

Abstract
Aberrant expression of histone deacetylases (HDACs) has been detected in a variety of cancers, which disrupts the balance between cell proliferation and apoptosis in favor of continuous growth. A previous study demonstrated that HDAC5 contributes to the proliferation of hepatocellular carcinoma (HCC) cells, but a clear understanding of the mechanism has not yet been provided. In the present work, we found that the levels of HDAC5 were significantly higher in HCC tissues and cells than in adjacent tissues and normal hepatic cells. In addition, knockdown of HDAC5 attenuated the proliferation of Hep3B and HepG2 cells. Through profiling the expressions of proliferation and apoptosis-related genes in Hep3B cells following HDAC5 knockdown, p63 and maspin were found obviously up-regulated in HDAC5-deprived cells compared with the control. Further investigations confirmed that HDAC5 knockdown induced TAp63 expression in HCC cells, accompanied with increased H3K9 acetylation at the TAp63 promoter. Overexpression of TAp63 led to proliferation inhibition by inducing cell cycle arrest. Additionally, TAp63 that was required for the maspin upregulation resulted from HDAC5 knockdown. Phenotype experiments showed that interrupting either TAp63 or maspin recovered the proliferative and tumorigenic capabilities of HCC cells with HDAC5 knockdown. Clinical analysis showed that HDAC5 was negatively correlated with TAp63 and maspin in HCC tissues. In addition, a high level of HDAC5 as well as a low level of TAp63 or maspin predicted poor survival in HCC patients. Taken together, this study proposes the existence of an aberrant HDAC5-TAp63-maspin pathway conferring HCC progression through proliferation induction, which suggests novel intervention targets for the disease.
Keywords: Histone deacetylase 5 (HDAC5), p63, mammary serpin (maspin), hepatocellular carcinoma (HCC), proliferation
Introduction
Diverse histone modifications are involved in controlling the transcriptional process, of which reversible acetylation plays a vital role. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are responsible for regulating the acetylation of histones. In general, histone acetylation is associated with transcriptional activation, whereas histone deacetylation blocks transcription. As the critical epigenetic regulators, HDACs were found to drive the initiation and progression of human diseases, including cancers, through activating oncogenes or inhibiting specific tumor suppressor genes [1]. In addition, aberrant expression of the oncogenic or tumor suppressive HDACs has been detected in various human cancers, such as gastric cancer, lung cancer, and hepatocellular carcinoma (HCC) [2-4]. Our previous work demonstrated that HDAC5, an overexpressed HDAC in HCC cells, enhanced the migration and invasion of hepatocellular carcinoma via relieving the HIPK2-mediated suppression of HIF-1α [5]. In addition, HDAC5 has been shown to promote proliferation and reduce apoptosis in HCC cells [6], but the mechanisms of which remain uncertain.
P63 belongs to the p53 protein family constituted by p53, p63 and p73, which shares high-sequence and structural similarities. Because of that, p63 and p53 can regulate similar sets of genes and possess some partially redundant functions [7]. The P63 gene encodes multiple isoforms that can be classified into two categories arising from different promoters; one with an acidic transactivation domain (TA isoforms) and the other lacking this domain (ΔN isoforms). Due to the loss of its transactivation domain, ΔNp63 acts as a negative regulator of the TAp63 function. TAp63 and ΔNp63 exhibit opposite functions, as shown by TAp63-induced cell death and ΔNp63-promoted survival. Therefore, TAp63 generally behaves as a tumor suppressor [8], whereas ΔNp63 has oncogenic potential [9].
Maspin (SERPINB5), a member of the serpin (serine protease inhibitor) superfamily, was discovered as a tumor suppressor in many cancers, including HCC. Increased clinical and in vitro evidence highlighted its role in inhibiting tumor growth, invasion and metastasis [10-12]. Maspin is often downregulated during tumor progression and its depletion is associated with poor prognosis [13,14]. Regulation of maspin transcription is central for controlling its activity, which seems to determine maspin protein quantity [15]. As a direct target of the tumor suppressor p53, maspin expression is rapidly induced by wild type p53. However, the function of p53 is often lost in many tumors due to mutation or deletion of its gene. However, as a member of the p53 family, p63 is rarely found mutated or deleted in human cancers, and it can substitute for p53 in inducing the expression of maspin [16,17]. Therefore, the regulation of the p63-maspin pathway needs to be deeply investigated, especially in the tumor with mutated p53.
Here, we described that HDAC5 could block the p63-maspin pathway in HCC cells through repressing TAp63 expression. Knockdown of HDAC5 restored the expression of TAp63 and maspin, consequently attenuating HCC cell proliferation. In conclusion, the clinical analysis showed that HDAC5 was negatively correlated with TAp63 and maspin in HCC tissues, and high expression of HDAC5 as well as low levels of TAp63 and maspin indicated a poor prognosis in HCC patients.
Materials and methods
Cell culture and transfection
Human HCC cell lines, Hep3B (p53-null) and HepG2 (p53-WT) were purchased from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). The normal hepatocyte, THLE-3, was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). All the cells were cultured in RPMI 1640 medium containing 10% fetal calf serum, 100 units/mL penicillin, and 100 μg/mL streptomycin in 5% CO2 and 95% air at 37°C. Transfection of plasmids and small interfering RNAs (siRNAs) was performed with X-tremeGENE HP DNA Transfection Reagent (Roche Applied Science, Mannheim, Germany) according to the manufacturer’s protocol. The Hep3B and HepG2 cells with shHDAC5 were constructed as described previously [5].
Quantitative real-time PCR (RT-PCR)
Total RNA was extracted using the RNAisoTM Plus reagent (Takara, Otsu, Japan) and reverse-transcribed using a PrimeScriptTM RT reagent kit (Takara). Quantitative PCR (qPCR) was performed with SYBR Green Mix (Takara) according to the manufacturer’s instructions. β-Actin served as loading control.
