

Abstract
This study aimed to investigate the exact function of RGC-32 in kidney diseases and explore the potential mechanism of RGC-32 in regulating cell cycle. RGC-32 knockout (RGC-32-/-) mice were generated from C57BL/6 embryonic stem cells. Differentially expressed proteins in the kidney were investigated with the isobaric tags for relative and absolute quantification (iTRAQ) technique. Gene ontology analyses (GO), Kyoto encyclopedia of genes and genomes (KEGG) pathway mapping analysis and functional network analysis were also performed. The expressions of Smc3, Smad 2-3, DNA-PK were further confirmed by qPCR. Results showed that 4690 proteins were quantified on the basis of 25165 unique peptides. Comparative proteomic analysis revealed 361 differentially expressed proteins in RGC-32-/- mice (knockout/wild ratio >+/- 1.2 and P<0.05). GO and KEGG pathway mapping analyses showed differentially expressed proteins were involved in spliceosome, fluid shear stress and atherosclerosis protein processing in endoplasmic reticulum, pathways in cancer, viral carcinogenesis, epithelial cell signaling in Helicobacter pylori infection, HTLV-I infection, PI3K-Akt signaling pathway, ubiquitin mediated proteolysis, Parkinson’s disease, MAPK signaling pathway, carbon metabolism, Alzheimer’s disease, NOD-like receptor signaling pathway, tight junction, Proteoglycans in cancer, phagosome, ribosome, mTOR signaling pathway, and AMPK signaling pathway. Differentially expressed proteins Smc3 (0.821), DNA-PK (0.761), Smad 2-3 (0.631) were involved in cell cycle regulation. mRNA expression of Smad2-3, DNA-PK, and Smc3 was consistent with that from iTRAQ. It is concluded that RGC-32 may affect the expression of many proteins (76 up-regulated and 285 down-regulated) in the kidney, and may regulate the expression of Smc3, DNA-PK and Smad 2-3 to affect the cell cycle.
Keywords: RGC-32, RGC-32 knockout mice, iTRAQ, cell cycle, renal tubular epithelial cell injury and repair
Introduction
Response gene to complement 32 (RGC-32), also known as the regulator of cell cycle (RGCC), was first cloned in the rat by Badea et al [1]. It’s expressed in a variety of tissues and organs including the kidney, skeletal muscle, placenta, pancreas, liver, etc [2]. Studies have shown that RGC-32 plays an important role in the cell proliferation, cell differentiation of inflammation and cancer [2-8]. It has been shown that RGC-32 may affect the cell cycle progression, especially the G2/M phase [9-12]. Over-expression of RGC-32 may increase the DNA synthesis and cause cell cycle progression from G1/G0 phase to G2/M phase, and RGC-32 gene silencing inhibits cell cycle progression and induces cell arrest in G2/M phase [4,13,14]. However, the exact mechanism underlying the regulation of RGC-32 on cell cycle remains still unclear and needs to be further elucidated.
Our previous study showed RGC-32 was involved in the pathogenesis of some renal diseases [6]. RGC-32 was critical for the transforming growth factor β-induced epithelial-mesenchymal transition of human renal proximal tubular cells. There was a Smad binding element in the upstream of the transcription start site of RGC-32 gene. Smad2 and PEA3 cooperatively activated the promoter of RGC-32 gene [15]. In children with immunoglobulin A nephropathy (IgAN), RGC-32 was expressed in the renal tubular epithelial cells and related to the degree of renal pathology [16]. In addition, RGC-32 expression following renal ischemia reperfusion (IRI) injury decreased significantly in a time-dependent manner in a rat model [17]. In vitro, RGC-32 gene silencing was found to arrest TNF-α (Tumor Necrosis Factor-α) treated NRK-52E cells in G2/M phase [18]. On the basis of these findings, we hypothesized that RGC-32 might be involved in the renal tubular injury and repair.
The present study aimed to investigate the protein expression profile in RGC-32 knockout (RGC-32-/-) mice as compared to wild type (RGC-32+/+) mice and explore the potential mechanism of RGC-32 regulating the cell cycle.
Materials and methods
Animal care
The mice were housed in animal cages (5 mice/cage) with suitable ventilation, 12 h light dark cycle, temperature of 22-25°C, and free access to food and water, in the Medical Genetic Research Institute of Shanghai. All experimental protocols were previously reviewed and approved by the Ethics Committee for Animal Experimentation of Shanghai Children’s Hospital.
