

Abstract
NK1.1-CD4+NKG2D+ cells exert their immune-regulatory function in tumor as an unconventional regulatory T cell subset through the production of TGF-β1; however, the molecular mechanisms involving with the activation of nuclear factors for TGF-β1 transcription remain unclear. Here we determined that the PI3K-p85α subunit was specifically activated in NK1.1-CD4+NKG2D+ cells following an 8-hour stimulation by sRAE-1 or α-CD3/sRAE-1, subsequently leading to the activation of PI3K-p110, Akt, and JNK. On the contrary, α-CD3/α-CD28 stimulation did not induce the activation of PI3K-p85 and JNK. Consequently, activation of the nuclear transcription factor AP-1 as a consequence of JNK activation regulated TGF-β1 expression in NK1.1-CD4+NKG2D+ cells. Furthermore, activation of NF-κB in NK1.1-CD4+NKG2D+ cells resulted from both protein kinase C activation downstream of TCR/CD3 signaling and PI3K activation induced by NKG2D engagement. The STAT3-Y705 phosphorylation, as activated by PI3K, under stimulations of the sRAE-1 or α-CD3/sRAE-1 also contributed to the TGF-β1 expression in NK1.1-CD4+NKG2D+ cells. Moreover, ChIP assay confirmed that STAT3 was capable of binding with the promoter regions of TGF-β1. In conclusion, our data showed that the TGF-β1 transcription in NK1.1-CD4+NKG2D+ cells induced by sRAE-1 or α-CD3/sRAE-1 was involved with the AP-1, NF-κB, and STAT3 signaling pathways; therefore, regulation of AP-1, NF-κB, and STAT3 activation may play important roles in the development and function of NK1.1-CD4+NKG2D+ cells.
Keywords: TGF-β1, expression, regulation, NK1.1-CD4+NKG2D+ cells, PI3K
Introduction
Regulatory T cells (Tregs) are important lymphocytes that maintain immune homeostasis. Besides conventional Tregs (CD4+CD25+Foxp3+), other non-conventional Tregs, including TGF-β1- and/or IL-10-producing regulatory T subsets, such as CD4+CD25- LAP (latency-associated peptide)+ [1], CD4+CD25-CD69+ [2], and CD4+CD25-CD45RO- LAG3 (lymphocyte activation gene 3)- cells [3]. CD4+NKG2D+ T cells with TGF-β1 production capacity have a role in patients bearing with NKG2D ligand-positive tumors [4] and in pediatric SLE patients [5]. We previously confirmed that increased CD4+NKG2D+ T cell numbers in the pCD86-RAE-1 transgenic mice could downregulate the function of NK cells [6]. We further divided CD4+NKG2D+ cells into two subsets: NK1.1-CD4+NKG2D+ and NK1.1+CD4+NKG2D+ cells. The NK1.1-CD4+NKG2D+ T cells also have a protective function in mice against DSS-induced colitis. Specifically, NK1.1-CD4+NKG2D+ cells highly express TGF-β1 and Fas ligand with low capacity to secrete IFN-γ, and lack the expression of IL-17A, IL4, Foxp3, granzyme, and perforin [7]. Induction of NK1.1-CD4+NKG2D+ T cells majorly depends on the costimulation of TCR/CD3 and NKG2D receptors [4]. However, the detailed molecular mechanisms of TGF-β1 transcription in this non-conventional Treg subset remain elusive.
After engagement with the MHC-antigenic peptide complex, TCR/CD3 crosslinks and leads to activation of ZAP70 (Zeta-chain associated protein 70) and the adapter protein (LAT). As a consequence, phospholipase C-γ1 (PLC-γ1) is activated to hydrolyze phosphatidylinositol diphosphate (PIP2) to inositol-1,4,5-trisphosphate (IP3) and diacyl glycerol (DAG). Upon activation of calcineurin by IP3, the transcription factor NF-AT becomes dephosphorylated and rapidly translocates to the nucleus for initiation of downstream gene expression. Meanwhile, DAG activates protein kinase C (PKC) and the downstream NF-κB pathway. Furthermore, ZAP70 can also activate Ras and mitogen-activated protein kinase (MAPK) and promote the generation of the nuclear transcription factor, AP-1 (c-Jun and c-Fos complex). Thus, the TCR/CD3 signaling pathway generally promotes T cell activation dependent on NF-AT, NF-κB, and AP-1 nuclear factors [8,9].
NKG2D is an important receptor for NK cell activation [10-12]. NKG2D is also expressed in CD8+, γδ+, and some CD4+ T cells [12,13]. Human NKG2D ligands are MHC class I chain-related protein A/B (MICA/MICB) and cytomegalovirus (CMV) UL16-binding proteins (ULBPs), which are generally expressed in stressed or tumor cells. Mouse NKG2D binds to retinoic acid early inducible-1 protein (RAE-1), H60, and murine UL16-binding protein-like transcript (MULT)-1 [14]. NKG2D transduces activated signals mainly via the adaptor protein DAP10 and/or DAP12. Phosphorylation of adaptor proteins can recruit growth factor binding protein-2 (Grb-2) and VAV-1. PLC-γ2, which is activated by Grb-2/VAV-1, similarly hydrolyzes PIP2 to generate IP3 and DAG. The YINM motif of DAP10 also binds to the p85 subunit of phosphatidylinositol 3-kinase (PI3K) upon activation, which in turn activates downstream molecules, such as Akt, Erk, JNK, and p38, and initiates expression of downstream genes though nuclear transcriptional factors [15,16].
Transcription of TGF-β gene can be regulated by AP-1 [17,18], SP-1 [19], NF-κB [18,20], Egr-1 (early growth response-1) [21], STAT3 [22,23], and ZF9/core promoter binding protein [24]. Here, we sought to determine whether TGF-β1 transcription in NK1.1-CD4+NKG2D+ cells after TCR/CD3 and NKG2D co-engagement was involved with AP-1, NF-κB, and STAT3. Additionally, the preceding molecular events of these nuclear factors were also analyzed.
Materials and methods
Cell line and mice
The pCD86-RAE-1ε transgenic mice were generated as previously described [6]. All animal procedures were approved by the Animal Care and Use Committee of Yangzhou University. The murine colon cancer cell line (MC38) was kindly gifted by Dr. Hursting (University of Texas-Austin).
