

Abstract
Asthma is a wide-spread disease that significantly impacts health throughout the world. A key aspect of the pathology of the disease is the remodeling of the airways by airway smooth muscle cells (ASMCs). MicroRNAs play an important role in post-transcriptional gene regulation and are involved in numerous biological functions, including those linked to asthma. A large number of microRNAs have been identified and investigated in various cell types to assess their function. In the present study, the role and potential mechanisms of miR-223 in ASMCs were investigated. Overexpression of miR-223 was found to induce a phenotypic switch in ASMCs that led to decreased expression of proteins involved in the extracellular matrix, such as α-SMA (ACTA2), and type I and III collagens. Inhibition of miR-223 caused the opposite result. However, unlike mast cells, neither overexpression nor inhibition of miR-223 affected cell viability or apoptosis in ASMCs. To further understand the effects of miR-223 on ASMCs, we applied bioinformatics analysis using predictive software, in combination with western blotting, to reveal that insulin-like growth factor-1 receptor (IGF-1R) was the functional target of miR-223 that leads to the phenotypic switch of ASMCs. Suppression of luciferase activity in a reporter containing the 3’-untranslated region (3’-UTR) of IGF-1R confirmed that this region is the target for the miRNA. Finally, we showed that miR-223 suppressed IGF-1R expression and decreased downstream phosphorylation of Akt (AKT1) in ASMCs. In conclusion, our data demonstrate that miR-223 exerts an inhibitory effect on the fibrotic phenotypes of ASMCs via the PI3K/Akt signaling pathway and IGF-1R is the likely functional target of the microRNA.
Keywords: microRNA-223, insulin-like growth factor-1 receptor, airway smooth muscle cell, extracellular matrix
Introduction
Asthma is a significant medical issue that leads to a substantial burden of disease across the world [1]. It is a relatively common respiratory disease that manifests through eosinophilic inflammation, airway hyperresponsiveness, and airway remodeling. The interactions between these various symptoms are unclear [2]. Within asthmatic airways, chronic inflammation cycles are maintained by inflammatory cells, although airway remodeling also contributes to pathology [3]. Several independent pathologic events result in this remodeling of the airway wall, including increased proliferation of mesenchymal cells, such as airway smooth muscle cells (ASMC) and fibroblasts, modified differentiation of mesenchymal cells, and de novo synthesis and deposition of pro-inflammatory extracellular matrix (ECM) components [4]. There have been several studies showing that ECM proteins can shift non-asthmatic derived ASMCs towards a proliferative phenotype [5] and the expression of type I/III collagens, fibrin, hyaluronate, polysaccharide, and laminin are all increased in the airways of patients with asthma [6]. It is therefore likely that the deposition of ECM proteins, such as type I and type III collagen by ASMCs is involved in airway remodeling. A further factor affecting treatment outcome for patients with asthma is that even though glucocorticoids and bronchodilators can relieve and control asthma attacks, they cannot prevent nor reverse airway remodeling. Thus, controlling deposition of ECM proteins by ASMCs could be a potential way to target airway remodeling and may serve as a therapeutic intervention for asthma.
MicroRNAs (miRNAs) are small, non-coding single-stranded RNAs between 19 and 24 nucleotides (nt) in length that have been highly conserved during evolution. Typically, miRNAs bind the 3’-untranslated regions (3’-UTRs) of specific mRNAs and mediate deadenylation and degradation of the transcript. This leads to translational repression of target proteins and is essential to the control of many genes [7,8]. Genes regulated by miRNAs are involved in many biological processes, such as differentiation, proliferation, maturation, apoptosis, and tumorigenesis [9]. Since miRNAs were first reported in 1993, there have many thousands of miRNAs identified in animals, plants, and viruses (http://www.mirbase.org) [10]. Additionally, several studies have shown that a wide variety of miRNAs are involved in the symptoms of asthma. For example, mice deficient in bic/microRNA-155 are immunodeficient and display increased lung airway remodeling [11]. Increased miRNA-146a expression negatively regulates the release of the pro-inflammatory chemokines IL-8 and RANTES [12]. Finally, miRNA-223 overexpression promotes mast cell apoptosis [9] and down-regulation promotes the degranulation of mast cells [13].
