

Abstract
Many stimuli including lipopolysaccharide (LPS) could activate microglial cells to subsequently cause inflammatory nerve injury. However, the mechanism of LPS-induced neuroinflammation in microglial cells is still elusive. Thus, the present study was undertaken to examine the role of COX-2 in mediating the activation of Stat3 and the production of IL-6 in BV2 cells challenged with LPS. After 24 h treatment, LPS dose-dependently enhanced COX-2 expression at both mRNA and protein levels. Meanwhile, IL-6 with other inflammatory cytokines including IL-1β, TNF-α, and MCP-1 were similarly enhanced by LPS. Then a specific COX-2 inhibitor (NS-398) was administered to BV2 before LPS treatment. Significantly, COX-2 inhibition suppressed the upregulation of IL-6 at both mRNA and protein levels in line with the trend blockade on IL-1β, TNF-α, and MCP-1. Stat3 drives proinflammatory signaling pathways and contributes to IL-6 production via a transcriptional mechanism in many diseases. Here we found that inhibition of COX-2 entirely blocked LPS-induced Stat3 phosphorylation, which might contribute to the blockade of IL-6 production to some extent. Meanwhile, COX-2 siRNA approach largely reproduced the phenotypes shown by specific COX-2 inhibitor in LPS-treated BV2 cells. Together, these findings suggested that COX-2 might contribute to LPS-induced IL-6 production possibly through activating Stat3 signaling pathway in microglial cells.
Keywords: Microglial cells, LPS, COX-2, IL-6, Stat3
Introduction
Microglial cell are innate immune cells in the central nervous system (CNS), and are related to the inflammatory response in the brain. Under the normal state, microglial cells play an important role in immune surveillance, maintenance of the homeostasis of CNS environment, clearance of damaged neurons and debris, and tissue repair [1,2]. When exposed to the diverse stimuli including infection, lipopolysaccharide (LPS), and neuron damage, microglial cells could be activated [3-5]. The activated microglial cells release pro-inflammatory mediators and cytokines, such as prostaglandin E2 (PGE2), IL-6, nitric oxide (NO), monocyte chemotactic protein-1 (MCP-1), interleukin (IL)-1β, and tumor necrosis factor-α (TNF-α) [2,4,6], leading to subsequent inflammation. Excessive production of pro-inflammatory mediators by activated microglia play a critical role in the pathogenesis of neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), cerebral ischemia, and multiple sclerosis [7,8]. IL-6 is involved in the pathogenesis of artery atherosclerosis via promoting local inflammatory lesions [9]. Several studies have reported that IL-6 plays an important role in blood-brain barrier (BBB) injury such as subarachnoid hemorrhage (SAH), excessive erythrocytosis, and cerebral ischemia [10-12]. BBB injury is a mark of neurological disorders and brain injury [13,14]. Therefore, targeting the excessive pro-inflammatory cytokines produced by microglial cells and the associated signaling pathways to find the potential targets for the treatment of neural diseases is of importance.
Cyclooxygenase (COX)-2, an inducible inform of COXs, is reported to participate in many inflammatory diseases [15,16]. Several studies have indicated that COX-2 is involved in many neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and prion diseases [17-20]. Under the pathological conditions, prostaglandin E2 (PGE2) is an important inflammatory mediator produced by COX-2 [21]. The secretion of PGE2 can be enhanced in response to various stimuli [2,4,6]. Stat3 is an important transcription factor in the immune system, participating in many inflammatory responses in CNS [22]. In macrophages, Stat3 is reported to participate in inflammation by regulating the production of pro-inflammatory cytokines [23]. In glial cells, LPS-induced neuroinflammation was associated with the activation of Stat3 [22]. It’s also reported that an increase of COX-2/PGE2 offsets the repressive activity of Berberin on inhibiting the invasion and metastasis of colorectal cancer cells via JAK2/Stat3 signaling pathway [24]. However, the role of COX-2/PGE2/Stat3 signaling pathway in LPS-induced microglial inflammation is unknown.
In the present study, employing a specific COX-2 inhibitor and COX-2 siRNA, we investigated the activation and contribution of COX-2 in LPS-induced IL-6 production and Stat3 activation in microglial cells.
Materials and methods
Reagents and antibodies
LPS was purchased from Sigma (St. Louis, USA). COX-2 inhibitor NS-398 was bought from Beyotime (Shanghai, China). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin, and trypsin solution (EDTA) were bought from Gibco (Invitrogen, Grand island, NY). The COX-2 antibody was purchased from Cayman Chemicals (Ann Arbor, MI). β-actin, Stat3, and p-Stat3 antibodies were obtained from Cell Signaling Technology (Dan-vers, MA). The PGE2 enzyme immunoassay kit was purchased from Cayman Chemicals (Ann Arbor, MI). The IL-6 enzyme immunoassay kit was bought from Boster (Wuhan, China).