Antibodies and reagents
The functional grade purified anti-CD3ε antibody (145-2C11) was purchased from BioLegend (San Diego, CA, USA) and anti-CD28 antibody (37.51) was obtained from eBioscience (San Diego, CA, USA). The recombinant mouse Rae-1ε protein (1135-RA) was obtained from R&D systems (Minneapolis, MN, USA). The following antibodies used for flow cytometry were obtained from BioLegend: anti-CD4 (GK1.5), anti-NKG2D (CX5), and anti-TGF-β1 (TW7-16B4). 7-Amino-Actinomycin D (7-AAD) (BD Biosciences) was used for exclusion of death cells. All antibodies we used for Western blot were purchased from Cell Signaling Technology (Boston, MA, USA), except p-STAT3 (Ser727) primary antibody (Merck & Millipore; Billerica, MA, USA). SR11302 (TOCRIS; Minneapolis, MN), stattic (Selleck Chemicals; Boston, MA, USA), cryptotanshinone (Selleck Chemicals), bortezomib (Cell Signaling Technology), and LY294002 (Cell Signaling Technology) were dissolved in dimethyl sulfoxide (DMSO) and stored at -20°C until use. SR11032, cryptotanshinone, and Bortezomib were all used at 50 μM, while the concentrations of LY294002 and stattic were respectively 20 μM and 10 μM.
T cell isolation
The pCD86-RAE-1 transgenic mice were injected subcutaneously with MC38 cells (2×106/mice) at day 0. At day 21, mice were sacrificed and splenic mononuclear cells were prepared. Splenic NK1.1-CD4+ T cells were enriched from splenic single cell suspension by mouse CD4+ T lymphocyte enrichment set (BD Biosciences) following the manufacturer’s instructions, and then NKG2D+ T cells were isolated by indirectly labeled the cells with PE-conjugated anti-mouse NKG2D antibody (CX5) and anti-PE MicroBeads (Miltenyi Biotec GmbH; Bergisch Gladbach, Germany). Cells were passed through a LS column, and the double-positive T cells were collected. The purity of NK1.1-CD4+NKG2D+ T cells were typically more than 90% as identified by flow cytometry.
Cell stimulation
A round bottom 96-well plate was coated with anti-CD3 antibody (10 μg/ml) or anti-CD28 antibody (5 μg/ml) in PBS at 4°C overnight. NK1.1-CD4+NKG2D+ T cells obtained from MACS were resuspended in serum-free 1640 medium in pre-coated wells without antibiotics. As indicated in several groups, recombinant mouse sRAE protein (50 ng/ml) was added into the culture system. NK1.1-CD4+NKG2D+ T cells were harvested respectively for further analysis after 0.5-, 2-, 8-, and 16-hour stimulation.
Western blot analysis
NK1.1-CD4+NKG2D+ T cells were harvested and lysed by a whole-cell lysis kit (KeyGEN BioTECH; Jiangsu, China). Immunoblots were performed routinely. Western Blots were probed with antibodies to PI3K p85 (Lot number: 4292) (1:1000), PI3K p110α (C73F8) (1:1000), Akt (5G3) (1:1000), p-Akt (Ser473) (D9W9U) (1:1000), NF-κB p105/50 (D7H5M) (1:1000), NF-κB p65 (D14E12) (1:1000), NF-κB p-p65 (Ser536) (1:1000), STAT3 (79D7) (1:2000), p-STAT3 (Tyr705) (D3A7) (1:2000), p-STAT3 (Ser727)(07-703) (1:500), p44/42 MAPK (Erk1/2) (137F5) (1:1000), p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) (1:2000), p38 MAPK (D13E1) (1:1000), p-p38 MAPK (Thr180/Tyr182) (D3F9) (1:1000), and p-SAPK/JNK (Thr183/Tyr185) (81E11) (1:1000). Blots were subsequently incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (1:5000) and detected by chemiluminescence. GAPDH (D16H11) (1:1000) was used as a positive control.
Intracellular staining
Intracellular staining kit was purchased from eBioscience (San Diego, CA, USA). NK1.1-CD4+NKG2D+ T were cultured under various stimulations for 8 hours. Brefeldin A was added to the culture system after 4-h stimulating. Cells were then stained by surface antibodies for 30 min at 4°. Cells were then fixed, permeabilized, stained with APC- conjugated anti-TGF-β1 antibody or isotype antibody and analyzed by FCM.
Real-time PCR
RNA was extracted from NK1.1-CD4+NKG2D+ T cells using TRIzol reagent (Life Technologies; Carlsbad, CA, USA) and cDNA was generated by a QuantiTect® reverse transcription kit (QIAGEN GmbH; Hilden, Germany). The amplification of cDNA was conducted by the QuaniNovaTM SYBR® Green PCR kit (QIAGEN) on ABI 7500 (PE applied Biosystems, Carlsbad, CA, USA). Primers for TGF-1 and GAPDH were designed by Primer Premier 5.0 software. The primer pairs were 5’-GGCGGTGCTCGCTTTGTA-3’ and 5’-CACTGCTTCCCGAATGTCT-3’ (TGF-β1); 5’-CAAAATGGTGAAGGTCGGTGTG-3’ and 5’-TGATGTTAGTGGGGTCTC GCTC-3’ (GAPDH). Relative RNA expression was calculated by the 2-ΔΔCt method after normalizing expression levels of TGF-β1 mRNA to GAPDH mRNA.
Chromatin immunoprecipitation assay
NK1.1-CD4+NKG2D+ T cells were collected following the stimulation of anti-CD3ε and sRAE for 8 hours. Chromatin immunoprecipitation (ChIP) was performed by a SimpleChIP® enzymatic chromatin IP Kit (Magnetic Beads) (Cell Signaling Technology) following standard protocols. Immunoprecipitation was performed with STAT3 antibody (79D7) (Cell Signaling Technology), as well as the positive control antibody (H3) and the negative control antibody (normal rabbit IgG) included in the ChIP kit. The quantitative PCR analysis of IP samples was performed with following primer pairs: 5’-CAGGCTAGCCTTGAACT-3’ and 5’-AGCAGCTGAAGGAAGCT-3’ (STAT3-1); 5’-AGTGAGTGGGAGATGAGAACC-3’ and 5’-TGGAGTCGAGGCCAACCTG-3’ (STAT3-2); 5’-AAGGGCTGTGGGTTGGAG-3’ and 5’-GGGCAGACTTTGCGGATG-3’ (STAT3-3).
