critical for TRIM59-STAT3 interaction, we constructed the TRIM59 vector with specific point mutations, substituting tyrosine (Y) to phenylalanine (F) 218 and Glutamine (Q) to Alanine (A) 221. Re-expression of shRNA-resistant TRIM59 wild type (WT*) rescued EGFRvIII-induced binding of TRIM59 with STAT3 (Fig. 5A), STAT3 phosphorylation (Fig. 5A), and transcription of SOCS3 and c-FOS (Fig. 5B), in both U87/EGFRvIII/shT59 and LN229/EGFRvIII/shT59 GBM cells. However, re-expression of shRNA-resistant TRIM59 Y218F/Q221A mutant did not restore EGFRvIII-induced STAT3 phosphorylation or target gene expression (Fig. 5A and 5B), suggesting that the Y218/Q221 sites are critical for TRIM59 interaction with STAT3.
Figure 5. Y218/Q221 sites of TRIM59 are critical for TRIM59-STAT3 interaction and EGFRvIII-driven tumor growth.
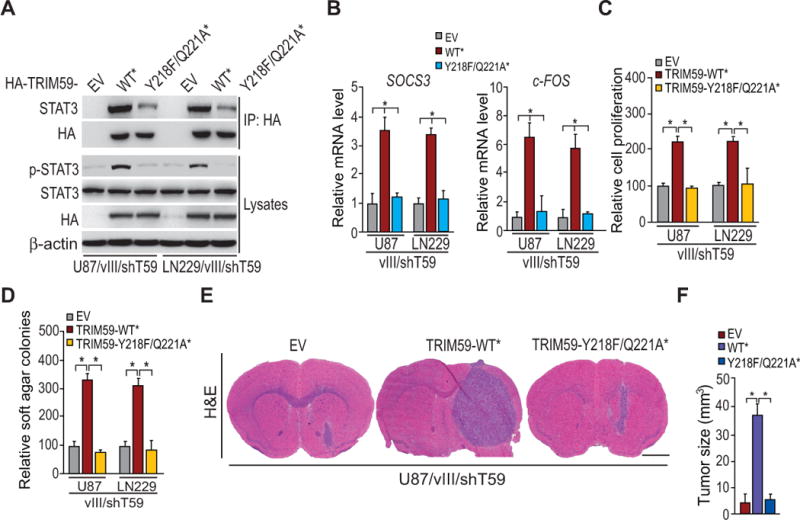
A, IP/WB analyses of the effect of TRIM59 Y218F/Q221A mutant on the association of TRIM59 with STAT3. shRNA-resistant TRIM59 WT and Y218F/Q221A mutant were re-expressed in U87/EGFRvIII/shTRIM59 and LN229/EGFRvIII/shTRIM59 GBM cells, respectively. B, Effects of re-expression of shRNA-resistant TRIM59 WT and Y218F/Q221A mutant on SOCS3 and c-FOS expression in A. C to E, Effects of re-expression of shRNA-resistant TRIM59WT and TRIM59Y218F/Q221A mutant on in vitro cell proliferation (C), cell colony formation (D), and tumor growth in the brain (E). Data in E, representative images of H&E analyses of brain sections. Scale bars: 1 mm Data. F, Quantification of tumor size in E. Data were from stained brain sections of 5 mice per group. Error bars±SD. *P < 0.05. Data and images are representative of two to three independent experiments.
To further reveal the roles of TRIM59Y218/Q221 in EGFR/EGFRvIII-driven glioma tumorigenesis, we re-expressed shRNA-resistant TRIM59WT* and TRIM59Y218F/Q221A* in U87/EGFRvIII/shT59 and LN229/EGFRvIII/shT59 cells. Compared to the controls, re-expressing TRIM59WT* rescued TRIM59 knockdown-suppressed cell proliferation and colony formation in soft agar stimulated by EGFRvIII, whereas re-expressing TRIM59Y218F/Q221A* did not (Fig. 5C and 5D). Moreover, re-expressing TRIM59WT* significantly restored TRIM59 depletion-inhibited tumor growth relative to the control xenografts, whereas re-expressing TRIM59Y218F/Q221A* showed no enhanced tumorigenic growth (Fig. 5E and 5F). Collectively, these findings suggest that Y218/Q221 sites are critical for TRIM59 functions in EGFR/EGFRvIII-driven tumorigenesis in gliomas.