Cell culture
The mouse microglia cell line BV2 was obtained from China Infrastructure of Cell Line Resources (Beijing, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), penicillin (100 U/ml) and streptomycin (100 μg/ml), and maintained at 37°C in a humidified 5% CO2 atmosphere. After BV2 were cultivated to 60%-70% confluence, cells were treated with LPS for 24 h at different doses (0.5, 1, 2 μg/ml) with or without a pretreatment of NS-398 (COX-2 inhibitor). In another experiment, COX-2 siRNA was applied to silence COX-2 in LPS-treated BV2 cells.
Quantitative real-time PCR (qRT-PCR) analysis
Total RNA was extracted using Trizol reagent (TaKaRa), and cDNA was prepared using a PrimeScript RT reagent Kit (TaKaRa) according to the manufacturer’s protocol. Oligo nucleotides were designed using Primer 5 software (available at http://frodo.wi.mit.edu/) and the sequences are shown in Table 1. Real-time PCR amplification was performed using the ABI 7500 Real-Time PCR Detection System (Foster City, CA) by using SYBR Premix Ex Taq (TaKaRa). The cycling program consisted of a preliminary denaturation (95°C for 10 min), followed by 40 cycles (95°C for 15 s and 60°C for 1 min). Relative gene expression of mRNA was normalized to GAPDH and calculated using the ΔΔCt method from the threshold cycle numbers.
Table 1.
Sequences of primers for quantitative real-time PCR
| Gene symbol | Primer sequence | Accession number |
|---|---|---|
| COX-2 | 5’-AGGACTCTGCTCACGAAGGA-3’ | YP_001686701.1 |
| 5’-TGACATGGATTGGAACAGCA-3 | ||
| IL-6 | 5’-GCTGGTGACAACCACGGCCT-3’ | NM_001314054.1 |
| 5’-AGCCTCCGACTTGTGAAGTGGT-3’ | ||
| IL-1β | 5’-ACTGTGAAATGCCACCTTTTG-3’ | NM_008361.4 |
| 5’-TGTTGATGTGCTGCTGTGAG-3’ | ||
| TNF-α | 5’-TCCCCAAAGGGATGAGAAG-3’ | NM_001278601.1 |
| 5’-CACTTGGTGGTTTGCTACGA-3’ | ||
| MCP-1 | 5’-GCTCTCTCTTCCTCCACCAC-3’ | NM_011333.3 |
| 5’-ACAGCTTCTTTGGGACACCT-3’ | ||
| GAPDH | 5’-GTCT TCACTACCATGGAGAAGG-3’ | M32599 |
| 5’-TCATGGATGACCTTGGCCAG-3’ |
Western blotting
Cells were rapidly washed with ice-cold PBS and lysed on ice in lysis buffer containing protease inhibitors. An equal amount of protein was separated on 10% SDS-PAGE, and transferred onto PVDF membrane (Bio-Rad) which was blocked with 5% nonfat milk in TBST for an hour. Membrane was then incubated with primary antibodies against COX-2 (1:500), Stat3 (1:500), p-Stat3 (1:1000), and β-actin (1:1000) overnight at 4°C. After washing, membranes were incubated with HRP-labeled secondary antibodies at room temperature for 1 h. β-actin was used as an internal standard control. Band intensity was measured using Image J software (NIH, Bethesda, MD, USA).
ELISA assay
The cell culture medium was collected and centrifuged for 10 min at 12,000× g. The IL-6 level in the medium was measured by ELISA kit (Boster). A ELISA kit from Cayman Chemicals was used to detect PGE2 in the medium according to the manufacturer’s instructions.
Statistical analysis
Data are presented as means ± SD. Comparisons among multiple groups were carried out using the one-way analysis variance (ANOVA) followed by Bonferroni’s comparison test. Statistical calculations were performed by GraphPad Prism (GraphPad Software, San Diego, CA, USA). P<0.05 was considered significant.
Results
LPS treatment upregulated IL-6 and other inflammatory cytokines in BV2 cells
At first, we observed the expressions of IL-6 and other inflammatory cytokines including IL-1β, TNF-α and MCP-1 in response to LPS stimulation at different doses in BV2 cells. As shown by the data, after LPS treatment at 0.5, 1 and 2 μg/ml for 24 h, the mRNA expressions of IL-6, IL-1β, TNF-α and MCP-1 were significantly increased in a dose-dependent manner (Figure 1A-D). These data demonstrated that LPS successfully induced the inflammatory response in BV2 microglial cells.
Figure 1.