Generation and mating of RGC-32-deficient mice
RGC-32-deficient (RGC32-/-) mice were generated from C57BL/6 embryonic stem cells with deletion of exon 2 and 3 of the RGC-32 gene. The RGC-32 gene has only one transcriptomes coding protein in mice. L1L2_Bact_P transgene vector was built and then introduc-ed into embryonic stem cells. 9311 bases from 79300967 to 79291656 on chromosome 14, which were exons 2 and 3 of RGC-32 gene were replaced by homologous recombination, aiming to make target gene disabled. Recombined embryonic stem cell was injected into blastomere of C57BL/6 mice, and then implanted into mice, which became RGC-32+/- mice. RGC-32-/- mice was then generated trough copulation. RGC-32-/- embryonic stem (ES) cells with C57BL/6 background were purchased from the Knockout Mouse Project (KOMP) Repository (https://www.komp.org/geneinfo.php?project = CSD69150, UC Davis, CA, USA). RGC-32-/- ES cell-mouse chimeras were also produced by the KOMP Repository. Germ line transmission was obtained by breeding the male chimeras with C57BL/6 female mice. RGC-32-/- mice were acquired by mating RGC-32-/+ male mice with RGC-32-/+ female mice.
Genotyping of mice by PCR
The tail tissues (0.5-1.0 cm) were obtained mice aged 4-6 weeks and total DNA was extracted for PCR. To genotype the mice, two pairs of primers were provided by KOMP Repository. 1st pair of primers (product size: 750 bp): 5’-CAG CAT CTC TGC TAC GCG TGT CAC TGA G-3’ (forward); 5’-CAC AAC GGG TTC TTC TGT TAG TCC-3’ (reverse); 2nd pair of primers (product size: 285 bp): 5’-AGG TAG CCA GTG GAC TTG GGC ACA CAC-3’ (forward); 5’-CAG GAG AGC TGG AGA GGA GTT GGT TGG-3’ (reverse).
Sample collection and protein extraction
Male mice were sacrificed at the age of 12-16 weeks. One kidney of each mouse was collected and stored at -80°C for protein processing. 15 RGC-32-/- mice and 15 RGC-32+/+ mice were randomly divided to 3 groups independently (5 mice in each group). Lysis buffer (4% SDS, 1 mM DTT, 150 mM Tris-HCl pH 7.6, and protease inhibitor) with protease inhibitor (Thermo Fisher, Germany) was added to the renal tissues of each group, followed by homogenization. The mixture was sonicated for 5 min on ice. The homogenate was then centrifuged at 14000 g for 10 min at 4°C. The supernatant was collected, transferred into a new tube and stored at -80°C. The protein content was determined with the BCA protein assay kit (Thermo Fisher, Germany).
Protein processing and iTRAQ labeling
Protein processing was performed according to the filter-aided sample preparation procedure described by Wisniewski et al [19]. The resultant peptide mixture was labeled using the 8-plex iTRAQ reagent (AB SCIEX, Framingham, MA, USA) according to the manufacturer’s instructions. Briefly, 200 mg of proteins from each sample was added into 30 mL of STD buffer (4% SDS, 100 mM DTT, 150 mM Tris-HCl, pH 8.0). The detergent, DTT and other low-molecular-weight components were removed using UA buffer (8 M urea, 150 mM Tris-HCl, pH 8.0), followed by repeated ultrafiltration (Microcon units, 30 kDa). 100 mL of 0.05 M iodoacetamide in UA buffer was then added to block reduced cysteine residues, followed by incubation for 20 min in dark. Finally, the protein suspension was digested with 2 mg of sequencing-grade trypsin (Promega, Madison, WI, USA) in 40 mL of DS buffer at 37°C for 16 h and the resultant peptides were collected as a filtrate. The peptide content was determined by detecting the optical density at 280 nm with an UV spectrophotometer, and an extinction coefficient of 1.1 of 0.1% (g/l) solution was calculated on the basis of the frequency of tryptophan and tyrosine in vertebrate proteins [20]. For labeling, each iTRAQ reagent was dissolved in 70 μl of ethanol and then added to the peptide mixture. The samples were labeled with iTRAQ reagent 113, 114, 115, 116, 117, 118, 119 and 121, respectively, multiplexed and then dried in vacuum.
Peptide fractionation with strong cation exchange (SCX) chromatography
iTRAQ-labeled peptides were fractionated by SCX chromatography using the AKTA purifier system (GE Healthcare, Fairfield, CT, USA). The dried peptide mixture was reconstituted and acidified with 2 ml of buffer A (10 mM KH2PO4 in 25% of ACN, pH 2.7) and subjected to a polysulfoethyl A column (100 mm × 4.6 mm, 200 A pore size, 5 mm particle size) (PolyLC, Columbia, MD, USA) on a Waters Delta 600 HPLC unit (Waters, Milford, MA, USA). The peptides were eluted at a flow rate of 1 ml/min-1 with 0-10% buffer B (500 mM KCl, 10 mM KH2PO4 in 25% of ACN, pH 2.7) for 2 min, 10-20% buffer B for 25 min, 20-45% buffer B for 5 min, and 50-100% buffer B for 5 min. Elution was monitored by detecting the absorbance at 214 nm, and the fractions were collected once every 1 min. The collected fractions (about 30 fractions) were integrated into 10 pools and desalted on C18 cartridges (Sigma-Aldrich Chemical Co, St. Louis, MO, USA). Each fraction was concentrated by vacuum centrifugation and reconstituted in 40 l of 0.1% (v/v) trifluoroacetic acid. All samples were stored at -80°C.