TGF-β1 ELISA
Concentrations of mouse TGF-β1 in cell culture supernatants were measured using sandwich ELISA kits (R&D systems) according to the manufacturer’s protocols.
Statistical analysis
Differences between groups were analyzed by Student’s t-test. Data were evaluated by one-way ANOVA followed by Dunnett’s test between control and various stimulation groups. Significance of differences was indicated when *P<0.05, **P<0.01, ***P<0.001.
Results
TGF-β1 production of NK1.1-CD4+NKG2D+ cells
We initially determined the level of TGF-β1 expression in NK1.1-CD4+NKG2D+ cells, purified from spleen of MC38 tumor-bearing transgenic mice, under various stimulatory conditions, namely α-CD3, sRAE-1, α-CD3/sRAE-1, and α-CD3/α-CD28 each at 0.5 h, 2 h, 8 h, or 16 h. As shown in Figure 1A and 1B, the highest expression level of TGF-β1 in NK1.1-CD4+NKG2D+ cells was obtained following 8-h stimulated by α-CD3/sRAE-1, but not by α-CD3/α-CD28. Moreover, sRAE-1 had a similar effect as α-CD3/sRAE-1 on TGF-β1 expression at the 8-h time-point. However, only α-CD3/sRAE-1 significantly promoted TGF-β1 expression after NK1.1-CD4+NKG2D+ cells were stimulated for 2-h or 16-h. There was no obvious expression of TGF-β1 after the 0.5-h stimulation.
Figure 1.
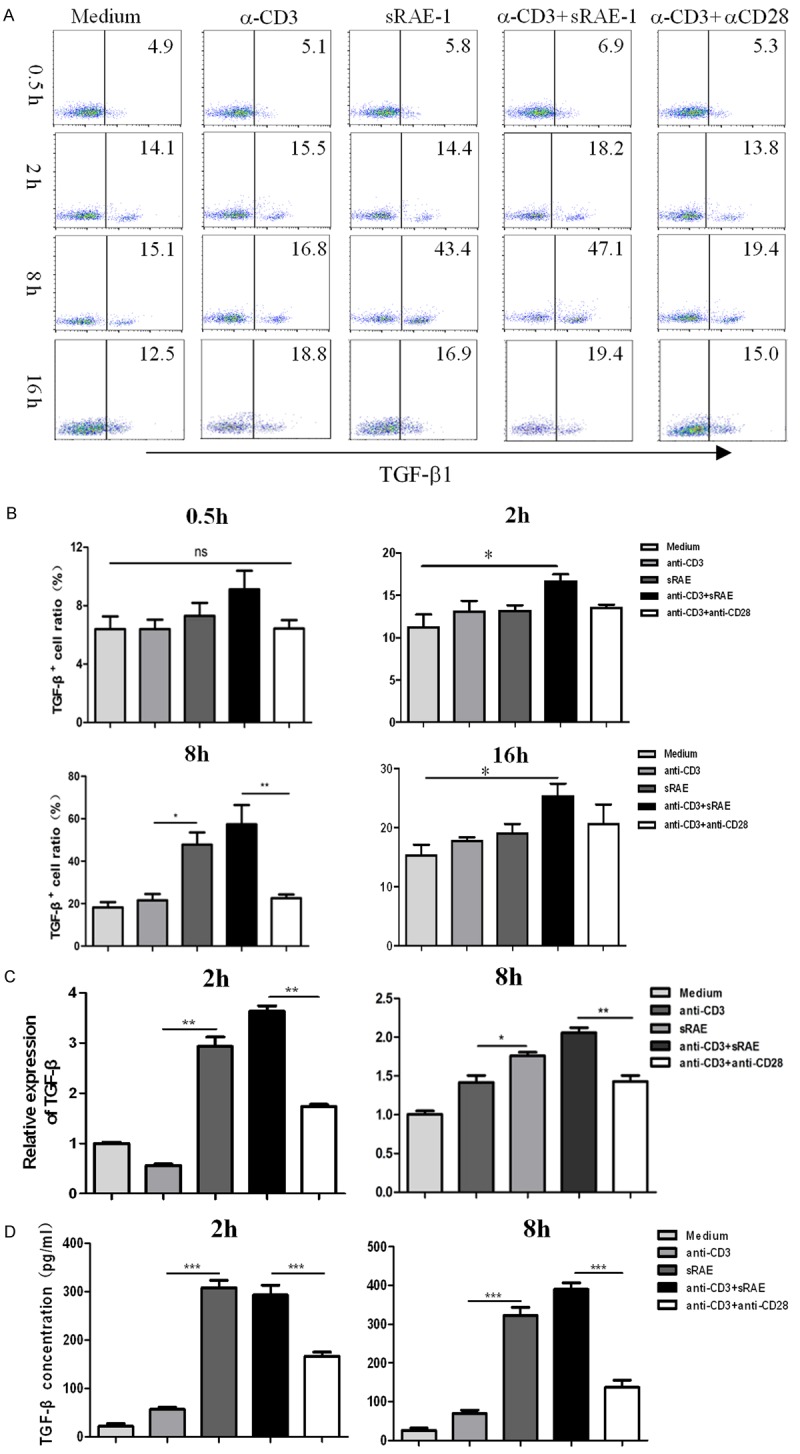
TGF-β1 production of NK1.1-CD4+NKG2D+ cells upon various stimulations at different times. A. TGF-β1 of NK1.1-CD4+NKG2D+ cells detected by flow cytometry at 0.5-, 2-, 8-, 16-hour stimulation. B. Statistical analysis of TGF-β1 variations at different time points. C. TGF-β1 transcription of NK1.1-CD4+NKG2D+ cells detected by real-time PCR. D. Concentration of TGF-β1 in supernatants of NK1.1-CD4+NKG2D+ cells upon indicated stimulations measured by ELISA. Each experiment was repeated at least thrice. *, P<0.05; **, P<0.01; ***, P<0.001; ns, no significance.