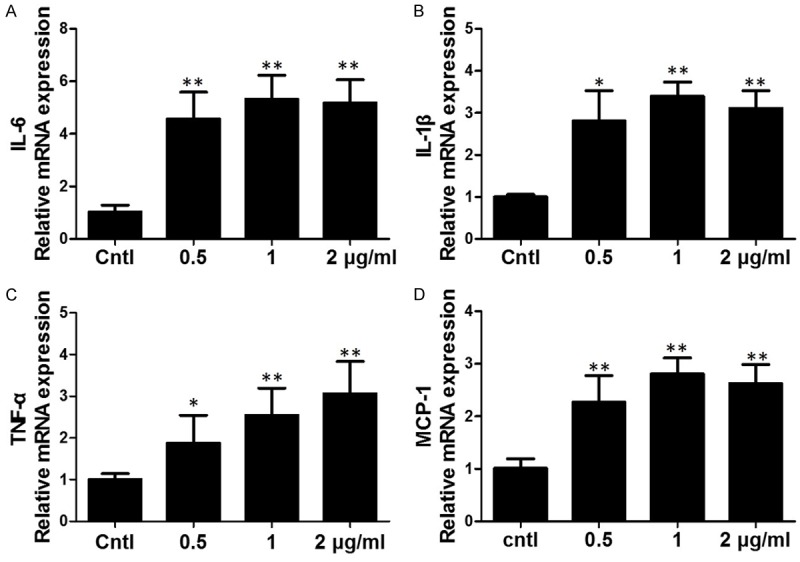
Effects of LPS on the expressions of inflammatory cytokines in BV2 cells. The mRNA levels of IL-6 (A), IL-1β (B), TNF-α (C) and MCP-1 (D) were upregulated by LPS treatment for 24 h at different doses. All values are means ± SD; n = 6 in each group. *P < 0.05 versus control, **P < 0.01 versus control.
LPS upregulated COX-2 expression in BV2 cells
In order to investigate the effect of LPS on COX-2 expression, we observed the protein and mRNA levels of COX-2 after LPS treatment in BV2 cells. By qRT-PCR, we detected that LPS enhanced the protein expression of COX-2 with the highest upregulation at the dose of 1 μg/ml (Figure 2A, 2B). Next, we further examined the COX-2 mRNA expression via qRT-PCR. As shown in Figure 2C, COX-2 mRNA was significantly upregulated following LPS treatment. These data suggested that LPS could directly upregulate COX-2 expression in BV2 microglial cells.
Figure 2.
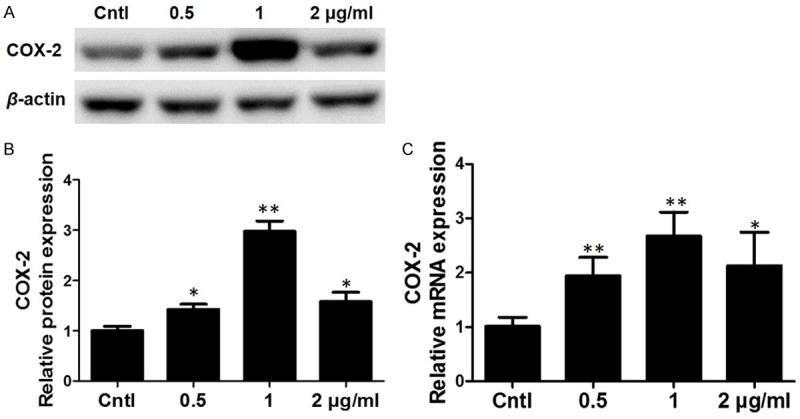
Effects of LPS on the expression of COX-2 in BV2 cells. A. Western blotting analysis of COX-2 in BV2 cells. B. Quantitative analysis of COX-2 Western blots. C. qRT-PCR analysis of COX-2 in a dose-dependent experiment. All values are means ± SD; n = 6 in each group. *P < 0.05 versus control, **P < 0.01 versus control.
COX-2 inhibitor attenuated LPS-stimulated PGE2 production
To evaluate the role of COX-2 in LPS-induced microglial inflammation, a specific COX-2 inhibitor (NS-398) was applied to BV2 cells before LPS administration. As shown by Figure 3A-C, COX-2 inhibitor at a dose of 10 μM significantly decreased COX-2 expression at both mRNA and protein levels. To further examine the efficacy of COX-2 inhibition, we measured PGE2 production in the medium. As shown by Figure 3D, LPS (1 μg/ml) treatment strikingly increased PGE2 level by 2.5 folds, which was significantly blocked by COX-2 inhibitor treatment. The results demonstrated a COX-2-dependent induction of PGE2 in response to LPS treatment.
Figure 3.
