

Abstract
Arsenic disulfide, a major effective component of realgar, has been investigated for its anti-cancer potential and shown to have therapeutic efficacies in hematological and some solid tumors. However, its effect against breast cancer is rarely reported. In this study, we investigated the anti-cancer effects of As2S2 in human breast cancer cell lines MCF-7 and MDA-MB-231, and further elucidated its underlying mechanisms. As2S2 significantly inhibited cell viabilities, induced apoptosis, and led to cell cycle arrest in both cell lines with a dose- and time-dependent manner. As2S2 upregulated pro-apoptotic proteins like p53 and PARP in MCF-7 cells. Besides, As2S2 downregulated anti-apoptotic proteins like Bcl-2 and Mcl-1, as well as cell cycle-related proteins cyclin A2 and cyclin D1 in both cell lines. Of note, the expression level of cyclin B1 was downregulated in MCF-7 cells, whereas, upregulated in MDA-MB-231 cells. Moreover, As2S2 significantly inhibited the pro-survival signals in PI3K/Akt pathway in both cell lines. In conclusion, As2S2 inhibited cell viabilities, induced apoptosis and cell cycle arrest in both MCF-7 and MDA-MB-231 cell lines by regulating the expression of key proteins involved in related pathways. These results provide fundamental insights into the clinical application of As2S2 for treatment of patients with breast cancer.
Keywords: Arsenic disulfide, MCF-7, MDA-MB-231, cell viability, apoptosis, cell cycle
Introduction
Breast cancer is the most common malignancy among women, with an estimated 400,000 cancer death annually throughout the world [1,2]. Current conventional therapies, including chemotherapy, surgery and radiation treatment, are insufficient to overcome drug side effects and drug resistance [3,4]. Herein, the development of new therapeutic agents with effectiveness and mild side effects is critically needed for the treatment of breast cancer.
Arsenic compounds, the component of traditional Chinese medicine, have been used widely and successfully for the treatment of various diseases in China for thousands of years and also in the western world [5-7]. In particular, arsenic trioxide, one of the trivalent arsenics, has been approved as a standard agent for the treatment of relapsed and refractory acute promyelocytic leukemia (APL) [8,9] for its significant reputation of therapeutic effects. However, adverse side effects including liver dysfunction, acute toxicity, and potential carcinogenicity limit its wide clinical applications [10,11]. Compared with arsenic trioxide, arsenic disulfide (As2S2), as a less toxic arsenic compound, possesses the advantages of oral administration safety and abundant resources [12]. It has been demonstrated that As2S2 has a similar anti-tumor potential but with less adverse effects than other arsenic compounds [13]. Emerging evidence has revealed the antitumor effects of As2S2 in various malignancies, which included hematopoietic tumors and solid malignancies [14-18]. Evidence from animal experiments further confirmed the safety and efficacy of As2S2 administrated orally in normal mice or xenograft mice cancer model [19-22]. More importantly, many clinical studies demonstrated the safety and effectiveness of the clinical application of As2S2 especially in respect to the oral administration of As2S2 [23-25] to patients with APL. Nevertheless, a limited number of studies have reported the potential antitumor activity of As2S2 in human breast cancer [26]. In addition, there is even more rare study investigating the underlying mechanisms of the effects of As2S2 against breast carcinoma.
Apoptosis induction and cell cycle arrest are the two important causes to inhibit cancer cell growth and proliferation [27]. Apoptosis is a genetically programmed cell death, which has been taken as a major mechanism of chemotherapy-induced cell death [28,29]. There are two main established pathways that lead to apoptosis: the extrinsic (cell death receptor) pathway and the intrinsic (mitochondrial) pathway [29,30]. The intrinsic pathway is mainly involved in the apoptosis induced by small molecule anti-cancer drugs in cancer cells [31]. Recent studies have shown that As2S2 could induce apoptosis in hematopoietic and some solid tumor cell lines [32-34]. However, the pro-apoptotic effect of As2S2 in breast cancer cells has been rarely reported. Cell cycle is a process characterized by a series of organized events, which involves a set of sequential phases (G1, S, G2 and M phases) to promote cell proliferation. Previous studies suggest that the development of cancer can be considered as a dysregulation of cell cycle [35], with the dysfunctions implicated in cell cycle checkpoints in most human tumors [36]. Previous studies reported that As2S2 can inhibit cell proliferation by inducing certain cell-cycle phase arrest in some human solid tumor cell lines, such as osteosarcoma [18], hepatocellular carcinoma HepG2 [17], and melanoma [37] cells. Thus, we assumed that As2S2 might have effects in regulating cell cycle phases in breast cancer cells.
The present study aimed to investigate the effects of As2S2 on different breast cancer cell lines by focusing on apoptosis induction and cell cycle arrest, with various drug concentrations and different incubation time courses. Our results demonstrated that As2S2 exerted an inhibitory effect on cell viabilities of breast cancer cells, in both dose- and time-dependent manners, along with apoptosis induction and cell cycle arrest. Moreover, the results indicated the remarkable effect of As2S2 in regulating cell survival-, apoptosis-, and cell cycle-related proteins.
Materials and methods
Reagents
Cell counting kit-8 (CCK-8) was purchased from DOJINDO Laboratories (Tokyo, Japan). Calcein-AM was purchased from Molecular Probes (Eugene, Oregon, USA). FITC Annexin V Apoptosis Detection Kit was obtained from BD Biosciences (San Diego, CA, USA). Arsenic disulfide, PI and RNase A solution were purchased from Sigma (St. Louis, MO, USA). ECLTM Western Blotting Analysis System and ECLTM Prime Western Blotting Detection Reagent were purchased from GE Healthcare (Buckinghamshire, UK). Pro-Survival B-cell lymphoma 2 (Bcl-2) Family Antibody Sampler Kit, rabbit anti-human p53, rabbit-anti human poly (ADP-ribose) polymerase (PARP), rabbit anti-human phosphatidylinositol 3-kinase (PI3K), rabbit anti-human Akt, mouse anti-human cyclin A2, mouse anti-human cyclin B1, and rabbit anti-human cyclin D1 were obtained from Cell Signaling (Danvers, MA, USA).
Cell line and cell culture
The human breast cancer cell lines MCF-7 and MDA-MB-231 were purchased from the American Type Culture Collection (Manassas, VA, USA). MCF-7 cells and MDA-MB-231 cells were cultured in MEM-alpha medium (Gibco, Grand Island, NY, USA) supplemented with penicillin, streptomycin, fetal bovine serum (FBS) (10% FBS for MCF-7 and 15% FBS for MDA-MB-231, respectively) (Sigma, St. Louis, MO, USA), and maintained as attached cells at 37°C in 5% carbon dioxide in a humidified atmosphere.
Cell culture assays and drug treatment
Both MCF-7 and MDA-MB-231 cells were seeded at a density of 10,000 cells per well in 500 μl of cell culture media into 48-well plates (IWAKI micro-plates), followed by overnight incubation. Reagents, including vehicle as controls (cell culture media) and As2S2 stock solutions (0.6 mM), were subsequently added into the corresponding wells to adjust the final drug concentrations of As2S2 to be 0-24 μM. Both MCF-7 and MDA-MB-231 cells were allowed to grow for 24, 48 and 72 h in the presence of vehicle or As2S2, respectively, followed by cytotoxicity assay.
Cytotoxicity assay
Cell cytotoxicity was analyzed by CCK-8 assay. For both cell lines, 1 × 104 cells per well were seeded into 48-well plates (IWAKI micro-plates), and As2S2 was subsequently added into the corresponding wells to adjust the final drug concentrations of 0-24 μM, respectively. Then, the plates were incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2 for 24, 48 and 72 h, respectively. After incubation, 25 µl CCK-8 reagent was added into each well, followed by additional incubation for 3 h at 37°C. The OD value of each well was measured by a micro-plate reader (Corona MT P-32; Corona Co., Hitachi, Ibaraki, Japan) at 570 nm. The cell viability rate was calculated according to the following formula:
Cell viability rate = (OD sample value-OD blank value)/(OD control value-OD blank value) × 100%.