TRIM59 promotes STAT3 activity by inhibiting STAT3-TC45 binding
Protein tyrosine phosphatase, TC45, can negatively regulate STAT3 activity by directly dephosphorylating STAT3 in the nucleus, thereby repressing oncogenic signaling (11, 12). We performed immunoprecipitation and Western blotting analysis to examine whether TRIM59 affects the dephosphorylation process of STAT3. As shown in Fig. 6A, TC45 could form a complex with STAT3 in both LN229/EGFRvIII and U87/EGFRvIII cells. However, we found that TRIM59 overexpression significantly suppressed the interaction of TC45 and STAT3, but increased STAT3 phosphorylation stimulated by EGFRvIII (Fig. 6A). This data suggests that TRIM59 may promote STAT3 activity through disruption of STAT3-TC45 interaction, which inhibits dephosphorylation of p-STAT3 in the nucleus.
Figure 6. TRIM59 promotes STAT3 activity through disruption of STAT3-TC45 interaction.
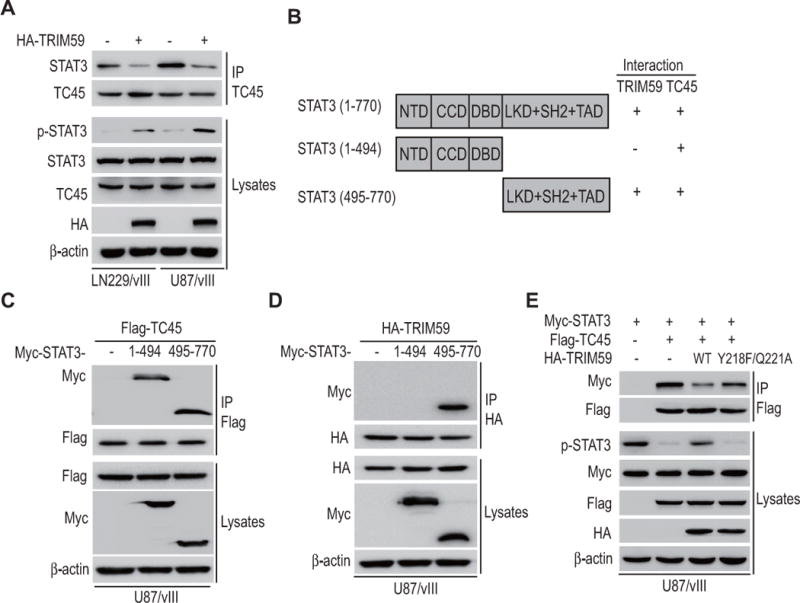
A, Effects of TRIM59 overexpression on TC45-STAT3 interaction and STAT3 phosphorylation in LN229/EGFRvIII and U87/EGFRvIII GBM cells. B, A schematic presentation of full-length STAT3 and its truncation mutants. C and D, Domain mapping of STAT3 demonstrates the domains that interact with TC45 (C) or TRIM59 (D). E, Effects of TRIM59Y218F/Q221A mutant on STAT3 phosphorylation and the interaction between TC45 and STAT3. Flag-TC45 and Myc-STAT3 with or without HA-TRIM59WT or HA-TRIM59Y218F/Q221A were separately cotransfected into U87/EGFRvIII cells. Data are representative of two or three independent experiments with similar results.
To validate this observation, we determined the domain of STAT3 binding with TRIM59 and TC45 in glioma cells. We generated an N-terminus and a C-terminus deleted mutants lacking various functional binding domains as indicated in Fig. 6B, and then separately transfected them into U87/EGFRvIII cells. As shown in Fig. 6C and 6D, it was revealed that both TRIM59 and TC45 interacted with the C-terminal fragment of STAT3, suggesting that TRIM59 may compete with TC45 to interact with STAT3.
To support that TRIM59 mediates STAT3 activity through disruption of STAT3-TC45 interaction, we co-expressed Myc-tagged STAT3 and Flag-tagged TC45 with or without HA-tagged TRIM59WT or TRIM59Y218F/Q221A into U87/EGFRvIII cells. Consistent with our findings, the level of p-STAT3 was markedly diminished in cells overexpressing TC45, a trend that was reversed by overexpressing TRIM59WT but not TRIM59Y218F/Q221A (Fig. 6E). Moreover, overexpressing TRIM59WT, but not TRIM59Y218F/Q221A, attenuated the interaction between TC45 and STAT3 (Fig. 6E). These data show that TRIM59 competes with TC45 to interact with STAT3, resulting in the disassociation of the STAT3-TC45 complex, preventing STAT3-inactivation by TC45 dephosphorylation, and stabilizing STAT3 transcriptional activation.