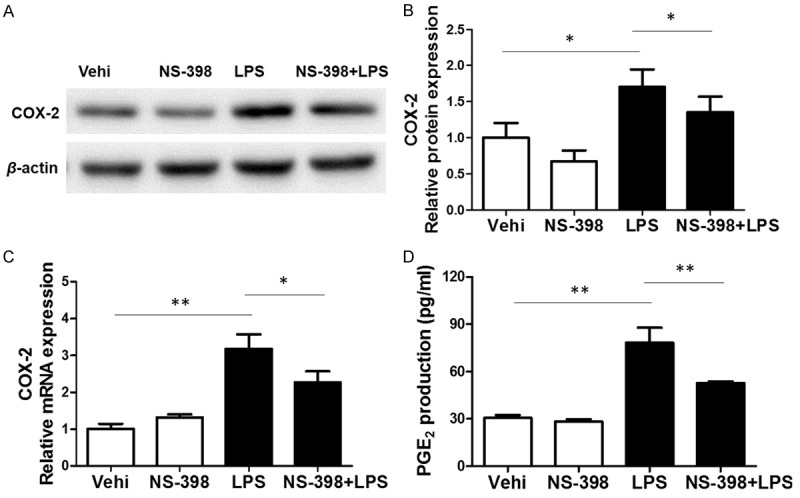
Effects of COX-2 inhibitor (NS-398) on COX-2 expression and PGE2 production in BV2 cells challenged with LPS. A. Western blotting analysis of COX-2 in BV2 cells. B. Quantitative analysis of COX-2 Western blots. C. COX-2 mRNA levels were elevated by LPS treatment. D. The levels of PGE2 in cell culture media were measured via ELISA assay. All values are means ± SD; n = 6 in each group. *P < 0.05 versus control or LPS group, **P < 0.01 versus control or LPS group.
Effects of COX-2 inhibition on LPS-induced upregulation of inflammatory cytokines in BV2 cells
Furthermore, we examined the role of COX-2 inhibition in LPS-induced inflammation. The BV2 cells were pretreated with COX-2 inhibitor for 24 h followed by LPS treatment for another 24 h. LPS exposure significantly increased the production of IL-6, IL-1β, TNF-α and MCP-1 at mRNA levels, whereas only IL-6 was significantly blocked by COX-2 inhibitor with a trend blockade on other inflammatory cytokines (Figure 4A, 4C-E). Furthermore, we measured IL-6 protein production in the medium by ELISA and found that LPS-induced IL-6 release was also significantly blocked by COX-2 inhibitor (Figure 4B).
Figure 4.
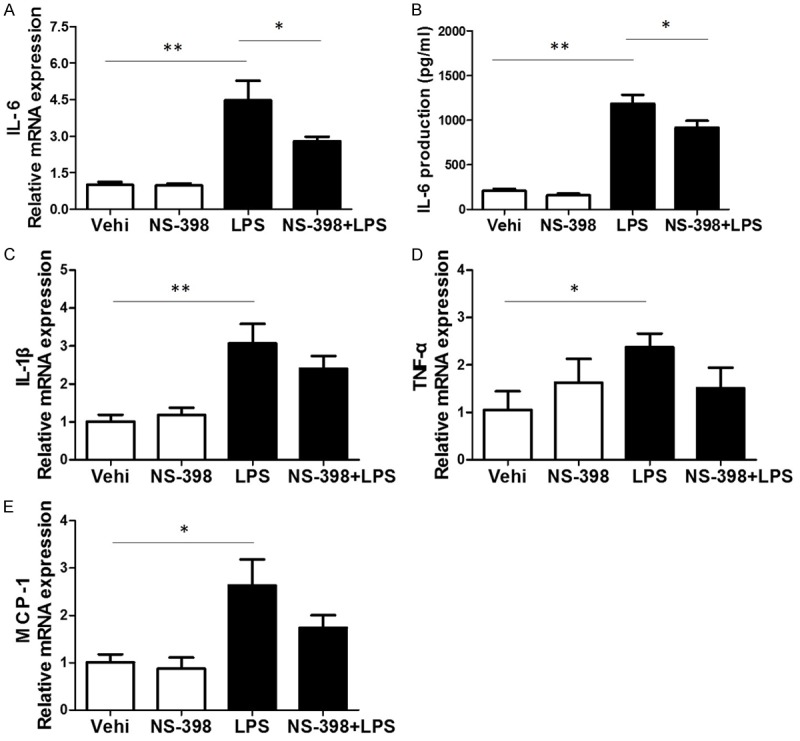
Effects of COX-2 inhibitor on the mRNA expressions of inflammatory cytokines in BV2 cells challenged with LPS. A. qRT-PCR analysis of IL-6. B. ELISA assay of IL-6 in the medium. C. qRT-PCR analysis of IL-1β. D. qRT-PCR analysis of TNF-α. E. qRT-PCR analysis of MCP-1. All values are means ± SD; n = 6 in each group. *P < 0.05 versus control or LPS group, **P < 0.01 versus control or LPS group.
