

Abstract
Circular RNAs (circRNAs) are novel noncoding RNAs with a wide range of physiological and pathological activities. However, the expression profile and roles in lung squamous cell carcinoma (LSCC) remain largely unknown. Therefore, we investigated the expression profile of circRNAs in three LSCC and matched adjacent normal tissues using microarray. Total 216 differentially expressed circRNAs were identified, including 135 upregulated and 81 downregulated ones in LSCC tissues. Bioinformatics analysis revealed that these differentially expressed circRNAs were potentially implicated in carcinogenesis using Gene ontology (GO) and KEGG pathway analyses. By constructing miRNA-circRNA interaction network, a total of ten key circRNAs, including 6 upregulated and 4 downregulated circRNAs were further screened and then confirmed using qRT-PCR analysis in another 40 paired of LSCC tissues and adjacent normal tissues. In addition, Kaplan-Meier survival analysis demonstrated that the overall survival time of LSCC patients with high hsa_circRNA_103827 expression and low hsa_circRNA_000122 was significantly shorter (P<0.001). In conclusion, this study provides evidence that circRNAs are differentially expressed in LSCC and closely related to the carcinogenesis of LSCC. Among these, hsa_circRNA_103827 and hsa_circRNA_000122 might be served as potential prognostic biomarkers and therapeutic target for LSCC.
Keywords: Lung squamous cell carcinoma, circRNA, microarray analysis, circRNA-miRNA network, biomarker
Introduction
Lung cancer is the most frequent cause of cancer death, accounting for about 22% of all cancer-related deaths in China in 2015 [1], which histologically classified as two major types of lung cancers, including small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) [2]. NSCLC is mainly divided into lung adenocarcinoma, lung squamous cell carcinoma (LSCC), and large cell carcinoma [3]. Among these, LSCC, the main type of NSCLC, occurred at the highest incidence among all lung cancer, remaining the leading risk factor for lung cancer progression [4,5]. Currently, the 5-year overall survival (OS) rate of lung cancer patients, including LSCC is less than 20% unless they receive treatment at an early stage [6]. Therefore, identifying effective biomarkers with early diagnostic value is urgently needed to improve therapeutic approaches for SCC.
Circular RNAs (circRNAs) are newly-discovered type of endogenous non-coding RNA (ncRNA) molecules with covalently joined 3’-and 5’-ends formed by back-splicing events [7], which makes them highly stable and largely resistant to RNA degradation [8]. Currently, circRNAs has caused great interest in the field of RNA research, along with microRNAs (miRNAs) and long noncoding RNA (IncRNA) [9]. CircRNAs can also function as microRNAs (miRNAs) sponges, thus contribute to post-transcriptional regulation of gene expression [10]. Recent studies indicate that circRNAs play an important role in the development of atherosclerotic vascular disease [11] and Alzheimer’s disease [12]. Likewise, circRNAs have been involved in the progression of several cancers. Li et al. have found that hsa_circ_002059, a typical circular RNA, was significantly down-regulated in gastric cancer tissues compared with paired adjacent non-tumorous tissues [13]. CircRNA 100876 has been shown to be closely related to the carcinogenesis of NSCLC by Yao et al. [14]. Using circRNA microarray, a growing number of circRNAs were reported to be aberrantly expressed in pancreatic ductal adenocarcinoma [15]. Notably, some novel circRNAs were identified and probably involved in tumorigenesis in squamous cell carcinoma, such as cutaneous squamous cell carcinoma [16] and hypopharyngeal squamous cell carcinoma [17]. However, there have been relatively few reports describing circRNAs in human LSCC, even though these promising circRNA findings across various diseases.
Therefore, here we profiled the circRNA expression profile in three pairs of LSCC samples tumors compared with matched adjacent normal tissues. After identifying differentially expression circRNAs in LSCC, we performed bioinformatics analysis to these differentially expressed circRNAs to screen key circRNAs. Our results may help improve our understanding of the pathogenesis of LSCC and identify potential circRNA biomarkers for LSCC.
Materials and methods
Patient samples
Our screening recruited 43 paired of patients’ tumor (T) and corresponding non-tumor lung (N) tissues from the First Affiliated Hospital of Nanjing Medical University (Jiangsu, China) following informed consent. All of the patients underwent neither chemotherapy nor radiotherapy before operation. After underwent radical resection, all tissues were histologically identified, diagnosed as LSCC, and graded according to the guidelines of American Joint Committee on Cancer (AJCC). Three paired samples were used for the circRNA microarray analysis (for basic clinical data, see Table 1), and the remained 40 paired tissues were used for verification with quantitative reverse transcription PCR (qRT-PCR). Each sample was snap-frozen in liquid nitrogen immediately after resection and subsequently was stored at -80°C untiluse. This study was approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University and the protocol was approved by the ethics review board of our hospital.
Table 1.
The clinical characteristics of patients with SQCC subjected to circRNA expression profile chip assay
| Sample ID | Group | Sex | Age | Histologic differentiation |
|---|---|---|---|---|
| LU-130611-1-N03 | Adjacent normal lung tissues | Male | 69 | Moderately |
| LU-130712-1-N04 | Adjacent normal lung tissues | Male | 78 | Moderately |
| LU-140301-1-N14 | Adjacent normal lung tissues | Male | 74 | High-moderately |
| LU-130611-1-T03 | SQCC | Male | 69 | Moderately |
| LU-130712-1-T04 | SQCC | Male | 78 | Moderately |
| LU-140301-1-T14 | SQCC | Male | 74 | High-moderately |
SQCC, squamous cell lung carcinoma.
Sample labeling and hybridization
According to the manufacturer’s protocol (Arraystar Inc.), total RNAs were digested with Rnase R (Epicentre, Inc.) to remove linear RNAs and enrich circular RNAs. Utilizing a random priming method (Arraystar Super RNA Labeling Kit; Arraystar), the enriched circular RNAs were amplified and transcribed into fluorescent cRNA. The labeled cRNAs were purified by RNeasy Mini Kit (Qiagen) and its concentration and specific activity were measured by NanoDrop ND-1000. The samples were then hybridized with Arraystar Human circRNA Array (8x15K, Arraystar). The hybridized arrays were washed, fixed and scanned using the Agilent Scanner G2505C.