Western blot
Western blot was carried out according to a previous study [18]. The whole cell lysate was examined using the antibodies against HDAC5 (sc-133225; Santa Cruz Biotechnology, Santa Cruz, CA, USA); maspin (sc-166260; Santa Cruz); CyclinD1 (sc-8396; Santa Cruz); GAPDH (sc-32233; Santa Cruz); H3K9ac (ab4441; Abcam, Cambridge, UK); H3 (ab1791; Abcam); p53 (ab26; Abcam); ΔNp63 (ab167612; Abcam); TAp63 (ab53039; Abcam); p73 (ab40658; Abcam); CDK1 (ab18; Abcam); CDK2 (ab32147; Abcam); CDK4 (ab108357; Abcam). IRDye 800CW- or IRDye 680-conjugated secondary antibodies (LI-COR Biosciences, Lincoln, NE, USA) were used for staining and then detected by an Odyssey infrared imaging system (LI-COR).
qPCR array
The high-throughput profiling of proliferation and apoptosis-related genes were analyzed by the ExProfileTM Gene qPCR Array (GeneCopoeia) according to the instructions. In 96-well plates, there are 93 pairs of qPCR primers for the studied genes and 3 pairs for controls, which are used to monitor the efficiency of the entire experimental process (from reverse transcription to qPCR reaction). A cDNA pool that contained reverse transcript products from the total RNA of the indicated cells was used as the qPCR validation template.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed according to the previous study [19]. Briefly, chromatin was crosslinked using 1% formaldehyde for 10 min and sonicated to obtain DNA fragments of 200-500 bp. After centrifugation, the supernatants were subjected to immunoprecipitation overnight at 4°C with antibodies against H3K9ac (ab4441; Abcam). Protein A/G PLUS-Agarose (sc-2003; Santa) was used to isolate the chromatin-antibody complexes. The crosslinking was reversed, and the precipitated DNA fragments were purified and analyzed by qPCR with the following primers (TAp63 promoter): -157 CAGCTGGTAAGAATCGAGTG -138 (Forward); -48 GGATTTGAAGATACAGACTAT -27 (Reverse).
Cell proliferation assay
Cells in logarithmic growth were plated and transfected with the indicated siRNAs and expression plasmids. After an indicated time of culture, CCK-8 (Cell Counting Kit-8) (Dojindo Molecular Technologies, Maryland, USA) was added, and the OD450 was measured using an automatic plate reader.
EdU incorporation assay
The EdU (5-ethynyl-2-deoxyuridine) incorporation assay was used to represent DNA synthesis in cells. Cells were transfected with the indicated siRNAs and expression plasmids. Next, cells were washed 3 times with PBS and then incubated in serum-free RPMI 1640 with 10 mM EdU for 2 h. After extensive washing with PBS, cells were blocked with 10% FBS in PBS for 30 min. Incorporated EdU was detected by the fluorescent azide coupling reaction (Invitrogen). Images of the cells were captured with a fluorescence microscope (Nikon, Tokyo, Japan) and analyzed by ImageJ (National Institutes of Health, Bethesda, MD).
Flow cytometry
Approximately 1×106 Hep3B or HepG2 cells were re-suspended and fixed with 70% ethanol at -20°C for at least an hour. Before analysis, cells were re-suspended in PBS containing 100 mg/ml RNaseA (Roche) and 50 mg/ml PI (Sigma) for at least half an hour. The staining cells were analyzed immediately on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA) using the CellQuest 3.0 software system.
Immunofluorescence
Hep3B monolayer on glass coverslips was fixed for 15 min using 4% paraformaldehyde, permeabilized for 20 min in PBS containing 0.2% Triton X-100 and then blocked for 2 h with PBS containing 1% BSA and 0.5% goat serum at 37°C. The cells were incubated with primary antibody at 4°C overnight. After rinsing with PBS, they were probed with fluorescein isothiocyanate (FITC)- or tetramethylrhodamine-5-(and 6)-isothiocyanate (TRITC)-conjugated secondary antibodies (Santa Cruz) for 1 h at 37°C. The nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI; Sigma, St Louis, MO, USA) for 15 min. The slides were mounted and visualized by fluorescence microscope.
Immunohistochemistry
The study was performed with the approval of the Ethics Committee of Sanmen People’s Hospital of Zhejiang. In total, 164 human HCC samples were collected at Sanmen People’s Hospital of Zhejiang, and informed consent was obtained from all patients. The immunohistochemistry was performed using an EnVision Detection System (DAKO, Carpinteria, CA, USA) according to the manufacturer’s instructions. To estimate the score for each slide, at least 10 individual fields at 100× were chosen, and 100 cancer cells were counted in each field. The immunostaining intensity was divided into four grades: 0, negative; 1, weak; 2, moderate; and 3, strong. The proportion of positive staining cells was divided into five grades: 0, <5%; 1, 6%-25%; 2, 26%-50%; 3, 51%-75%; and 4, >75%. The staining results were evaluated and confirmed by two independent investigators blinded to the clinical data. The percentage of positivity of the tumor cells and the staining intensities were then multiplied to generate the IHC score and graded as low expression (score 0-6) and high expression (score 7-12). Cases with a discrepancy in scores were discussed to obtain a consensus.
Statistics
A database was created and transferred to SPSS 22.0 and GraphPad Prism 5.0 for Windows. Statistical data analysis was performed using the two-tailed Student’s t-test, chi-squared, and ANOVA. Kaplan-Meier plots and log-rank tests were used for survival analysis. The Spearman test was used in analyzing the correlation. The results are presented as the mean ± S.E of three separate experiments. A P value <0.05 was considered statistically significant.
Results
High expression of HDAC5 in HCC tissues indicates a poor prognosis
To characterize the expression of HDAC5 in HCC, immunohistochemical analysis of the HCC tissues from 164 cases was performed. As shown in Figure 1A, a higher level of HDAC5 immunostaining was detected in the HCC tissue compared with the adjacent normal tissue. Consistently, HDAC5 mRNA was also aberrantly overexpressed in most HCC tissues (Figure 1B). Further investigations confirmed that the HDAC5 staining was positively correlated with tumor size, intrahepatic metastasis, and distant metastasis (Table 1). In addition, the overall survival (OS) rate of the HCC patients with HDAC5 high expression was significantly lower than that of the patients with low expression (32.94% vs 50.63%, P=0.0371) (Figure 1C). These results support that the abnormal high expression of HDAC5 may lead to a poor survival in HCC patients.
Figure 1.