COX-2 inhibitor blocked LPS-induced Stat3 phosphorylation in BV2 microglia
Stat3 is a key transcription factor and contributes to the IL-6 production at a transcription level [25,26]. We previously found that PGE2 could activate the Stat3 in podocytes [27]. Thus, we examined whether COX-2 inhibitor could attenuate LPS-induced Stat3 phosphorylation. As expected, LPS treatment strikingly enhanced Stat3 phosphorylation in BV2 microglial cells, which was largely normalized by COX-2 inhibitor (Figure 5A, 5B). These data suggested that LPS-stimulated Stat3 phosphorylation is through a COX-2-mediated mechanism, which might contribute to the IL-6 production to some extent.
Figure 5.
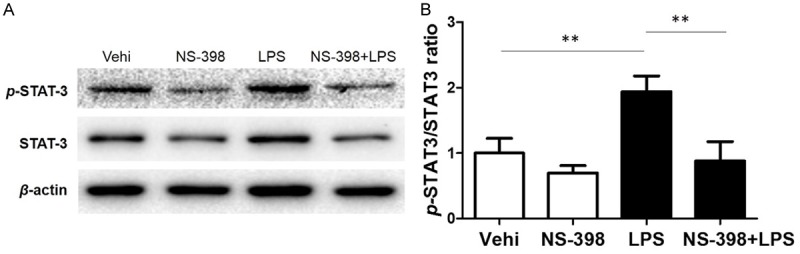
Effect of COX-2 inhibition on LPS-induced Stat3 phosphorylation. A. Western blotting analysis of p-Stat3 and Stat3 in BV2 cells. B. The ratio of p-Stat3 to Stat3. All values are means ± SD; n = 6 in each group. *P < 0.05 versus control or LPS group, **P < 0.01 versus control or LPS group.
Silencing COX-2 blunted LPS-induced Stat3 phosphorylation and IL-6 upregulation in BV2 cells
In order to further confirm the COX-2 effect in this experimental setting, we silenced COX-2 using a siRNA approach. As shown by the data, both COX-2 protein and PGE2 secretion were blocked by COX-2 siRNA (Figure 6A-D), suggesting the efficacy of COX-2 siRNA in silencing COX-2. Meanwhile, we observed a significant blockade of Stat3 phosphorylation and IL-6 production induced by LPS (Figure 7A-D), while other inflammatory cytokines like IL-1β, TNF-α, and MCP-1 were unaffected (Figure 7E-G). These data indicated a specific role of COX-2 in modulating Stat-3 activation and IL-6 expression in BV2 cells.
Figure 6.
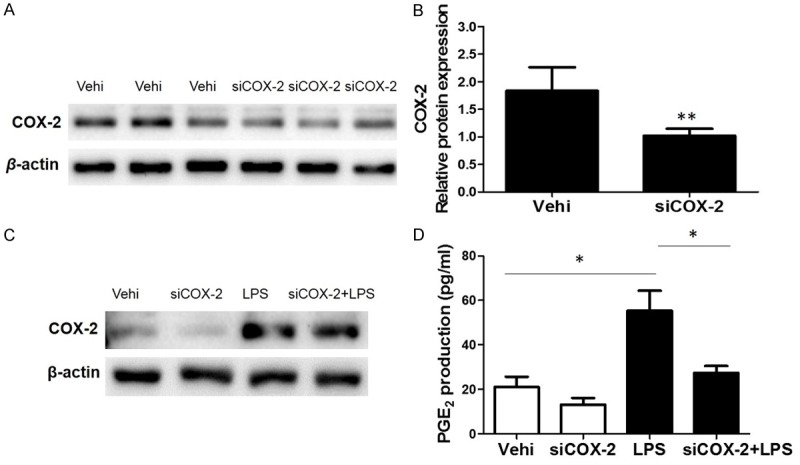
Silencing COX-2 ameliorated LPS-induced PGE2 production. To examine the role of COX-2 in LPS-induced PGE2 production, COX-2 siRNA was applied to BV2 cells. A. Protein levels of COX-2 in BV2 cells treated with COX-2 siRNA or negative control treatment. B. mRNA levels of COX-2 in BV2 cells treated with COX-2 siRNA or negative control treatment. C. Representative images of Western blots of COX-2 with or without COX-2 silencing in response to LPS treatment. D. EIA assay of PGE2 in medium. All values are means ± SD; n = 6 in each group. *P < 0.05 versus control or LPS group, **P < 0.01 versus control or LPS group.
Figure 7.