Differential expression levels of circRNAs from microarrays
Signals were scanned by Agilent G2565C Microarray Scanner. Array images were introduced into Agilent Feature Extraction software (version 11.0.1.1) to obtain raw data (v10.7). Quantile normalization of raw data and subsequent data processing were performed using the R software package limma package (version 2.7.10). Acquired circRNA expression profile data were classified into two groups, including tumor (T) and normal (N) groups. Subsequently, differentially expressed circRNAs between the two groups were evaluated using t-test, and the P-values were corrected for False Discovery Rate (FDR) by Benjamini-Hochberg (BH) procedure. Statistical significance was defined as an absolute value of fold change |logFC| >2.0 and FDR P-value <0.05.
Scatter plots were used to assess differentially expressed circRNAs with statistical significance between two groups. Volcano Plot filtering was used to visualize the significantly differential circRNAs between each pairwise comparison. Hierarchical Clustering was performed to show the distinguishable circRNAs expression pattern among samples.
Bioinformatics analysis
The GO analysis was divided into molecular function (MF), biological process (BP) and cellular component (CC). CircRNA targets identified with profiling data were subjected to gene ontology (GO) based on their correlated mRNAs using Gene Ontology (http://www.geneongoloty.org/). The -log10 (P-value) yields an enrichment score representing the significance of GO term enrichment among differentially expressed genes. KEGG analysis was performed to determine the involvement of target genes in different biological pathways using KOBAS software (KEGG Orthology-Based Annotation System). Here, the -log10 (P-value) yields an enrichment score indicating the significance of pathway correlations.
Prediction of miRNA-circRNA interactions
CircRNAs have been reported to function as miRNA sponge to regulate the gene expression [16] and can also bind cancer-associated miRNAs to be involved in cancer-associated pathways. Therefore, Arraystar’s home-made miRNA target prediction software based on TargetScan and miRanda were applied to predict putative miRNA/circRNA interaction and search for miRNA response elements (MREs) for the differentially expressed circRNAs identified from the microarray. To get a better understanding of selected differentially expressed circRNAs in miRNA-circRNA interaction network, the degree was calculated and key target miRNAs with the degree of larger than 5 were selected. The differentially expressed circRNAs were further screened in key target miRNAs. On the basis of these findings, Cytoscape software was then used to construct the miRNA-circRNA network [18].
Validation of key circRNAs using qRT-PCR
Using the Trizol reagent, total RNA was extracted from fresh frozen samples as described previously [19]. The primers are listed in Table 2. The qPCR was performed on the ABI 7300 PCR instrument using the SYBR Green (Takara Bio Inc., Dalian, China) detection method. The cycle parameters for the PCR reaction were 94°C for 2 min, 40 cycles of 94°C for 20 s, and 58°C for 20 s. The relative gene expression levels were analyzed by the 2-ΔΔCt method [20]. GAPDH was used as an internal control to reduce errors caused by the RNA concentration and transcription efficiency.
Table 2.
Primers used for qRT-PCR analysis of circRNA and mRNA levels
| Target ID | Primer sequence 5’-3’ | PS (bp) |
|---|---|---|
| hsa_circRNA_103827 | F: CCCACTCCAAATGATGACAC | 120 |
| R: GCATATCAGGCTTGTAAAAATCA | ||
| hsa_circRNA_103829 | F: TCAGCAATTAGCAGGGAAGAGA | 123 |
| R: TGTTGCATATGTGTCCCACG | ||
| hsa_circRNA_026195 | F: ATGATCCCCTCCATTGTTGT | 158 |
| R: TTGAGAAAAGCCAGAGCTGA | ||
| hsa_circRNA_104852 | F: AGAGAAGCCTGCAGAAAAGC | 140 |
| R: GGTCACCATAACCACCACAA | ||
| hsa_circRNA_103565 | F: TATCTTAGCCGGAGGACCTG | 141 |
| R: GTATCTGGCTGGAGATGCTG | ||
| hsa_circRNA_103831 | F: CTAGCTCGGATGTTGCTGAA | 122 |
| R: AAAAATCATAGGCATGTTGCAT | ||
| hsa_circRNA_000122 | F: TGTTCATTCCTGATGGGCGG | 148 |
| R: CAGGGGTATTGACATCCACCA | ||
| hsa_circRNA_102878 | F: CCTGGTTCCTGAAGATGAGG | 124 |
| R: GCAATGGTTGCAATGATGAA | ||
| hsa_circRNA_002131 | F: AGTGGAGCCATGAAGAAAGG | 150 |
| R: TTGCCATTATCATTGCCATT | ||
| hsa_circRNA_102556 | F: TGAGAGTCTAGCTGACCGTG | 128 |
| R: CTCATGACGTTGGGATGGTC | ||
| GAPDH | F: TGTTCGTCATGGGTGTGAAC | 154 |
| R: ATGGCATGGACTGTGGTCAT |
F, forward; R, reverse; PS, product size.
Statistical analysis
Quantile normalization and subsequent data processing were performed using the R software package. The fold-change of each circRNA was computed from the profile difference between the cancer and control groups, and the significance was analyzed with a t-test. The survival analysis was conducted using the Kaplan-Meier method (log-rank test) with R software version 3.3.1 (https://www.r-project.org/). Statistical analyses were performed by GraphPad Prism 5 (GraphPad Software, La Jolla, CA).
Results
Screening of differentially expressed circRNAs
To study the expression profile of circRNAs in LSCC, we performed the circRNA expression profiles in human LSCC tumor and matched normal tissues using the microarray analysis. Unsupervised hierarchical clustering of circRNA expression patterns obviously discriminated tumor from matched normal tissue (Figure 1). The box plot is a convenient way to quickly visualize the dataset distributions. As shown in Figure 2A, the distributions of log2 ratios among six samples are nearly the same after normalization. The scatter plot of circRNA expression profile was used to assess the variations between the two groups (Figure 2B). The variations of circRNA expression between tumor and normal samples were additionally identified using volcano plot filtering (Figure 2C). Overall, 216 circRNAs were found to be significantly differentially expressed (|logFC|-value >2.0, P-value <0.05). Compared to control samples, 135circRNAs, including 1 antisense, 124 exonic, 5 intronic and 5 sense overlapping were up-regulated and 81 circRNAs, containing 2 antisense, 71 exonic, 7 intronic and 1 sense overlapping were down-regulated in tumor tissue samples (Figure 2D). The top 40 differentially expressed circRNAs were listed in Table 3.