Next, mRNA levels of TGF-β1 were measured by various stimulations at 2- or 8-hour. As expected, α-CD3/sRAE-1 induced higher TGF-β1 transcription than α-CD3/α-CD28, or α-CD3 alone (Figure 1C), either at 2-h or 8-h. Simultaneously, sRAE-1 stimulation resulted in higher level of TGF-β1 mRNA than α-CD3/α-CD28, or α-CD3 alone. Secretion of TGF-β1 in supernatants of NK1.1-CD4+NKG2D+ cell culture systems was determined at 2-h and 8-h. Changes of TGF-β1 concentration by different stimulations showed similar patterns as TGF-β1 transcription (Figure 1D). Therefore, NK1.1-CD4+NKG2D+ cells produced the highest level of TGF-β1 upon stimulation with α-CD3/sRAE-1 or sRAE-1 for 8 hours.
The PI3K-p85/JNK, NF-κB and STAT3 activation of NK1.1-CD4+NKG2D+ cells
To investigate whether NKG2D initiates PI3K/Akt activating for promoting TGF-β1 transcription in NK1.1-CD4+NKG2D+ T cells, expressional changes of two subunits of PI3K (p110 and p85α) and its downstream Akt/pAkt were detected upon various stimulations. At 2-hour culture, PI3K-p85α, PI3K-p110, total Akt, and phosphorylated Akt (pAkt) were significantly increased by stimulations of either sRAE-1 or α-CD3/sRAE-1. CD3 antibody alone did not induce PI3K or Akt activation. Although there was no difference in PI3K-p110 expression between α-CD3/sRAE-1 and α-CD3/α-CD28, the PI3K-p85α and Akt/pAkt expression following the two stimulations was significantly increased (Figure 2A and 2B). With 8-h stimulation, PI3K-p85α and -p110, Akt and pAkt displayed similar variations to those of the 2-h stimulation, but the stimulatory effects were more significant (Figure 2C and 2D). We did not observe any changes of all above proteins in NK1.1-CD4+NKG2D+ cells with 0.5-h treatment (Figure S1). No difference in PI3K-p85α expression was seen at 16-h stimulation, but variations of Akt and pAkt were maintained at that time-point (Figure S2), which confirmed that the PI3K-p85α activation is an early molecular event.
Figure 2.

Activation of PI3K, MAPK, STAT3 and NF-κB signaling pathways. Western blot analysis of PI3K-p85, -p110, Akt, pAkt in NK1.1-CD4+NKG2D+ cells upon different stimulations at 2-hour (A) or 8-hour (C). Statistical analysis of expression variations of PI3K-p85, -p110, Akt, pAkt at 2-hour (B) or 8-hour (D). Analysis of JNK, Erk, p38 (E, F), NF-κB, STAT3 (G, H) and their phosphorylation in NK1.1-CD4+NKG2D+ cells upon stimulations for 8 hours. Each experiment was repeated at least thrice. *, P<0.05; **, P<0.01; ***, P<0.001; ns, no significance.
The activation of downstream signaling MAPK, STAT3 and NF-κB were also detected. JNK, Erk, and p38 are three key molecules of the MAPK signaling pathway [25]. As shown in Figure 2E and 2F, after 8-h culture, α-CD3/sRAE-1 significantly resulted in the phosphorylation of JNK in NK1.1-CD4+NKG2D+ cells, as compared to α-CD3/α-CD28. There was no significant difference of JNK activation upon stimulation of α-CD3 or sRAE-1. Phosphorylated Erk and p38 levels were enhanced after being stimulated by sRAE-1, α-CD3/sRAE-1, or α-CD3/-CD28, compared with culture medium or α-CD3 alone. However, there were no significant differences of pErk and pp38 between treatments of α-CD3/sRAE-1 and α-CD3/α-CD28.
After 0.5-h stimulation, we did not observe any differences of NF-κB activation by all treatments (Figure S3). With 2-h culture, only NF-κB p105 was increased by stimulations of α-CD3, sRAE-1, or α-CD3/sRAE-1 (Figure S4). At 8-h culture, NF-κB p50, p65, pp65 were all enhanced in groups stimulated by α-CD3, sRAE-1, or α-CD3/sRAE-1. Particularly, expression levels of NF-κB p65 and pp65 were higher with the treatment of α-CD3/sRAE-1 than α-CD3/α-CD28 (Figures 2G, 2H and S5). However, NF-κB p105 and p50 expression levels did not significantly vary at the same time. When 16-h culture was used, the stimulatory differences between α-CD3/sRAE-1 and α-CD3/α-CD28 disappeared, but sRAE-1 still had better effects on promoting NF-κB p65 and pp65 expression than α-CD3 (Figure S6), which confirmed that the NKG2D engagement alone resulted in NF-κB p65 activation.
Expression of STAT3 and its activated form (pY705 and pS727) had no significant changes after NK1.1-CD4+NKG2D+ cells were differently stimulated at 0.5- or 2-h (Figures S7 and S8). With 8-h culture, STAT3-pY705 was significantly increased by stimulations of sRAE-1 or α-CD3/sRAE-1, compared with those by culture medium, α-CD3, or α-CD3/α-CD28. STAT3-pS727 was activated by α-CD3 alone or α-CD3/sRAE-1 (Figure 2G and 2H), which was different from STAT3-pY705. There was a similar trend of STAT3-pY705 and pS727 expression with 16-h stimulation (Figure S9).
Inhibition of PI3K/JNK/AP-1 on TGF-β1 expression
To investigate whether the TGF-β1 expression depends on the PI3K activation, LY294002 (a PI3K inhibitor) was used to treat with NK1.1-CD4+NKG2D+ cells. As shown in Figure 3A and 3B, expression of TGF-β1 in NK1.1-CD4+NKG2D+ cells pre-treated with sRAE-1 or α-CD3/sRAE-1 was significantly inhibited by LY294002. As expected, there were no significant differences in other stimulatory conditions before or after LY294002 treatment. Thus, Akt, which was activated by sRAE-1 or α-CD3/sRAE-1 due to the upstream PI3K-p85α and -p110 activation, played a key role for TGF-β1 expression. We further confirmed that whether the phosphorylation of JNK is initiated by PI3K activation. Upon inhibition of PI3K using LY294002, pJNK was almost downregulated to the level before stimulation. This data inferred that JNK is activated by PI3K in NK1.1-CD4+NKG2D+ cells (Figure 3C and 3D).
Figure 3.