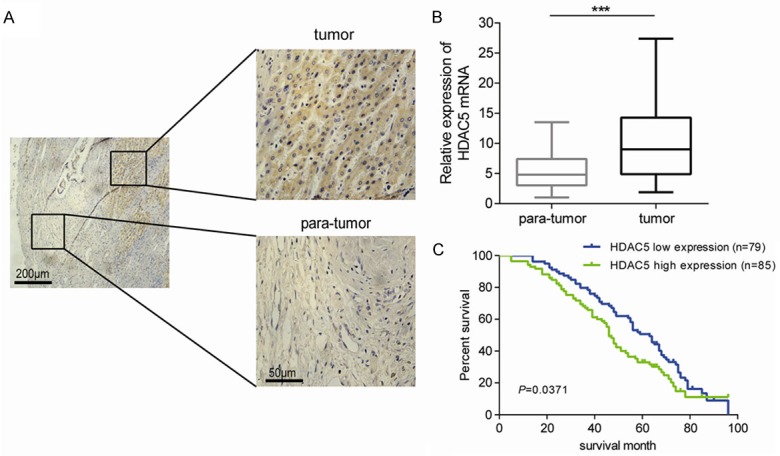
High expression of HDAC5 in HCC tissues indicates a poor prognosis. A. A representative image of HDAC5 immunostaining in HCC tissue and its adjacent normal tissue. B. Relative mRNA levels of HDAC5 in HCC tissues compared with adjacent tissues. ***P<0.001. C. The overall survival of HCC patients with low and high expression of HDAC5 was shown.
Table 1.
Correlation of the expression of HDAC5, TAp63 and maspin with clinicopathological features in HCC
| Cases | HDAC5 expression | TAp63 expression | Maspin expression | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Low | High | P Value | Low | High | P Value | Low | High | P Value | ||
|
|
|
|
||||||||
| Cases | Cases | Cases | Cases | Cases | Cases | |||||
| 164 | 79 | 85 | 111 | 53 | 103 | 61 | ||||
| Gender | 0.1986 | 0.6559 | 0.8653 | |||||||
| Male | 123 | 65 | 58 | 83 | 40 | 75 | 48 | |||
| Female | 41 | 14 | 27 | 28 | 13 | 28 | 13 | |||
| Age | 0.5226 | 0.4999 | 0.5510 | |||||||
| <60 | 93 | 46 | 47 | 61 | 32 | 58 | 35 | |||
| ≥60 | 71 | 33 | 38 | 50 | 21 | 45 | 26 | |||
| Tumor size | 0.0478* | 0.0315* | 0.0413* | |||||||
| <5 cm | 65 | 35 | 30 | 34 | 31 | 33 | 32 | |||
| ≥5 cm | 99 | 44 | 55 | 77 | 22 | 70 | 29 | |||
| AFP | 0.8482 | 0.6417 | 0.9750 | |||||||
| ≤20 U/L | 56 | 27 | 29 | 37 | 19 | 31 | 25 | |||
| >20 U/L | 108 | 52 | 56 | 74 | 34 | 72 | 36 | |||
| HBsAg | 0.7631 | 0.6359 | 0.8470 | |||||||
| Positive | 147 | 71 | 76 | 100 | 47 | 93 | 54 | |||
| Negative | 17 | 8 | 9 | 11 | 6 | 10 | 7 | |||
| Liver cirrhosis | 0.0750 | 0.8622 | 0.7283 | |||||||
| Yes | 121 | 53 | 68 | 81 | 40 | 78 | 43 | |||
| No | 43 | 26 | 17 | 30 | 13 | 25 | 18 | |||
| TNM | 0.1127 | 0.2255 | 0.2933 | |||||||
| I/II | 69 | 36 | 33 | 43 | 26 | 39 | 30 | |||
| III/IV | 95 | 43 | 52 | 68 | 27 | 64 | 31 | |||
| Edmondson stage | 0.9008 | 0.8260 | 0.1169 | |||||||
| I/II | 86 | 42 | 44 | 56 | 30 | 50 | 36 | |||
| III/IV | 78 | 37 | 41 | 55 | 23 | 53 | 25 | |||
| Intrahepatic metastasis | 0.0069* | 0.0007* | 0.0025* | |||||||
| Yes | 51 | 16 | 35 | 46 | 5 | 45 | 6 | |||
| No | 113 | 63 | 50 | 65 | 48 | 58 | 55 | |||
| Distant metastasis | 0.0027* | 0.0399* | 0.0314* | |||||||
| Yes | 39 | 12 | 27 | 30 | 9 | 27 | 12 | |||
| No | 125 | 67 | 58 | 81 | 44 | 76 | 49 | |||
AFP: alpha-fetoprotein.
P<0.05.
Stable knockdown of HDAC5 attenuates the proliferation of HCC cells
To analyze the expression of HDAC5 in HCC cells, we examined the levels of HDAC5 protein in an immortalized liver cell line (THLE-3) and two HCC cell lines (Hep3B and HepG2). The results showed an evidently higher expression of HDAC5 in Hep3B and HepG2 cells than in the THLE-3 cells (Figure 2A). Then, Hep3B and HepG2 cells with stable knockdown of HDAC5 were constructed. As expected, the level of acetylated H3 in both Hep3B and HepG2 cells increased after knocking down HDAC5 (Figure 2B). Furthermore, it was noticed that knockdown of HDAC5 reduced the proliferation of Hep3B and HepG2 cells (Figure 2C). Due to the observed effect of HDAC5 knockdown on Hep3B growth, we studied the change in the expression profiles of proliferation and apoptosis-related genes in Hep3B after HDAC5 knockdown (Table S1). As described in the heatmap, the mRNA levels of p63 and maspin (SERPINB5) significantly increased after stable knockdown of HDAC5, while the cell cycle drivers, CDK1/2/4 and cyclinD1, decreased to some extent (Figure 2D).
Figure 2.