Figure 1.
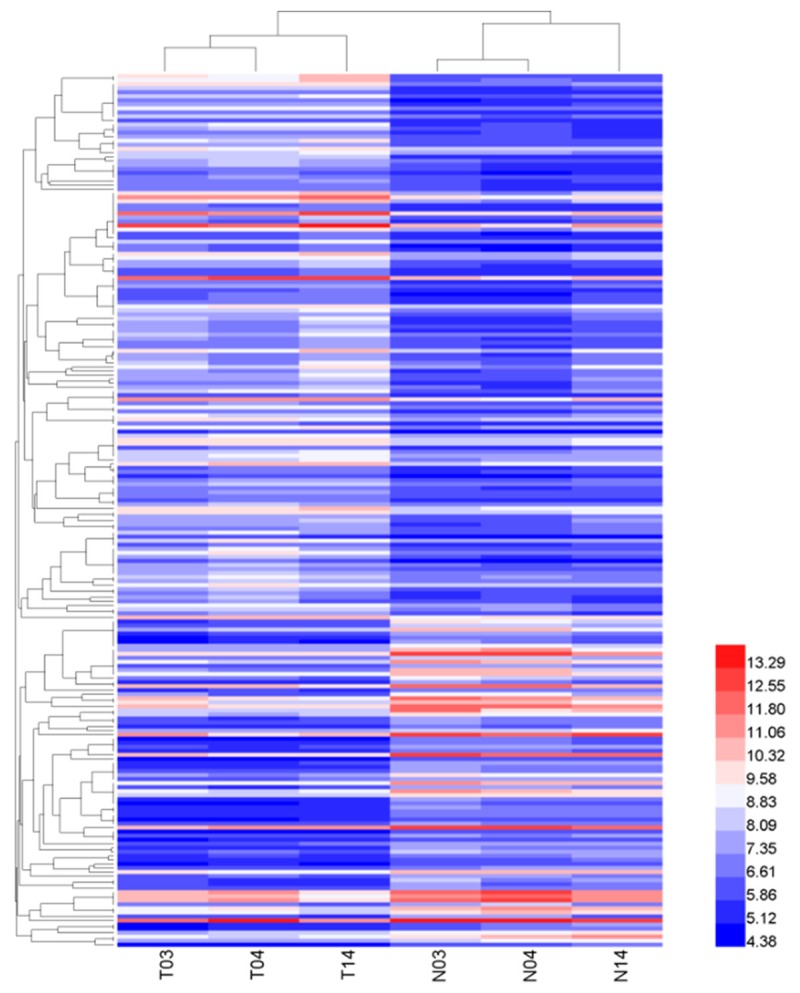
Hierarchical clustering of the differentially expressed circRNA expression data in tissues of 3 tumors (T) and 3 normal controls (N). Expression values (fold change >1.0, P<0.05) were represented in different colors, indicating expression levels above and below the median expression level across all samples.
Figure 2.
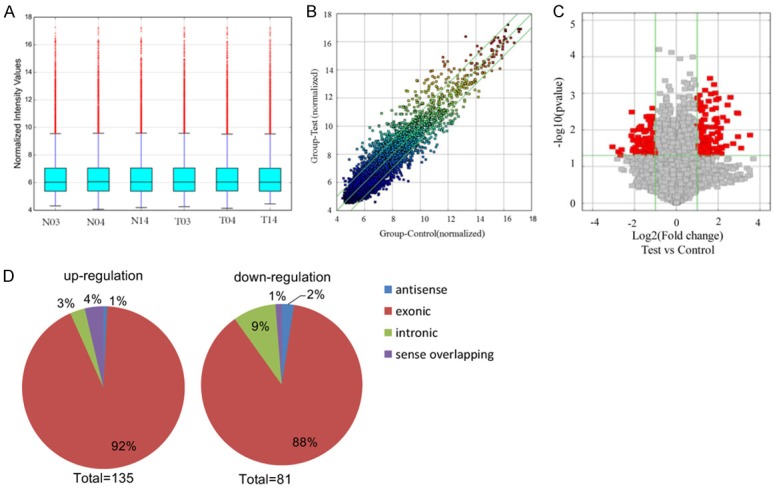
Overview of the microarray signatures. A. The box plot is used to quickly visualize the distributions of a dataset for the circRNAs profiles. After normalization, the distributions of log2 ratios among 6 samples are nearly the same (T: tumor tissue; N: normal tissue). B. The scatter plot shows the circRNA expression variation between the cancer and normal samples. The axis represents the mean normalized circRNA signal values for each comparator group (log2 scaled). The green fold-change lines represent 2.0× fold-changes, so the circRNAs lying above and below these green lines displayed greater than a 2.0-fold upregulation or downregulation. C. Volcano plot of the differentially expressed circRNAs. The vertical lines correspond to 2-fold up and down, respectively, and the horizontal line represents P=0.05. The red point in the plot represents the differentially expressed circRNAs with statistical significance. D. Classification of dysregulated circRNAs. “Exonic” represents circRNA arising from the exons of the linear transcript; “Intronic” represents the circRNA arising from an intron of the linear transcript; “antisense” represents circRNA whose gene locus overlap with the linear RNA, but transcribed from the opposite strand; “sense overlapping” represents circRNA transcribed from same gene locus as the linear transcript.
Table 3.