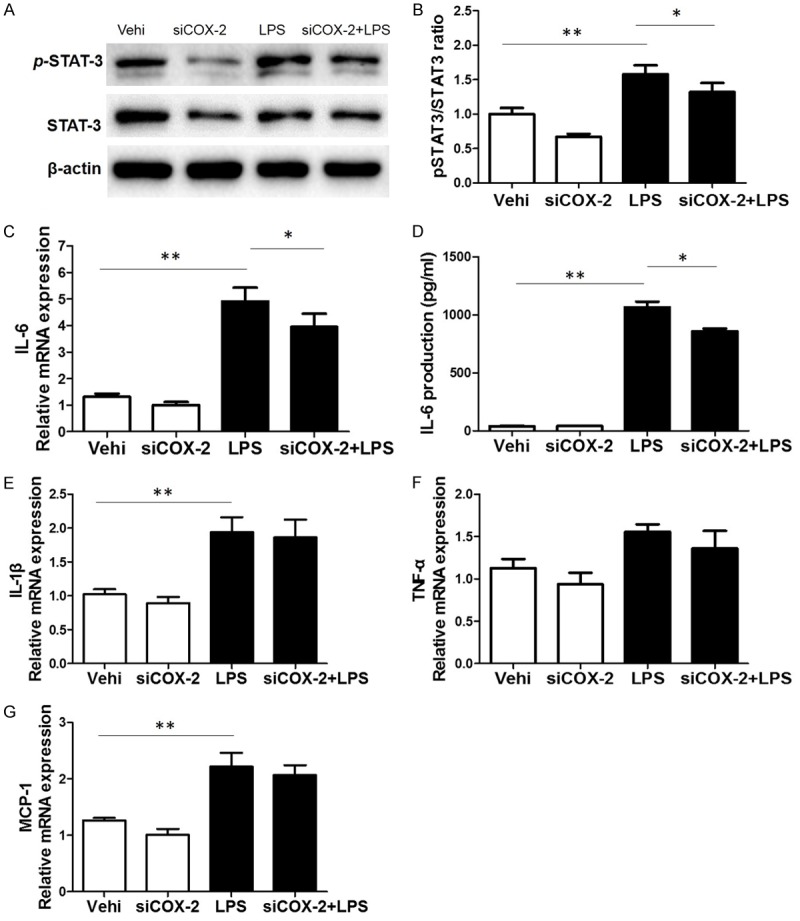
Silencing COX-2 blunted LPS-induced Stat3 phosphorylation and IL-6 upregulation. A. Western blotting analysis of p-Stat3 and Stat3 in BV2 cells. B. The ratio of p-Stat3 to Stat3. C. The mRNA level of IL-6 was measured by qRT-PCR. D. IL-6 release in cell culture medium were measured by using a ELISA kit. E. qRT-PCR analysis of IL-1β. F. qRT-PCR analysis of TNF-α. G. qRT-PCR analysis of MCP-1. All values are means ± SD; n = 6 in each group. *P < 0.05 versus control or LPS group, **P < 0.01 versus control or LPS group.
Discussion
Microglial cells play a critical role in neurodegenerative diseases such as AD, PD, cerebral ischemia, and multiple sclerosis. LPS is a component of the outer membrane of gram-negative bacteria. Exposure to LPS and LPS-induced inflammatory response could lead to septic shock and sepsis [28]. However, the pathological mechanisms of LPS-induced microglial inflammation still need in-depth investigation.
Among a number of proinflammatory cytokines, IL-6 is an important one with multiple functions. Using either anti-IL-6 neutralizing antibody or IL-6 siRNA attenuated TNF-α-dependent generation of reactive oxygen species (ROS) and brain microvascular endothelial cell (HBMvEC) hyperpermeability [29]. Some studies also reported that IL-6 was involved in the pathogenesis of artery atherosclerosis via promoting local inflammatory lesions [9]. In our study, LPS enhanced the production of IL-6, which was partially but significantly blocked by specific COX-2 inhibitor or COX-2 siRNA in line with the trend reduction of other inflammatory cytokines including IL-1β, TNF-α and MCP-1. These results suggested that COX-2 inhibition ameliorated LPS-induced inflammatory response in microglial cells with a specific effect on blocking IL-6. It was established that COX-2 played important roles in many inflammatory diseases [9,10], including the cytotoxicity in brain injuries and many neurodegenerative diseases such as AD, PD, and prion diseases [17-20]. Our data suggested such a COX-2-medicated pro-inflammatory effect in neural diseases could be through a IL-6-associated mechanism to some extent.