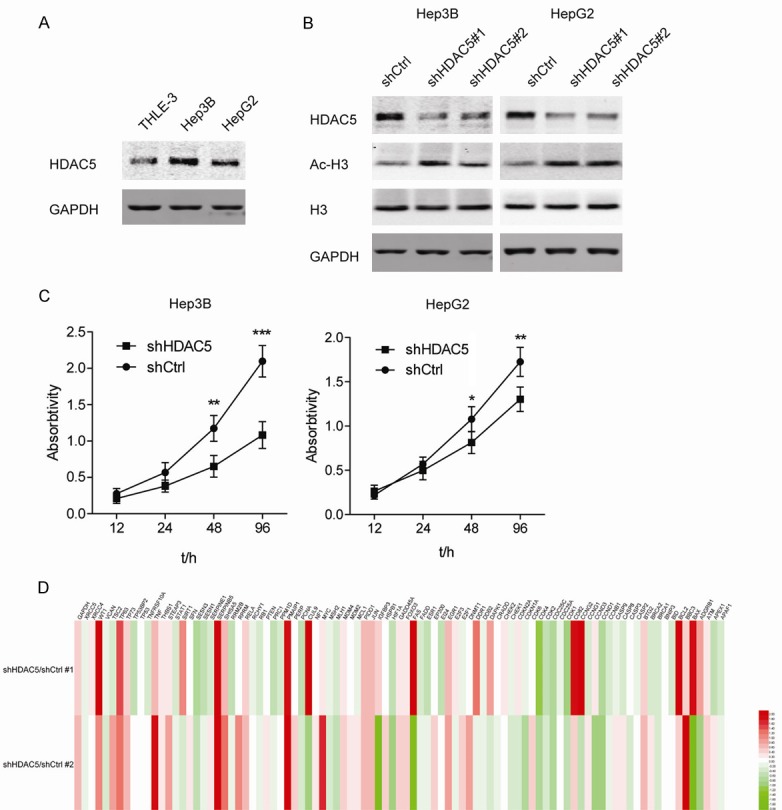
Stable knockdown of HDAC5 attenuates the proliferation of HCC cells. A. Western blot analyses of HDAC5 in THLE-3, Hep3B, and HepG2 cells. B. Western blot analyses of HDAC5, Ac-H3K9 and total H3 in Hep3B and HepG2 cells stably transfected with shHDAC5#1 and #2. C. Cell viability was measured in Hep3B and HepG2 cells with stable knockdown of HDAC5. *P<0.05; **P<0.01; ***P<0.001. D. Heatmap of qPCR arrays for proliferation and apoptosis-related genes in Hep3B with or without stable knockdown of HDAC5. Each row represents an experiment. Each column represents a gene. Red color indicates an increased expression. Green color indicates a decreased expression.
Stable knockdown of HDAC5 induces TAp63 expression which inhibits the proliferation of HCC cells
Considering the different p53 status in Hep3B and HepG2 cells, we focused on the p53 family members including p53, p63 (TAp63 and ΔNp63) and p73. In p53-null Hep3B cells, knockdown of HDAC5 upregulated the mRNA and protein levels of TAp63, ΔNp63, and p73 (Figure 3A and 3B). However, in p53-WT HepG2 cells, the expressions of p53, TAp63, and p73 were induced by HDAC5 knockdown (Figure 3A and 3B). Notably, ΔNp63 could not be detected in HepG2 cells by Western blot. In addition, as exogenous HDAC5 increased, the mRNA and protein level of TAp63 gradually decreased in THLE-3 cells (Figure 3C and 3D). In view of the mechanism for HDAC-mediated gene silence, the level of acetylated H3 binding to the TAp63 gene promoter was determined by ChIP. As shown in Figure 3E, a higher relative enrichment of active histone modification H3K9ac at the TAp63 promoter was observed in HDAC5-deprived Hep3B cells than in the control, suggesting that overexpressed HDAC5 in HCC cells may suppress the transcription of TAp63. Similarly, with HDAC5 knockdown, overexpression of TAp63 inhibited the proliferation of Hep3B and HepG2 cells (Figure 3F). In addition, the exogenous TAp63 markedly reduced the protein levels of CDK2, CDK4, and CyclinD1 in Hep3B and HepG2 cells (Figure 3G). Cell cycle analysis showed that TAp63-overexpressed Hep3B cells underwent arrest in G1 phases, with a strong depletion of cells entering the S phase, however, HepG2 cells with TAp63 overexpression went through a relatively slight S phase arrest (Figure 3H). Altogether, these findings indicate that HDAC5 may promote the proliferation of HCC cells via repressing the transcription of TAp63.
Figure 3.

Stable knockdown of HDAC5 induces TAp63 expression, which inhibits the proliferation of HCC cells. A. The mRNA level of p53, TAp63, ΔNp63 and p73 in Hep3B and HepG2 cells with stable knockdown of HDAC5 was analyzed by RT-PCR. *P<0.05. B. Western blot analyses of p53, TAp63, ΔNp63 and p73 in Hep3B and HepG2 cells with stable knockdown of HDAC5. C. Western blot analyses of HDAC5 and TAp63 in THLE-3 cells transfected with different amounts of HDAC5 expression plasmids. D. The mRNA level of TAp63 in THLE-3 cells transfected with different amounts of HDAC5 expression plasmids. E. The binding of H3 and acetylated H3 proteins to TAp63 gene promoter was detected by ChIP-PCR in Hep3B cells with stable knockdown of HDAC5. *P<0.05. F. Cell viability was measured in Hep3B and HepG2 cells with overexpression of TAp63. *P<0.05; **P<0.01; ***P<0.001. G. Western blot analyses of CDK1, CDK2 CDK4 and CyclinD1 in Hep3B and HepG2 cells with overexpression of TAp63. H. Cell cycle profiles of Hep3B and HepG2 with overexpression of TAp63 by flow cytometry.
Stable knockdown of HDAC5 increased the expression of maspin in a TAp63-dependent manner
In consideration of the dramatic upregulation of maspin observed in the heatmap, we next explored the maspin expression in HCC cells with HDAC5 knockdown. The results showed that knockdown of HDAC5 upregulated maspin at both mRNA and protein levels in Hep3B and HepG2 cells (Figure 4A and 4B). Immunofluorescence also confirmed the maspin expression induced by HDAC5 knockdown in HCC cells (Figure 4C). Since TAp63 directly regulates the expression of maspin [16], it is necessary to determine whether TAp63 mediates the upregulation of maspin after knocking down HDAC5. As shown in Figure 4D, silencing TAp63 by siRNA impaired the induction of maspin that arose from HDAC5 knockdown in Hep3B cells. In addition, we found that overexpression of maspin retarded the growth of Hep3B and HepG2 cells (Figure 4E). These data reveal that TAp63 is required for the regulation of maspin by HDAC5 in HCC cells.
Figure 4.