Top 40 differently expressed circRNAs in squamous cell lung carcinoma
| CircRNA | P-value | FC (abs) | Regulation | circRNA_type | Chrom | Strand | Gene symbol |
|---|---|---|---|---|---|---|---|
| hsa_circRNA_015879 | 0.01386628 | 11.5283772 | Up | Exonic | Chr1 | + | PKP1 |
| hsa_circRNA_026428 | 0.031485562 | 8.5694066 | Up | Exonic | Chr12 | - | KRT6A |
| hsa_circRNA_103830 | 0.003522181 | 7.7518175 | Up | Exonic | Chr5 | - | HMGCS1 |
| hsa_circRNA_104811 | 0.01528389 | 7.7173547 | Up | Exonic | Chr9 | + | NTRK2 |
| hsa_circRNA_104870 | 0.003397307 | 7.4855021 | Up | Exonic | Chr9 | - | PTGR1 |
| hsa_circRNA_026358 | 0.023629284 | 6.5706933 | Up | Exonic | Chr12 | + | KRT7 |
| hsa_circRNA_103827 | 0.001281935 | 6.0012377 | Up | Exonic | Chr5 | - | HMGCS1 |
| hsa_circRNA_001716 | 0.002075333 | 5.4783997 | Up | Antisense | Chr7 | - | LIMK1 |
| hsa_circRNA_103829 | 0.005658113 | 5.3675678 | Up | Exonic | Chr5 | - | HMGCS1 |
| hsa_circRNA_026195 | 0.027202535 | 5.2398221 | Up | Exonic | Chr12 | - | RACGAP1 |
| hsa_circRNA_087856 | 0.004488697 | 4.6929911 | Up | Exonic | Chr9 | + | RAD23B |
| hsa_circRNA_001586 | 0.03168581 | 4.5146028 | Up | Sense overlapping | Chr6 | - | HIST1H3D |
| hsa_circRNA_400446 | 0.044576345 | 4.4676052 | Up | Exonic | Chr1 | + | KIAA1804 |
| hsa_circRNA_001880 | 0.008970229 | 4.3776375 | Up | Exonic | Chr9 | + | RAD23B |
| hsa_circRNA_001681 | 0.044916399 | 4.339333 | Up | Exonic | Chr7 | - | RAPGEF5 |
| hsa_circRNA_105041 | 0.029236425 | 4.2849807 | Up | Exonic | ChrX | - | G6PD |
| hsa_circRNA_104852 | 0.016202792 | 4.2686317 | Up | Exonic | Chr9 | + | RAD23B |
| hsa_circRNA_103565 | 0.014032939 | 4.2594858 | Up | Exonic | Chr3 | - | DLG1 |
| hsa_circRNA_103499 | 0.033015781 | 4.1795048 | Up | Exonic | Chr3 | + | RSRC1 |
| hsa_circRNA_103831 | 0.03312385 | 4.1121241 | Up | Exonic | Chr5 | - | HMGCS1 |
| hsa_circRNA_001640 | 0.029103156 | 8.3301093 | Down | Exonic | Chr6 | - | EPB41L2 |
| hsa_circRNA_005536 | 0.03934785 | 6.8276119 | Down | Exonic | Chr2 | - | RFX8 |
| hsa_circRNA_105055 | 0.047690013 | 6.4173918 | Down | Antisense | ChrX | + | CDR1 |
| hsa_circRNA_001729 | 0.034586505 | 6.142103 | Down | Antisense | Chr16 | - | ZNF646 |
| hsa_circRNA_406549 | 0.014367194 | 4.4334509 | Down | Exonic | Chr4 | - | NR3C2 |
| hsa_circRNA_405974 | 0.003242037 | 4.3796355 | Down | Intronic | Chr2 | - | ACVR1 |
| hsa_circRNA_000122 | 0.037309993 | 4.2534161 | Down | Intronic | Chr1 | + | NBPF10 |
| hsa_circRNA_102878 | 0.019240939 | 4.1755922 | Down | Exonic | Chr2 | + | SLC39A10 |
| hsa_circRNA_002131 | 0.010388138 | 4.171018 | Down | Exonic | Chr8 | + | BNIP3L |
| hsa_circRNA_103383 | 0.032687391 | 4.1524014 | Down | Exonic | Chr3 | + | MAPKAPK3 |
| hsa_circRNA_104169 | 0.029684253 | 3.8717715 | Down | Exonic | Chr6 | + | SOBP |
| hsa_circRNA_103730 | 0.045580688 | 3.8610618 | Down | Exonic | Chr4 | - | PRDM5 |
| hsa_circRNA_101911 | 0.038859021 | 3.7748384 | Down | Exonic | Chr16 | - | FANCA |
| hsa_circRNA_020624 | 0.020144053 | 3.6887455 | Down | Exonic | Chr11 | + | IFITM1 |
| hsa_circRNA_102051 | 0.023760037 | 3.5624094 | Down | Exonic | Chr17 | + | TADA2A |
| hsa_circRNA_102556 | 0.015587946 | 3.5538688 | Down | Exonic | Chr19 | + | AXL |
| hsa_circRNA_007443 | 0.041903051 | 3.5493882 | Down | Exonic | Chr8 | - | RUNX1T1 |
| hsa_circRNA_103908 | 0.01913262 | 3.5444991 | Down | Exonic | Chr5 | - | EDIL3 |
| hsa_circRNA_403691 | 0.009785705 | 3.4970096 | Down | Exonic | Chr6 | + | LOC101927768 |
| hsa_circRNA_100789 | 0.038176191 | 3.456096 | Down | Exonic | Chr11 | + | CAPRIN1 |
FDR, false discover rate; FC, fold change
CircRNAs gene symbols GO analysis and pathway analysis
In order to investigate exposures gene expression on a more functional level, we conducted GO analysis and pathway analysis for circRNAs gene symbols to speculate circRNA potential functions. These differentially expressed circRNAs were firstly classified into GO terms, including BP, CC and MF. The count number larger than 2 and FDR less than 0.05 were chosen as cut-off criteria. The results showed that the most significant enriched GO term in the BP was cell morphogenesis involved in differentiation (GO: 0000904, P=4.26E-07) (Figure 3A); the most significant enriched GO term in the CC was membrane-bounded organelle (GO: 0043227, P=2.37E-06) (Figure 3B) and tRNA binding was the most significant enriched GO term in MF (GO: 0000049, P=1.32E-04) (Figure 3C). In addition, total 13 pathways were screened to be involved in the progression of LSCC (Figure 3D).
Figure 3.