Co-expression of p-EGFRY1173, TRIM59 and p-STAT3Y705 correlates with a worse prognosis of GBM
Previous studies have shown that high expression of EGFR, EGFRvIII, and p-STAT3 are highly correlated with a poor prognosis for glioma patients (8, 37–40). To further assess the clinical relevance of our findings in this study, we examined expression of p-EGFRY1173, TRIM59, and p-STAT3Y705 in clinical glioma samples. Using antibodies with validated specificities against these proteins, we performed IHC analyses on serial sections of 107 GBM specimens (Fig. 7A). Co-expression of TRIM59 and p-STAT3Y705 was found in the majority of p-EGFR positive tumors (Fig. 7A). Spearman’s rank correlation analysis, based on quantification of the IHC staining, revealed statistically significant positive correlations between p-EGFRY1173, TRIM59, and p-STAT3Y705 (Fig. 7B). Moreover, Kaplan-Meier analyses of survival showed that co-expression levels of p-EGFR/TRIM59 and TRIM59/p-STAT3 were correlated with a significantly shorter survival in glioma patients (Fig. 7C). Taken together, these data support the role of EGFR/TRIM59/STAT3 signaling in the pathophysiology, clinical progression, and aggressiveness of human gliomas. These results also suggest that TRIM59, in conjunction with other clinical markers, could improve the assessment of clinical outcomes in GBM with EGFR activation.
Figure 7. Co-expression of p-EGFRY1173, TRIM59, and p-STAT3Y705 is correlated with a worse prognosis of gliomas.
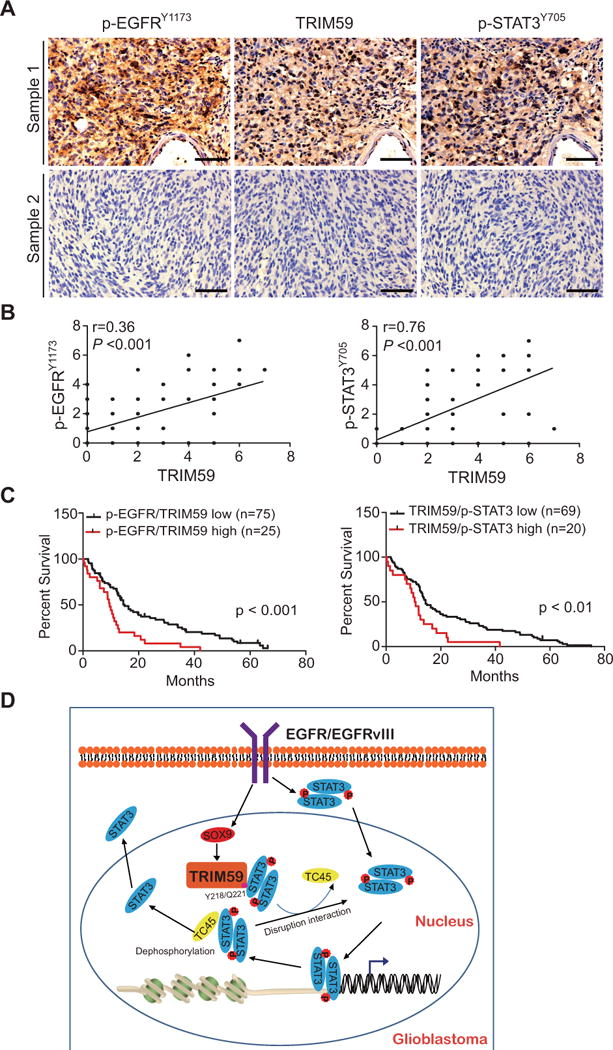
A, In total, 107 GBM specimens were analyzed by IHC. Representative images of two GBM specimens that were IHC stained positive or negative by the indicated antibodies. Scale bars: 50 μm. B, Correlation of expression between p-EGFRY1173 and TRIM59, or TRIM59 and p-STAT3Y705. C, Kaplan-Meier analyses of patients with high p-EGFRY1173/high TRIM59-expressing tumors (red line) versus low EGFRY1173/low TRIM59-expressing tumors (black line), or high TRIM59/high p-STAT3Y705-expressing tumors (red line) versus low TRIM59/low p-STAT3Y705-expressing tumors (black line) based on IHC analyses in A. Median survival (in months) in left: low, 14.90; high, 9.53. Median survival (in months) in right: low, 14.51; high, 10.13. P values were calculated by the log-rank test. Black tick markers indicate censored data. D, A working model of EGFR/TRIM59/STAT3 signaling pathway in glioma tumorigenesis.