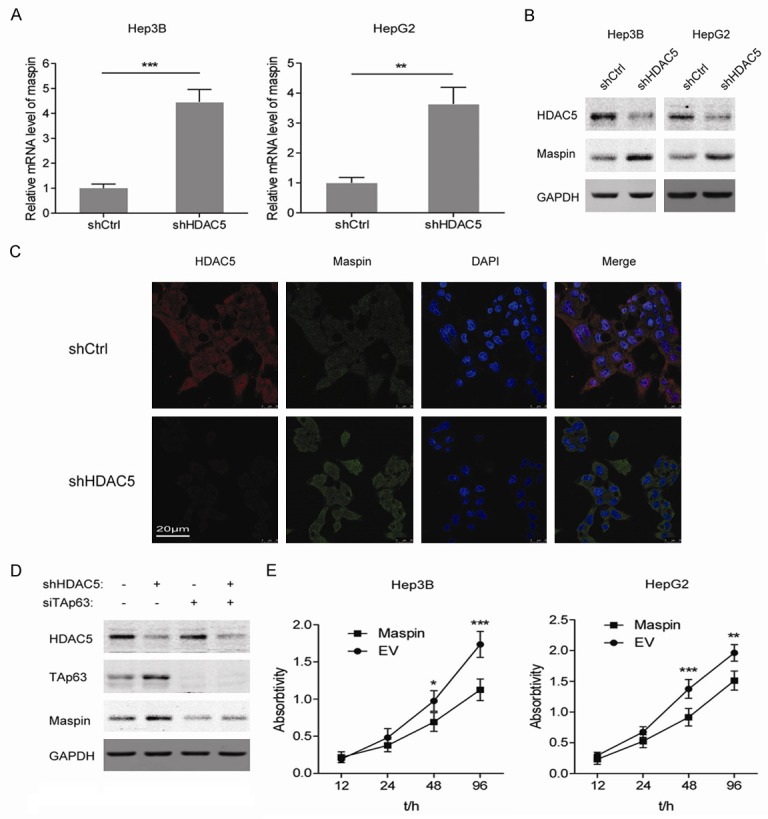
Stable knockdown of HDAC5 increased the expression of maspin in a TAp63-dependent manner. A. The mRNA level of maspin in Hep3B and HepG2 cells with stable knockdown of HDAC5 was analyzed by RT-PCR. **P<0.01; ***P<0.001. B. Western blot analyses of maspin in Hep3B and HepG2 cells with stable knockdown of HDAC5. C. Immunofluorescence analysis of HDAC5 and maspin proteins in Hep3B cells with stable knockdown of HDAC5. D. Western blot analyses of HDAC5, TAp63 and maspin in Hep3B cells with stable knockdown of HDAC5 and transient knockdown of TAp63. E. Cell viability was measured in Hep3B and HepG2 cells with overexpression of maspin. *P<0.05; **P<0.01; ***P<0.001.
Stable knockdown of HDAC5 suppresses the proliferation and tumorigenesis of HCC cells through recovering the TAp63-maspin pathway
To further elucidate the role of HDAC5-TAp63-maspin regulatory axis in HCC progression, the proliferation and colony-formation capabilities of HCC cells were determined. As shown in Figure 5A and 5B, compared with the control, a significant proliferation inhibition appeared in Hep3B and HepG2 cells with stable knockdown of HDAC5, while silencing TAp63 or maspin partially restored the proliferation. Consistently, knockdown of HDAC5 decreased the colony formation numbers of Hep3B and HepG2 cells, and deprivation of TAp63 or maspin rescued the attenuated tumorigenesis to a certain extent (Figure 5C). The above results indicate that the TAp63-maspin pathway is essential for inhibiting HCC proliferation and tumorigenesis by HDAC5 knockdown.
Figure 5.
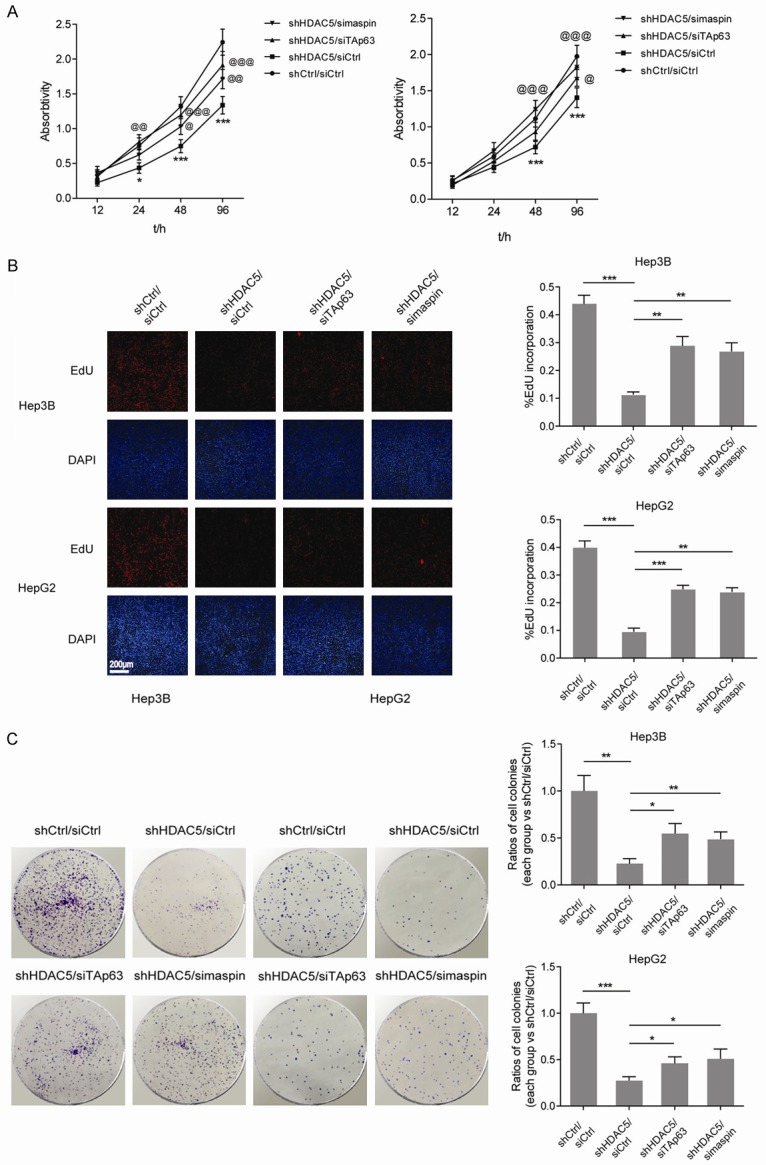
Stable knockdown of HDAC5 suppresses the proliferation and tumorigenesis of HCC cells through recovering the TAp63-maspin pathway. A. Cell viability was measured in Hep3B and HepG2 cells with stable knockdown of HDAC5 and transient knockdown of TAp63 or maspin. *,@P<0.05; **,@@P<0.01; ***,@@@P<0.001 (* vs shCtrl/siCtrl; @ vs shHDAC5/siCtrl). B. DNA synthesis was measured by EdU incorporation assays in Hep3B and HepG2 cells with stable knockdown of HDAC5 and transient knockdown of TAp63 or maspin. **P<0.01; ***P<0.001. C. Surviving colonies of Hep3B and HepG2 cells with stable knockdown of HDAC5 and transient knockdown of TAp63 or maspin. *P<0.05; **P<0.01; ***P<0.001.