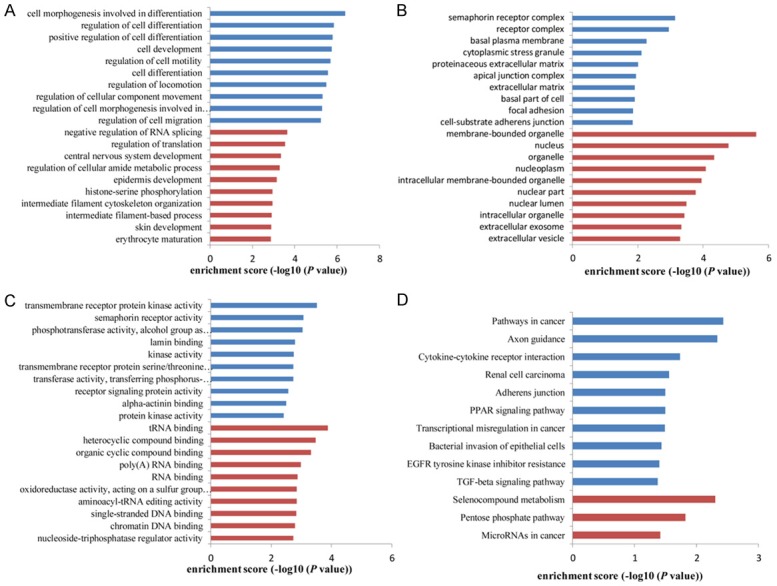
GO enrichment and pathway analysis for dysregulated circRNAs gene symbols. Most significantly enriched GO (-log10 (P value)) terms of circRNAs gene symbols according to (A) biological process, (B) cellular component and (C) molecular function. (D) Top 10 classes of KEGG pathway enrichment terms (-log10 (P value)). Red bar represents upregulated circRNA gene symbol; blue bar represents downregulated circRNA gene symbol.
miRNA prediction of differentially expressed circRNAs
The miRNA prediction of differentially expressed circRNAs was performed based on TargetScan and miRanda. A total of 1070 pairs of regulatory relationships between miRNA and circRNA were identified. To further investigate which miRNA was more significant, we calculated the degree and 17 miRNAs (containing 114 regulatory relationship pairs) with the degree of larger than 5 were screened (Table 4). Using Cytoscape, the regulatory relationships were integrated and visualized in Figure 4. Moreover, we found these 17 miRNAs could regulate 55 upregulated differentially expressed circRNAs and 23 downregulated differentially expressed circRNAs.
Table 4.
The results for miRNA-circRNA prediction with the number of circRNA greater than 5
| miRNA | Degree |
|---|---|
| hsa-miR-138-5p | 11 |
| hsa-miR-627-3p | 9 |
| hsa-miR-766-5p | 9 |
| hsa-miR-129-5p | 8 |
| hsa-miR-22-5p | 7 |
| hsa-miR-511-5p | 7 |
| hsa-miR-512-3p | 7 |
| hsa-miR-637 | 7 |
| hsa-miR-140-3p | 5 |
| hsa-miR-197-3p | 5 |
| hsa-miR-325 | 5 |
| hsa-miR-532-5p | 5 |
| hsa-miR-544a | 5 |
| hsa-miR-593-3p | 5 |
| hsa-miR-625-3p | 5 |
| hsa-miR-765 | 5 |
| hsa-miR-889-5p | 5 |
Figure 4.

Network map of miRNA-circRNA interactions in LSCC tissues (miRNAs with the degree of larger than 5). Gray circular node: miRNA; red triangle node: upregulated differentially expressed circRNA; blue V type node: downregulated differentially expressed circRNA.
Screening of key circRNAs
After further analysis, total 10 differentially expressed circRNAs, including 6 upregulated hsa_circRNA_103827, hsa_circRNA_103829, hsa_circRNA_026195, hsa_circRNA_104852, hsa_circRNA_103565, hsa_circRNA_103831 and 4 downregulated hsa_circRNA_000122, hsa_circRNA_102878, hsa_circRNA_002131 and hsa_circRNA_102556 were regulated by hsa-miR-138-5p, hsa-miR-627-3p, hsa-miR-766-5p and hsa-miR-129-5p with the degree of larger than 7, thus were identified as key circRNAs. Furthermore, we searched for putative MREs through Arraystar’s circRNA target prediction software. The predicted MREs for the 10 key circRNAs were listed in Table 5.
Table 5.
Significantly up- and down-regulated circRNAs in squamous cell lung carcinoma versus benign lung tissue
| CircRNA | Gene symbol | P-value | FC-change | Predicted miRNA response elements (MREs) | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| MRE1 | MRE2 | MRE3 | MRE4 | MRE5 | ||||
| hsa_circRNA_103827 | HMGCS1 | 0.0013 | ↑6.001 | hsa-miR-411-5p | hsa-miR-625-3p | hsa-miR-129-5p | hsa-miR-205-5p | hsa-miR-532-5p |
| hsa_circRNA_103829 | HMGCS1 | 0.0057 | ↑5.367 | hsa-miR-625-3p | hsa-miR-129-5p | hsa-miR-532-5p | hsa-miR-548c-3p | hsa-let-7c-5p |
| hsa_circRNA_026195 | RACGAP1 | 0.0272 | ↑5.239 | hsa-miR-129-5p | hsa-miR-1183 | hsa-miR-616-3p | hsa-miR-5197-3p | hsa-miR-6833-3p |
| hsa_circRNA_104852 | RAD23B | 0.0162 | ↑4.269 | hsa-miR-138-5p | hsa-miR-325 | hsa-miR-593-3p | hsa-miR-512-3p | hsa-miR-766-5p |
| hsa_circRNA_103565 | DLG1 | 0.0140 | ↑4.259 | hsa-miR-627-3p | hsa-miR-138-5p | hsa-miR-155-3p | hsa-miR-653-5p | hsa-miR-651-3p |
| hsa_circRNA_103831 | HMGCS1 | 0.0331 | ↑4.112 | hsa-miR-625-3p | hsa-miR-129-5p | hsa-miR-548c-3p | hsa-let-7c-5p | hsa-let-7b-5p |
| hsa_circRNA_000122 | NBPF10 | 0.0373 | ↓4.253 | hsa-miR-627-3p | hsa-miR-510-5p | hsa-miR-552-3p | hsa-miR-4695-3p | hsa-miR-503-3p |
| hsa_circRNA_102878 | SLC39A10 | 0.0192 | ↓4.176 | hsa-miR-335-3p | hsa-miR-130b-5p | hsa-miR-302b-5p | hsa-miR-329-5p | hsa-miR-627-3p |
| hsa_circRNA_002131 | BNIP3L | 0.0104 | ↓4.171 | hsa-miR-136-5p | hsa-miR-627-3p | hsa-miR-942-5p | hsa-miR-6500-5p | hsa-miR-5003-3p |
| hsa_circRNA_102556 | AXL | 0.0156 | ↓3.554 | hsa-miR-764 | hsa-miR-346 | hsa-miR-584-3p | hsa-miR-654-3p | hsa-miR-766-5p |
Verification of the expression changes of key circRNAs
To verify the microarray data, we selected the above 10 key circRNAs, including six up-regulated circRNAs and four down-regulated circRNAs. We validated their expression levels via qRT-PCR in 40 sets of LSCC tissues and adjacent normal tissues. We found that the expression patterns of ten key circRNAs were consistent with the microarray data (Figure 5). These results further validated our findings and implied that these key circRNAs may play important roles in LSCC carcinogenesis.