Discussion
Multiple cancers have frequent aberrant activation of EGFR/STAT3 signaling, including GBM, and cancers of the lung, breast, head and neck, and bladder, resulting in enhanced tumorigenesis, invasion, metastasis, and immunosuppression in these malignant tumors (8, 12, 41–44). Oncogenic EGFR/STAT3 signaling is activated by gene amplification, overexpression, or activating mutations through several different mechanisms, including direct activation of STAT3 by EGFR binding (8, 45) and indirect activation of STAT3 through Src- or JAK (46, 47). In addition, EGFR/STAT3 signaling is activated by IL-6 (41). Recently, EGFR-induced STAT3 phosphorylation in breast cancer was found to be dependent on suppressing TC45 dephosphorylation of nuclear STAT3 (12). In this study, we describe a novel function of TRIM59, acting as a new downstream effector of EGFR signaling to mediate EGFR/STAT3-driven tumorigenesis. We demonstrate that TRIM59 expression is associated with glioma progression and a poor survival of glioma patients. TRIM59 is transcriptionally regulated by EGFR/EGFRvIII through SOX9. Moreover, EGFR/EGFRvIII promotes TRIM59 interaction with STAT3 in nucleus. The association of TRIM59 with STAT3 suppresses TC45-mediated dephosphorylation of STAT3 in the nucleus, thereby promoting STAT3 activation and glioma tumorigenesis in vitro and in vivo (Fig. 7D). Taken together, our study provides clinical and mechanistic evidence demonstrating that TRIM59 upregulation is critical for EGFR/STAT3-driven tumorigenesis in human cancers.
TRIM59 was initially identified to have proto-oncogenic function in SV40 Tag and Ras signaling pathways in mouse prostate cancer models (15). However, the mechanisms that mediate TRIM59 expression and/or activation remain poorly characterized (15). Recently, TRIM59 was found to be upregulated, and its expression was correlated with gastric cancer progression and patient survival. TRIM59 promoted gastric tumorigenesis through enhancing ubiquitination and degradation of p53 (16). In lung cancers, TRIM59 is also highly expressed and promotes cell proliferation and migration. However, TRIM59 regulates expression of cell cycle proteins CDC25C and CDK1 but not the p53 signaling pathway in lung cancer cells (18). Moreover, TRIM59 acts as an adaptor protein to interact with ECSIT in innate immune response (20). In the present study, we described a distinct mechanism by which TRIM59 mediates EGFR/EGFRvIII/STAT3-driven tumorigenesis. Association of TRIM59 with nuclear STAT3 is enhanced by EGFR/EGFRvIII, and the Y218/Q221 sites of TRIM59 are required for their binding. TRIM59 does not regulate STAT3 expression but attenuates TC45-STAT3 interaction, preventing STAT3 dephosphorylation by TC45 in the nucleus, thereby maintaining STAT3 signaling and enhanced tumorigenesis. Our data also demonstrate that TRIM59 is important for proliferative or classical and mesenchymal subtype gliomas. Although EGFR amplification and the EGFRvIII mutation occurs with greater frequency in the GBM classical subtype, we found that TRIM59 is important for tumorigenicity of patient-derived mesenchymal subtype glioma stem cells (GSCs) with EGFRvIII overexpression. Moreover, TRIM59 expression is positively correlated with SOX9, and SOX9 is co-expressed with EGFR in mesenchymal and classical subtype gliomas in the Chinese Glioma Genome Atlas (CGGA) RNA-Seq datasets. Together, this investigation identifies a previously unrecognized mechanism, in which TRIM59 functions as a signaling relay in mediating EGFR/EGFRvIII stimulation of oncogenic STAT3 signaling, thereby promoting tumorigenesis in human gliomas. Additionally, our results and those of the aforementioned studies (15, 16, 18) also provide substantial evidence demonstrating the context-dependent role of TRIM59 in modulating distinct oncogenic pathways in multiple cancers.
