

Abstract
Myosin X is an unconventional actin-based molecular motor involved in filopodial formation, microtubule-actin filament interaction, and cell migration. Myosin X is an important component of filopodia regulation, localizing to tips of growing filopodia by an unclear targeting mechanism. The native α-helical dimerization domain of myosin X is thought to associate with antiparallel polarity of the two amino acid chains, making myosin X the only myosin that is currently considered to form antiparallel dimers. This study aims to determine if antiparallel dimerization of myosin X imparts selectivity toward actin bundles by comparing the motility of parallel and antiparallel dimers of myosin X on single and fascin-bundled actin filaments. Antiparallel myosin X dimers exhibit selective processivity on fascin-bundled actin and are only weakly processive on single actin filaments below saturating [ATP]. Artificial forced parallel dimers of myosin X are robustly processive on both single and bundled actin, exhibiting no selectivity. To determine the relationship between gating of the reaction steps and observed differences in motility, a mathematical model was developed to correlate the parameters of motility with the biochemical and mechanical kinetics of the dimer. Results from the model, constrained by experimental data, suggest that the probability of binding forward, toward the barbed end of the actin filament, is lower in antiparallel myosin X on single actin filaments compared to fascin-actin bundles and compared to constructs of myosin X with parallel dimerization.
Introduction
Myosin X is an actin-based molecular motor that is widely expressed in vertebrate cells (1) and has a diverse set of functions, primarily acting to mediate traction between actin and other filament and structural systems (2). Myosin X contains a MyTH4 domain that binds directly to microtubules and in some organisms is a critical component for maintaining mitotic/meiotic spindle integrity and orientation (3, 4). The FERM domain of myosin X binds to β-integrin and connects the actin cytoskeleton to the extracellular matrix, facilitating cell traction and mechanosignaling (2).
Myosin X is a necessary component for filopodia formation, and the level of myosin X expression influences the number and length of filopodia generated by a cell (5, 6, 7, 8, 9). Within these structures, myosin X facilitates localization of integrin at filopodial tips, which may enhance filopodial stabilization and growth through integrin-extracellular matrix binding (10). On the other hand, it was shown that myosin X induces filopodia upon dimer formation without the tail domain, suggesting that the motor activity of the myosin X dimer is critical for the initiation of filopodia formation (7). Supporting this notion, it was shown that PIP3-induced myosin X dimer formation facilitates filopodia formation (11). Myosin X is also involved in the formation and stabilization of other actin-based protrusions, such as invadopodia, that promote cancer metastasis (12). Myosin X’s role in invadopodia formation makes it a target for cancer therapy, as silencing myosin X disrupts invadopodia formation and reduces their invasive character (13).
The actin cross-linking protein fascin is another necessary component for filopodial formation (14, 15). Fascin binds along the length of the actin filament, linking filaments together into tightly packed hexagonal parallel bundles with an ∼11 nm spacing (15, 16). In filopodia, fascin-bundled actin aligns with the minus ends of the filaments pointing toward the cell center. Although myosin X and fascin show no direct interaction, the accumulation of myosin X at the distal ends of filopodia (17, 18) suggests that myosin X may be recruited to filopodia by a preferential interaction with fascin-bundled actin. Alternatively, a recent study suggested that focal adhesion components, including β-integrin, are involved in the recruitment of myosin X at the base of nascent filopodia, thus producing myosin-X-dependent filopodia formation (9).
Increased processivity of myosin X on actin bundles compared to that on single actin filaments has been reported (19, 20, 21, 22), but other studies have shown little selectivity (23) or myosin X constructs that are highly processive on single actin filaments (24). A consensus regarding bundle selectivity by myosin X has not been realized, in part because of the use of constructs with different dimerization domains and a lack of mechanistic understanding as to how myosin might selectively interact with bundled actin.
Recently, Zhang and co-workers (25) discovered that the native coiled-coil (CC) domain of myosin X forms an antiparallel dimer with a weak 0.6 μM affinity, and the antiparallel dimerization has been confirmed in a crystal structure (22). This would make myosin X the only known antiparallel dimer in the myosin superfamily. Antiparallel dimerization introduces an extra hinge and additional 4.7 nm spacing to the protein contour length between the motor heads (25). These structural features might correlate with selectivity of myosin X for bundled actin and explain why groups using forced parallel dimers of myosin X have observed strong processivity on single filaments (23, 24).
Herein, the processivity of a forced antiparallel construct of myosin X (APCCMX) is compared to a parallel construct, and the mechanistically relevant properties of motility are compared. We hypothesize that antiparallel dimerization may reduce the processivity of myosin, likely through a mechanism that disrupts gating of biochemical or mechanical reaction steps. Total internal reflection fluorescence (TIRF) microscopy is utilized to track the motility of myosin X dimers formed by different dimerization domains on both single actin filaments and fascin-bundled actin. We find that antiparallel myosin X is less processive than parallel myosin X. On single actin filaments, antiparallel myosin X is only weakly processive at ATP concentrations below 5 μM, whereas on fascin-bundled actin, antiparallel myosin X displays robust processivity. Parallel myosin X is found to be processive on both single actin filaments and fascin-bundled actin, showing no significant differences in motility. These findings suggest that differences in motor dimerization can alter the preferred track of a molecular motor and may explain how myosin X becomes concentrated in regions of the cell rich in fascin-bundled actin.