Figure 5.

Total ten key differentially expressed circRNAs, including six up-regulated and four down-regulated were validated by qRT-PCR in 40 LSCC tissues compared with paired adjacent normal tissues, which was consistent with the microarray data; ***P<0.001.
Clinical impact of circRNAs in LSCC patients
Among 10 key circRNAs, hsa_circRNA_103827 and hsa_circRNA_000122 were selected as a model for prognostic analysis, mainly due to their relatively large differential expression in LSCC tissues. Within 40 LSCC tissues, hsa_circRNA_103827 and hsa_circRNA_000122 were displayed higher and lower expression relative to normal tissues, respectively. We thus further divided LSCC patients into a high expression group and a low expression group according to the median value of hsa_circRNA_103827 and hsa_circRNA_000122 ex-pression levels, respectively. Kaplan-Meier analysis with log-rank test was performed to study the prognostic significance ofhsa_circRNA_103827 and hsa_circRNA_000122. As shown in Figure 6A, patients with low hsa_circRNA_103827 expression had higher overall survival rate than those with high expression (P<0.001). Consistently, the survival of the hsa_circRNA_000122-high group was significantly longer than that of the hsa_circRNA_000122-low group (Figure 6B). Thus, hsa_circRNA_103827 and hsa_circRNA_000122 might be potential biomarkers for clinical diagnosis and evaluation.
Figure 6.
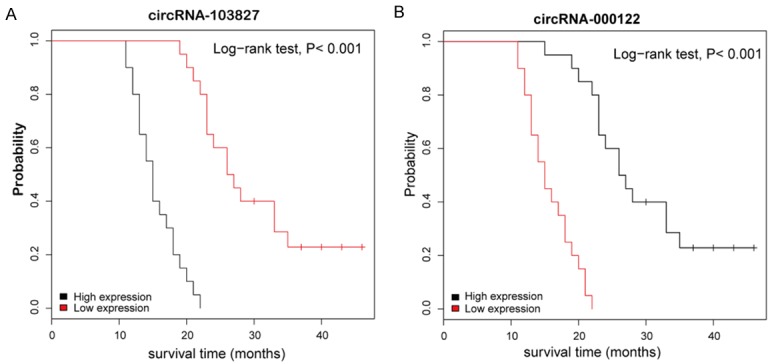
Kaplan-Meier curves for overall survival based on hsa_circRNA_103827 and hsa_circRNA_000122 expression in group of 40 LSCC patients; P<0.001, log-rank test.
Discussion
LSCC, as the main type of NSCLC, is associated with higher incidence relative to other lung cancer types, remaining the leading risk factor for lung cancer progression. More recently, noncoding circRNAs have become more attention as new diagnostic markers for diseases, especially cancer, which mainly ascribed to highly conserved sequences and high degree of stability in mammalian cells compared with other noncoding miRNAs and lncRNAs [9,11,21]. Currently, although an increasing number of researchers have begun tostudy potential functions of circRNAs, however, very little is known regarding circRNAs dysregulation in LSCC.
In this study, we utilized circRNA microarray to acquire circRNAs expression profile of LSCC for the first time. Hierarchical clustering analysis further confirmed the different expression patterns of circRNAs in LSCC tissues and adjacent normal tissues. A total of 216 differentially expressed circRNAs were screened out, including 135 upregulated and 81 downregulated ones (|logFC|-value >2.0, P-value <0.05) in three paired LSCC samples. Furthermore, these differentially expressed circRNAs were performed GO analysis and pathway analysis. Among the GO terms found in this study, negative regulation of RNA splicing had been reported to be prominent drivers of cancer [22], of which polypyrimidine tract-binding protein (PTBP1) can act as a repressive or as an enhancing alternative splicing regulator [23,24]. In addition, “PPAR signaling pathway” [25,26] and “TGF-beta signaling pathway” [27,28] have been demonstrated to participate the progression of lung cancer. We thus guess that these differentially expressed circRNAs might be closely associated with the development and progression of LSCC.
As miRNA sponges, circRNAs can interact cancer-associated miRNAs to be involved in cancer-associated pathways [29-31]. Considering miRNAs play important roles in the progression of lung cancer [32,33], we performed the miRNA prediction of these above differentially expressed circRNAs and selected 4 miRNAs (the degree of larger than 7), including hsa-miR-138-5p, hsa-miR-627-3p, hsa-miR-766-5p and hsa-miR-129-5p. We further screened 10 differentially expressed circRNAs, including 6 upregulated hsa_circRNA_103827, hsa_circRNA_-103829, hsa_circRNA_026195, hsa_circRNA_104852, hsa_circRNA_103565, hsa_circRNA_103831 and 4 downregulated hsa_circRNA_000122, hsa_circRNA_102878, hsa_circRNA_002131 and hsa_circRNA_102556 have potential interactions with these four miRNAs. For example, hsa_circRNA_103827 is potentially able to bind hsa-miR-129-5p. Moreover, miR-129-5p could inhibit NSCLC cell proliferation and invasion [34]. Hsa-miR-129-5p, interacted with hsa_circRNA_103831, has been reported to reverse gefitinib resistance in NSCLC cells [35]. However, additional studies are still required to confirm the relation between these screened circRNAs and miRNAs in LSCC.