Liquid chromatography (LC) - electrospray ionization (ESI) Tandem MS (MS/MS) analysis
Experiments were performed on a Q Exactive mass spectrometer that was coupled to Easy nLC (Thermo Fisher Scientific Inc., San Jose, CA, USA). 10 mL of each fraction was injected for nano LC-MS/MS. The peptide mixture (5 mg) was loaded onto a C18-reversed-phase column (Thermo Scientific Easy Column, 10 cm long, 75 mm inner diameter, 3 mm resin) in buffer A (0.1% formic acid) and separated using a linear gradient of buffer B (80% acetonitrile and 0.1% formic acid) at a flow rate of 250 nl/min controlled by IntelliFlow technology over 140 min. MS data were acquired using a data-dependent top 10 method, and the most abundant precursor ions were dynamically chosen from the survey scan (300-1800 m/z) for higher-energy collisional dissociation fragmentation. Determination of the target value was based on predictive automatic gain control. The dynamic exclusion duration was 60 s. Survey scans were acquired at a resolution of 70 000 at the m/z of 200, and the resolution for higher-energy collisional dissociation spectra was set to 17500 at the m/z of 200. The normalized collision energy was 30 eV and the underfill ratio, which specifies the minimum percentage of the target value likely to reach the maximum fill time, was defined as 0.1%. The instrument was run with the peptide recognition mode enabled.
Protein identification and quantification
MS/MS spectra were searched using the MASCOT engine (Matrix Science, London, UK; version 2.2) in the Proteome Discoverer 1.3 (Thermo Fisher Scientific Inc., San Jose, CA, USA) against uniprot_mouse_82199_20170105.fasta (82199 sequences, download at January 5, 2017) and the decoy database. For protein identification, the following options were used: peptide mass tolerance = 20 ppm, MS/MS tolerance = 0.1 Da, enzyme = trypsin, missed cleavage = 2, fixed modification: carbamidomethyl (C), iTRAQ8-plex (K), iTRAQ8-plex (N-term), variable modification: oxidation (M), FDR r≤0.01.
Quantitative real-time PCR of target proteins
Total RNA was extracted from the renal tissues using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Subsequently, the total RNA was quantified by detecting the ratio 260/280 nm using an UV spectrophotometer (Thermo Fisher Scientific, Waltham, MA), and then reversely transcribed to single-stranded cDNA with PrimeScript RT Reagent Kit (Takara Bio, Kyoto, Japan). Quantitative real-time PCR was performed on a Light Cycler 96 (Roche, Basel, Switzerland) with SYBR Select Master Mix (Life Technology, Grand Island, NY). Detection was done in duplicate, and reaction was performed at 95°C for 10 min followed by 45 cycles of denaturation at 95°C for 10 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal reference. The primers used for PCR are shown in Table 1. The specificity of each primer was verified by melting curve analysis. Relative mRNA expression was calculated using the 2-ΔΔct method.
Table 1.
Primers used for real-time PCR
| Gene | Sequences | Size |
|---|---|---|
| DNA-PK | Forward: 5’-AGTTCGCCTGATGAAGCACT-3’ | 117 bp |
| Reverse: 5’-CCTTTCTCCGGATGTAGCTG-3’ | ||
| Smad2-3 | Forward: 5’-CACAGCCACCATGAATTACG-3’ | 120 bp |
| Reverse: 5’-TGGAGGTAGAACTGGCGTCT-3’ | ||
| Smc3 | Forward: 5’-CAAGGCATGCTGTTGAAGAA-3’ | 111 bp |
| Reverse: 5’-CAACTGCTTCAGGCTCAGTG-3’ |
Statistical analyses and bioinformatic analysis
The statistical significance of the differential expression levels on proteins was tested by t-test. A protein with KO/WT ratio >1.20 or <0.83 and with a corrected P value of <0.05 was considered the differentially expressed one. The method of false discovery rate (FDR) was used for addressing the multiple-testing adjustment, and adjusted P<0.01 was used as the significant criterion. Functional protein analyses were performed using the AmiGO tool in the gene ontology platform (http://www.geneontology.org/). Pathway analyses were performed using the Search pathway tool in the Kegg Mapper platform (http://www.genome.jp/kegg/mapper.html). To investigate the direct (physical) and/or indirect (functional) interactions among the identified genes, the search tool was used for the retrieval of interacting genes (STRING) database (http://string.embl.de/) and the functional network was analyzed.