Insulin-like growth factors (IGFs) are hormones that are involved in growth and survival and suppress apoptosis and promote cell cycle progression, angiogenesis, and have also been linked with the metastatic activities of various cancers. Specifically, insulin-like growth factor 1 receptor (IGF-1R) has been shown to be involved in cell transformations induced by tumors and mediates the activity of IGFs [14]. IGF-1R is a transmembrane receptor that belongs to a large class of tyrosine kinase receptors. Structurally, two alpha subunits and two beta subunits make up IGF-1R, with the former inducing the tyrosine autophosphorylation of the latter. This triggers a cascade of intracellular signaling involved in cell survival and proliferation [15,16]. Due to this, there have been several investigations into the potential interactions between microRNAs and IGF-1R. According to these prior studies, miR-133a and miR-122 regulate hepatocellular carcinoma growth and sensitivity to the kinase inhibitor sorafenib by targeting IGF-1R [17,18]. There is also evidence that miR-7 and miR-320a modulate glioma cell proliferation, apoptosis, migration, invasion, and tumorigenesis [19,20]. Overexpression of miR-150, miR-630, and miR-497 can also influence pancreatic cancer cell proliferation and apoptosis [21,22]. Finally, we have previously shown that miR-223 has an important role in several cellular functions, across many different cell types, by targeting IGF-1R. For example, miR-223 can promote mast cell apoptosis and affect degranulation [9,23], induce endothelial cell apoptosis, and suppress endometrial carcinoma cell proliferation by targeting IGF-1R [24,25].
To further understand the functions of miRNAs and IGF-1R during asthma, particularly the role of ASMCs and their deposition of the ECM, we delivered a miRNA-223 mimics and inhibitors into ASMCs to alter their expression of miRNA-223. We also hypothesized that miR-223 may promote the apoptosis or inhibit proliferation of ASMCs. To assess this hypothesis, the direct effects of miRNA-223 on ASMCs, including apoptosis and proliferation, were assessed. We also examined potential mechanisms by which miR-223 induces a phenotypic switch in ASMCs. Our data suggests that miR-223 had no effect on apoptosis nor proliferation of ASMCs but exerted an inhibitory effect on ECM deposition by ASMCs. This suggests that miR-223 is a promising target for the treatment of asthma and airway remodeling.
Materials and methods
Cell culture and identification
Airway smooth muscle cells (ASMCs) were isolated from Sprague Dawley (SD) rats weighing approximately 150 g each (Laboratory Animal Center, Nanjing Medical University, Jiangsu, China). These were digested with collagenase type I and papain at 37°C in a humidified atmosphere supplemented with 5% CO2 for 25 min. They were then treated with Dulbecco’s modified Eagle’s medium (DMEM) to terminate digestion and centrifuged at 1000 rpm for 5 min. The supernatant was then removed and the remaining pellet further digested with 0.25% pancreatin at 37°C for 15 min, followed by digestion termination and centrifugation as previously described. The remaining sediment of ASMCs was incubated at 37°C in DMEM supplemented with 20% fetal bovine serum (FBS; Wisent, Inc., Quebec, Canada) under a humidified atmosphere supplemented with 5% CO2 for 3 days. After cell passage, ASMCs were maintained in DMEM supplemented with 10% FBS. Morphological confirmation of rat ASMCs in the primary culture was performed using an inverted phase contrast microscope (Olympus, Inc., Japan) and α-Actin immunohistochemistry (Affinity biosciences Inc., Shanghai, China) was used to assess ASMC differentiation.
Transfection
ASMCs were transfected for 24 h using Lipofectamine 2000 (Thermo Fisher Scientific, Inc., Waltham, MA, US) one day after seeding at. For assessing the effects of miR-223 overexpression and inhibition, a miR-223 mimic (GenePharma Co., Ltd., Shanghai, China), miR-223 inhibitor (GenePharma Co., Ltd. , Shanghai, China), and their respective negative controls (NC; GenePharma Co., Ltd., Shanghai, China) were transfected at a final concentration of 50 nM. At 48 h post-transfection, cells were harvested and the total RNA was extracted for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis. Lysates were also prepared for western blotting. Finally, cell viability and apoptosis assays were performed 48 h after transfection.
RT-qPCR
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used to purify total RNA from ASMCs and a TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used to reverse transcribe RNA into cDNA, following the manufacturer’s instructions. qPCR was performed using an Applied Biosystems 7500 Fast Real-Time PCR system (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and a TaqMan MicroRNA Assay (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). All qPCR reactions used the thermal conditions of an amplification step at 95°C for 5 min, then 40 cycles of 95°C for 10 s, 60°C for 30 s, and 72°C for 10 s. All reactions were performed in triplicate, with a final volume of 20 µL (10 µL TaqMan master mix, 1.33 µL cDNA, 1 µL miR-223/U6RNA probe, and 7.67 µL diethylpyrocarbonate). The 2-ΔΔCt method was used to determine miRNA expression [26], relative to U6RNA (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) as an endogenous control.