High HDAC5 expression is associated with a suppressed TAp63-maspin pathway and a poor prognosis
To study the relationships among HDAC5, TAp63, and maspin in HCC, immunohistochemical (IHC) analysis of HCC tissues with serial sections was performed. The representative images showed that the case with a low expression of HDAC5 had stronger TAp63 and maspin stainings than that with high expression (Figure 6A). Clinical relevance analysis showed that the expressions of TAp63 and maspin were negatively associated with tumor size, intrahepatic metastasis, and distant metastasis (Table 1). Furthermore, statistical analysis of IHC negatively correlated the expression levels of HDAC5 with the degree of TAp63 and maspin (Figure 6B). The potential associations between immunostaining and overall survival (OS) were retrospectively evaluated in these patients. Kaplan-Meier analysis showed that OS was worse in patients with low TAp63 or maspin staining than in those with high staining (Figure 6C). These results suggest that highly expressed HDAC5 is closely related with the downregulation of TAp63 and maspin in HCC tissues, which is involved in HCC development.
Figure 6.
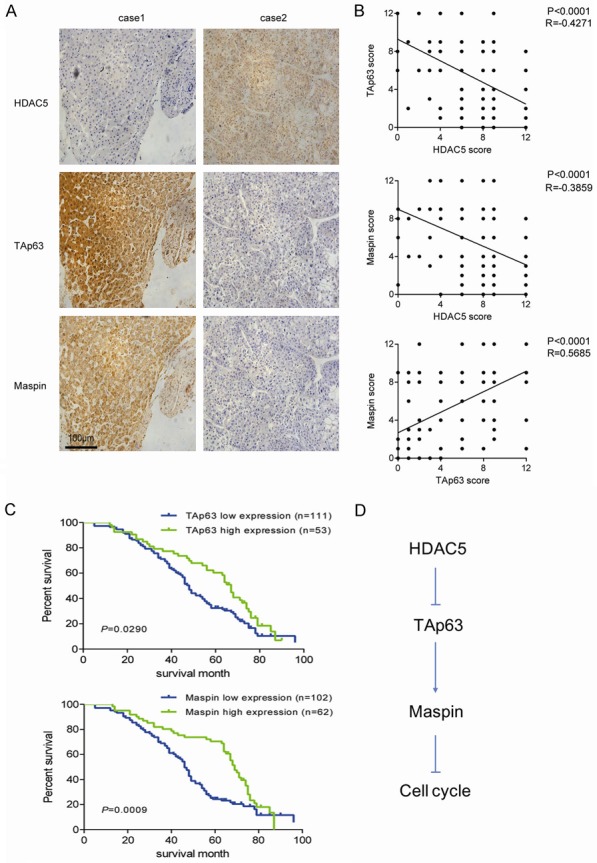
High HDAC5 expression is associated with a suppressed TAp63-maspin pathway and a poor prognosis. A. Representative images of HDAC5, TAp63, and maspin immunostaining in human HCC tissues. B. The correlation among concurrent immunostaining scores of HDAC5, TAp63, and maspin in HCC tissues. C. The overall survival of HCC patients with low and high expression of TAp63 or maspin was shown. D. The schema indicating that highly expressed HDAC5 accelerates cell cycle by disrupting the TAp63-maspin pathway in HCC cells.
Discussion
Emerging evidence indicated that normal expression of HDACs was remarkably disrupted in many human cancers. Lachenmayer et al. reported that expression of a subset of HDACs was increased in liver cancer compared to normal liver tissues [20]. In this circumstance, HDAC5 DNA copy numbers were altered, and its expression level was significantly upregulated in HCC [21]. Fan et al. found that inhibiting the anomalous expression of HDAC5 repressed growth of HCC cell lines by inducing apoptotic cell death and G1/S cell cycle arrest [6]. In the present study, we noticed that knockdown of HDAC5 suppressed the proliferation of both Hep3B and HepG2 cells, which was consistent with the report of Fan et al. However, a stronger proliferation inhibition by HDAC5 knockdown appeared in Hep3B cells than in HepG2 cells.
Through analyzing the expression profiles of proliferation and apoptosis-related genes in Hep3B, we found that p63, a p53 family member, increased significantly after knocking down HDAC5. Further investigations revealed that the expression of TAp63 but not ΔNp63 was largely influenced by HDAC5 silence. Even though HDAC5 knockdown also upregulated TAp63 in HepG2 cells, neither qPCR or Western blot detected ΔNp63. These results were consistent with the findings of Petitjean et al. who demonstrated that the activities of TAp63 and ΔNp63 promoters were differentially regulated, according to the p53 protein status of the cell [22]. The promoter of the ΔNp63 isoform was inhibited by the presence of wild-type p53, whereas activity of the promoter used to generate TAp63 appeared to be p53-independent. Therefore, we propose that knockdown of HDAC5 may induce TAp63 expression by activating the promoter of the TAp63 isoform in Hep3B and HepG2 cells.
Current studies regarding the regulation of TAp63 expression have mainly focused on the post-translational events, including the degradation pathway through the ubiquitin-proteosome system. However, the transcriptional regulation of the TAp63 gene has been poorly understood. An OCT4 binding site within the TAp63 promoter activated its expression, suggesting the involvement of TAp63 in stem cell regulation [23]. Yao et al. reported that overexpression of c-jun enhanced TAp63 transcription in Hep3B cells [24]. Our present work suggested that highly expressed HDAC5 in HCC cells may suppress the transcription of TAp63 through decreasing the acetylation level of H3K9 at the TAp63 promoter. Consistently, the HDAC inhibitor, valproic acid, showed a strong promotive effect on TAp63 expression in HCC cells [25]. However, Sayan et al. discovered that induction of TAp63 expression by the HDAC inhibitor TSA in HCT116 cells depended on p53, as p53-positive but not negative HCT116 cells succeeded in inducing significant TAp63 expression following treatment with TSA [26]. According to our findings, inhibition of HDAC5 resulted in an increased expression of TAp63 in HepG2 and Hep3B cells. Since HepG2 and Hep3B cells express with and without wild-type p53 respectively, HDAC5-mediated regulation of TAp63 seems to be independent of p53 in HCC cells.