Results
Genotyping and characterization of RGC-32-/- mice
The mice were genotyped by PCR (Figure 1). Approximately 17% of mice born after RGC-32+/- x RGC-32+/- crosses were found to be RGC-32-/-, indicating some embryonic lethality. These RGC-32-/- mice were indistinguishable from their RGC-32+/+ and RGC-32+/- littermates according to the appearance. RGC-32-/- mice did not have any obviously anatomical or histological defects, and their life expectancy was normal. RGC-32-/- mice did not display any significant phenotypic change as compared to RGC-32+/+ mice.
Figure 1.
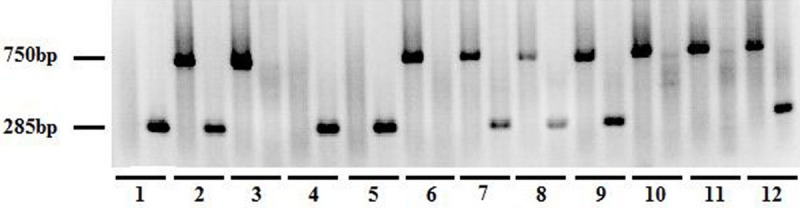
Genotyping of wild-type (RGC32+/+), RGC32+/-, and RGC32-/- mice by PCR using genomic DNA isolated from mouse tails. 285 bp product alone indicates wild type. Presence of both 750 and 285 bp fragments indicate heterozygous, whereas 750 bp alone indicates the homozygous knockout. The genotype of No. 1, 4 and 5 mouse was RGC32+/+; the genotype of No. 2, 7, 8, 9, 12 mouse was RGC32+/-; the genotype of No. 3, 6, 10, 11 mouse was RGC32-/-.
Protein identification and quantification
Soluble proteins extracted from the kidneys of RGC-32-/- mice and wild mice were compared by iTRAQ-based proteomic analysis. In total, 4690 proteins were identified on the basis of 25165 unique peptides. All of the identified proteins were categorized into biological processes, cellular components, and molecular functions according to their GO annotation (Figure 2).
Figure 2.
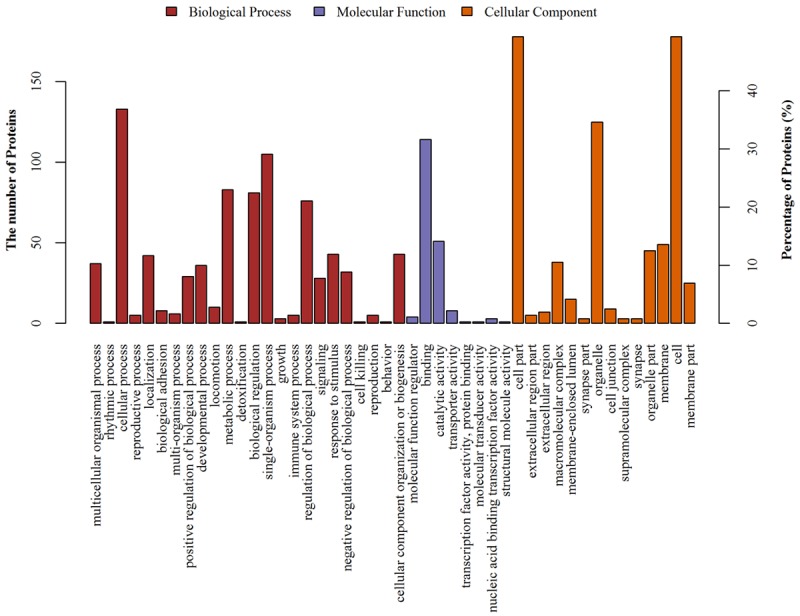
Protein classification based on the GO analysis of biological processes, molecular functions and cellular components. Each protein is characterized in terms of three ontologies: molecular function, cellular component and the involved biological process. Using the GO database, the involved proteins were classified in order to gain an overall picture of potential functions of the differentially expressed proteins in this study.
GO analysis showed that the most common biological processes related to these proteins were cellular process, single-organism process, metabolic process, biological regulation, and regulation of biological processes. The major functions of these proteins were blinding activity and catalytic activity. The majority of these proteins located in cell compartment and organelles; remaining proteins located in the membrane, organelle part, macromolecular complex, membrane-enclosed lumen, extracellular region, extracellular region part, cell junction, synapse part, supramolecular part and synapse.
Differentially expressed proteins
Of 4690 proteins, the expression of 361 proteins (76 up-regulated and 285 down-regulated) was statistically different between RGC-32-/- mice and RGC-32+/+ mice. The top 5 up-regulated proteins were fructose-bisphosphate aldolase (3.83 folds), sulfotransferase family cytosolic 2B member 1 (3.05 folds), peripheral-type benzodiazepine receptor-associated protein 1 (2.32 folds), protein Gm10073 (2.24 folds), and testis-expressed sequence 10 protein (1.96 folds). The top 5 down-regulated proteins were ephexin-1 (0.27 folds), rotatin (0.29 folds), ATP synthase subunit alpha (0.32 folds), NADPH oxidase 3 (0.33 folds), GTP-binding protein Di-Ras1 (0.35 folds).