Stat3 is an inflammation-associated transcription factor and regulates many pro-inflammatory or anti-inflammatory cytokines [23]. A study showed that berberine inhibited colorectal cancer cell invasion and metastasis via the down-regulation of COX-2/PGE2-JAK2/Stat3 signaling pathway [24]. Our group also reported that PGE2 stimulated Stat3 to promote the inflammatory response in podocytes [27]. In agreement with above findings, here we observed that LPS-induced Stat3 phosphorylation can be striking blocked by a specific COX-2 inhibitor or COX-2 siRNA. Considering that IL-6 is one of the cytokines driven by Stat3 signaling [25,26], we could speculate that the effect of COX-2 on IL-6 production in microglial cells challenged with LPS might be through a Stat3-associated mechanism to some degree. In general, inflammation-associated factors could activate each other to form a complex network to further promote the inflammation. Indeed, IL-6 also can activate Stat3 to magnify the inflammatory response [30,31]. Therefore, in the present study, the reduction of IL-6 after COX-2 inhibition in LPS-treated microglial cells might also contribute to the regulation on Stat3 phosphorylation in theory.
Taken together, our study proposed a possible role of COX-2/PGE2/Stat3 cascade in LPS-induced upregulation of IL-6 and inflammatory response in microglial cells and central nervous system. Therapies by targeting COX-2/PGE2/Stat3/IL-6 signaling pathway in microglia and central nervous system might be beneficial for the treatment of neuroinflammatory diseases.
Acknowledgements
This work was supported by Grants from the National Natural Science Foundation of China (nos. 81600557, 81600532, 81600352, 81370802, 81300591, 81670647, and 81570616) and the Natural Science Foundation of Jiangsu Province (no. BK20130077).
Disclosure of conflict of interest
None.
References
- 1.Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. 2013;35:601–612. doi: 10.1007/s00281-013-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weng L, Zhang H, Li X, Zhan H, Chen F, Han L, Xu Y, Cao X. Ampelopsin attenuates lipopolysaccharide-induced inflammatory response through the inhibition of the NF-kappaB and JAK2/STAT3 signaling pathways in microglia. Int Immunopharmacol. 2017;44:1–8. doi: 10.1016/j.intimp.2016.12.018. [DOI] [PubMed] [Google Scholar]
- 3.Xia CY, Zhang S, Gao Y, Wang ZZ, Chen NH. Selective modulation of microglia polarization to M2 phenotype for stroke treatment. Int Immunopharmacol. 2015;25:377–382. doi: 10.1016/j.intimp.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 4.Okorji UP, Velagapudi R, El-Bakoush A, Fiebich BL, Olajide OA. Antimalarial drug artemether inhibits neuroinflammation in BV2 microglia through Nrf2-dependent mechanisms. Mol Neurobiol. 2016;53:6426–6443. doi: 10.1007/s12035-015-9543-1. [DOI] [PubMed] [Google Scholar]
- 5.Moidunny S, Matos M, Wesseling E, Banerjee S, Volsky DJ, Cunha RA, Agostinho P, Boddeke HW, Roy S. Oncostatin M promotes excitotoxicity by inhibiting glutamate uptake in astrocytes: implications in HIV-associated neurotoxicity. J Neuroinflammation. 2016;13:144. doi: 10.1186/s12974-016-0613-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh JH, Kwon TK. Withaferin A inhibits tumor necrosis factor-alpha-induced expression of cell adhesion molecules by inactivation of Akt and NF-kappaB in human pulmonary epithelial cells. Int Immunopharmacol. 2009;9:614–619. doi: 10.1016/j.intimp.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 9.Kanda T, Takahashi T. Interleukin-6 and cardiovascular diseases. Jpn Heart J. 2004;45:183–193. doi: 10.1536/jhj.45.183. [DOI] [PubMed] [Google Scholar]
- 10.Zhou N, Xu T, Bai Y, Prativa S, Xu JZ, Li K, Han HB, Yan JH. Protective effects of urinary trypsin inhibitor on vascular permeability following subarachnoid hemorrhage in a rat model. CNS Neurosci Ther. 2013;19:659–666. doi: 10.1111/cns.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogunshola OO, Moransard M, Gassmann M. Constitutive excessive erythrocytosis causes inflammation and increased vascular permeability in aged mouse brain. Brain Res. 2013;1531:48–57. doi: 10.1016/j.brainres.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 12.Siren AL, McCarron R, Wang L, Garcia-Pinto P, Ruetzler C, Martin D, Hallenbeck JM. Proinflammatory cytokine expression contributes to brain injury provoked by chronic monocyte activation. Mol Med. 2001;7:219–229. [PMC free article] [PubMed] [Google Scholar]
- 13.Alves JL. Blood-brain barrier and traumatic brain injury. J Neurosci Res. 2014;92:141–147. doi: 10.1002/jnr.23300. [DOI] [PubMed] [Google Scholar]