Cell viability assay
ASMCs were plated onto 96-well plates at a density of 5,000 cells/well. These were then transfected with 50 nM miR-223 mimic, miR-223 inhibitor, or negative controls. At 48 h post-transfection, Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) assays were used to determine cell viability. Briefly, 10 µL CCK-8 solution was added to each well and incubated for 1 h in DMEM supplemented with 10% FBS. Absorbance at 450 nm was then measured for each well using a microplate reader (DNM-9602; Beijing Perlong New Technology Co., Ltd., Beijing, China) and OD values calculated based on the results of three independent experiments.
Apoptosis assay
ASMCs were seeded onto 6-well plates in DMEM supplemented with 10% FBS until cells reached 70% confluence. These were then transfected with 50 nM miR-223 mimic, miR-223 inhibitor, or negative controls using Lipofectamine 2000. Forty-eight hours after transfection, ASMCs were harvested and a FITC Annexin V Apoptosis Detection Kit I (BD Biosciences; Franklin Lakes, NJ, USA) was used to analyze the apoptosis of ASMCs, according to the manufacturer’s instructions. PI- and Annexin V-positive cells were gated on a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) to distinguish apoptotic cells and Cell Quest Pro software (version 4.01; BD Biosciences, San Jose, CA, USA) was used to analyze the distribution of cells. All experiments were performed in triplicate.
Luciferase reporter assay
In order to determine if miR-223 functions by targeting IGF-1R, a luciferase reporter assay was performed using HEK-293T cells (Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Science, Shanghai, China). Cells were co-transfected with a IGF-1R-3’UTR fusion vector and either a miR-223 mimic or miR-223 inhibitor. Luciferase levels were measured after 48 h using a Dual-Luciferase Reporter Assay System (Promega Corp., Madison, WI, USA), according to the manufacturer’s instructions. Results were calculated based on data from three independent experiments.
Western blot analysis
ASMCs were seeded onto 6-well plates with 10% FBS DMEM and transfected after 24 h. These were then harvested after reaching 90% confluence and washed three times with ice-cold PBS buffer. Cells were lysed in ice-cold radioimmunoprecipitation assay buffer (Beyotime Institute of Biotechnology, Nantong, China) and total protein content assessed using an enhanced BCA protein assay kit (Beyotime Institute of Biotechnology). Equivalent amounts of protein were then separated on 8% or 10% sodium dodecyl sulfate-polyacrylamide gels by electrophoresis and transferred to a nitrocellulose membrane (Merck Millipore, Billerica, MA, USA). Membranes were blocked with 5% non-fat milk in PBS with Tween 20 (PBST) for 2 h at room temperature Blots were next incubated with anti-COL3A1, anti-COL1A2 (Bioword Technology, Inc., St Louis Park, MN, USA), anti-α-Actin/α-SMA (Affinity Biosciences Inc., Shanghai, China), anti-phospho-AKT, anti-AKT (Cell Signaling Technology Inc., Danvers, MA, USA), anti-IGF-1R, and anti-GAPDH (Santa Cruz, Dallas, TX, USA) overnight at 4°C. Afterwards, the membranes were washed five times with PBST and then incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse antibodies (Zhongshan Golden Bridge Biotechnology Co., Ltd., Zhongshan, China) for 2 h at room temperature. Protein bands were detected using a Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Statistical analysis
SPSS Version 20.0 (IBM, Armonk, NY, USA) was used to assess statistical differences between independent groups with Student’s t-tests. Results are presented as means ± SEM and P < 0.05 was used as the threshold of statistical significance.
Results
Culture and identification of rat ASMCs
ASMCs were isolated from SD rats. To confirm their presence, cells were observed under an inverted phase contrast microscope. ASMCs were distinguished as cells with spindles and an ovoid nucleus that grew at distinctive rates with peaks and valleys (Figure 1A). α-Actin-specific staining was used to immunohistochemically identify cells that were ASMCs or had differentiated into other cells types (Figure 1B).
Figure 1.
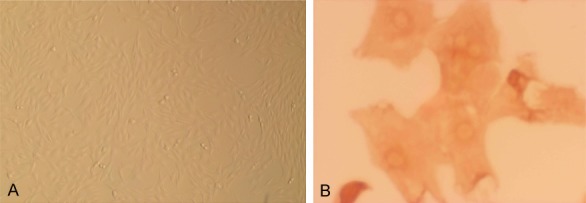
The identification of ASMCs. A. The morphological characteristics of cultured cells were observed by under an inverted phase contrast microscope (×10). B. ASMCs were identified by immunohistochemistry (×20), demonstrating.