Because different proteins interact and cooperate to involve biochemical reactions, a KEGG pathway-based analysis was performed to identify pathways that would be potentially affected by differential expressed proteins in RGC-32-/- mice. The KEGG pathway analysis showed that the top 20 pathways with involvement of differentially expressed proteins were spliceosome, fluid shear stress and atherosclerosis, protein processing in endoplasmic reticulum, pathways in cancer, viral carcinogenesis, epithelial cell signaling in Helicobacter pylori infection, HTLV-I infection, PI3K-Akt signaling pathway, ubiquitin mediated proteolysis, Parkinson’s disease, MAPK signaling pathway, carbon metabolism, Alzheimer’s disease, NOD-like receptor signaling pathway, tight junction, proteoglycans in cancer, phagosome, ribosome, mTOR signaling pathway, and AMPK signaling pathway (Figure 3 and Table 2).
Figure 3.
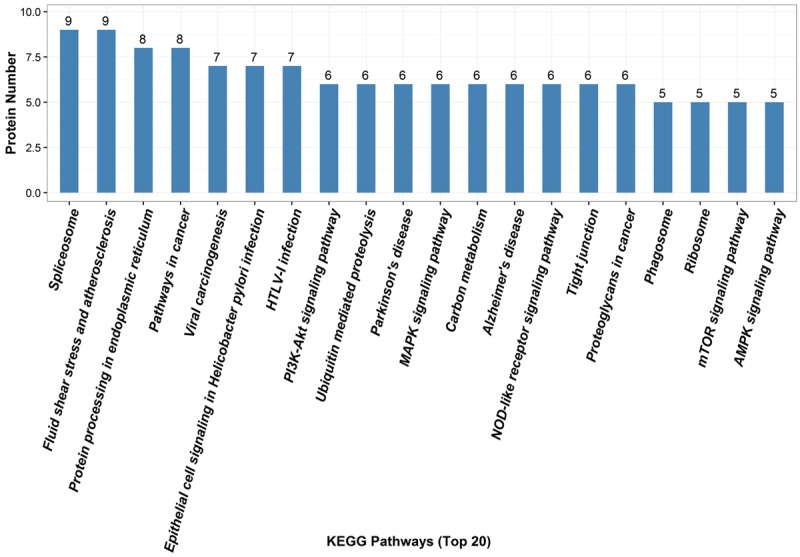
The top 20 pathways related to the differentially expressed proteins by KEGG database analysis. Numbers refer to assigned proteins in each pathway.
Table 2.
Top 20 pathways related to the differentially expressed proteins (KEGG pathway analysis)
| Pathway | Differentially Expressed Proteins |
|---|---|
| Spliceosome | PRPF19 SF3B14 LSM3 SF3A2 DDX42 SART1 HNRNPU PRPF31 SFRS4_5_6 |
| Fluid shear stress and atherosclerosis | SUMO trxA TRPV4 SRC NOX CYBA argG IKBKG RELA |
| Protein processing in endoplasmic reticulum | HERPUD1 SYVN1 MARCH6 SEC61B UFD1 OS9 UBE2G2 BAX |
| Pathways in cancer | TPM3 BCR1 RALBP1 SMAD2_3 IKBKG BAX ARHGEF1 RELA |
| Viral carcinogenesis | SRC H2B DDX3X IKBKG MAPKAPK2 BAX RELA |
| Epithelial cell signaling in Helicobacter pylori infection | SRC ATPeV1G ADAM10 IKBKG F11R JAM3 RELA |
| HTLV-I infection | RRAS SMAD2_3 PPP3C IKBKG BAX RELA TSPO |
| PI3K-Akt signaling pathway | TSC2 EIF4B PPP2R5 IKBKG TSC1 RELA |
| Ubiquitin mediated proteolysis | PRPF19 UBE2Z SYVN1 UBE2L3 UBE3C UBE2G2 |
| Parkinson’s disease | UCHL1 COX5A ATPeF0F6 UBE2L3 QCR9 UBE2G2 |
| MAPK signaling pathway | SRC RRAS MAPT PPP3C IKBKG MAPKAPK2 RELA |
| Carbon metabolism | ALDO ENO OGDH PHGDH atoB ADPGK |
| Alzheimer’s disease | COX5A ATPeF0F6 ADAM10 MAPT QCR9 PPP3C |
| NOD-like receptor signaling pathway | TrxA NOX CYBA TRIP6 IKBKG RELA |
| Tight junction | PRKAB SRC SYNPO TJP3 JAM1 JAM3 |
| Proteoglycans in cancer | SRC PPP1R12B RRAS EIF4B PDCD4 ARHGEF1 |
| Phagosome | SEC61B NOX CYBA ATPeV1G MBL |
| Ribosome | RP-L28 RP-LP1 RP-S21 RP-L21 RP-L34 |
| mTOR signaling pathway | TSC2 ATPeV1G RRAGA_B EIF4B TSC1 |
| AMPK signaling pathway | PRKAB TSC2 PPP2R5 TSC1 HNF4A |
Protein-protein interaction network
The analysis of protein-protein interaction and protein-protein interaction network is important to reveal the functions of proteins. To further analyze the biological functions of differentially expressed proteins, the STRING was used to construct a functionally associated network (Figure 4). It is indicated that Ywhae localized at the center of the network.