Microscopy images
MCF-7 and MDA-MB-231 cells were seeded in a 96-well plate at the density of 5 × 103 cells per well in 100 µl culture medium, followed by the exposure to different concentrations of As2S2 (0, 4, 8, 12, and 16 µM), for 24, 48 and 72 h, respectively. Then the cells were stained for 15 min in the dark at 37°C with the specific live probe Calcein-AM, followed by images measured and analyzed by a fluorescence micro-plate reader and Harmony software (Operetta CLS, PerkinElmer, Japan).
Assessment of apoptosis
MCF-7 and MDA-MB-231 cells with the density of 4 × 105 cells per well were seeded in 6-well plates (IWAKI micro-plates) respectively, and treated with serial concentrations of As2S2 (0, 4, 8, 12 and 16 µM), and followed by an additional incubation for 24, 48 and 72 h, respectively. The apoptotic rates of both cell lines were detected by using FITC Annexin V Apoptosis Detection Kit. The staining procedure was performed according to the manufacturer’s instructions. Approximate 1 × 104 cells were analyzed using a flow cytometer (BD Biosciences, CA, US). The cells were subsequently assessed for total apoptotic cells composed by early apoptotic (Annexin V+/PI-) and late apoptotic (Annexin V+/PI+) cells.
Cell cycle analysis
Both MCF-7 and MDA-MB-231 cells were seeded at a density of 4 × 105 cells per well in 6-well plates (IWAKI micro-plates), followed by overnight incubation. Cells were treated with 0, 4, 8, 12 and 16 μM As2S2 for 24, 48 and 72 h, respectively. Then, the cells were harvested and washed with PBS, subsequently, fixed in 70% ethanol overnight at -20°C, and stained with PI and RNase A solution (5 μg/ml PI, and 0.5 μg/μl RNase A). The DNA content was determined by flow cytometry (BD Biosciences, CA, US), and data were analyzed by CellQuest analysis software. ModFit LTTM Ver.3.0 (Verity Software House, Topsham, ME, USA) was used to calculate the number of cells at each G0/G1, S and G2/M phase fraction.
Western blot analysis
Western blot was performed in order to evaluate the protein levels of Bcl-2, Mcl-1, p53, PARP, PI3K, Akt, cyclin A2, cyclin B1, and cyclin D1 in both MCF-7 and MDA-MB-231 cells. The total protein was extracted from both cell lines treated by As2S2 with various concentrations (0, 4, 8 and 16 µM) in different time courses (24, 48 and 72 h) respectively. Briefly, cell lysates were separated by SDS-PAGE and transferred into a polyvinylidene difluoride transfer membrane (Immobilon-P, Darmstadt, Germany). Membranes were blocked with 5% milk for 1 h. The membranes were washed by Tris buffered saline with Tween (TBST) and then incubated overnight at 4°C with anti-rabbit p53 specific antibody (1:1000, Cell Signaling, #9282), anti-rabbit PARP specific antibody (1:1000, Cell Signaling, #9542), anti-rabbit Bcl-2 specific antibody (1:1000, Cell Signaling, #4223), anti-rabbit Mcl-1 specific antibody (1:1000, Cell Signaling, #5453), anti-mouse cyclin A2 specific antibody (1:1000, Cell Signaling, #4656), anti-mouse cyclin B1 specific antibody (1:1000, Cell Signaling, #4135), anti-rabbit cyclin D1 specific antibody (1:1000, Cell Signaling, #2922), anti-rabbit PI3K p85 specific antibody (1:1000, Cell Signaling, #4228), and anti-rabbit Akt specific antibody (1:1000, Cell Signaling, #4691), respectively. Membranes were also probed with anti-beta-actin (abcam, ab49900) at 1:3000 dilutions as the internal control. The membranes were incubated with respective primary antibodies listed above at 4°C overnight. Then, the membranes were incubated with anti-mouse or anti-rabbit specific polyclonal secondary antibodies for 1 h at room temperature, and followed by three-time washes with TBST. Signals were detected using ECL Western Blot detection kit in a luminescent image analyzer (Fujifilm, LAS-3000, Tokyo, Japan).
Statistical analysis
Data processing was carried out using software GraphPad Prism version 6.0 and results were presented as means ± SEM of at least three independent experiments. Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test for multiple comparisons, and the Student’s t-test for comparison of two groups. P < 0.05 was considered to be statistically significant.
Results
As2S2 inhibits cell proliferation of breast cancer cells
To investigate the cytotoxicity of As2S2 against breast cancer cells, MCF-7 and MDA-MB-231 cells were exposed to serial concentrations of As2S2 from 0 to 24 μM for 24, 48 and 72 h, and the cell viability was evaluated by CCK-8 assay. As shown in Figure 1, As2S2 inhibited the cell proliferation of breast cancer cell lines MCF-7 and MDA-MB-231 both in dose- and time-dependent manner.
Figure 1.

Effects of As2S2 on the viability of breast cancer cells. (A) MCF-7 and (B) MDA-MB-231 cells were treated with various concentrations of As2S2 (0, 4, 8, 12, 16, 20 and 24 µM) for 24 h (●), 48 h (□) and 72 h (▲), respectively, and the cell viability was assessed by CCK-8 assay procedures. All of the data were expressed as the mean ± SEM (n ≥ 3). Asterisks indicate significant differences between the control and the drug treatment groups (*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001).
In MCF-7 cells, as shown in Figure 1A, a significant decrease in cell viability was observed in a dose- and time-dependent manner after treatment with different concentrations of As2S2. In detail, compared to the control group (0 µM As2S2), the cell viability significantly reduced to 84.95±3.81 (P = 0.3837), 62.93±2.17 (P = 0.0009) and 50.80±4.22% (P < 0.0001) after exposure to 4 µM As2S2 for 24, 48 and 72 h, respectively. Exposure of cells to 24 µM As2S2 for 24, 48 and 72 h significantly reduced the cell viability to 36.31±3.26 (P = 0.0001), 26.38±3.78 (P < 0.0001), and 14.68±1.27% (P < 0.0001), respectively.
In MDA-MB-231 cells, as shown in Figure 1B, a significant decrease in cell viability was also observed in a dose- and time-dependent manner after treatment with different concentrations of As2S2. In detail, compared to the control group (0 µM As2S2), cell viability significantly reduced to 73.57±4.17 (P = 0.1819), 70.49±6.80 (P = 0.0102), and 62.42±0.30% (P < 0.0001) after exposure to 4 µM As2S2 for 24, 48 and 72 h, respectively. Exposure to 24 µM As2S2 for 24, 48 and 72 h further reduced cell viability to 48.03±2.64 (P = 0.0019), 21.15±1.52 (P < 0.0001) and 8.49±0.25% (P < 0.0001).
The half-maximal inhibitory concentrations (IC50 values) of As2S2 on MCF-7 and MDA-MB-231 cells in different time courses were listed in Table 1. The mean of IC50 values of As2S2 in MDA-MB-231 cells were relatively higher than that in MCF-7 cells when treated with As2S2 for 24 and 48 h. A significant difference was further observed in the IC50 values between two cell lines after exposure to As2S2 for 72 h (P = 0.03).
Table 1.
IC50 values of As2S2 in human breast cancer cell lines exposed for different time
| IC50 (µM as As2S2) | |||
|---|---|---|---|
|
| |||
| 24 h | 48 h | 72 h | |
| MCF-7 | 15.27±0.49 | 8.13±1.11## | 3.69±0.47##,& |
| MDA-MB-231 | 25.5±4.00 | 9.18±1.78## | 5.37±0.12##,$ |
Data are the means ± SEM from at least three independent experiments.
P < 0.01 vs. 24 h.
P < 0.05 vs. 48 h.
P < 0.05 vs. MCF-7 with the same exposure time.
The results indicated that As2S2 inhibits cell proliferation of breast cancer cells in dose- and time-dependent manners, and the MCF-7 cells were relatively more sensitive to As2S2 in comparison with MDA-MB-231 cells.