TGF-β1 stimulation lowers the expression of miR-223
To determine whether miR-223 expression was affected by TGF-β1 stimulation in ASMCs, we used RT-qPCR to analyze expression in cells exposed to TGF-β1 for 48 h t. As shown in Figure 2A, stimulation with TGF-β1 significantly decreased miR-223 expression compared to control cells (P < 0.05). To further explore the role of miR-223 in ASMCs, gain- and loss-of-function experiments were conducted by transfecting ASMCs with a miR-223 mimic or a miR-223 inhibitor, and their respective controls. The expression of miR-223 increased after transfection with the miR-223 mimic but not with the negative control (Figure 2B). Conversely, ASMCs showed lower levels of miR-223 after transfection with the miR-223 inhibitor and there was no change in the negative control. Both of these results indicated successful transfection (P < 0.05; Figure 2B).
Figure 2.
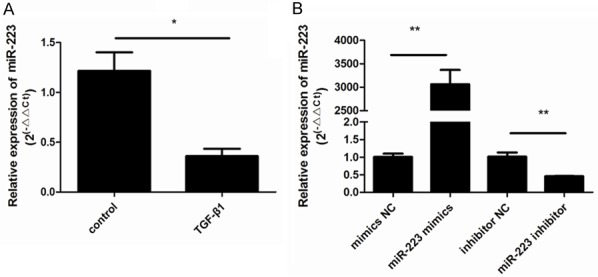
TGF-β1 stimulation downregulates the expression of miR-223. A. The effects of stimulation with TGF-β1 on the expression of miR-223 in ASMCs (**P < 0.01; *P < 0.05; n = 6). B. ASMCs were transfected with a miR-223 mimic and negative control (NC), and a miR-223 inhibitor and NC. miR-223 expression was then assessed. This demonstrated that the expression of miR-223 increased after transfection with the miR-223 mimic and decreased after transfection with the miR-223 inhibitor. There were no changes in the negative controls. Data shown are means ± SD from three experiments (**P < 0.01; *P < 0.05; n = 6).
MiRNA-223 overexpression or inhibition does not affect the viability or apoptosis of ASMCs
We next explored the role of miR-223 in AMSCs by assessing any effects on cell viability or apoptosis. We induced either overexpression or inhibition of miR-223 in ASMCs and then investigated cell viability using a CCK-8 assay. This demonstrated that neither transfection with a miR-223 mimic nor miR-223 inhibitor affected cell viability within 48 h of transfection when compared to the control groups (Figure 3). We also used a cytofluorometric apoptosis assay to show that the apoptosis of ASMCs was also not significantly affected following transfection with the miR-223 mimic nor miR-223 inhibitor, compared to controls (Figure 4). These data suggest that miR-223 does not affect cell viability nor apoptosis, unlike mast cells.
Figure 3.
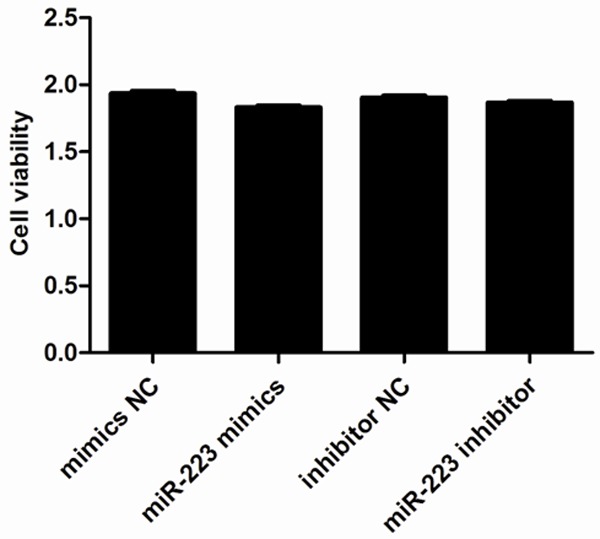
Effect of miR-223 on ASMC viability. Cells were transfected with 50 nM miR-223 mimic or negative control (NC), and 50 nM miR-223 inhibitor or miR-223 inhibitor NC. Cell viability was then assessed 48 h post-transfection using a CCK-8 kit. Data are means ± SD from three independent experiments (**P < 0.01; *P < 0.05; n = 5).
Figure 4.