Figure 4.

Differentially expressed protein interaction network analysis. Yellow nodes represent target proteins and green nodes represent the related proteins. Ywhae (Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, epsilon) localized at the center of the network.
Validation of iTRAQ data by qPCR
In order to confirm the results of iTRAQ-based proteomics analysis and to further investigate the mechanism of RGC-32 regulating the cell cycle, the mRNA expression of three genes (Smad2-3, DNA-PK, and Smc3) related to the regulation of cell cycle was detected by quantitative real-time PCR (qPCR) and compared with that from iTRAQ. Results showed the mRNA expression of Smad2-3, DNA-PK, and Smc3 was consistent with that from iTRAQ, which suggests that the expression of these differentially expressed proteins was regulated at the transcription level (Figure 5 and Table 3).
Figure 5.
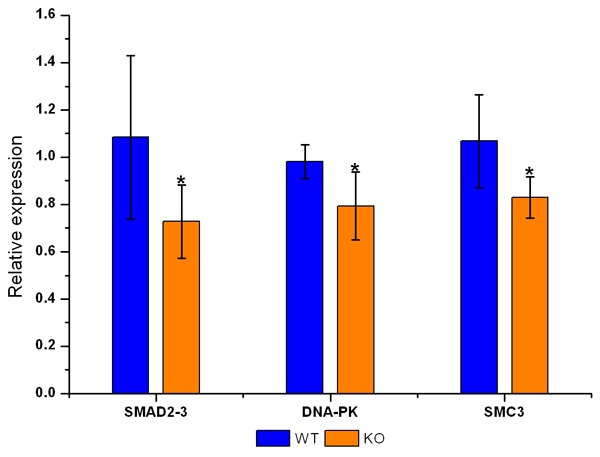
mRNA expression of Smad2-3, DNA-PK and Smc3 in the kidney of RGC-32-/- mice and RGC-32+/+ mice. (*P<0.05 vs RGC-32+/+ mice). Results showed the mRNA expression of Smad2-3, DNA-PK, and Smc3 was consistent with that from iTRAQ.
Table 3.
Fold change of mRNA expression of three proteins related to cell cycle regulation
| Methods | Proteins | ||
|---|---|---|---|
|
| |||
| Smad2-3 | DNA-PK | Smc3 | |
| iTRAQ | 0.631 | 0.761 | 0.821 |
| qPCR | 0.671 | 0.808 | 0.776 |
Discussion
RGC-32, expressed in many tissues and organs, plays an important role in maintaining normal physiological functions. It has been confirmed that RGC-32 is an important checkpoint prote-in in the cell cycle [1,21]. Our previous studies showed RGC-32 expression decreased in the rat kidney after I/R injury and played an important role in the renal tubular epithelial cell injury and repair by regulating the G2/M checkpoint in cell cycle [17,18]. However, the exact mechanism is still poorly understood.
In this study, a quantitative proteomic analysis was done to investigate the differentially expressed proteins in the kidney of RGC-32-/- mice as compared to RGC-32+/+ mice. Some differentially expressed proteins were found in the kidney of RGC-32-/- mice, which may provide new-insights into the function of RGC-32 and its role in renal tubular epithelial cell injury and repair. KEGG pathway-based analysis revealed the differently expressed proteins in RGC-32-/- mice involved in some important pathways, which may provide information on the potential function of RGC-32 in vivo.
In this study, pathway in cancer was the 2nd common pathway related to the differently expressed proteins (Figure 3). The RGC-32 expression has been detected in a wide variety of human cancers. In colon cancer [4], breast cancer [4,22], prostate cancer [4] and cutaneous T cell lymphoma [4], RGC-32 expression is increased, but it is decreased in advanced stages of primary astrocytoma, invasive prostate cancer, multiple myeloma and nonsmall cell lung cancer [11,23]. The role of RGC-32 in cancer is still not fully understood. The role of RGC-32 in cancer is ascribed to its regulation on cell proliferation and cell cycle. The investigation of differentially expressed proteins may provide a new way to explore the role of RGC-32 in cancer.