In general, many functional parallels are found between TAp63 and p53. Overexpression of TAp63 closely mimics the transcriptional activity and the biological function of p53. Reporter assays showed that TAp63 was capable of transactivating a gene set, including many p53 targets involved in anti-proliferative and pro-apoptotic progression, such as Bax, p21, 14-3-3σ, and so on [9]. Indeed, TAp63 could induce cell cycle arrest and apoptosis [27,28]. Yao et al. found that doxorubicin increased the expression of TAp63 and knockdown of the acquired TAp63 attenuated doxorubicin-induced cell growth arrest in Hep3B cells, suggesting that TAp63 may play a compensatory role in cell cycle regulation, especially in p53-deficient cancer cells [29]. Here, our results showed that TAp63 overexpression reduced the proliferation of both Hep3B and HepG2 cells with a remarkable decrease in S phase. In accord with HDAC5 knockdown, ectopic expression of TAp63 caused a stronger inhibition on Hep3B than HepG2 cells, indicating that TAp63 plays a greater anti-proliferative function in p53-deficient HCC cells.
Except for TAp63, maspin (SERPINB5) was found elevated in qPCR arrays. Further study showed that TAp63 was dispensable for the induction of maspin after HDAC5 knockdown. In addition, exogenous maspin aroused a growth inhibition of HCC cells. The above findings suggest that HDAC5 promotes the proliferation of HCC cells by repressing the TAp63-maspin pathway. Phenotype experiments also indicated that interrupting the TAp63-maspin pathway by knockdown of TAp63 or maspin partially recovered the proliferative and tumorigenic abilities undermined by HDAC5 knockdown. In addition, Fujisawa et al. reported that five HCC cell lines including Hep3B and HepG2 exhibited extensive hypoacetylation at the maspin promoter, and treatment with TSA activated maspin expression with increased histone acetylation at the maspin promoter [30]. Thus, epigenetic regulation of maspin may also confer its loss in HCC cells, and the mechanism requires further definition.
A large number of published studies with human specimens indicated that maspin expression predicted a better prognosis for several types of carcinomas, including breast, prostate, colon, and oral squamous cell carcinoma [31]. In this study, clinical analysis showed that the patients with a lower level of maspin in HCC tissues had a shorter survival than those with a higher level. Additionally, the characteristics of clinicopathology, not only tumor size but intrahepatic and distant metastasis, were negatively related with maspin expression, suggesting that the role of maspin in HCC may not be limited to proliferation inhibition.
In conclusion, our findings reveal a HDAC5-induced inactivation of the TAp63-maspin pathway in HCC cells. More details suggest that highly expressed HDAC5 in HCC cells decreases the histone acetylation at the TAp63 promoter and inhibits the transcription of TAp63, consequently resulting in a downregulation of maspin, which functions as a cell-cycle suppressor (Figure 6D). Understanding the mechanism underlying the silence of the TAp63-maspin pathway may help to provide a more effective treatment for HCC patients, especially with loss of p53.
Acknowledgements
This work was supported by Zhejiang Medical and Health Science and Technology Plan (2018ZD055) and Science Technology Department of Zhejiang Province (2015c33262).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: what are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 2.Kim JK, Noh JH, Eun JW, Jung KH, Bae HJ, Shen Q, Kim MG, Chang YG, Kim SJ, Park WS, Lee JY, Borlak J, Nam SW. Targeted inactivation of HDAC2 restores p16INK4a activity and exerts antitumor effects on human gastric cancer. Mol Cancer Res. 2013;11:62–73. doi: 10.1158/1541-7786.MCR-12-0332. [DOI] [PubMed] [Google Scholar]
- 3.Liu C, Lv D, Li M, Zhang X, Sun G, Bai Y, Chang D. Hypermethylation of miRNA-589 promoter leads to upregulation of HDAC5 which promotes malignancy in non-small cell lung cancer. Int J Oncol. 2017;50:2079–2090. doi: 10.3892/ijo.2017.3967. [DOI] [PubMed] [Google Scholar]
- 4.Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ, Chang YG, Kim MG, Park WS, Lee JY, Lee SY, Chu IS, Nam SW. Histone deacetylase 6 functions as a tumor suppressor by activating c-Jun NH2-terminal kinase-mediated beclin 1-dependent autophagic cell death in liver cancer. Hepatology. 2012;56:644–657. doi: 10.1002/hep.25699. [DOI] [PubMed] [Google Scholar]
- 5.Ye M, Fang Z, Gu H, Song R, Ye J, Li H, Wu Z, Zhou S, Li P, Cai X, Ding X, Yu S. Histone deacetylase 5 promotes the migration and invasion of hepatocellular carcinoma via increasing the transcription of hypoxia-inducible factor-1alpha under hypoxia condition. Tumour Biol. 2017;39:1010428317705034. doi: 10.1177/1010428317705034. [DOI] [PubMed] [Google Scholar]
- 6.Fan J, Lou B, Chen W, Zhang J, Lin S, Lv FF, Chen Y. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumour Biol. 2014;35:11523–11532. doi: 10.1007/s13277-014-2358-2. [DOI] [PubMed] [Google Scholar]
- 7.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 8.Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, Leung ML, El-Naggar A, Creighton CJ, Suraokar MB, Wistuba I, Flores ER. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986–990. doi: 10.1038/nature09459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Candi E, Dinsdale D, Rufini A, Salomoni P, Knight RA, Mueller M, Krammer PH, Melino G. TAp63 and DeltaNp63 in cancer and epidermal development. Cell Cycle. 2007;6:274–285. doi: 10.4161/cc.6.3.3797. [DOI] [PubMed] [Google Scholar]