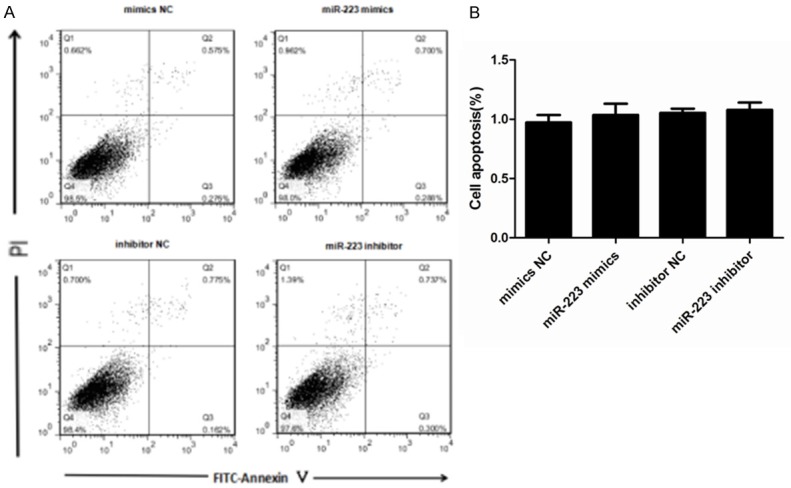
Effects of miR-223 on the apoptosis of ASMCs. A. The effects on ASMC apoptosis were determined using a cytofluorometric apoptosis kit and subsequent flow cytometry to distinguish PI- and Annexin V-positive cells. This showed that apoptosis was not affected by transfection with a miR-223 mimic or miR-223 inhibitor. B. The mean proportion of apoptotic cells in each treatment group (both early and late apoptosis cells) from three independent experiments. Data are means ± SD (**P < 0.01; * P < 0.05; n = 5).
miRNA-223 inhibits the deposition of the extracellular matrix by airway smooth muscle cells
As extracellular matrix (ECM) proteins have been shown to promote non-asthmatic derived ASMCs towards a proliferative phenotype [5]. we next determined whether miR-223 expression affected the deposition of the ECM by ASMCs. Cells were transfected with a miR-223 mimic, miR-223 inhibitor, or negative controls. We then investigated the expression of α-SMA (ACTA2), type I collagen (COL1A2), and type III collagen (COL3A1) protein in ASMCs by western blot after transfection. We found the abundances of these three proteins were altered post-transfection (Figure 5). Specifically, α-SMA, type I collagen, and type III collagen were all markedly lower in cells that had overexpression of miR-223, while miR-223 inhibition caused the opposite effect. These data indicate that miR-223 may inhibit ECM deposition by ASMCs and may serve as a possible intervention for this particular asthma pathology.
Figure 5.
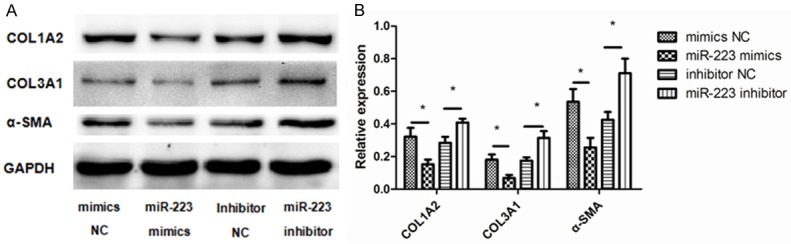
Effect of miR-223 on the expression of α-SMA (ACTA2), type I collagen (COL1A2), and type III collagen (COL3A1). A. ASMCs were transfected with 50 nM miR-223 mimic, miR-223 inhibitor, and negative controls. At 48 h post-transfection, western blots were used to detect the protein expression levels of α-SMA, type I collagen, and type III collagen. GAPDH was used as a loading control. B. Data were normalized to GAPDH expression and are presented as expression relative to untreated cellular controls. Data are means ± SD from three experiments (**P < 0.01; *P < 0.05).
IGF-1R/PI3K-Akt is a signaling pathway downstream of miR-223 in ASMCs
An increasing number of studies have shown that IGF-1R and miRNAs have a close functional relationship [17-22] and our own studies have shown that miR-223 acts through the targeting of IGF-1R [9,23-25]. To further investigate the underlying mechanisms through which miR-223 induces ASMC phenotypic switching, we assessed whether IGF-1R is the target gene for miR-223 in ASMCs and its putative role. To this end, the miR-223 mimic, miR-223 inhibitor, and negative controls were transfected into ASMCs. The levels of IGF-1R protein expression were then determined, revealing that the expression of IGF-1R was down-regulated in ASMCs after treatment with the miR-223 mimic (Figure 6A and 6B).