Inhibition of PI3K/JNK/AP-1 on TGF-β1 expression. NK1.1-CD4+NKG2D+ cells were treated with the PI3K inhibitor (LY294002) under different stimulations for 8 hours (A). (B) Statistical analysis of effects of LY294002 on TGF-β1 expression. (C, D) Effects on JNK/pJNK expression by the PI3K inhibitor (LY294002). Effects of JNK inhibitor (E, F), Erk or p38 inhibitor (G), and AP-1 inhibitor (H, I) on TGF-β1 expression of NK1.1-CD4+NKG2D+ cells. Each experiment was repeated at least thrice. *, P<0.05; **, P<0.01; ***, P<0.001; ns, no significance.
Inhibitors against JNK, Erk, p38, and AP-1 (c-Jun and c-Fos) were respectively used to treat NK1.1-CD4+NKG2D+ cells under various stimulations. As expected, inhibiting JNK activation by SU3327 almost blocked the TGF-β1 expression in the sRAE-1-, or α-CD3/sRAE-1-stimulating group, whereas no effects were observed in the rest groups (Figure 3E and 3F). Treatment with either Erk inhibitor (PD98059) or p38 inhibitor (VX-702) resulted in significantly decreased expression of TGF-β1 in all groups, which indicated that activations of Erk and p38 were not specific for TGF-β1 expression of NK1.1-CD4+NKG2D+ cells induced by sRAE-1 or α-CD3/sRAE-1 (Figure 3G). AP-1 inhibition also lead to wide inhibition of TGF-β1 expression (Figure 3H and 3I). However, AP-1 inhibition did not return TGF-β1 expression to the baseline level, which suggested other nuclear factors might be involved.
NF-κB inhibition decreases TGF-β1 expression
Treatment with Bortezomib, an inhibitor of NF-κB activation, resulted in the partial inhibition of TGF-β1 expression, which indicated that NF-κB was one of the regulators of TGF-β1 expression in NK1.1-CD4+NKG2D+ cells (Figure 4A and 4B). Next, we speculated whether NF-κB p65 was also activated by PI3K signaling as induced by NKG2D. After LY294002 treatment, NF-κB p65 activation induced by sRAE-1 or α-CD3/sRAE-1 was completely reversed (Figure 4C and 4D). Thus, the NF-κB activation in NK1.1-CD4+NKG2D+ cells could be induced by both TCR/CD3 and NKG2D signaling pathways and is involved with TGF-β1 transcription.
Figure 4.
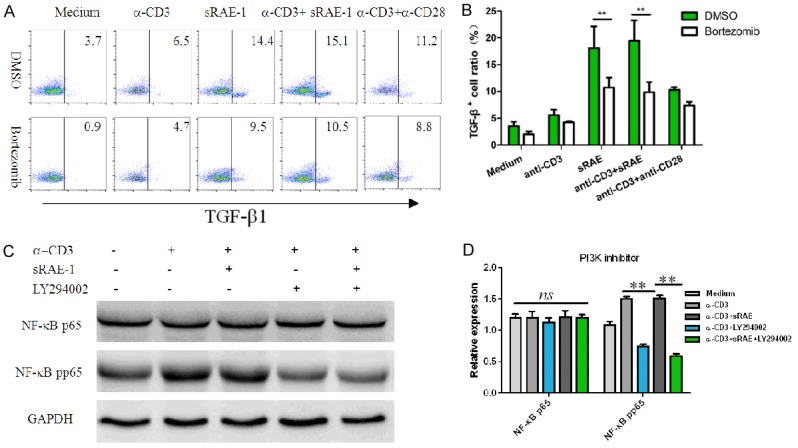
Inhibition of NF-κB on TGF-β1 expression. Effects of Bortezomib (NF-κB inhibitor) on TGF-β1 expression (A, B). Effects of LY294002 on NF-κB p65 activation in NK1.1-CD4+NKG2D+ cells for 8 hours (C, D). Each experiment was repeated at least thrice. *, P<0.05; **, P<0.01; ***, P<0.001; ns, no significance.
STAT3 inhibition suppresses TGF-β1 expression
Two STAT3 inhibitors (stattic and cryptotanshinone) were respectively treated with NK1.1-CD4+NKG2D+ cells under various stimulations. Both reagents resulted in significant inhibitions of TGF-β1 expression, which indicated that STAT3 was also a transcriptional factor for TGF-β1 expression in NK1.1-CD4+NKG2D+ cells (Figure 5A). Next, we sought to determine the upstream molecules for STAT3 activation in the unconventional regulatory T cells. PI3K could play a key role for STAT3 activation, because LY294002 almost blocked the phosphorylation of Y705 residue of STAT3 (Figure 5B and 5C). On the contrary, Bortezomib only downregulated STAT3-pY705 expression by 30% (Figure 5D and 5E). Therefore, co-ligation of TCR/CD3 and NKG2D in NK1.1-CD4+NKG2D+ cells could induce STAT3 activation for TGF-β1 expression via the PI3K signaling pathway.
Figure 5.