In our research, 3 differently expressed proteins (Smad2-3, DNA-PK, and Smc3) were involved in the regulation of cell cycle. Smad2-3, DNA-PK and Smc3 regulate the G1/S phase, S, G2/M phase and M phase, respectively. Yang et al [24] reported that G2/M arrest in epithelial cell cycle mediated renal fibrosis after injury and bypassing the G2/M arrest could rescue renal fibrosis in the ischemic kidney. Our previous studies also indicated that P53 inhibitor could improve the renal tubular epithelial cell injury and repair through regulating G2/M phase [25]. G2/M phase is closely related to the activity of the CyclinB1/Cdk1 complex. Up to now, the mechanism by which RGC-32 regulates the G2/M checkpoint in renal tubular epithelial cell injury and repair is still unclear. On the basis of our findings, we hypothesize that RGC-32 may regulate the activity of the CyclinB1/Cdk1 complex through DNA-PK-P53-14-3-3σ pathway. Although the role of this pathway has not been confirmed, the protein-protein interaction network may provide information on this pathway. In the present study, protein-protein interaction network analysis indicated that Ywhae localized at the center of the network. Ywhae is also known as 14-3-3ε and 14-3-3σ which are members of 14-3-3 protein family [26]. 14-3-3 protein family represents a family of highly homologous proteins and the mammalian 14-3-3 proteins have seven isoforms (β, γ, ε, η, σ, τ and ζ) [26]. Early studies implicated 14-3-3 proteins were a critical integration point for many protein kinases and phosphatases that control the transition from G2 phase into M phase [26,27]. 14-3-3σ regulates the G2/M phase by binding to CDC2, CDC25B, WEE1 and p53; Ywhae (14-3-3ε) binds to CDC25B, CDC25C, p53 and MDMX in the regulation of G2/M phase [26]. These findings suggest that RGC-32 may regulate the G2/M phase through DNA-PK-P53-14-3-3σ pathway.
Conclusion
In summary, iTRAQ proteomic analysis was applied to investigate the protein expression profile in the kidney of RGC-32 knockout mice and a total of 361 differentially expressed proteins (76 up-regulated and 285 down-regulated) were identified as compared to wide type mice. GO analysis revealed some biological functions were related to these differentially expressed proteins. KEGG pathway-based analysis showed important pathways in which the differently expressed proteins were involved. Ywhae (14-3-3ε) localizes at the center of the functionally associated network. These findings provide new insights into the functions of RGC-32 in the kidney and on its role in the renal tubular epithelial cell injury and repair.
Acknowledgements
This work was supported by the Chinese National Nature Science Foundation (No: 81370813 to Wen-Yan Huang) and Precision medical project of Shanghai Jiao Tong University School of Medicine (15ZH4008 to Wen-Yan Huang).
Disclosure of conflict of interest
None.
References
- 1.Badea TC, Niculescu FI, Soane L, Shin ML, Rus H. Molecular cloning and characterization of RGC-32, a novel gene induced by complement activation in oligodendrocytes. J Biol Chem. 1998;273:26977–26981. doi: 10.1074/jbc.273.41.26977. [DOI] [PubMed] [Google Scholar]
- 2.Badea T, Niculescu F, Soane L, Fosbrink M, Sorana H, Rus V, Shin ML, Rus H. RGC-32 increases p34CDC2 kinase activity and entry of aortic smooth muscle cells into S-phase. J Biol Chem. 2002;277:502–508. doi: 10.1074/jbc.M109354200. [DOI] [PubMed] [Google Scholar]
- 3.Blaxall BC, Tschannen-Moran BM, Milano CA, Koch WJ. Differential gene expression and genomic patient stratification following left ventricular assist device support. J Am Coll Cardiol. 2003;41:1096–1106. doi: 10.1016/s0735-1097(03)00043-3. [DOI] [PubMed] [Google Scholar]
- 4.Fosbrink M, Cudrici C, Niculescu F, Badea TC, David S, Shamsuddin A, Shin ML, Rus H. Overexpression of RGC-32 in colon cancer and other tumors. Exp Mol Pathol. 2005;78:116–122. doi: 10.1016/j.yexmp.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Fosbrink M, Cudrici C, Tegla CA, Soloviova K, Ito T, Vlaicu S, Rus V, Niculescu F, Rus H. Response gene to complement 32 is required for C5b-9 induced cell cycle activation in endothelial cells. Exp Mol Pathol. 2009;86:87–94. doi: 10.1016/j.yexmp.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang WY, Li ZG, Rus H, Wang X, Jose PA, Chen SY. RGC-32 mediates transforming growth factor-beta-induced epithelial-mesenchymal transition in human renal proximal tubular cells. J Biol Chem. 2009;284:9426–9432. doi: 10.1074/jbc.M900039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim HW, Lee JE, Shin SJ, Lee YE, Oh SH, Park JY, Seong JK, Park JS. Identification of differentially expressed mRNA during pancreas regeneration of rat by mRNA differential display. Biochem Biophys Res Commun. 2002;299:806–812. doi: 10.1016/s0006-291x(02)02741-9. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J. Clin. Oncol. 2005;23:9408–9421. doi: 10.1200/JCO.2005.01.5594. [DOI] [PubMed] [Google Scholar]