Figure 6.
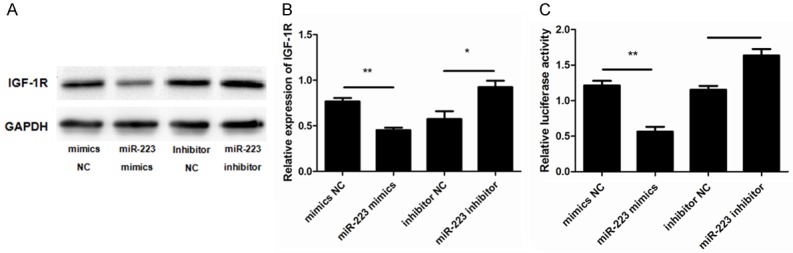
The effects of targeting IGF-1R by miR-223. A. Representative western blots show that overexpression of the miR-223 mimic lowered IGF-1R protein levels compared to the controls and inhibitor. GAPDH was used as a loading control. B. Data were normalized to the expression of GAPDH and presented as expression relative to untreated cellular controls. C. The 3’-UTR of IGF-1R was fused to a Luciferase reporter and activity was assessed after treatment with the miR-223 mimic, miR-223 inhibitor, or negative controls. Luciferase activity was lower in cells overexpressing miR-223 compared to controls. Data are means ± SD from three experiments (**P < 0.01; *P < 0.05).
To further examine whether miR-223 is able to directly bind to IGF-1R and inhibit its expression, a reporter assay was designed that used the 3’-UTR of IGF-1R fused to a luciferase reporter. As expected, cells transfected with the miR-223 mimic showed a decrease in luciferase activity compared to the cells transfected with NC. Conversely, the miR-223 inhibitor enhanced luciferase activity (Figure 6C). This suggests that miR-223 directly binds to the 3’-UTR of the IGF-1R transcript, inhibiting IGF-1R protein expression. Finally, as PI3K-Akt is reported to be a signaling pathway downstream of IGF-1, we assessed the potential involvement of PI3K-Akt in the miR-223-mediated effects on ASMCs. The expression of Akt (AKT1), and of its active form (p-Akt), were measured after treatment with the miR-223 mimic, miR-223 inhibitor, and negative controls. This revealed that the expression of p-Akt was significantly lower in cells transfected with the miR-223 mimic compared to the control group. However, the total expression of Akt was unaffected (Figure 7A and 7B).
Figure 7.
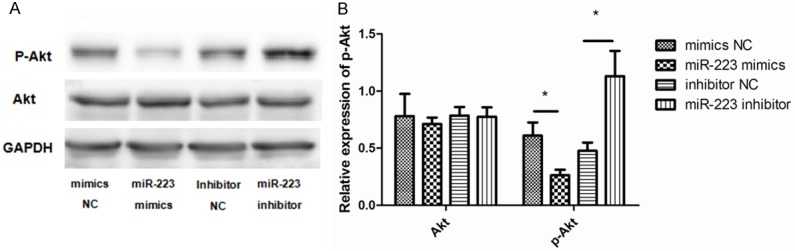
miR-223 inhibits the PI3K/Akt signaling pathway. A. Representative western blots show the levels of p-AKT and total AKT (t-AKT) from cells treated with miR-223 mimic, miR-223 inhibitor, or negative controls. GAPDH was used as a loading control. Overexpression of miR-223 decreased expression of p-AKT in ASMCs relative to other groups. B. Data were normalized to GAPDH expression and presented as expression relative to untreated cellular controls. Data are means ± SD from three experiments (**P < 0.01; *P < 0.05).
Discussion
Although miR-223 has been suggested to play a role in apoptosis and proliferation in several cell types [9,24,25], its role in airway smooth muscle cells (ASMCs) remains unknown. Our data has demonstrated that miR-223 overexpression inhibits the deposition of the ECM by ASMCs, one of the more important functions of these cells in the airway. ECM proteins can also shift non-asthmatic derived ASMCs towards a more proliferative phenotype [5]. It is therefore likely that the study of ASMCs, and their role in the deposition of the ECM, will provide new avenues for the treatment of asthma. Prior studies have already shown an important role for miR-223 in mast cells, a population that are involved in several diseases, such as bronchial asthma and chronic skin inflammation [9,23]. We have also demonstrated in the present study that the expression of miR-223 was lower in ASMCs stimulated with TGF-β1.