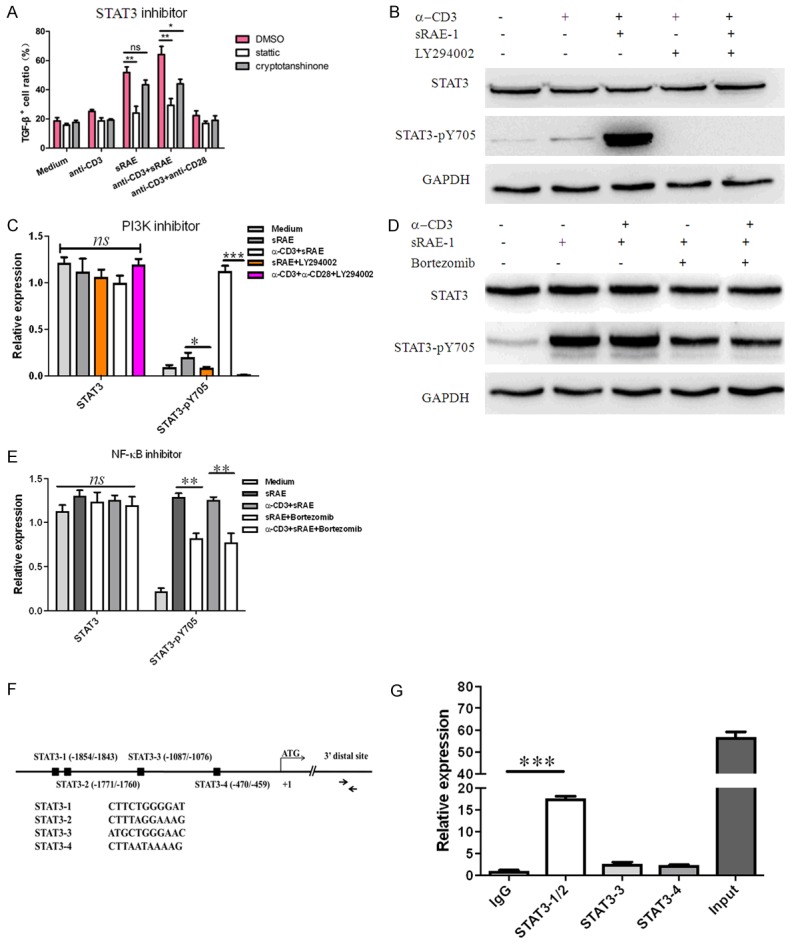
Inhibition of STAT3 on TGF-β1 expression. Effects of STAT3 inhibitors on TGF-β1 expression in NK1.1-CD4+NKG2D+ cells for 8 hours (A). (B, C) Effects of LY294002 on STAT3-pY705 phosphorylation. (D, E) Effects of Bortezomib on STAT3-pY705 phosphorylation. (F) The predicted binding sites of the TGF-β promoter by STAT3. (G) STAT3 engagement measured by a ChIP assay. Each experiment was repeated at least thrice. *, P<0.05; **, P<0.01; ***, P<0.001; ns, no significance.
STAT3 has been shown to regulate TGF-β1 expression in many human cancer cells, but not in murine cells [26]. We therefore investigated whether STAT3 directly bound to the TGF-β1 promoter in mouse NK1.1-CD4+NKG2D+ T cells by the ChIP assay. Four STAT3 binding sites of TGF-β1 promoter (STAT3-1-4) were predicted (www.ncbi.nlm.nih.gov/mapview/index.html) (Figure 5F). As STAT3-2 is just 72 bp away from STAT3-1, we synthesized three pairs of primers for quantitative RT-PCR. Our data showed that STAT3 was selectively engaged to the STAT3-1 or STAT3-2 binding site (Figure 5G). Thus, we concluded that upon exposure of NK1.1-CD4+NKG2D+ T cells to NKG2D ligands (MICA, ULBPs, and RAE-1), PI3K is activated to induce JNK-AP-1, NF-κB and STAT3 activation for TGF-β1 transcription.
Discussion
NK1.1-CD4+NKG2D+ regulatory T cells have distinct transcriptional profiles from NK1.1+CD4+NKG2D+ T cells, as previously demonstrated by our group [7]. It is currently unclear why the crosslinking of TCR/CD3 and NKG2D results in TGF-β1 expression, whereas co-stimulation of TCR/CD3 and CD28 leads to full activation of effector T cells. In this study, NK1.1-CD4+NKG2D+ cells co-stimulated by α-CD3/sRAE-1 or sRAE-1 increased the activation of the PI3K-p85α subunit, in contrast with stimulation by α-CD3/α-CD28. JNK and STAT3 were also specifically phosphorylated by PI3K when stimulate by α-CD3/sRAE-1 or sRAE-1. In addition, TCR/CD3 and NKG2D synergistically promoted NF-κB activation in NK1.1-CD4+NKG2D+ cells. Thus, our data suggest that all those three nuclear factors (AP-1, NF-κB, and STAT3) were involved in the TGF-β1 expression of NK1.1-CD4+NKG2D+ cells as co-stimulated by TCR/CD3 and NKG2D. The schematic diagram is summarized in Figure 6.
Figure 6.
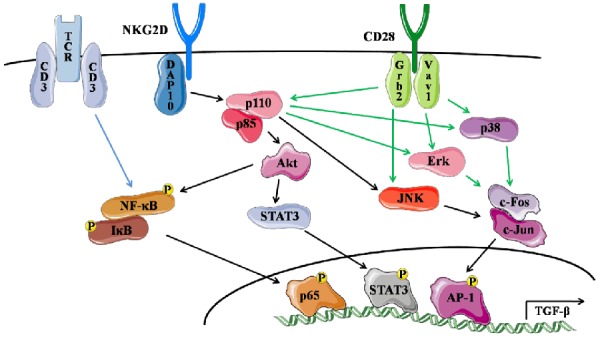
Diagram of the TGF-β1 transcription in NK1.1-CD4+NKG2D+ cells regulated by AP-1, NF-κB, and STAT3.
CD4+NKG2D+ T cells can also be divided into CD4+NKG2D+CD28+ and CD4+NKG2D+CD28null subsets. CD4+NKG2D+CD28null cells have the capacity of IFN-γ or IL-17 production, even perforin secretion [27,28]. Abnormal expressions of CD4+NKG2D+CD28null cells have been identified in cervical cancer [29], rheumatoid arthritis [30], granulomatosis with polyangiitis [31], and human CMV infection [28]. Some CD4+NKG2D+ T cells with immunostimulatory activity also contribute to Crohn’s disease, neuroinflammation, and the mediation of anti-tumor activity [27,32]. As suggested by our previous study, NK1.1-CD4+NKG2D+ cells positively express CD28, which are obviously different from the CD4+NKG2D+CD28null subset. NK1.1-CD4+NKG2D+ cells also differed from Th2 or Th17 cells because the former did not express GATA-3 and ROR-γt [7].
Human NKG2D associates with DAP10, whereas murine NKG2D associates with DAP10 or DAP12. A YINM motif in DAP10 could recruit PI3K-p85α and result in PI3K activation. Additionally, Vav1 interacts with the YXNM motif of DAP10 through the adaptor protein Grb2 and is required for activation of PI3K-dependent Akt signaling. DAP12 recruits Syk/ZAP70 and activates PLCγ2 [15,16]. Although we did not detect activation of the Syk/ZAP70 pathway, co-engagement of TCR/CD3 and NKG2D should be involved in the activation of Syk/ZAP70. NKG2D or CD3/NKG2D engagement could stimulate phosphorylation of JNK, whereas α-CD3/α-CD28 engagement does not have the same effect. The specific inhibition of JNK almost blocked the TGF-β1 expression by NK1.1-CD4+NKG2D+ cells, which indicated that JNK was a key molecule of the MAPK pathway for regulation of TGF-β1.