STAT3 activity is regulated by positive activators, such as receptor tyrosine kinases (RTKs) (8), interleukin-6 (IL-6) family cytokines, G-protein-coupled receptors (GPCRs), and Toll-like receptors (TLRs) (48). Aberrant activation of STAT3 is inhibited by negative regulators, including TC45 (11–13), PIAS3 (49), and SHP-1 (50). EGFR/EGFRvIII can activate STAT3 directly or through positive regulators (8, 46, 47). However, the mechanism by which EGFR/EGFRvIII overcomes these endogenous STAT3 inhibitory factors remains unclear. Recently, it was shown that EGF-stimulated STAT3 activation can be suppressed by overexpression of Gdx/UBL4A in breast cancer cells, which specifically bridges and stabilizes TC45-STAT3 binding, and promotes dephosphorylation of STAT3, leading to the suppression of tumorigenesis (12). Here, we demonstrate that knockdown of TRIM59 inhibited STAT3 activation stimulated by EGFR/EGFRvIII, whereas re-expression of shRNA resistant TRIM59 restored STAT3 activation. Furthermore, TRIM59 interacted with STAT3 in the nucleus. Overexpression of TRIM59 inhibited TC45-STAT3 association and promoted STAT3 activation. Further investigation indicated that EGFR/EGFRvIII-upregulated TRIM59 could disrupt the interaction between TC45 and STAT3 by competing with TC45 to associate with STAT3. Our results identify a novel mechanism of EGFR-activated STAT3, in which TRIM59 maintains oncogenic signaling by inhibiting STAT3-TC45 complex formation to keep STAT3 phosphorylation and activation.
In conclusion, our findings reveal a previously unknown signal relay by which TRIM59 mediates EGFR stimulation of STAT3 by inhibiting TC45 dephosphorylation of STAT3 in the nucleus, thereby enhancing the oncogenic activity of the EGFR/STAT3 signaling pathway in human gliomas. The newly established roles of TRIM59 in EGFR-driven tumorigenesis provide a rationale for TRIM59 as a novel prognostic marker for glioma patients and a potential target for further therapeutic investigation.
Supplementary Material
Acknowledgments
Grant Support: We thank Ichiro Nakano for providing patient-derived glioma stem cells. This work was supported in part by National Natural Science Foundation of China (No. 81372704, 81572467 to H. Feng; No.81470315, 81772663 to Y. Li); the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (No. 2014024), Shanghai Municipal Education Commission—Gaofeng Clinical Medicine Grant Support (No. 20161310), New Hundred Talent Program (Outstanding Academic Leader) at Shanghai Municipal Health Bureau (2017BR021), Technology Transfer Project of Science & Technology Dept. at Shanghai Jiao Tong University School of Medicine (ZT201701) to H. Feng; Shanghai Jiao Tong University Medical Engineering Cross Fund (No. YG2017MS32) to Y. Li; US NIH grants (NS093843, NS95634 and CA209345) to S.-Y. Cheng; NIH/NCI training grant T32 CA070085 and NIH LRP award (L32 MD010147) to A.A.A.; JSPS KAKENHI Grant Number 24112006, 15H04690 to S. Hatakeyama.