In light of the anti-proliferative and apoptosis-inducing roles of miR-223 in endothelial cells and mast cells [9,24,25], we hypothesized that miR-223 may promote the apoptosis or inhibit proliferation of ASMCs. Conversely, our results were at odds with this hypothesis and ASMCs transfected with a miR-223 mimic or inhibitor showed no difference in cell viability or apoptosis. This demonstrates that miRNAs can have different effects depending on the cell type they are expressed in. It is also possible that this discrepancy may be due to the fact that the miR-223 concentrations used in our study (50 nM) were not sufficient to directly regulate apoptosis or proliferation and levels of miR-223 were only partially decreased. The remaining small amount of miR-223 may have been enough to prevent ASMCs from undergoing apoptosis or proliferation. However, cells transfected with the miR-223 mimic did show decreased expression of several key ECM proteins, including α-SMA, type I collagen, and type III collagen protein. Cells transfected with a miR-223 inhibitor had higher expression of these three proteins. This demonstrates that the miRNA was able to elicit some effects and that the observed decrease in α-SMA, type I collagen, and type III collagen expression is not due to decreased numbers of ASMCs. Finally, IGF-1R and the PI3K/Akt signaling pathway have also been suggested to be involved in the effects of miR-223 [9,23-25,27] and we have demonstrated that miR-223 overexpression significantly decreased the expression of IGF-1R and p-Akt.
A further limitation of our study is a lack of in vivo data derived from human or animal studies. Distinguishing expression of miR-223 is more often easier between patients with asthma and controls in both humans and animal models. Also, to confirm without doubt that IGF-1R is the target of miR-223 in ASMCs, knocking out the gene in ASMCs in vivo and then assessing any changes to the deposition of the ECM would be the most convincing experiment. Although out of the scope of the current study, these are both directions for our future research.
In conclusion, our study has demonstrated that miR-223 exerts an inhibitory effect on the deposition of the ECM by ASMCs isolated from SD rats. This is likely through targeting IGF-1R, subsequently affecting the PI3K/Akt signaling pathway. Our work strongly suggests that miR-223 is a promising target for the future treatment of asthma and airway remodeling.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (grant no. 81200012; awarded to Feng Liu), National Natural Science Foundation of China (grand no. 81370132; awarded to Deyu Zhao), Nanjing Medical Science and Technique Development Foundation (grant no. JQX15008; awarded to Feng Liu), Jiangsu Provincial Medical Youth Talent (grant no. QNRC2016087; awarded to Feng Liu), and Cadre Talent Foundation (grant no. ETYYGG2014002; awarded to Feng Liu). The authors would like to thank the Nanjing Medical University for its technical support. Language editing services were provided by Editage, a division of Cactus Communications Inc.
Disclosure of conflict of interest
None.
References
- 1.Beasley R, Semprini A, Mitchell EA. Risk factors for asthma: is prevention possible? Lancet. 2015;386:1075–1085. doi: 10.1016/S0140-6736(15)00156-7. [DOI] [PubMed] [Google Scholar]
- 2.Pilecki B, Schlosser A, Wulf-Johansson H, Trian T, Moeller JB, Marcussen N, Aguilar-Pimentel JA, de Angelis MH, Vestbo J, Berger P. Microfibrillar-associated protein 4 modulates airway smooth muscle cell phenotype in experimental asthma. Thorax. 2015;70:862–872. doi: 10.1136/thoraxjnl-2014-206609. [DOI] [PubMed] [Google Scholar]
- 3.Silva RA, Almeida FM, Olivo CR, Saraiva-Romanholo BM, Martins MA, Carvalho CR. Airway remodeling is reversed by aerobic training in a murine model of chronic asthma. Scand J Med Sci Sports. 2015;25:e258–266. doi: 10.1111/sms.12311. [DOI] [PubMed] [Google Scholar]