Nuclear transcription factor AP-1 is one of important members to regulate TGF-β1 transcription. AP-1 is generally composed of c-Fos and c-Jun subunits. The -1211--1202 site of the promoter region of the human TGF-β1 gene has an AP-1-binding sequence [18]. The activation of AP-1 is mainly regulated by the PI3K/MAPK signaling pathway, and continuous activation of Erk is associated with the expression of TGF-β1 on the surface of a group of CD4+CD25-CD69+ T cells [2]. The AP-1 inhibitor, SR11302, significantly downregulated TGF-β1 expression of NK1.1-CD4+NKG2D+ cells, which confirmed that AP-1 should have activity in the regulation of TGF-β1 transcription.
There are three NF-κB binding sites in the upstream promoter region of TGF-β1 gene, which are located at -2105 to -2096, -2104 to -2005 and -789 to -780. The ChIP experiment showed that the phosphorylated p65 could conjugate to the DNA sequence located at -789 to -780 [18]. Therefore, NF-κB has a direct effect on the transcriptional activation of TGF-β1 gene. In NK1.1-CD4+NKG2D+ T cells, the activation of NF-κB is potentially the result of protein kinase C activation downstream of TCR signaling and PI3K activated by NKG2D engagement.
Phosphorylated STAT3 protein directly combines with the SBE1 and SBE2 sequences to activate the gene transcription of TGF-β1. By using the STAT3 inhibitor (stattic or cryptotanshinone) in NK1.1-CD4+NKG2D+ T cells, TGF-β1 expression was blocked, which confirmed that STAT3 activation contributed to TGF-β1 expression in regulatory T cells. The phosphorylation of STAT3-Y705 was completely inhibited by the PI3K inhibitor but partly inhibited by the NF-κB inhibitor. Thus, STAT3-Y705 activation depended on PI3K activation preferentially.
In conclusion, TGF-β1 transcription in NK1.1-CD4+NKG2D+ T cells after stimulation with α-CD3/sRAE-1 or sRAE-1 was activated by AP-1, NF-κB, and STAT3. The activation of PI3K-p85 and JNK only occurred upon stimulation with α-CD3/sRAE-1 or sRAE-1. This study provides molecular mechanisms of nuclear factor regulation in TGF-β1 transcription of NK1.1-CD4+NKG2D+ T cells.
Acknowledgements
This work was supported by the National Natural Science Foundation (No.81471547, No.81671547, No.81273214, No.81172785) of China, the “Six peaks” Talent Project, and the “333” Talent Project in Jiangsu Province.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Ochi H, Abraham M, Ishikawa H, Frenkel D, Yang K, Basso AS, Wu H, Chen ML, Gandhi R, Miller A, Maron R, Weiner HL. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+CD25-LAP+ T cells. Nat Med. 2006;12:627–635. doi: 10.1038/nm1408. [DOI] [PubMed] [Google Scholar]
- 2.Han Y, Guo Q, Zhang M, Chen Z, Cao X. CD69+CD4+CD25- T cells, a new subset of regulatory T cells, suppress T cell proliferation through membrane-bound TGF-beta 1. J Immunol. 2009;182:111–120. doi: 10.4049/jimmunol.182.1.111. [DOI] [PubMed] [Google Scholar]
- 3.Sumitomo S, Fujio K, Okamura T, Morita K, Ishigaki K, Suzukawa K, Kanaya K, Kondo K, Yamasoba T, Furukawa A, Kitahara N, Shoda H, Shibuya M, Okamoto A, Yamamoto K. Transcription factor early growth response 3 is associated with the TGF-beta1 expression and the regulatory activity of CD4-positive T cells in vivo. J Immunol. 2013;191:2351–2359. doi: 10.4049/jimmunol.1202106. [DOI] [PubMed] [Google Scholar]
- 4.Groh V, Smythe K, Dai Z, Spies T. Fas-ligand-mediated paracrine T cell regulation by the receptor NKG2D in tumor immunity. Nat Immunol. 2006;7:755–762. doi: 10.1038/ni1350. [DOI] [PubMed] [Google Scholar]
- 5.Dai Z, Turtle CJ, Booth GC, Riddell SR, Gooley TA, Stevens AM, Spies T, Groh V. Normally occurring NKG2D+CD4+ T cells are immunosuppressive and inversely correlated with disease activity in juvenile-onset lupus. J Exp Med. 2009;206:793–805. doi: 10.1084/jem.20081648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin Z, Wang C, Xia H, Liu W, Xiao W, Qian L, Jia X, Ding Y, Ji M, Gong W. CD4(+) NKG2D(+) T cells induce NKG2D down-regulation in natural killer cells in CD86-RAE-1epsilon transgenic mice. Immunology. 2014;141:401–415. doi: 10.1111/imm.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian X, Hu C, Han S, Lin Z, Xiao W, Ding Y, Zhang Y, Qian L, Jia X, Zhu G, Gong W. NK1.1(-) CD4(+) NKG2D(+) T cells suppress DSS-induced colitis in mice through production of TGF-beta. J Cell Mol Med. 2017;21:1431–1444. doi: 10.1111/jcmm.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paul S, Schaefer BC. A new look at T cell receptor signaling to nuclear factor-kappaB. Trends Immunol. 2013;34:269–281. doi: 10.1016/j.it.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yabe T, McSherry C, Bach FH, Fisch P, Schall RP, Sondel PM, Houchins JP. A multigene family on human chromosome 12 encodes natural killer-cell lectins. Immunogenetics. 1993;37:455–460. doi: 10.1007/BF00222470. [DOI] [PubMed] [Google Scholar]
- 11.Houchins JP, Yabe T, McSherry C, Bach FH. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J Exp Med. 1991;173:1017–1020. doi: 10.1084/jem.173.4.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 2002;17:19–29. doi: 10.1016/s1074-7613(02)00333-3. [DOI] [PubMed] [Google Scholar]