- 14.Borlongan CV, Rodrigues AA Jr, Oliveira MC. Breaking the barrier in stroke: what should we know? A mini-review. Curr Pharm Des. 2012;18:3615–3623. doi: 10.2174/138161212802002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia Z, Wang N, Aoyagi T, Wang H, Liu H, Yang T. Amelioration of cisplatin nephrotoxicity by genetic or pharmacologic blockade of prostaglandin synthesis. Kidney Int. 2011;79:77–88. doi: 10.1038/ki.2010.331. [DOI] [PubMed] [Google Scholar]
- 16.Frolich S, Olliges A, Kern N, Schreiber Y, Narumiya S, Nusing RM. Temporal expression of the PGE2 synthetic system in the kidney is associated with the time frame of renal developmental vulnerability to cyclooxygenase-2 inhibition. Am J Physiol Renal Physiol. 2012;303:F209–219. doi: 10.1152/ajprenal.00418.2011. [DOI] [PubMed] [Google Scholar]
- 17.Giovannini MG, Scali C, Prosperi C, Bellucci A, Pepeu G, Casamenti F. Experimental brain inflammation and neurodegeneration as model of Alzheimer's disease: protective effects of selective COX-2 inhibitors. Int J Immunopathol Pharmacol. 2003;16:31–40. [PubMed] [Google Scholar]
- 18.Lukiw WJ, Bazan NG. Cyclooxygenase 2 RNA message abundance, stability, and hypervariability in sporadic Alzheimer neocortex. J Neurosci Res. 1997;50:937–945. doi: 10.1002/(SICI)1097-4547(19971215)50:6<937::AID-JNR4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 19.Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc Natl Acad Sci U S A. 2003;100:5473–5478. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh DT, Perry VH, Minghetti L. Cyclooxygenase-2 is highly expressed in microglial-like cells in a murine model of prion disease. Glia. 2000;29:392–396. [PubMed] [Google Scholar]
- 21.Hein AM, O’Banion MK. Neuroinflammation and memory: the role of prostaglandins. Mol Neurobiol. 2009;40:15–32. doi: 10.1007/s12035-009-8066-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhat SA, Goel R, Shukla R, Hanif K. Angiotensin receptor blockade modulates NFkappaB and STAT3 signaling and inhibits glial activation and neuroinflammation better than angiotensin-converting enzyme inhibition. Mol Neurobiol. 2016;53:6950–6967. doi: 10.1007/s12035-015-9584-5. [DOI] [PubMed] [Google Scholar]
- 23.Bode JG, Ehlting C, Haussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 2012;24:1185–1194. doi: 10.1016/j.cellsig.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Ji Q, Ye N, Sui H, Zhou L, Zhu H, Fan Z, Cai J, Li Q. Berberine inhibits invasion and metastasis of colorectal cancer cells via COX-2/PGE2 mediated JAK2/STAT3 signaling pathway. PLoS One. 2015;10:e0123478. doi: 10.1371/journal.pone.0123478. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Ansari RA, Husain K, Rizvi SA. Role of transcription factors in steatohepatitis and hypertension after ethanol: the epicenter of metabolism. Biomolecules. 2016:6. doi: 10.3390/biom6030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X, Su X, Xu S, Wang H, Han D, Li J, Huang M, Cao X. MicroRNA in vivo precipitation identifies miR-151-3p as a computational unpredictable miRNA to target Stat3 and inhibits innate IL-6 production. Cell Mol Immunol. 2018;15:99–110. doi: 10.1038/cmi.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, Wu Y, Wang L, Zhang W, Xu M, Song J, Fu Y, Cui Y, Gong W, Li S, Xia W, Huang S, Zhang A, Jia Z. mPGES-1-derived prostaglandin E2 stimulates Stat3 to promote podocyte apoptosis. Apoptosis. 2017;22:1431–1440. doi: 10.1007/s10495-017-1418-7. [DOI] [PubMed] [Google Scholar]
- 28.Min KJ, Choi K, Kwon TK. Withaferin A down-regulates lipopolysaccharide-induced cyclooxygenase-2 expression and PGE2 production through the inhibition of STAT1/3 activation in microglial cells. Int Immunopharmacol. 2011;11:1137–1142. doi: 10.1016/j.intimp.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 29.Rochfort KD, Collins LE, McLoughlin A, Cummins PM. Tumour necrosis factor-alpha-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J Neurochem. 2016;136:564–572. doi: 10.1111/jnc.13408. [DOI] [PubMed] [Google Scholar]
- 30.Miao Z, Yu F, Ren Y, Yang J. d,l-Sulforaphane induces ROS-dependent apoptosis in human gliomablastoma cells by inactivating STAT3 signaling pathway. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu MJ, Feng D, Wu H, Wang H, Chan Y, Kolls J, Borregaard N, Porse B, Berger T, Mak TW, Cowland JB, Kong X, Gao B. Liver is the major source of elevated serum lipocalin-2 levels after bacterial infection or partial hepatectomy: a critical role for IL-6/STAT3. Hepatology. 2015;61:692–702. doi: 10.1002/hep.27447. [DOI] [PMC free article] [PubMed] [Google Scholar]