Footnotes
Conflict of interest
All authors declare no conflict of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat reviews cancer. 2015;15:302–10. doi: 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature reviews cancer. 2010;10:319–31. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- 5.Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clinical cancer research. 2005;11:1462–6. doi: 10.1158/1078-0432.CCR-04-1737. [DOI] [PubMed] [Google Scholar]
- 6.Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. The Journal of clinical investigation. 2017;127:415–26. doi: 10.1172/JCI89587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer cell. 2013;23:839–52. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer cell. 2013;24:438–49. doi: 10.1016/j.ccr.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi He K, Chan Q, Xiao CB, Liu G, Tucker-Burden XC, et al. Blockade of glioma proliferation through allosteric inhibition of JAK2. Science signaling. 2013;6:ra55. doi: 10.1126/scisignal.2003900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nature reviews molecular cell biology. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto T, Sekine Y, Kashima K, Kubota A, Sato N, Aoki N, et al. The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochemical and biophysical research communications. 2002;297:811–7. doi: 10.1016/s0006-291x(02)02291-x. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Ning H, Ren F, Zhang Y, Rong Y, Wang Y, et al. GdX/UBL4A specifically stabilizes the TC45/STAT3 association and promotes dephosphorylation of STAT3 to repress tumorigenesis. Molecular cell. 2014;53:752–65. doi: 10.1016/j.molcel.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Current biology: CB. 2002;12:446–53. doi: 10.1016/s0960-9822(02)00697-8. [DOI] [PubMed] [Google Scholar]
- 14.Hatakeyama S. TRIM proteins and cancer. Nature reviews Cancer. 2011;11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- 15.Valiyeva F, Jiang F, Elmaadawi A, Moussa M, Yee SP, Raptis L, et al. Characterization of the oncogenic activity of the novel TRIM59 gene in mouse cancer models. Molecular cancer therapeutics. 2011;10:1229–40. doi: 10.1158/1535-7163.MCT-11-0077. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Z, Ji Z, Wang Y, Li J, Cao H, Zhu HH, et al. TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology. 2014;147:1043–54. doi: 10.1053/j.gastro.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 17.Liang J, Xing D, Li Z, Shen J, Zhao H, Li S. TRIM59 is upregulated and promotes cell proliferation and migration in human osteosarcoma. Molecular medicine reports. 2016;13:5200–6. doi: 10.3892/mmr.2016.5183. [DOI] [PubMed] [Google Scholar]
- 18.Zhan W, Han T, Zhang C, Xie C, Gan M, Deng K, et al. TRIM59 Promotes the Proliferation and Migration of Non-Small Cell Lung Cancer Cells by Upregulating Cell Cycle Related Proteins. PloS one. 2015;10:e0142596. doi: 10.1371/journal.pone.0142596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aierken G, Seyiti A, Alifu M, Kuerban G. Knockdown of Tripartite-59 (TRIM59) Inhibits Cellular Proliferation and Migration in Human Cervical Cancer Cells. Oncology research. 2017;25:381–8. doi: 10.3727/096504016X14741511303522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondo T, Watanabe M, Hatakeyama S. TRIM59 interacts with ECSIT and negatively regulates NF-kappaB and IRF-3/7-mediated signal pathways. Biochemical and biophysical research communications. 2012;422:501–7. doi: 10.1016/j.bbrc.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 21.Allen M, Bjerke M, Edlund H, Nelander S, Westermark B. Origin of the U87MG glioma cell line: Good news and bad news. Science translational medicine. 2016;8:354re3. doi: 10.1126/scitranslmed.aaf6853. [DOI] [PubMed] [Google Scholar]
- 22.Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8644–9. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Zhang W, Li Y, Alvarez A, Li Z, Wang Y, et al. SHP-2-upregulated ZEB1 is important for PDGFRalpha-driven glioma epithelial-mesenchymal transition and invasion in mice and humans. Oncogene. 2016;35:5641–52. doi: 10.1038/onc.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dasgupta M, Unal H, Willard B, Yang J, Karnik SS, Stark GR. Critical role for lysine 685 in gene expression mediated by transcription factor unphosphorylated STAT3. The Journal of biological chemistry. 2014;289:30763–71. doi: 10.1074/jbc.M114.603894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng H, Lopez GY, Kim CK, Alvarez A, Duncan CG, Nishikawa R, et al. EGFR phosphorylation of DCBLD2 recruits TRAF6 and stimulates AKT-promoted tumorigenesis. The Journal of clinical investigation. 2014;124:3741–56. doi: 10.1172/JCI73093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. Rna. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faustino-Rocha A, Oliveira PA, Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG, et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab animal. 2013;42:217–24. doi: 10.1038/laban.254. [DOI] [PubMed] [Google Scholar]