As2S2 affects cell morphology of breast cancer cells
To better understand the cell growth inhibition induced by As2S2 in breast cancer cells, the morphological features of MCF-7 and MDA-MB-231 cells were observed following As2S2 exposure in different time courses. The fluorescent images of treated and untreated cells were examined after staining the cells with a fluorescent dye Calcein-AM, which restricts to label living cells with green fluorescent [38,39]. Consistent with cell viability assays (Figure 1), a similar dose- and time-dependent decrease in the cell density was observed in both cell lines (Figure 2).
Figure 2.

Assessment of cell viability by calcein-AM staining. MCF-7 and MDA-MB-231 cells were seeded at the density of 5,000 cells per well. Cells were treated with a serial concentrations of As2S2 (0, 4, 8, 12 and 16 µM) for 24, 48 and 72 h, respectively. Viable cells exposed to calcein-AM showed bright green fluorescence. Images were taken and analyzed by a fluorescence Micro-plate reader (Operetta CLS, PerkinElmer, Japan) with 10 × objective (original magnifications 100 ×).
As2S2 triggers cell cycle arrest in breast cancer cells
To investigate the correlation of cell growth inhibition and cell cycle arrest, MCF-7 and MDA-MB-231 cells were treated with serial concentrations of As2S2 (0, 4, 8, 12 and 16 µM) for 24, 48 and 72 h, respectively, and the cell cycle distribution was analyzed by flow cytometry.
In MCF-7 cells, treatment with As2S2 mainly induced cell cycle arrest in the G0/G1 and G2/M phases.
As shown in Figure 3A, 3B, in MCF-7 cells after 24 h of As2S2 treatment, the percentage of cells at G2/M phase significantly increased with As2S2 at concentrations of 12 µM (P = 0.0021) and 16 µM (P = 0.0110). Whereas, the percentage of MCF-7 cells at S phase significantly decreased after exposure to As2S2 at concentrations of 12 µM (P = 0.0078) and 16 µM (P = 0.0125) for 24 h.
Figure 3.

As2S2 triggers cell cycle arrest in MCF-7 cells. (A) MCF-7 cells were treated with various concentrations of As2S2 (0, 4, 8, 12 and 16 µM) for 24, 48 and 72 h. The peaks in the figure represent the G0/G1, S, and G2/M phases in the cell cycle, respectively. (B) The percentages of cell numbers in the cell cycle of MCF-7 cells after 24 h, (C) 48 h, and (D) 72 h of drug treatment. All data were expressed as the mean ± SEM (n ≥ 3). Asterisks indicate significant differences between the control (0 µM) and the drug treatment groups (*P < 0.05, **P < 0.01).
As shown in Figure 3A, 3C, in MCF-7 cells after 48 h of As2S2 treatment, the percentage of cells at G0/G1 phase significantly increased with As2S2 at concentrations of 4 µM (P < 0.0001) and 8 µM (P < 0.0001). The percentage of MCF-7 cells at G2/M phase significantly increased 48 h after treatment with As2S2 at concentrations of 8 µM (P < 0.0001), 12 µM (P < 0.0001) and 16 µM (P < 0.0001), respectively. In contrast, the percentage of MCF-7 cells at S phase significantly decreased after exposure to As2S2 for 48 h at concentrations ranging from 4 to 16 µM (P < 0.0001 respectively).
As shown in Figure 3A, 3D, in MCF-7 cells after 72 h of As2S2 treatment, the percentage of cells at G0/G1 phase significantly increased with As2S2 at concentrations of 4 and 8 µM, but significantly decreased at concentrations of 12 and 16 µM (P < 0.0001, respectively). The percentage of MCF-7 cells at G2/M phase significantly increased with As2S2 at concentrations of 8 µM (P = 0.0008), 12 µM (P < 0.0001) and 16 µM (P < 0.0001) for 72 h. In contrast, the percentage of MCF-7 cells at S phase significantly decreased after exposure to As2S2 for 72 h at concentrations of 4 µM (P < 0.0001), 8 µM (P < 0.0001) and 12 µM (P = 0.0006) for 72 h, whereas significantly increased at 16 µM (P = 0.0159).
In MDA-MB-231 cells, treatment with As2S2 mainly induced cell cycle arrest in G2/M and S phases in a dose- and time-dependent manner.
As shown in Figure 4A, 4B, in MDA-MB-231 cells after 24 h of As2S2 treatment, the percentage of cells at G0/G1 phase significantly decreased with As2S2 at concentrations of 8, 12, and 16 µM (P < 0.0001, respectively). The percentage of MDA-MB-231 cells at G2/M phase significantly increased with As2S2 at concentrations ranging from 8 to 16 µM (P < 0.0001 respectively).
Figure 4.

As2S2 triggers cell cycle arrest in MDA-MB-231 cells. (A) MDA-MB-231 cells were treated with serial concentrations of As2S2 (0, 4, 8, 12 and 16 µM) for 24, 48 and 72 h. The peaks in the figure represent the G0/G1, S, and G2/M phases in the cell cycle, respectively. (B) The percentages of cell numbers in the cell cycle of MDA-MB-231 cells after 24 h, (C) 48 h, and (D) 72 h of drug treatment. All data were expressed as the mean ± SEM (n ≥ 3). Asterisks indicate significant differences between the control (0 µM) and the drug treatment groups (*P < 0.05, **P < 0.01).
As shown in Figure 4A, 4C, in MDA-MB-231 cells after 48 h of As2S2 treatment, the percentage of cells at G0/G1 phase significantly decreased with As2S2 at concentrations ranging from 4 to 16 µM (P = 0.0093 at 4 µM, P < 0.0001 at 8, 12, and 16 µM, respectively). The percentage of cells at G2/M phase significantly increased with As2S2 at concentrations ranging from 4 to 16 µM (P = 0.0020 at 4 µM, P < 0.0001 at 8, 12 and 16 µM, respectively). The percentage of MDA-MB-231 cells at S phase significantly decreased after exposure to As2S2 for 48 h at concentrations of 4 µM (P = 0.0157) and 8 µM (P = 0.0216), whereas significantly increased at 16 µM (P < 0.0001).
As shown in Figure 4A, 4D, in MDA-MB-231 cells after 72 h of As2S2 treatment, the percentage of cells at G0/G1 phase significantly decreased with As2S2 at concentrations ranging from 4 to 16 µM (P < 0.0001 respectively). The percentage of MDA-MB-231 cells at G2/M phase significantly increased with As2S2 at concentrations ranging from 4 to 16 µM (P < 0.0001 respectively) for 72 h. The percentage of MDA-MB-231 cells at S phase significantly increased with As2S2 at concentrations ranging from 8 to 16 µM (P = 0.0001 at 8 µM, P < 0.0001 at 12 and 16 µM, respectively) for 72 h.
As2S2 regulates cell cycle-related proteins
Cell cycle progression is controlled by cyclins and cyclin-dependent kinases (CDKs) which become the rational targets for drug treatment to be blocked in order to induce cell death [40,41]. To elucidate the underlying mechanisms, we measured the expression of selected cell-cycle related proteins so as to correlate the cell cycle arrests in different phases with actual alterations of these protein levels. As shown in Figures 5 and 6, in both MCF-7 and MDA-MB-231 cells, As2S2 regulated cell cycle-related proteins, such as cyclin A2, cyclin B1, and cyclin D1 in dose- and time-dependent manners.
Figure 5.

Effects of As2S2 on cell cycle regulators in MCF-7 cells. MCF-7 cells were cultured with various concentrations of As2S2 (0, 4, 8 and 16 µM) for 24, 48 and 72 h, and Western blot assays were carried out to examine the effects of As2S2 on the expressions of key proteins cyclin A2, cyclin B1 and cyclin D1 in MCF-7 cells after 24, 48 and 72 h of drug treatment. Protein β-actin was used as internal control. All images are representative of three independent analyses from three independent cellular preparations. Asterisks indicate significant differences between the control (0 µM) and the drug treatment groups (*P < 0.05, **P < 0.01).
Figure 6.