Moreover, we selected the above ten dysregulated circRNAs from the microarray data to validate their expression levels using qRT-PCR in another 40 LSCC tissues and paired adjacent normal tissues. As expect, the results of qRT-PCR analysis were consistent with the microarray data, confirming that the microarray data are reliable. Importantly, we performed prognostic analysis of hsa_circRNA_103827 and hsa_circRNA_000122 in these 40 paired tissues using Kaplan-Meier analysis. The results indicated that elevated hsa_circRNA_103827 was always accompanied by a decreased cumulative survival rate, while decreased hsa_circRNA_000122 had higher overall survival rate. According the previous studies, hsa_circRNA_103827 could be used to predict in vitro fertilization prognosis [36,37]. Hsa_circRNA_000122 was reported to provide a new therapeutic line of approach to glioblastoma multiforme [38] and micropapillary carcinoma [39]. Thus, we inferred that hsa_circRNA_103827 and hsa_circRNA_000122 might be correlated with poor prognosis in LSCC.
In summary, this study provided a preliminary landscape of circRNA differential expression in LSCC tissues. After a series of bioinformatics analysis, we found elevated expression of hsa_circRNA_103827 and decreased expression of hsa_circRNA_000122 were closely related to the development of LSCC. Altogether, these findings provide potential targets for the future treatment of LSCC and novel insights into LSCC. However, further studies are still required to explore their potential as biomarkers for LSCC as well as their pathologicrole.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No: 81502513); The Jiangsu Province Natural Science Foundation of China (Grant No: BK2015-1028) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (JX10231801). The funding organizations had no role in study design, data collection, analysis, decision to publish and manuscript preparation.
Disclosure of conflict of interest
None.
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Zhang F, Chen X, Wei K, Liu D, Xu X, Zhang X, Shi H. Identification of key transcription factors associated with lung squamous cell carcinoma. Med Sci Monit. 2017;23:172–206. doi: 10.12659/MSM.898297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ettinger DS, Wood DE, Akerley W, Bazhenova LA, Borghaei H, Camidge DR, Cheney RT, Chirieac LR, D’Amico TA, Demmy TL, Dilling TJ, Dobelbower MC, Govindan R, Grannis FW Jr, Horn L, Jahan TM, Komaki R, Krug LM, Lackner RP, Lanuti M, Lilenbaum R, Lin J, Loo BW Jr, Martins R, Otterson GA, Patel JD, Pisters KM, Reckamp K, Riely GJ, Rohren E, Schild SE, Shapiro TA, Swanson SJ, Tauer K, Yang SC, Gregory K, Hughes M National comprehensive cancer network. Non-small cell lung cancer, version 6.2015. J Natl Compr Canc Netw. 2015;13:515–524. doi: 10.6004/jnccn.2015.0071. [DOI] [PubMed] [Google Scholar]
- 4.Heist RS, Sequist LV, Engelman JA. Genetic changes in squamous cell lung cancer: a review. J Thorac Oncol. 2012;7:924–933. doi: 10.1097/JTO.0b013e31824cc334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drilon A, Rekhtman N, Ladanyi M, Paik P. Squamous-cell carcinomas of the lung: emerging biology, controversies, and the promise of targeted therapy. Lancet Oncol. 2012;13:e418–426. doi: 10.1016/S1470-2045(12)70291-7. [DOI] [PubMed] [Google Scholar]
- 6.Hou Z, Xu C, Xie H, Xu H, Zhan P, Yu L, Fang X. Long noncoding RNAs expression patterns associated with chemo response to cisplatin based chemotherapy in lung squamous cell carcinoma patients. PLoS One. 2014;9:e108133. doi: 10.1371/journal.pone.0108133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu HJ, Zhang CY, Zhang S, Chang M, Wang HY. Microarray expression profile of circular RNAs in heart tissue of mice with myocardial infarction-induced heart failure. Cell Physiol Biochem. 2016;39:205–216. doi: 10.1159/000445617. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Li C, Tan C, Liu X. Circular RNAs: a new frontier in the study of human diseases. J Med Genet. 2016;53:359–365. doi: 10.1136/jmedgenet-2016-103758. [DOI] [PubMed] [Google Scholar]
- 9.Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, Sun W, Dou K, Li H. Circular RNA: a new star of noncoding RNAs. Cancer Lett. 2015;365:141–148. doi: 10.1016/j.canlet.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: identification, biogenesis and function. Biochim Biophys Acta. 2016;1859:163–168. doi: 10.1016/j.bbagrm.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233. doi: 10.1371/journal.pgen.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lukiw WJ. Circular RNA (circRNA) in Alzheimer’s disease (AD) Front Genet. 2013;4:307. doi: 10.3389/fgene.2013.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peifei L, Shengcan C, Huilin C, Xiaoyan M, Tianwen L, Yongfu S, Bingxiu X, Junming G. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clinica Chimica Acta. 2015;444:132–136. doi: 10.1016/j.cca.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 14.Yao JT, Zhao SH, Liu QP, Lv MQ, Zhou DX, Liao ZJ, Nan KJ. Over-expression of CircRNA_100876 in non-small cell lung cancer and its prognostic value. Pathol Res Pract. 2017;213:453–456. doi: 10.1016/j.prp.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Hao X, Wang H, Liu Z, He Y, Pu M, Zhang H, Yu H, Duan J, Qu S. Circular RNA expression profile of pancreatic ductal adenocarcinoma revealed by microarray. Cell Physiol Biochem. 2016;40:1334–1344. doi: 10.1159/000453186. [DOI] [PubMed] [Google Scholar]