- 29.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 30.Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. The Journal of biological chemistry. 1997;272:2927–35. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- 31.Liu F, Hon GC, Villa GR, Turner KM, Ikegami S, Yang H, et al. EGFR Mutation Promotes Glioblastoma through Epigenome and Transcription Factor Network Remodeling. Molecular cell. 2015;60:307–18. doi: 10.1016/j.molcel.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Azare J, Leslie K, Al-Ahmadie H, Gerald W, Weinreb PH, Violette SM, et al. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Molecular and cellular biology. 2007;27:4444–53. doi: 10.1128/MCB.02404-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang T, Alvarez AA, Pangeni RP, C MH, Lu S, Kim SH, et al. A regulatory circuit of miR-125b/miR-20b and Wnt signalling controls glioblastoma phenotypes through FZD6-modulated pathways. Nature communications. 2016;7:12885. doi: 10.1038/ncomms12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda O, Miyasaka Y, Sekine Y, Mizushima A, Muromoto R, Nanbo A, et al. STAP-2 is phosphorylated at tyrosine-250 by Brk and modulates Brk-mediated STAT3 activation. Biochemical and biophysical research communications. 2009;384:71–5. doi: 10.1016/j.bbrc.2009.04.076. [DOI] [PubMed] [Google Scholar]
- 35.Abe K, Hirai M, Mizuno K, Higashi N, Sekimoto T, Miki T, et al. The YXXQ motif in gp 130 is crucial for STAT3 phosphorylation at Ser727 through an H7-sensitive kinase pathway. Oncogene. 2001;20:3464–74. doi: 10.1038/sj.onc.1204461. [DOI] [PubMed] [Google Scholar]
- 36.Minoguchi M, Minoguchi S, Aki D, Joo A, Yamamoto T, Yumioka T, et al. STAP-2/BKS, an adaptor/docking protein, modulates STAT3 activation in acute-phase response through its YXXQ motif. The Journal of biological chemistry. 2003;278:11182–9. doi: 10.1074/jbc.M211230200. [DOI] [PubMed] [Google Scholar]
- 37.Chua CY, Liu Y, Granberg KJ, Hu L, Haapasalo H, Annala MJ, et al. IGFBP2 potentiates nuclear EGFR-STAT3 signaling. Oncogene. 2016;35:738–47. doi: 10.1038/onc.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeom SY, Nam DH, Park C. RRAD promotes EGFR-mediated STAT3 activation and induces temozolomide resistance of malignant glioblastoma. Molecular cancer therapeutics. 2014;13:3049–61. doi: 10.1158/1535-7163.MCT-14-0244. [DOI] [PubMed] [Google Scholar]
- 39.Zheng Q, Han L, Dong Y, Tian J, Huang W, Liu Z, et al. JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII-expressing glioblastoma. Neuro-oncology. 2014;16:1229–43. doi: 10.1093/neuonc/nou046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clinical cancer research. 2008;14:6042–54. doi: 10.1158/1078-0432.CCR-07-4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. The Journal of clinical investigation. 2007;117:3846–56. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaib I, Karachaliou N, Pilotto S, Codony Servat J, Cai X, Li X, et al. Co-activation of STAT3 and YES-Associated Protein 1 (YAP1) Pathway in EGFR-Mutant NSCLC. Journal of the National Cancer Institute. 2017;109 doi: 10.1093/jnci/djx014. djx014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vlaicu P, Mertins P, Mayr T, Widschwendter P, Ataseven B, Hogel B, et al. Monocytes/macrophages support mammary tumor invasivity by co-secreting lineage-specific EGFR ligands and a STAT3 activator. BMC cancer. 2013;13:197. doi: 10.1186/1471-2407-13-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sen M, Joyce S, Panahandeh M, Li C, Thomas SM, Maxwell J, et al. Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clinical cancer research. 2012;18:4986–96. doi: 10.1158/1078-0432.CCR-12-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson H, Del Rosario AM, Bryson BD, Schroeder MA, Sarkaria JN, White FM. Molecular characterization of EGFR and EGFRvIII signaling networks in human glioblastoma tumor xenografts. Molecular & cellular proteomics: MCP. 2012;11:1724–40. doi: 10.1074/mcp.M112.019984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quesnelle KM, Boehm AL, Grandis JR. STAT-mediated EGFR signaling in cancer. Journal of cellular biochemistry. 2007;102:311–9. doi: 10.1002/jcb.21475. [DOI] [PubMed] [Google Scholar]
- 47.Gao SP, Chang Q, Mao N, Daly LA, Vogel R, Chan T, et al. JAK2 inhibition sensitizes resistant EGFR-mutant lung adenocarcinoma to tyrosine kinase inhibitors. Science signaling. 2016;9:ra33. doi: 10.1126/scisignal.aac8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nature reviews cancer. 2014;14:736–46. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 49.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–5. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 50.Han Y, Amin HM, Franko B, Frantz C, Shi X, Lai R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood. 2006;108:2796–803. doi: 10.1182/blood-2006-04-017434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.