Effects of As2S2 on cell cycle regulators in MDA-MB-231 cells. MDA-MB-231 cells were cultured with serial concentrations of As2S2 (0, 4, 8 and 16 µM) for 24, 48 and 72 h. Western blot assays were carried out to examine the effects of As2S2 on the expressions of key proteins cyclin A2, cyclin B1 and cyclin D1 in MDA-MB-231 cells after 24, 48 and 72 h of drug treatment. Protein β-actin was used as internal control. All images are representative of three independent analyses from three independent cellular preparations. Asterisks indicate significant differences between the control (0 µM) and the drug treatment groups (*P < 0.05, **P < 0.01).
Compared to control, the expression levels of cyclin A2 in MCF-7 cells and MDA-MB-231 significantly decreased with As2S2 at concentrations of 8 µM (P = 0.0273 in MCF-7, P = 0.0137 in MDA-MB-231) and 16 µM (P = 0.0003 in MCF-7, P = 0.0001 in MDA-MB-231) after 48 h of As2S2 treatment. After exposure to As2S2 for 72 h, the statistically significant decrease in the amount of cyclin A2 expression started to occur in MCF-7 cells with As2S2 at concentrations of 4 µM (P = 0.0021) and in MDA-MB-231 cells with As2S2 at concentrations of 8 µM (P = 0.0241). Compared to control, the expression levels of cyclin D1 in MCF-7 cell significantly decreased by 16 µM of As2S2 at all of the three time courses (P = 0.0086 in 24 h, P = 0.0084 in 48 h, and P = 0.0010 in 72 h). In contrast, the significant downregulation of cyclin D1 expression in MDA-MB-231 cells merely occurred after 72 h of As2S2 treatment (16 µM, P = 0.0049). These results are congruent with G2/M and S phases arrest caused by As2S2 in both breast cancer cell lines.
Interestingly, the protein levels of cyclin B1 significantly decreased in MCF-7 cells after 48 h and 72 h of As2S2 treatments with final concentrations of 4 µM (P = 0.0072 in 48 h, P = 0.0006 in 72 h), 8 µM (P = 0.0020 in 48 h, P < 0.0001 in 72 h) and 16 µM (P = 0.0002 in 48 h, P < 0.0001 in 72 h), whereas the protein levels of cyclin B1 significantly increased in MDA-MB-231 cells after 72 h of As2S2 treatment (16 µM, P = 0.0227). These data suggested that As2S2 induced G2/M arrests by inhibiting cyclin B1 expression in MCF-7 cells, whereas by activating cyclin B1 levels in MDA-MB-231 cells, respectively.
As2S2 induces apoptosis in breast cancer cells
Apoptotic dysfunction is central to tumorigenesis and tumor progression which are in part characterized by imbalance between cell growth and programmed cell death. Numerous drug treatments act through inducing apoptosis in cancerous cells [42,43]. To confirm whether the inhibition of cell proliferation in breast cancer cells induced by As2S2 was attributed to apoptotic induction, we performed Annexin V/PI double staining assay followed by flow cytometry analysis. The apoptotic index was defined as the ratio of total apoptotic cell percentages between As2S2 treatment groups (with concentrations of As2S2 as 4, 8, 12 and 16 µM) and the control (with the concentration of As2S2 as 0 µM).
As shown in Figure 7A and Table 2, the total apoptotic indices in MCF-7 cells treated with 4, 8, 12 and 16 µM of As2S2 increased in a dose- and time-dependent manner after 24, 48 and 72 h drug treatments. After exposure to As2S2 for 24 h, the total apoptotic indices elevated to a certain degree by As2S2 at 4-16 µM, though the difference was not statistically significant. In contrast, significant increases of apoptotic indices were observed in the cells after treatment with 12 and 16 µM As2S2 for 48 and 72 h, respectively (P < 0.05).
Figure 7.

As2S2 induces apoptosis in breast cancer cells. (A) MCF-7 and (B) MDA-MB-231 cells were treated with different concentrations of As2S2 (0, 4, 8, 12 and 16 µM) for 24, 48 and 72 h, followed by staining with Annexin V/PI, and then analyzed by flow cytometry. The cells were assessed for total apoptotic cells composed by early apoptotic (Annexin V+/PI-) and late apoptotic (Annexin V+/PI+) cells. The apoptotic index was defined as the ratio of total apoptotic cell percentages between As2S2 treatment groups (with concentrations of As2S2 as 4, 8, 12 and 16 µM) and the control (As2S2 as 0 µM). All data were expressed as the mean ± SEM (n ≥ 3). Asterisks indicate significant differences between the control and the drug treatment groups (*P < 0.05, **P < 0.01).
Table 2.
Apoptotic indices following exposure to As2S2 in human breast cancer cell lines
| Apoptotic Index (folds of control) | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Breast cancer cell lines | Exposure time | As2S2 concentrations (µM) | ||||
|
| ||||||
| 0 | 4 | 8 | 12 | 16 | ||
| MCF-7 | 24 h | 1 | 1.18±0.12 | 1.34±0.16 | 1.45±0.13 | 1.40±0.11 |
| 48 h | 1 | 2.03±0.28 | 2.76±0.27 | 3.70±0.53** | 3.54±0.67* | |
| 72 h | 1 | 1.69±0.36 | 2.60±0.32 | 3.80±0.61** | 4.33±0.47** | |
| MDA-MB-231 | 24 h | 1 | 1.08±0.04 | 1.22±0.09 | 1.33±0.06* | 1.21±0.08 |
| 48 h | 1 | 1.48±0.08 | 1.77±0.17* | 1.99±0.10** | 2.08±0.21** | |
| 72 h | 1 | 2.91±0.15 | 4.17±0.18* | 4.33±0.06* | 5.68±1.31** | |
Data are the means ± SEM from at least three independent experiments.
P < 0.05 vs. control (As2S2 0 µM);
P < 0.01 vs. control (As2S2 0 µM).
In case of MDA-MB-231 cells (Figure 7B and Table 2), after 24 h exposure to As2S2, the total apoptotic index significantly increased by 1.33±0.06 (P = 0.0257) folds in the presence of 12 µM As2S2, when compared with control (1.00). Statistically significant increases of apoptotic indices began to appear 48 and 72 h after the exposure of cells to 8 µM of As2S2, with significant increase by 1.77±0.17 (P = 0.0159) and 4.17±0.18 (P = 0.0234) folds, respectively, when compared with control (1.00).
Taken together, the results indicated that apoptotic indices of both MCF-7 and MDA-MB-231 cells increased in a dose- and time-dependent manner after exposure to As2S2 for 24, 48 and 72 h.
As2S2 regulates apoptosis-related proteins
To further delineate and validate the apoptotic induction by As2S2 in breast cancer cells, the expression of apoptosis-related proteins were explored by western blot analysis.
As shown in Figure 8, in case of MCF-7 cells with wild-type p53, an increase in pro-apoptotic p53 and cleavage PARP expressions were observed after treatment by As2S2 in dose- and time-dependent manners. Compared to control, the expression levels of p53 in MCF-7 cells significantly increased by As2S2 (with concentrations of 4, 8 and 16 µM) at all of three time courses (with 4 µM, P = 0.0167 in 24 h, P = 0.0024 in 48 h, and P = 0.0392 in 72 h). The statistically significant increases of cleavage PARP expression started to occur in MCF-7 cells with As2S2 at concentrations of 8 µM (P = 0.0155) and 16 µM (P = 0.0001) after 48 h and 72 h of As2S2 treatments, respectively. By contrast, in case of MDA-MB-231 cells with mutated p53, the expressions of p53 neither changed by any concentrations of As2S2 nor altered by any incubation time course in the presence of As2S2 (Figure 9). In addition, the expressions of cleavage PARP were little altered by As2S2 treatment in MDA-MB-231 cells as well (Figure 9).
Figure 8.