- 13.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 14.Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res. 2015;3:575–582. doi: 10.1158/2326-6066.CIR-15-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Upshaw JL, Leibson PJ. NKG2D-mediated activation of cytotoxic lymphocytes: unique signaling pathways and distinct functional outcomes. Semin Immunol. 2006;18:167–175. doi: 10.1016/j.smim.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Serrano-Pertierra E, Cernuda-Morollon E, Lopez-Larrea C. NKG2D- and CD28-mediated costimulation regulate CD8+ T cell chemotaxis through different mechanisms: the role of Cdc42/N-WASp. J Leukoc Biol. 2014;95:487–495. doi: 10.1189/jlb.0613316. [DOI] [PubMed] [Google Scholar]
- 17.Kim SJ, Glick A, Sporn MB, Roberts AB. Characterization of the promoter region of the human transforming growth factor-beta 1 gene. J Biol Chem. 1989;264:402–408. [PubMed] [Google Scholar]
- 18.Lee KY, Ito K, Hayashi R, Jazrawi EP, Barnes PJ, Adcock IM. NF-kappaB and activator protein 1 response elements and the role of histone modifications in IL-1beta-induced TGF-beta1 gene transcription. J Immunol. 2006;176:603–615. doi: 10.4049/jimmunol.176.1.603. [DOI] [PubMed] [Google Scholar]
- 19.Peralta-Zaragoza O, Bermudez-Morales V, Gutierrez-Xicotencatl L, Alcocer-Gonzalez J, Recillas-Targa F, Madrid-Marina V. E6 and E7 oncoproteins from human papillomavirus type 16 induce activation of human transforming growth factor beta1 promoter throughout Sp1 recognition sequence. Viral Immunol. 2006;19:468–480. doi: 10.1089/vim.2006.19.468. [DOI] [PubMed] [Google Scholar]
- 20.Lin W, Tsai WL, Shao RX, Wu G, Peng LF, Barlow LL, Chung WJ, Zhang L, Zhao H, Jang JY, Chung RT. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology. 2010;138:2509–18. 2518.e1. doi: 10.1053/j.gastro.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoo YD, Chiou CJ, Choi KS, Yi Y, Michelson S, Kim S, Hayward GS, Kim SJ. The IE2 regulatory protein of human cytomegalovirus induces expression of the human transforming growth factor beta1 gene through an Egr-1 binding site. J Virol. 1996;70:7062–7070. doi: 10.1128/jvi.70.10.7062-7070.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogata H, Chinen T, Yoshida T, Kinjyo I, Takaesu G, Shiraishi H, Iida M, Kobayashi T, Yoshimura A. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene. 2006;25:2520–2530. doi: 10.1038/sj.onc.1209281. [DOI] [PubMed] [Google Scholar]
- 23.Hosui A, Kimura A, Yamaji D, Zhu BM, Na R, Hennighausen L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-{beta} and STAT3 activation. J Exp Med. 2009;206:819–831. doi: 10.1084/jem.20080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim Y, Ratziu V, Choi SG, Lalazar A, Theiss G, Dang Q, Kim SJ, Friedman SL. Transcriptional activation of transforming growth factor beta1 and its receptors by the Kruppel-like factor Zf9/core promoter-binding protein and Sp1. Potential mechanisms for autocrine fibrogenesis in response to injury. J Biol Chem. 1998;273:33750–33758. doi: 10.1074/jbc.273.50.33750. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence MC, Jivan A, Shao C, Duan L, Goad D, Zaganjor E, Osborne J, McGlynn K, Stippec S, Earnest S, Chen W, Cobb MH. The roles of MAPKs in disease. Cell Res. 2008;18:436–442. doi: 10.1038/cr.2008.37. [DOI] [PubMed] [Google Scholar]
- 26.Luwor RB, Baradaran B, Taylor LE, Iaria J, Nheu TV, Amiry N, Hovens CM, Wang B, Kaye AH, Zhu HJ. Targeting Stat3 and Smad7 to restore TGF-beta cytostatic regulation of tumor cells in vitro and in vivo. Oncogene. 2013;32:2433–2441. doi: 10.1038/onc.2012.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruck T, Bittner S, Gross CC, Breuer J, Albrecht S, Korr S, Gobel K, Pankratz S, Henschel CM, Schwab N, Staszewski O, Prinz M, Kuhlmann T, Meuth SG, Wiendl H. CD4+NKG2D+ T cells exhibit enhanced migratory and encephalitogenic properties in neuroinflammation. PLoS One. 2013;8:e81455. doi: 10.1371/journal.pone.0081455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saez-Borderias A, Guma M, Angulo A, Bellosillo B, Pende D, Lopez-Botet M. Expression and function of NKG2D in CD4+ T cells specific for human cytomegalovirus. Eur J Immunol. 2006;36:3198–3206. doi: 10.1002/eji.200636682. [DOI] [PubMed] [Google Scholar]
- 29.Romero AI, Chaput N, Poirier-Colame V, Rusakiewicz S, Jacquelot N, Chaba K, Mortier E, Jacques Y, Caillat-Zucman S, Flament C, Caignard A, Messaoudene M, Auperin A, Vielh P, Dessen P, Porta C, Mateus C, Ayyoub M, Valmori D, Eggermont A, Robert C, Zitvogel L. Regulation of CD4(+)NKG2D(+) Th1 cells in patients with metastatic melanoma treated with sorafenib: role of IL-15Ralpha and NKG2D triggering. Cancer Res. 2014;74:68–80. doi: 10.1158/0008-5472.CAN-13-1186. [DOI] [PubMed] [Google Scholar]
- 30.Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2003;100:9452–9457. doi: 10.1073/pnas.1632807100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Menthon M, Lambert M, Guiard E, Tognarelli S, Bienvenu B, Karras A, Guillevin L, Caillat-Zucman S. Excessive interleukin-15 transpresentation endows NKG2D+CD4+ T cells with innate-like capacity to lyse vascular endothelium in granulomatosis with polyangiitis (Wegener’s) Arthritis Rheum. 2011;63:2116–2126. doi: 10.1002/art.30355. [DOI] [PubMed] [Google Scholar]
- 32.Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, Caillat-Zucman S, Paul P, Gornet JM, Douay C, Ravet S, Tamouza R, Charron D, Lemann M, Mayer L, Toubert A. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology. 2007;132:2346–2358. doi: 10.1053/j.gastro.2007.03.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.