Materials and Methods
Actin preparation
Globular-actin (G-actin) was obtained from rabbit muscle as described (26) and stored in G-actin buffer (2 mM Tris buffer (pH 8.0), 0.2 mM CaCl2, 0.2 mM ATP, and 0.5 mM dithiothreitol (DTT)). Rhodamine-phalloidin-labeled F-actin was prepared by mixing 1 μM G-actin with 4× F-actin buffer (300 mM KCl, 10 mM MgCl2, 40 mM HEPES (pH 7.0)) and 1.1 μM rhodamine-phalloidin (R415; Molecular Probes, Eugene, OR). The mixture was incubated at room temperature for 10 min, transferred onto ice, stored at 4°C, and used within 1 month. M5 buffer solution (25 mM KCl, 20 mM HEPES (pH 7.6), 2 mM MgCl2, 1 mM EGTA (E4378; Sigma-Aldrich, St. Louis, MO)) was mixed with 100 μg mL−1 calmodulin (CaM) and 100 mM DTT (M5+) on each experimental day. Motility assay buffer was prepared by adding the appropriate concentration of ATP, an additional 100 μg mL−1 CaM and 100 mM DTT, and ∼20 nM of the Myosin-QD525 conjugate to M5+ buffer. To delay photo bleaching of the labeled F-actin, 3 mg/mL glucose, 100 μg/mL glucose oxidase, and 40 μg/mL catalase (G0543; Sigma-Aldrich) were added to the motility assay buffer immediately before each experiment.
Fascin (a gift from Dr. Dan Safer) was expressed in Escherichia coli and purified as previously described (27). Fascin-bundled actin was prepared by mixing 3 μM fascin and 8 μM phalloidin-stabilized F-actin in F-actin buffer and incubating on ice for 24 h.
Myosins
Three recombinant constructs of bovine myosin X were expressed in and purified from SF9 cells (see below). To promote the formation of stable dimers with the native orientation, the antiparallel coiled coil was extended by inserting a repeat dimerization motif (two heptads, e.g., 14 residues, that are critical for antiparallel CC formation) into the sequence following the single stable α-helical segment and the antiparallel coiled coil (Fig. 1 A, construct APCCMX). The tandem antiparallel segment of 14 residues should add an extra ∼2 nm spacing between the heads. The C-terminal tail domains of myosin X were not included, but a c-Myc target sequence was added at the C-terminus of the constructs for purification and labeling with fluorescent quantum dots. Two parallel dimers of myosin X were similarly created (Fig. 1, B and D): one containing a leucine zipper (LZIPMX) CC motif, and one with the mouse myosin V coiled coil (MVCCMX) following the α-helical segment domain. Cartoons of APCCMX and LZIPMX are shown in Fig. 1, C and D. More detailed sequence schematics are shown in Fig. S1.
Figure 1.

Schematics of the different constructs of myosin X. In (A) and (B), CC and α-helical domains are shown as cylinders, the motor head is shown as a green ellipse, and the CaMs are shown as orange ellipses with white circles. Yellow cylinders indicate native helical domains, and the following nonyellow cylinders indicate nonnative domains. Dark blue and red segments are c-Myc and biotin acceptor antigens, respectively, for quantum dot labeling. The cartoons below depict the antiparallel (C) and parallel (D) myosin X dimers bound to actin, color-coded as in (A) and (B). SAH, single α-helical segment.
To confirm the stabilized antiparallel orientation of the APCCMX construct and stable parallel orientation of LZIPMX, custom 44-residue peptides of the CC portion of LZIPMX and APCCMX with single cysteine mutations located toward the outside of the putative coiled coils were generated for single-molecule Förster resonance energy transfer (FRET) experiments (Fig. S2 A). Peptides were labeled with a 1:1 ratio of Cy3 maleimide (donor) and Cy5 maleimide (acceptor) (Sigma-Aldrich) to obtain ∼50% of dimers with complementary FRET pairs. Details of this experiment are in the Supporting Material. As anticipated, the parallel LZIPMX coiled coil showed a peak FRET efficiency of ∼1.0 consistent with the close proximity of the donor and acceptor because of the parallel orientation of the coiled coil (Fig. S2 B). The APCCMX showed a peak FRET intensity at 0.6, consistent with a donor and acceptor separation of ∼5.1 nm, which is consistent with the anticipated separation for antiparallel dimerization (Fig. S2 B). Some heterogeneity of FRET efficiency was observed for both sequences, possibly because of fraying at the short peptide ends or some misregistration.