Effects of As2S2 on the expression levels of pro-apoptotic proteins in MCF-7 cells. MCF-7 cells were treated with different concentrations of As2S2 (0, 4, 8 and 16 µM) for 24, 48 and 72 h. Western blot assays were carried out to examine the effects of As2S2 on the expressions of pro-apoptotic proteins p53 and PARP in MCF-7 cells after 24, 48 and 72 h of drug treatment. Protein β-actin was used as internal control. All images are representative of three independent analyses from three independent cellular preparations. Asterisks indicate significant differences between the control (0 µM) and the drug treatment groups (*P < 0.05, **P < 0.01).
Figure 9.

Effects of As2S2 on the expression levels of pro-apoptotic proteins in MDA-MB-231 cells. MDA-MB-231 cells were treated with different concentrations of As2S2 (0, 4, 8 and 16 µM) for 24, 48 and 72 h. Western blot assays were carried out to examine the effects of As2S2 on the expressions of pro-apoptotic proteins p53 and PARP in MDA-MB-231 cells after 24, 48 and 72 h of drug treatment. Protein β-actin was used as internal control. All images are representative of three independent analyses from three independent cellular preparations.
As shown in Figures 10 and 11, the protein expressions of anti-apoptotic markers Bcl-2 and Mcl-1 were inhibited by As2S2 in both MCF-7 and MDA-MB-231 cells, in dose- and time-dependent manners. Significant decreases of Bcl-2 expression were observed in MCF-7 cells after treatment with 16 µM of As2S2 for 48 h (P = 0.0014), as well as 8 µM (P = 0.0424) and 16 µM (P = 0.0054) of As2S2 for 72 h, respectively (Figure 10). Compared to control, the expression levels of Mcl-1 in MCF-7 cells significantly decreased by As2S2 with concentrations of 8 µM (P = 0.0147) and 16 µM (P = 0.0005) after 72 h of treatment (Figure 10). In case of MDA-MB-231, significant decreases of Bcl-2 (P = 0.0045) and Mcl-1 (P = 0.0487) expressions were observed after treatment with 16 µM of As2S2 for 72 h (Figure 11).
Figure 10.

Effects of As2S2 on the expression levels of anti-apoptotic proteins in MCF-7 cells. MCF-7 cells were treated with different concentrations of As2S2 (0, 4, 8 and 16 µM) for 24, 48 and 72 h. Western blot assays were carried out to examine the effects of As2S2 on the expressions of anti-apoptotic proteins Bcl-2 and Mcl-1 in MCF-7 cells after 24, 48 and 72 h of drug treatment. Protein β-actin was used as internal control. All images are representative of three independent analyses from three independent cellular preparations. Asterisks indicate significant differences between the control (0 µM) and the drug treatment groups (*P < 0.05, **P < 0.01).
Figure 11.
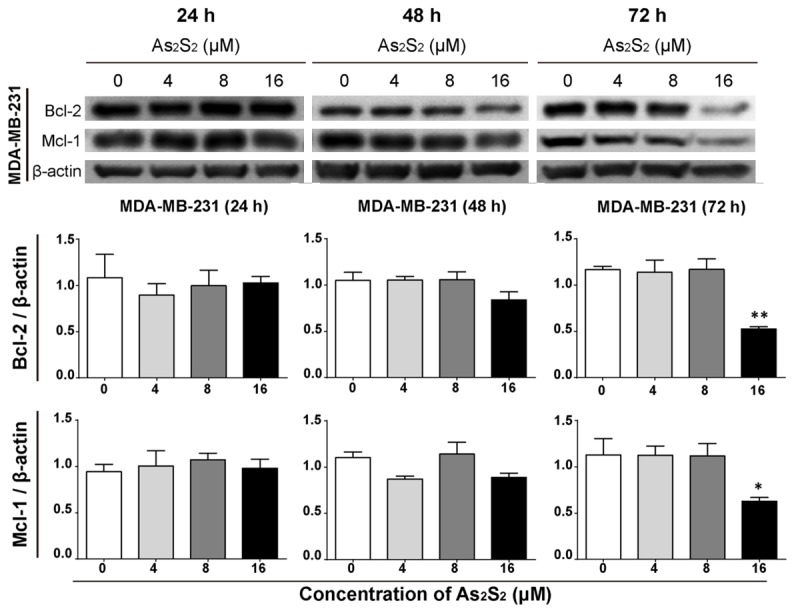
Effects of As2S2 on the expression levels of anti-apoptotic proteins in MDA-MB-231 cells. MDA-MB-231 cells were treated with different concentrations of As2S2 (0, 4, 8 and 16 µM) for 24, 48 and 72 h. Western blot assays were carried out to examine the effects of As2S2 on the expressions of anti-apoptotic proteins Bcl-2 and Mcl-1 in MDA-MB-231 cells after 24, 48 and 72 h of drug treatment. Protein β-actin was used as internal control. All images are representative of three independent analyses from three independent cellular preparations. Asterisks indicate significant differences between the control (0 µM) and the drug treatment groups (*P < 0.05, **P < 0.01).
As2S2 inhibits cell survival-related proteins
To further investigate the molecular mechanism underlying the inhibitory effects of As2S2 on cell survival and growth in breast cancer cells, we evaluated the effect of As2S2 on pro-survival PI3K/Akt pathway. Abundant evidence demonstrates the essential role of this cell survival pathway in cell growth, proliferation, and survival [44,45]. The protein expressions of PI3K and Akt were detected in breast cancer cells by western blot analysis.
As shown in Figures 12 and 13, in both MCF-7 and MDA-MB-231 cells, the protein expressions of PI3K and Akt were effectively downregulated by As2S2 in dose- and time-dependent manners. Compared to control, the expression levels of PI3K in MCF-7 (Figure 12) and MDA-MB-231 (Figure 13) cells significantly decreased by As2S2 at all of three time courses (P < 0.05). Significant decreases of Akt expression were observed in both MCF-7 (Figure 12) and MDA-MB-231 (Figure 13) cells after treatment with 16 µM of As2S2 for 48 h (P = 0.0006 in MCF-7, P = 0.0181 in MDA-MB-231) and 72 h (P = 0.0424 in MCF-7, P = 0.0281 in MDA-MB-231), respectively.
Figure 12.

Effects of As2S2 on the expression levels of pro-survival proteins in MCF-7 cells. MCF-7 cells were cultured with various concentrations of As2S2 (0, 4, 8 and 16 µM) for 24, 48 and 72 h. Western blot assays were carried out to examine the effects of As2S2 on the expressions of key proteins PI3K and Akt in MCF-7 cells after 24, 48 and 72 h of drug treatment. Protein β-actin was used as internal control. All images are representative of three independent analyses from three independent cellular preparations. Asterisks indicate significant differences between the control (0 µM) and the drug treatment groups (*P < 0.05, **P < 0.01).
Figure 13.

Effects of As2S2 on the expression levels of pro-survival proteins in MDA-MB-231 cells. MDA-MB-231 cells were cultured with serial concentrations of As2S2 (0, 4, 8 and 16 µM) for 24, 48 and 72 h. Western blot assays were carried out to examine the effects of As2S2 on the expressions of key proteins PI3K and Akt in MDA-MB-231 cells after 24, 48 and 72 h of drug treatment. Protein β-actin was used as internal control. All images are representative of three independent analyses from three independent cellular preparations. Asterisks indicate significant differences between the control (0 µM) and the drug treatment groups (*P < 0.05, **P < 0.01).
Collectively, the results indicated that the blockade of PI3K/Akt signals by As2S2 in breast cancer cells may contribute to the induction of apoptosis as well as the inhibition of cell viability following As2S2 exposure.
Discussion
Numerous studies on breast carcinomas are based on in vivo and in vitro researches focusing on breast cancer cell lines [38,46]. In this study, we took two typically distinctive subtypes of breast cancer cell lines as the research subjects, which can represent different categories of breast cancer disease, namely MCF-7 and MDA-MB-231 cell lines. MCF-7 is relatively a non-aggressive breast cancer cell line, characterized by estrogen-dependent, higher differentiated, ER-positive, and weakly invasive features. By contrast, MDA-MB-231 is an aggressive breast cancer cell line, characterized by estrogen-independent, undifferentiated, ER-negative, and highly invasive features [47-49].