- 4.Roth M, Zhao F, Zhong J, Lardinois D, Tamm M. Serum IgE Induced airway smooth muscle cell remodeling is independent of allergens and is prevented by omalizumab. PLoS One. 2015;10:e0136549. doi: 10.1371/journal.pone.0136549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirst SJ, Twort CH, Lee TH. Differential effects of extracellular matrix proteins on human airway smooth muscle cell proliferation and phenotype. Am J Respir Cell Mol Biol. 2000;23:335–344. doi: 10.1165/ajrcmb.23.3.3990. [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto H, Niimi A, Takemura M, Ueda T, Tabuena R, Yamaguchi M, Matsuoka H, Hirai T, Muro S, Ito Y. Prognosis of cough variant asthma: a retrospective analysis. J Asthma. 2006;43:131–135. doi: 10.1080/02770900500498477. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DE. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;16:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 8.Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 9.Gao H, Deng H, Xu H, Yang Q, Zhou Y, Zhang J, Zhao D, Liu F. MicroRNA-223 promotes mast cell apoptosis by targeting the insulin-like growth factor 1 receptor. Exp Ther Med. 2016;11:2171–2176. doi: 10.3892/etm.2016.3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRbase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA. Requirement of bic/microRNA-155 for normal immune function. Science. 2001;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA. Rapid changes in microRNA-146a expression negatively regulate the IL-1beta-induced inflammatory response in human lung alveolar epithelial cells. J Immunol. 2008;180:5689–5698. doi: 10.4049/jimmunol.180.8.5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q, Zhao DY, Xu H, Zhou H, Yang QY, Liu F, Zhou GP. Down-regulation of microRNA-223 promotes degranulation via the PI3K/Akt pathway by targeting IGF-1R in mast cells. PLoS One. 2015;10:e0123575. doi: 10.1371/journal.pone.0123575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brahmkhatri VP, Prasanna C, Atreya HS. Insulin-like growth factor system in cancer: novel targeted therapies. Biomed Res Int. 2015;2015:538019. doi: 10.1155/2015/538019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 16.LeRoith D, Werner H, Beitner-Johnson D, Roberts CT. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 17.Zhang W, Liu K, Liu S, Ji B, Wang Y, Liu Y. MicroRNA-133a functions as a tumor suppressor by targeting IGF-1R in hepatocellular carcinoma. Tumour Biol. 2015;36:9779–9788. doi: 10.1007/s13277-015-3749-8. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y, Huang J, Ma L, Shan J, Shen J, Yang Z, Liu L, Luo Y, Yao C, Qian C. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1R to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. 2016;371:171–181. doi: 10.1016/j.canlet.2015.11.034. [DOI] [PubMed] [Google Scholar]
- 19.Wang B, Sun F, Dong N, Sun Z, Diao Y, Zheng C, Sun J, Yang Y, Jiang D. MicroRNA-7 directly targets insulin-like growth factor 1 receptor to inhibit cellular growth and glucose metabolism in gliomas. Diagn Pathol. 2014;9:211. doi: 10.1186/s13000-014-0211-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo T, Feng Y, Liu Q, Yang X, Jiang T, Chen Y, Zhang Q. MicroRNA-320a suppresses in GBM patients and modulates glioma cell functions by targeting IGF-1R. Tumour Biol. 2014;35:11269–11275. doi: 10.1007/s13277-014-2283-4. [DOI] [PubMed] [Google Scholar]
- 21.Farhana L, Dawson MI, Murshed F, Das JK, Rishi AK, Fontana JA. Upregulation of miR-150* and miR-630 induces apoptosis in pancreatic cancer cells by targeting IGF-1R. PLoS One. 2013;8:e61015. doi: 10.1371/journal.pone.0061015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu JW, Wang TX, You L, Zheng LF, Shu H, Zhang TP, Zhao YP. Insulin-like growth factor 1 receptor (IGF-1R) as a target of MiR-497 and plasma IGF-1R levels associated with TNM stage of pancreatic cancer. PLoS One. 2014;9:e92847. doi: 10.1371/journal.pone.0092847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q, Zhao DY, Xu H, Zhou H, Yang QY, Liu F, Zhou GP. Down-regulation of microRNA-223 promotes degranulation via the PI3K/Akt pathway by targeting IGF-1R in mast cells. PLoS One. 2015;10:e0123575. doi: 10.1371/journal.pone.0123575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan Y, Liang H, Liu H, Li D, Chen X, Li L, Zhang CY, Zen K. Platelet-secreted microRNA-223 promotes endothelial cell apoptosis induced by advanced glycation end products via targeting the insulin-like growth factor 1 receptor. J Immunol. 2014;192:437–446. doi: 10.4049/jimmunol.1301790. [DOI] [PubMed] [Google Scholar]
- 25.Huang K, Dong X, Sui C, Hu D, Xiong T, Liao S, Zhang H. MiR-223 suppresses endometrial carcinoma cells proliferation by targeting IGF-1R. Am J Transl Res. 2014;6:841–849. [PMC free article] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Shan Z, Qin S, Li W, Wu W, Yang J, Chu M, Li X, Huo Y, Schaer GL, Wang S. An endocrine genetic signal between blood cells and vascular smooth muscle cells. J Am Coll Cardiol. 2015;65:2526–2537. doi: 10.1016/j.jacc.2015.03.570. [DOI] [PMC free article] [PubMed] [Google Scholar]