- 9.Saigusa K, Imoto I, Tanikawa C, Aoyagi M, Ohno K, Nakamura Y, Inazawa J. RGC32, a novel p53-inducible gene, is located on centrosomes during mitosis and results in G2/M arrest. Oncogene. 2007;26:1110–1121. doi: 10.1038/sj.onc.1210148. [DOI] [PubMed] [Google Scholar]
- 10.Tegla CA, Cudrici CD, Azimzadeh P, Singh AK, Trippe R 3rd, Khan A, Chen H, Andrian-Albescu M, Royal W 3rd, Bever C, Rus V, Rus H. Dual role of response gene to complement-32 in multiple sclerosis. Exp Mol Pathol. 2013;94:17–28. doi: 10.1016/j.yexmp.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Vlaicu SI, Cudrici C, Ito T, Fosbrink M, Tegla CA, Rus V, Mircea PA, Rus H. Role of response gene to complement 32 in diseases. Arch Immunol Ther Exp (Warsz) 2008;56:115–122. doi: 10.1007/s00005-008-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vlaicu SI, Tegla CA, Cudrici CD, Fosbrink M, Nguyen V, Azimzadeh P, Rus V, Chen H, Mircea PA, Shamsuddin A, Rus H. Epigenetic modifications induced by RGC-32 in colon cancer. Exp Mol Pathol. 2010;88:67–76. doi: 10.1016/j.yexmp.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlick SN, Wood CD, Gunnell A, Webb HM, Khasnis S, Schepers A, West MJ. Upregulation of the cell-cycle regulator RGC-32 in Epstein-Barr virus-immortalized cells. PLoS One. 2011;6:e28638. doi: 10.1371/journal.pone.0028638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tegla CA, Cudrici CD, Nguyen V, Danoff J, Kruszewski AM, Boodhoo D, Mekala AP, Vlaicu SI, Chen C, Rus V, Badea TC, Rus H. RGC-32 is a novel regulator of the T-lymphocyte cell cycle. Exp Mol Pathol. 2015;98:328–337. doi: 10.1016/j.yexmp.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang WY, Xie W, Guo X, Li F, Jose PA, Chen SY. Smad2 and PEA3 cooperatively regulate transcription of response gene to complement 32 in TGF-beta-induced smooth muscle cell differentiation of neural crest cells. Am J Physiol Cell Physiol. 2011;301:C499–506. doi: 10.1152/ajpcell.00480.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niu XL, Kuang XY, Zhang ZG, Liu XG, Zhao ZH, Zhang X, Xu H, Huang WY. Expression of response gene to complement-32 in renal tissue of children with immunoglobulin a nephropathy. Scand J Urol Nephrol. 2011;45:371–376. doi: 10.3109/00365599.2011.585624. [DOI] [PubMed] [Google Scholar]
- 17.Sun L, Shen YL, Liu HJ, Hu YJ, Kang YL, Huang WY. The expression of response gene to complement 32 on renal ischemia reperfusion injury in rat. Ren Fail. 2016;38:276–281. doi: 10.3109/0886022X.2015.1120118. [DOI] [PubMed] [Google Scholar]
- 18.Shen YL, Liu HJ, Sun L, Niu XL, Kuang XY, Wang P, Hao S, Huang WY. Response gene to complement 32 regulates the G2/M phase checkpoint during renal tubular epithelial cell repair. Cell Mol Biol Lett. 2016;21:19. doi: 10.1186/s11658-016-0021-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 20.Unwin RD, Griffiths JR, Whetton AD. Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC-MS/MS. Nat Protoc. 2010;5:1574–1582. doi: 10.1038/nprot.2010.123. [DOI] [PubMed] [Google Scholar]
- 21.Strom CC, Aplin M, Ploug T, Christoffersen TE, Langfort J, Viese M, Galbo H, Haunso S, Sheikh SP. Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. Febs J. 2005;272:2684–2695. doi: 10.1111/j.1742-4658.2005.04684.x. [DOI] [PubMed] [Google Scholar]
- 22.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 23.Kim DS, Lee JY, Lee SM, Choi JE, Cho S, Park JY. Promoter methylation of the RGC32 gene in nonsmall cell lung cancer. Cancer. 2011;117:590–596. doi: 10.1002/cncr.25451. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. doi: 10.1038/nm.2144. 531p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen YL, Sun L, Hu YJ, Liu HJ, Kuang XY, Niu XL, Huang WY. P53 inhibitor pifithrin-alpha prevents the renal tubular epithelial cells against injury. Am J Transl Res. 2016;8:4040–4053. [PMC free article] [PubMed] [Google Scholar]
- 26.Hermeking H, Benzinger A. 14-3-3 proteins in cell cycle regulation. Semin Cancer Biol. 2006;16:183–192. doi: 10.1016/j.semcancer.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Gardino AK, Yaffe MB. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin Cell Dev Biol. 2011;22:688–695. doi: 10.1016/j.semcdb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]