In this study, we have provided the first evidence that As2S2 exerts potent antitumor effects on both MCF-7 and MDA-MB-231 cell lines. The cytotoxicity of As2S2 against human breast cancer cells was measured by assessing different parameters. The results demonstrated that As2S2 inhibited the cell viabilities by inducing cell cycle arrest and cell apoptosis in both MCF-7 and MDA-MB-231 cells. These antitumor effects of As2S2 were correlated to the regulation of signals that related to cell cycle progression, apoptosis and cell survival pathway. Our results also showed that As2S2 triggered cell cycle arrest in MCF-7 and MDA-MB-231 cells via regulating the protein expressions of cyclin A2, B1 and D1. Furthermore, activation of pro-apoptotic signals such as p53 and PARP, and the inhibition of anti-apoptotic signals such as Bcl-2 and Mcl-1 were connected to the induction of apoptosis in the breast cancer cells. In addition, the blockage of the pro-survival PI3K/Akt pathway also contributed to the antitumor effect of As2S2 in MCF-7 and MDA-MB-231 cell lines. To our knowledge, the results herein showed for the first time the anti-cancer effects of As2S2 on different types of breast cancer cells, which especially involve cell cycle arrest and the programmed cell death pathway such as apoptosis induction, as well as the related underlying molecular mechanisms.
Since uncontrolled cell proliferation and the loss of regulated cell death are two major characteristics of cancer cells, accumulating anti-tumor strategies are emerging to develop therapies related to cell growth suppression by activating cell death pathways [29,50-51]. Previous studies have reported significant inhibitory effects of As2S2 on cell viability and proliferation in a number of cancer cell lines representing different cancer diseases including gastric, hepatocellular, melanoma, pancreatic, cervical and ovarian carcinoma [12,27,33,52], with little effect on normal control cells [12,33,53]. In the present study, we focused on the anti-tumor effects of As2S2 on human breast cancer cell lines, which has been rarely reported before. The results from WST assay showed a potent inhibitory action on cell viabilities exerted by As2S2 in both MCF-7 and MDA-MB-231 cells in dose- and time-dependent manners (Figure 1A, 1B). The viabilities of MCF-7 and MDA-MB-231 cells were confirmed and visualized by calcein staining test after exposure to As2S2 with different final concentrations (0, 4, 8, 12 and 16 µM) and in different time courses (24, 48 and 72 h after drug exposure) (Figure 2). The morphology results have shown that live cell numbers decreased by As2S2 treatment in both breast cancer cell lines in dose- and time-dependent manners, which is congruent with the cell viability results detected by WST assay. Moreover, the IC50 values of As2S2 in each cell line in different time courses exhibited culture time-dependent efficacy of As2S2 with the highest effect at 72 h after the treatment (Table 1). Furthermore, our results demonstrated that MCF-7 cells are more sensitive to As2S2 than MDA-MB-231 cells.
Abundant studies indicate that tumor cell response to chemotherapies is mainly through several cell death mechanisms, which include cell cycle arrest and apoptosis [54,55]. Cell cycle is a series of organized events that allow cells to grow and divide into two daughter cells [35,56]. This process consists of four sequential phases, namely G1, S, G2 and M (mitosis) phases. Since cell cycle dysfunction often contributes to tumorigenesis, it turns into one of the essential targets for current anticancer therapies to exert their actions by regulating cell cycle progression [36]. In the present study, we found that As2S2 treatment arrested the cell cycle progression at G2/M phase in both two breast cancer cell lines with dose- and time-dependent manners. In addition to the G2/M phase-arrest effect, As2S2 also blocked G0/G1 phase in MCF-7 cells, whereas the agent led to S phase arrest in MDA-MB-231 cells. The different cell cycle arrest observed in MCF-7 and MDA-MB-231 cells at different phases could be ascribed to the biological variation between the two cell lines. More importantly, the blockage of cell cycle progression by As2S2 treatment may facilitate the induction of apoptosis [57]. Apoptosis is the most common and well-orchestrated form of programmed cell death, which is essential for many biological processes [55]. Dysregulation of apoptosis can result in numerous pathological conditions, including different types of cancer [58]. Therefore, the induction of apoptosis became one of the principles to develop novel chemotherapeutic agents. In the present study, we found that As2S2 induced apoptosis in both MCF-7 and MDA-MB-231 breast cancer cell lines in both dose- and time-dependent manners. These results are consistent with the inhibited cell growth and cell cycle arrest triggered by As2S2 in both MCF-7 and MDA-MB-231 cells, suggesting the promising anti-cancer characteristics of As2S2 against breast cancer.
The molecular mechanisms underlying the anticancer actions of As2S2 in breast cancer cells in the present study involved various signals and pathways, including the activation of apoptosis pathways, regulation of specific proteins in cell cycle, as well as the suppression of pro-survival pathway. In terms of regulating apoptosis related proteins, we found that As2S2 activated the pro-apoptotic signals such as p53 and PARP in MCF-7 cells in dose- and time-dependent manners (Figure 8), while the agent showed little effect in MDA-MB-231 cells (Figure 9). The tumor suppressor p53 plays essential role in the prevention of human cancer through either cell cycle regulation or apoptosis induction [59,60]. It is found to be mutated in 50-55% of human cancers [61]. In this study, different genetic status of p53 in MCF-7 (with wide-type gene) and MDA-MB-231 (with mutant gene) cells may attribute to the discrepancy in p53 protein expression activated by As2S2 between these two different cell lines. Cleavage of PARP is a hallmark of apoptosis, which is commonly observed during apoptosis and mediated by the activation of caspases [62]. Our results showed that As2S2 promoted the expression of cleaved PARP in MCF-7 cells other than MDA-MB-231 cells, suggesting possible caspase-dependent apoptotic induction in MCF-7 cells by As2S2 treatment. Furthermore, the different status of these two key pro-apoptotic indicators after treatment by As2S2 may be partially responsible for the different sensitivities of MCF-7 and MDA-MB-231 cells to As2S2. Bcl-2 protein family has a critical role in regulating the intrinsic pathway of apoptosis [63]. The anti-apoptotic members in Bcl-2 family, such as Bcl-2 and Mcl-1 undergo down-regulation during apoptosis induction [31]. In this research, we found that As2S2 inhibited Bcl-2 and Mcl-1 expressions in both MCF-7 and MDA-MB-231 cells in dose- and time-dependent manners, suggesting that these proteins were related to As2S2 induced apoptosis in both cell lines (Figures 10 and 11). Cell cycle progression is regulated by protein kinase complexes, which consists of cyclins and CDKs [36]. Different cyclins are required at different phases of the cell cycle. In detail, cyclin D regulates the progression from G1 phase to S phase, while cyclin B and cyclin A regulate the progression from G2 to M phase [36,64]. The results of the present study indicated that the treatment of the cells by As2S2 decreased the protein expression of cyclin D1 and cyclin A2 in the two breast cancer cell lines, which is in agreement with the cell cycle arrests induced by As2S2 in S and G2/M phases. In addition, the protein expression of cyclin B1 was down-regulated by As2S2 in MCF-7 cells, whereas up-regulated in MDA-MB-231 cells. Both inhibition and activation of cyclin B1 could lead to cell cycle arrest in G2/M phase [65,66]. The opposite regulatory effect of As2S2 on cyclin B1 amounts may correspond to the biological variation between the two different breast cancer cell lines. PI3K/Akt signaling pathway implicates in tumorigenesis by regulating cell growth, cell cycle progression, cell apoptosis, and cell metabolism [44,45]. Disruption of PI3K/Akt signals contributes to inhibiting cell survival, restraining tumor growth, and promoting pro-apoptotic activities. Our results demonstrated that As2S2 treatments suppressed the PI3K/Akt signals in the two breast cancer cell lines with similar dose- and time-dependent manners. These observations suggest the possible inhibitory function of As2S2 to control cancer cell survival signals, which acts in accordance with the regulation of apoptotic signals as well as cell cycle related proteins.