- 16.Sand M, Bechara FG, Gambichler T, Sand D, Bromba M, Hahn SA, Stockfleth E, Hessam S. Circular RNA expression in cutaneous squamous cell carcinoma. J Dermatol Sci. 2016;83:210–218. doi: 10.1016/j.jdermsci.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 17.Cao S, Wei D, Li X, Zhou J, Li W, Qian Y, Wang Z, Li G, Pan X, Lei D. Novel circular RNA expression profiles reflect progression of patients with hypopharyngeal squamous cell carcinoma. Oncotarget. 2017;8:45367–45379. doi: 10.18632/oncotarget.17488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 19.Yuan D, Lan F, Ouyang X, Wang K, Lin Y, Yu Y, Wang L, Wang Y, Huang Q. Expression and clinical significance of long non-coding RNA HNF1A-AS1 in human gastric cancer. World J Surg Oncol. 2015;13:302. doi: 10.1186/s12957-015-0706-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai Y, Sui W, Lan H, Yan Q, Huang H, Huang Y. Comprehensive analysis of microRNA expression patterns in renal biopsies of lupus nephritis patients. Rheumatol Int. 2009;29:749–754. doi: 10.1007/s00296-008-0758-6. [DOI] [PubMed] [Google Scholar]
- 21.Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollander D, Donyo M, Atias N, Mekahel K, Melamed Z, Yannai S, Lev-Maor G, Shilo A, Schwartz S, Barshack I, Sharan R, Ast G. A network-based analysis of colon cancer splicing changes reveals a tumorigenesis-favoring regulatory pathway emanating from ELK1. Genome Res. 2016;26:541–553. doi: 10.1101/gr.193169.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon YS, Zhang C, Yeo G, Black DL, Sun H, Fu XD, Zhang Y. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keppetipola N, Sharma S, Li Q, Black DL. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol. 2012;47:360–378. doi: 10.3109/10409238.2012.691456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iitaka D, Moodley S, Shimizu H, Bai XH, Liu M. PKCdelta-iPLA2-PGE2-PPARgamma signaling cascade mediates TNF-alpha induced Claudin 1 expression in human lung carcinoma cells. Cell Signal. 2015;27:568–577. doi: 10.1016/j.cellsig.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 26.Reka AK, Goswami MT, Krishnapuram R, Standiford TJ, Keshamouni VG. Molecular cross-regulation between PPAR-γ and other signaling pathways: Implications for lung cancer therapy. Lung Cancer. 2011;72:154–159. doi: 10.1016/j.lungcan.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin LC, Hsu SL, Wu CL, Hsueh CM. TGFβ can stimulate the p 38/β-catenin/PPARγ signaling pathway to promote the EMT, invasion and migration of non-small cell lung cancer (H460 cells) Clin Exp Metastasis. 2014;31:881–95. doi: 10.1007/s10585-014-9677-y. [DOI] [PubMed] [Google Scholar]
- 28.He XR, Han SY, Li XH, Zheng WX, Pang LN, Jiang ST, Li PP. Chinese medicine Bu-Fei decoction attenuates epithelial-mesenchymal transition of non-small cell lung cancer via inhibition of transforming growth factor beta1 signaling pathway in vitro and in vivo. J Ethnopharmacol. 2017;204:45–57. doi: 10.1016/j.jep.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 29.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–8. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 30.Wang K, Long B, Liu F, Wang JX, Liu CY, Zhao B, Zhou LY, Sun T, Wang M, Yu T, Gong Y, Liu J, Dong YH, Li N, Li PF. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016;37:2602–2611. doi: 10.1093/eurheartj/ehv713. [DOI] [PubMed] [Google Scholar]
- 31.Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215. doi: 10.1038/ncomms11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shao Y, Liang B, Long F, Jiang SJ. Diagnostic MicroRNA biomarker discovery for non-small-cell lung cancer adenocarcinoma by integrative bioinformatics analysis. Biomed Res Int. 2017;2017:2563085. doi: 10.1155/2017/2563085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daugaard I, Veno MT, Yan Y, Kjeldsen TE, Lamy P, Hager H, Kjems J, Hansen LL. Small RNA sequencing reveals metastasis-related microRNAs in lung adenocarcinoma. Oncotarget. 2017;8:27047–27061. doi: 10.18632/oncotarget.15968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, An J, Lv W, Lou T, Liu Y, Kang W. miRNA-129-5p suppresses cell proliferation and invasion in lung cancer by targeting microspherule protein 1, E-cadherin and vimentin. Oncol Lett. 2016;12:5163–5169. doi: 10.3892/ol.2016.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Y, Fan X, Li W, Ping W, Deng Y, Fu X. miR-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124. Biochem Biophys Res Commun. 2014;446:179–186. doi: 10.1016/j.bbrc.2014.02.073. [DOI] [PubMed] [Google Scholar]
- 36.Cheng J, Huang J, Yuan S, Zhou S, Yan W, Shen W, Chen Y, Xia X, Luo A, Zhu D, Wang S. Circular RNA expression profiling of human granulosa cells during maternal aging reveals novel transcripts associated with assisted reproductive technology outcomes. PLoS One. 2017;12:e0177888. doi: 10.1371/journal.pone.0177888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura T, Iwase A, Bayasula B, Nagatomo Y, Kondo M, Nakahara T, Takikawa S, Goto M, Kotani T, Kiyono T, Kikkawa F. CYP51A1 induced by growth differentiation factor 9 and follicle-stimulating hormone in granulosa cells is a possible predictor for unfertilization. Reprod Sci. 2015;22:377–384. doi: 10.1177/1933719114529375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W, Li K, Zhao L, Zou H. Bioinformatics analysis reveals disturbance mechanism of MAPK signaling pathway and cell cycle in Glioblastoma multiforme. Gene. 2014;547:346–350. doi: 10.1016/j.gene.2014.06.042. [DOI] [PubMed] [Google Scholar]
- 39.Natrajan R, Wilkerson PM, Marchio C, Piscuoglio S, Ng CK, Wai P, Lambros MB, Samartzis EP, Dedes KJ, Frankum J, Bajrami I, Kopec A, Mackay A, A’Hern R, Fenwick K, Kozarewa I, Hakas J, Mitsopoulos C, Hardisson D, Lord CJ, Kumar-Sinha C, Ashworth A, Weigelt B, Sapino A, Chinnaiyan AM, Maher CA, Reis-Filho JS. Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast. J Pathol. 2014;232:553–565. doi: 10.1002/path.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]