- 10.Zhu H, Mao Q, Liu W, Yang Z, Jian X, Qu L, He C. Maspin suppresses growth, proliferation and invasion in cutaneous squamous cell carcinoma cells. Oncol Rep. 2017;37:2875–2882. doi: 10.3892/or.2017.5574. [DOI] [PubMed] [Google Scholar]
- 11.Wei X, Li J, Xie H, Wang H, Wang J, Zhang X, Zhuang R, Lu D, Ling Q, Zhou L, Xu X, Zheng S. Chloride intracellular channel 1 participates in migration and invasion of hepatocellular carcinoma by targeting maspin. J Gastroenterol Hepatol. 2015;30:208–216. doi: 10.1111/jgh.12668. [DOI] [PubMed] [Google Scholar]
- 12.Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, Cheresh DA, Karin M. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature. 2007;446:690–694. doi: 10.1038/nature05656. [DOI] [PubMed] [Google Scholar]
- 13.Denk AE, Bettstetter M, Wild PJ, Hoek K, Bataille F, Dietmaier W, Bosserhoff AK. Loss of maspin expression contributes to a more invasive potential in malignant melanoma. Pigment Cell Res. 2007;20:112–119. doi: 10.1111/j.1600-0749.2007.00363.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Wang Y, Li S, Dong B, Zheng Q, Yan S, Ma Y, Zhang J, Fang J, Wu N, Wu H, Yang Y. Decreased maspin combined with elevated vascular endothelial growth factor C is associated with poor prognosis in non-small cell lung cancer. Thorac Cancer. 2014;5:383–390. doi: 10.1111/1759-7714.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sager R, Sheng S, Pemberton P, Hendrix MJ. Maspin. A tumor suppressing serpin. Adv Exp Med Biol. 1997;425:77–88. [PubMed] [Google Scholar]
- 16.Kim S, Han J, Kim J, Park C. Maspin expression is transactivated by p63 and is critical for the modulation of lung cancer progression. Cancer Res. 2004;64:6900–6905. doi: 10.1158/0008-5472.CAN-04-1657. [DOI] [PubMed] [Google Scholar]
- 17.Spiesbach K, Tannapfel A, Mossner J, Engeland K. TAp63gamma can substitute for p53 in inducing expression of the maspin tumor suppressor. Int J Cancer. 2005;114:555–562. doi: 10.1002/ijc.20766. [DOI] [PubMed] [Google Scholar]
- 18.Fang Z, Gong C, Liu H, Zhang X, Mei L, Song M, Qiu L, Luo S, Zhu Z, Zhang R, Gu H, Chen X. E2F1 promote the aggressiveness of human colorectal cancer by activating the ribonucleotide reductase small subunit M2. Biochem Biophys Res Commun. 2015;464:407–415. doi: 10.1016/j.bbrc.2015.06.103. [DOI] [PubMed] [Google Scholar]
- 19.Fang Z, Lin A, Chen J, Zhang X, Liu H, Li H, Hu Y, Zhang X, Zhang J, Qiu L, Mei L, Shao J, Chen X. CREB1 directly activates the transcription of ribonucleotide reductase small subunit M2 and promotes the aggressiveness of human colorectal cancer. Oncotarget. 2016;7:78055–78068. doi: 10.18632/oncotarget.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A, Minguez B, Tsai HW, Ward SC, Thung S, Friedman SL, Llovet JM. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol. 2012;56:1343–1350. doi: 10.1016/j.jhep.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu LM, Yang Z, Zhou L, Zhang F, Xie HY, Feng XW, Wu J, Zheng SS. Identification of histone deacetylase 3 as a biomarker for tumor recurrence following liver transplantation in HBV-associated hepatocellular carcinoma. PLoS One. 2010;5:e14460. doi: 10.1371/journal.pone.0014460. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Petitjean A, Cavard C, Shi H, Tribollet V, Hainaut P, Caron de Fromentel C. The expression of TA and DeltaNp63 are regulated by different mechanisms in liver cells. Oncogene. 2005;24:512–519. doi: 10.1038/sj.onc.1208215. [DOI] [PubMed] [Google Scholar]
- 23.Ng WL, Chen G, Wang M, Wang H, Story M, Shay JW, Zhang X, Wang J, Amin AR, Hu B, Cucinotta FA, Wang Y. OCT4 as a target of miR-34a stimulates p63 but inhibits p53 to promote human cell transformation. Cell Death Dis. 2014;5:e1024. doi: 10.1038/cddis.2013.563. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Yao JY, Pao CC, Chen JK. Transcriptional activity of TAp63 promoter is regulated by c-jun. J Cell Physiol. 2010;225:898–904. doi: 10.1002/jcp.22300. [DOI] [PubMed] [Google Scholar]
- 25.Sun G, Mackey LV, Coy DH, Yu CY, Sun L. The histone deacetylase inhibitor vaproic acid induces cell growth arrest in hepatocellular carcinoma cells via suppressing notch signaling. J Cancer. 2015;6:996–1004. doi: 10.7150/jca.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sayan BS, Yang AL, Conforti F, Bernardini S, Tucci P, Vasa-Nicotera M, Knight RA, Melino G. Induction of TAp63 by histone deacetylase inhibitors. Biochem Biophys Res Commun. 2010;391:1748–1751. doi: 10.1016/j.bbrc.2009.12.147. [DOI] [PubMed] [Google Scholar]
- 27.Gressner O, Schilling T, Lorenz K, Schulze Schleithoff E, Koch A, Schulze-Bergkamen H, Lena AM, Candi E, Terrinoni A, Catani MV, Oren M, Melino G, Krammer PH, Stremmel W, Muller M. TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J. 2005;24:2458–2471. doi: 10.1038/sj.emboj.7600708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melino G, Lu X, Gasco M, Crook T, Knight RA. Functional regulation of p73 and p63: development and cancer. Trends Biochem Sci. 2003;28:663–670. doi: 10.1016/j.tibs.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Yao JY, Chen JK. TAp63 plays compensatory roles in p53-deficient cancer cells under genotoxic stress. Biochem Biophys Res Commun. 2010;403:310–315. doi: 10.1016/j.bbrc.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 30.Fujisawa K, Maesawa C, Sato R, Wada K, Ogasawara S, Akiyama Y, Takeda M, Fujita T, Otsuka K, Higuchi T, Suzuki K, Saito K, Masuda T. Epigenetic status and aberrant expression of the maspin gene in human hepato-biliary tract carcinomas. Lab Invest. 2005;85:214–224. doi: 10.1038/labinvest.3700214. [DOI] [PubMed] [Google Scholar]
- 31.Sheng S. The promise and challenge toward the clinical application of maspin in cancer. Front Biosci. 2004;9:2733–2745. doi: 10.2741/1432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.