Taken together, this study provides the first evidence that As2S2 has the ability to exert its anticancer effects against different human breast cancer cell lines, by inhibiting cell proliferation, blocking cell cycle progression, and inducing apoptosis. Such anticancer functions by As2S2 are suggested to be correlated with molecular alterations involving different cellular signaling pathways. The present research provides mechanistic bases for therapeutic potential of As2S2 in the application of treating breast cancer. However, the present study is confined to elucidate the antitumor effect of As2S2 against breast cancer in vitro. Further studies will be performed to investigate its therapeutic potentials in animal models of breast cancer.
Acknowledgements
This study was supported by China Scholarship Council (File No. 201709110064).
Disclosure of conflict of interest
None.
References
- 1.Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J. Clin. Oncol. 2006;24:2137–2150. doi: 10.1200/JCO.2005.05.2308. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, Pusztai L. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008;26:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 4.Huober J, von Minckwitz G, Denkert C, Tesch H, Weiss E, Zahm DM, Belau A, Khandan F, Hauschild M, Thomssen C, Högel B, Darb-Esfahani S, Mehta K, Loibl S. Effect of neoadjuvant anthracycline-taxane-based chemotherapy in different biological breast cancer phenotypes: overall results from the GeparTrio study. Breast Cancer Res Treat. 2010;124:133–140. doi: 10.1007/s10549-010-1103-9. [DOI] [PubMed] [Google Scholar]
- 5.Emadi A, Gore SD. Arsenic trioxide-an old drug rediscovered. Blood Rev. 2010;24:191–199. doi: 10.1016/j.blre.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Douer D, Tallman MS. Arsenic trioxide: new clinical experience with an old medication in hematologic malignancies. J. Clin. Oncol. 2005;23:2396–2410. doi: 10.1200/JCO.2005.10.217. [DOI] [PubMed] [Google Scholar]
- 7.Miller WH Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 8.Soignet SL, Frankel SR, Douer D, Tallman MS, Kantarjian H, Calleja E, Stone RM, Kalaycio M, Scheinberg DA, Steinherz P, Sievers EL, Coutré S, Dahlberg S, Ellison R, Warrell RP Jr. United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J. Clin. Oncol. 2001;19:3852–3860. doi: 10.1200/JCO.2001.19.18.3852. [DOI] [PubMed] [Google Scholar]
- 9.Cicconi L, Lo-Coco F. Current management of newly diagnosed acute promyelocytic leukemia. Ann Oncol. 2016;27:1474–1481. doi: 10.1093/annonc/mdw171. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Liu Y, Wang X, Liu D, Sun Z, Wang C, Jin G, Zhang B, Yu S. Randomized clinical control study of locoregional therapy combined with arsenic trioxide for the treatment of hepatocellular carcinoma. Cancer. 2015;121:2917–2925. doi: 10.1002/cncr.29456. [DOI] [PubMed] [Google Scholar]
- 11.Wu JZ, Ho PC. Comparing the relative oxidative DNA damage caused by various arsenic species by quantifying urinary levels of 8-hydroxy-2’-deoxyguanosine with isotope-dilution liquid chromatography/mass spectrometry. Pharm Res. 2009;26:1525–1533. doi: 10.1007/s11095-009-9865-7. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Kim S, Ding W, Tong Y, Zhang X, Pan M, Chen S. Arsenic sulfide inhibits cell migration and invasion of gastric cancer in vitro and in vivo. Drug Des Devel Ther. 2015;9:5579–5590. doi: 10.2147/DDDT.S89805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, Shao Y, Liu J, Chen G, Ho PC. The medicinal use of realgar (As4S4) and its recent development as an anticancer agent. J Ethnopharmacol. 2011;135:595–602. doi: 10.1016/j.jep.2011.03.071. [DOI] [PubMed] [Google Scholar]
- 14.Zhang QY, Mao JH, Liu P, Huang QH, Lu J, Xie YY, Weng L, Zhang Y, Chen Q, Chen SJ, Chen Z. A systems biology understanding of the synergistic effects of arsenic sulfide and Imatinib in BCR/ABL-associated leukemia. Proc Natl Acad Sci U S A. 2009;106:3378–3383. doi: 10.1073/pnas.0813142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, He P, Cheng X, Zhang M. Long-term outcome of 31 cases of refractory acute promyelocytic leukemia treated with compound realgar natural indigo tablets administered alternately with chemotherapy. Oncol Lett. 2015;10:1184–1190. doi: 10.3892/ol.2015.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Liu X, Li X, Lv X, Lu K, Chen N, Li P, Wang X. Arsenic disulfide induces apoptosis of human diffuse large B cell lymphoma cells involving Bax cleavage. Oncol Rep. 2013;30:2427–2434. doi: 10.3892/or.2013.2729. [DOI] [PubMed] [Google Scholar]
- 17.Song P, Chen P, Wang D, Wu Z, Gao Q, Wang A, Zhu R, Wang Y, Wang X, Zhao L, Duan Z, Zhu S, Cui P, Li Y, Li H. Realgar transforming solution displays anticancer potential against human hepatocellular carcinoma HepG2 cells by inducing ROS. Int J Oncol. 2017;50:660–670. doi: 10.3892/ijo.2016.3831. [DOI] [PubMed] [Google Scholar]
- 18.Wang G, Zhang T, Sun W, Wang H, Yin F, Wang Z, Zuo D, Sun M, Zhou Z, Lin B, Xu J, Hua Y, Li H, Cai Z. Arsenic sulfide induces apoptosis and autophagy through the activation of ROS/JNK and suppression of Akt/mTOR signaling pathways in osteosarcoma. Free Radic Biol Med. 2017;106:24–37. doi: 10.1016/j.freeradbiomed.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Liang SX, Lu YF, Miao JW, Wu Q, Shi JS. Realgar and realgar-containing Liu-Shen-Wan are less acutely toxic than arsenite and arsenate. J Ethnopharmacol. 2011;134:26–31. doi: 10.1016/j.jep.2010.11.052. [DOI] [PubMed] [Google Scholar]
- 20.Lu YF, Wu Q, Yan JW, Shi JZ, Liu J, Shi JS. Realgar, cinnabar and An-Gong-Niu-Huang Wan are much less chronically nephrotoxic than common arsenicals and mercurials. Exp Biol Med (Maywood) 2011;236:233–239. doi: 10.1258/ebm.2010.010247. [DOI] [PubMed] [Google Scholar]
- 21.Miao JW, Liang SX, Wu Q, Liu J, Sun AS. Toxicology evaluation of realgar-containing niu-huang-jie-du pian as compared to arsenicals in cell cultures and in mice. ISRN Toxicol. 2011;2011:250387. doi: 10.5402/2011/250387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Q, Wang C, Li X, Guo H, Meng J, Liu J, Xu H. Fabrication of water-soluble polymer-encapsulated As4S4 to increase oral bioavailability and chemotherapeutic efficacy in AML mice. Sci Rep. 2016;6:29348. doi: 10.1038/srep29348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu DP, Qiu JY, Jiang B, Wang Q, Liu KY, Liu YR, Chen SS. Tetra-arsenic tetra-sulfide for the treatment of acute promyelocytic leukemia: a pilot report. Blood. 2002;99:3136–3143. doi: 10.1182/blood.v99.9.3136. [DOI] [PubMed] [Google Scholar]
- 24.Zhu HH, Wu DP, Jin J, Li JY, Ma J, Wang JX, Jiang H, Chen SJ, Huang XJ. Oral tetra-arsenic tetra-sulfide formula versus intravenous arsenic trioxide as first-line treatment of acute promyelocytic leukemia: a multicenter randomized controlled trial. J. Clin. Oncol. 2013;31:4215–4221. doi: 10.1200/JCO.2013.48.8312. [DOI] [PubMed] [Google Scholar]
- 25.Lu DP, Wang Q. Current study of APL treatment in China. Int J Hematol. 2002;76:316–318. doi: 10.1007/BF03165273. [DOI] [PubMed] [Google Scholar]
- 26.Tian Y, Wang X, Xi R, Pan W, Jiang S, Li Z, Zhao Y, Gao G, Liu D. Enhanced antitumor activity of realgar mediated by milling it to nanosize. Int J Nanomedicine. 2014;9:745–757. doi: 10.2147/IJN.S56391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan KT, Meng FY, Li Q, Ho CY, Lam TS, To Y, Lee WH, Li M, Chu KH, Toh M. Cucurbitacin B induces apoptosis and S phase cell cycle arrest in BEL-7402 human hepatocellular carcinoma cells and is effective via oral administration. Cancer Lett. 2010;294:118–124. doi: 10.1016/j.canlet.2010.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Kadam CY, Abhang SA. Apoptosis markers in breast cancer therapy. Adv Clin Chem. 2016;74:143–193. doi: 10.1016/bs.acc.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11:342–357. doi: 10.1634/theoncologist.11-4-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wan J, Liu T, Mei L, Li J, Gong K, Yu C, Li W. Synergistic antitumour activity of sorafenib in combination with tetrandrine is mediated by reactive oxygen species (ROS)/Akt signaling. Br J Cancer. 2013;109:342–350. doi: 10.1038/bjc.2013.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng YX, Liu R, Wang Q, Li BS, Xu XX, Hu M, Chen L, Fu Q, Pu DM, Hong L. Realgar-induced apoptosis of cervical cancer cell line Siha via cytochrome c release and caspase-3 and caspase-9 activation. Chin J Integr Med. 2012;18:359–365. doi: 10.1007/s11655-011-0697-z. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Liu Z, Gou Y, Qin Y, Xu Y, Liu J, Wu JZ. Apoptosis and necrosis induced by novel realgar quantum dots in human endometrial cancer cells via endoplasmic reticulum stress signaling pathway. Int J Nanomedicine. 2015;10:5505–5512. doi: 10.2147/IJN.S83838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S, Fang Y, Ma L, Liu S, Li X. Realgar-induced apoptosis and differentiation in all-trans retinoic acid (ATRA)-sensitive NB4 and ATRA-resistant MR2 cells. Int J Oncol. 2012;40:1089–1096. doi: 10.3892/ijo.2011.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;138:255–271. doi: 10.1016/j.pharmthera.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 36.Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol Sci. 2003;24:139–145. doi: 10.1016/S0165-6147(03)00026-9. [DOI] [PubMed] [Google Scholar]
- 37.Pastorek M, Gronesova P, Cholujova D, Hunakova L, Bujnakova Z, Balaz P, Duraj J, Lee TC, Sedlak J. Realgar (As4S4) nanoparticles and arsenic trioxide (As2O3) induced autophagy and apoptosis in human melanoma cells in vitro. Neoplasma. 2014;61:700–709. doi: 10.4149/neo_2014_085. [DOI] [PubMed] [Google Scholar]
- 38.Oral AY, Cevatemre B, Sarimahmut M, Icsel C, Yilmaz VT, Ulukaya E. Anti-growth effect of a novel trans-dichloridobis[2-(2-hydroxyethyl) pyridine] platinum (II) complex via induction of apoptosis on breast cancer cell lines. Bioorg Med Chem. 2015;23:4303–4310. doi: 10.1016/j.bmc.2015.06.037. [DOI] [PubMed] [Google Scholar]
- 39.Wei C, Fan B, Chen D, Liu C, Wei Y, Huo B, You L, Wang J, Chen J. Osteocyte culture in microfluidic devices. Biomicrofluidics. 2015;9:1–10. doi: 10.1063/1.4905692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- 41.Dickson MA, Schwartz GK. Development of cell-cycle inhibitors for cancer therapy. Curr Oncol. 2009;16:36–43. doi: 10.3747/co.v16i2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parton M, Dowsett M, Smith I. Studies of apoptosis in breast cancer. BMJ. 2001;322:1528–1532. doi: 10.1136/bmj.322.7301.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:1–14. doi: 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 45.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 46.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat. 2004;83:249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- 47.Hunakova L, Macejova D, Toporova L, Brtko J. Anticancer effects of tributyltin chloride and triphenyltin chloride in human breast cancer cell lines MCF-7 and MDA-MB-231. Tumour Biol. 2016;37:6701–6708. doi: 10.1007/s13277-015-4524-6. [DOI] [PubMed] [Google Scholar]
- 48.Hasanpourghadi M, Pandurangan AK, Mustafa MR. Modulation of oncogenic transcription factors by bioactive natural products in breast cancer. Pharmacol Res. 2017;S1043-6618:30864–30872. doi: 10.1016/j.phrs.2017.09.009. [DOI] [PubMed] [Google Scholar]
- 49.Pasqualini JR, Schatz B, Varin C, Nguyen BL. Recent data on estrogen sulfatases and sulfotransferases activities in human breast cancer. J Steroid Biochem Mol Biol. 1992;41:323–329. doi: 10.1016/0960-0760(92)90358-p. [DOI] [PubMed] [Google Scholar]
- 50.Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, Iglehart JD. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004;101:10137–10142. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vermeulen K, Berneman ZN, Van Bockstaele DR. Cell cycle and apoptosis. Cell Prolif. 2003;36:165–175. doi: 10.1046/j.1365-2184.2003.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Du YH, Ho PC. Arsenic compounds induce cytotoxicity and apoptosis in cisplatin-sensitive and -resistant gynecological cancer cell lines. Cancer Chemother Pharmacol. 2001;47:481–490. doi: 10.1007/s002800100278. [DOI] [PubMed] [Google Scholar]
- 53.Ding W, Zhang L, Kim S, Tian W, Tong Y, Liu J, Ma Y, Chen S. Arsenic sulfide as a potential anti-cancer drug. Mol Med Rep. 2015;11:968–974. doi: 10.3892/mmr.2014.2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–237. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 55.Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer. 2004;4:592–603. doi: 10.1038/nrc1412. [DOI] [PubMed] [Google Scholar]
- 56.Barnum KJ, O’Connell MJ. Cell cycle regulation by checkpoints. Methods Mol Biol. 2014;1170:29–40. doi: 10.1007/978-1-4939-0888-2_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J. Clin. Oncol. 2005;23:9408–9421. doi: 10.1200/JCO.2005.01.5594. [DOI] [PubMed] [Google Scholar]
- 58.Bergamaschi G, Rosti V, Danova M, Lucotti C, Cazzola M. Apoptosis: biological and clinical aspects. Haematologica. 1994;79:86–93. [PubMed] [Google Scholar]
- 59.Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- 60.Colman MS, Afshari CA, Barrett JC. Regulation of p53 stability and activity in response to genotoxic stress. Mutat Res. 2000;462:179–188. doi: 10.1016/s1383-5742(00)00035-1. [DOI] [PubMed] [Google Scholar]
- 61.Wang X, Simpson ER, Brown KA. p53: Protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res. 2015;75:5001–5007. doi: 10.1158/0008-5472.CAN-15-0563. [DOI] [PubMed] [Google Scholar]
- 62.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pritchard JR, Gilbert LA, Meacham CE, Ricks JL, Jiang H, Lauffenburger DA, Hemann MT. Bcl-2 family genetic profiling reveals microenvironment-specific determinants of chemotherapeutic response. Cancer Res. 2011;71:5850–5858. doi: 10.1158/0008-5472.CAN-11-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yadav N, Kumar P, Chhikara A, Chopra M. Development of 1,3,4-oxadiazole thione based novel anticancer agents: design, synthesis and in-vitro studies. Biomed Pharmacother. 2017;95:721–730. doi: 10.1016/j.biopha.2017.08.110. [DOI] [PubMed] [Google Scholar]
- 66.Shaik SP, Vishnuvardhan MVPS, Sultana F, Subba Rao AV, Bagul C, Bhattacharjee D, Kapure JS, Jain N, Kamal A. Design and synthesis of 1,2,3-triazolo linked benzo[d] imidazo[2,1-b] thiazole conjugates as tubulin polymerization inhibitors. Bioorg Med Chem. 2017;25:3285–3297. doi: 10.1016/j.bmc.2017.04.013. [DOI] [PubMed] [Google Scholar]