Sf9 cells (6 × 108) were coinfected with two baculoviruses expressing M10 heavy chain and CaM, and the infected cells were cultured for 3 days at 28°C. After centrifugation, the cells were resuspended in a lysis buffer containing 0.1 M NaCl, 50 mM Tris-HCl (pH 8), 2 mM MgCl2, 5 mM EGTA, 1 mM ATP, 0.5 mM phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, and 1 mM DTT, and homogenized with a sonicator (Sonifier 250; Branson, Danbury, CT) at the output amplitude of ∼1 and the 50% duty cycle for 60 s. The homogenate was centrifuged at 126,000 × g (Type 70 Ti rotor; Beckman, Brea, CA) for 20 min, and the supernatant was incubated with anti-FLAG antibody-conjugated agarose (Sigma-Aldrich) with gentle rotation for 1 h at 4°C. After washing with a wash buffer containing 0.15 M NaCl, 20 mM MOPS-KOH (pH 7.5), 1 mM EGTA, 1 μg/mL leupeptin, and 1 mM DTT, the resin was packed into a column, and the protein was eluted with the wash buffer containing 0.1 mg/mL FLAG peptide and 12.5% sucrose. An aliquot of the purified protein was rapidly frozen by liquid N2 and stored at −80°C before use.
Stoichiometry of CaM binding to the myosin X constructs was measured by calibrated gel electrophoresis as described in (28), using recombinant human CaM (CALM2) expressed in E. coli and turkey gizzard myosin heavy chains as standards. Close to three CaM per heavy chain were bound (values in legend for Fig. S3). An SDS-PAGE gel confirms the same heavy chain:CaM ratio for the three purified constructs (Fig. S3).
Quantum dot attachment
QD525 quantum dots from Life Technologies (Q11041MP; Molecular Probes), conjugated with F(ab')2-Goat anti-Mouse IgG (H+L) secondary antibody, were incubated with c-Myc monoclonal primary antibody (Ab) (631206; Clontech, Mountain View, CA) at a 1:1 ratio and incubated at 4°C for 24 h. Myosin X constructs terminated in the c-Myc target sequence were then decorated with c-Myc Ab functionalized quantum dots. Myosin stocks were diluted to ∼500 nM with M5+ buffer and incubated at 0.167 μM with 0.67 μM of the Ab-coated quantum dot solution in 7 mg/mL bovine serum albumin (BSA) on ice for >2 h before each experiment. A final ratio of myosin:QDs (∼1:8) mol/mol was used to assure that few quantum dots bound more than one motor.
Motility assay
2 wt % poly(methyl methacrylate) (182265; Sigma Aldrich) films in methylene chloride (Sigma Aldrich) were spin-coated onto ethanol-washed cover slips at 5000 rotations per minute for 60 s (29). Flow cells were constructed creating ∼10 μL channels by attaching a poly(methyl methacrylate)-coated cover slip to an ethanol-washed glass slide with solvent-free double-sided tape (3 M, 9731). The channels were then used individually for motility assays by first flowing in 10 μL of 30 nM N-ethylmaleimide-inactivated myosin (30), waiting 5 min, and then rinsing with 10 μL high salt buffer (500 mM KCl, 10 mM HEPES (pH 7.0), 5 mM MgCl2) plus 10 mM DTT. Actin was then introduced into the chamber by flowing 20 μL of 250 nM phalloidin-stabilized F-actin in M5+ buffer into the channel. Filaments were allowed to attach for 5 min before the flow channel was rinsed twice with 10 μL of M5+ buffer and blocked with 10 μL BSA (20 mg/mL). BSA was incubated in the channel for 5 min, after which 20 μL of motility assay buffer was added to the flow channel at a final myosin concentration of ∼15 nM.
Quantum-dot-labeled myosin X was imaged using a 405 nm solid-state laser (Coherent, Santa Clara, CA) under TIRF illumination. The TIRF excitation field was rotated around the optical axis at 40 Hz to average out interference fringes in the illumination and create a uniform intensity field (31) that enhances tracking fidelity.
Video stacks were recorded at 50 ms frame interval for 100 or 150 s periods using a Cascade-512B electron multiplying-CCD camera (Photometrics, Huntington Beach, CA) and the In Vivo imaging software package (Thermo Fisher, Waltham, MA). Individually labeled myosin Xs were localized using FIESTA tracking software (32). Steps were then determined from the FIESTA position traces by fitting the location along the actin filament versus time with a step-finder algorithm (33).
Data analysis and display
Statistical significance was determined via a Student’s t-test when p-values were below 0.01 and indicated by ∗∗ for normally distributed data (step sizes), or a Mann-Whitney-Wilcoxon rank sum for nonnormally distributed data, the run length and dwell time distributions corrected for multiple comparisons where applicable. Box plots show mean (square), median (line), mean ± standard error (SE) (box), and standard deviation (SD) whiskers. Plots were generated in Origin 9.0 or QtiPlot. Values for run length were determined using maximal likelihood estimation, which accounts for minimal detectable run length (34). Mathcad V14 (PTC, Needham, MA) was used for kinetic modeling and Monte Carlo (MC) simulations.
Results
APCCMX: short strides and frequent backward steps
The step size and backward stepping rates of parallel and antiparallel myosin X constructs were compared on single actin filaments at 500 nM MgATP. On single actin filaments, the antiparallel construct (APCCMX Fig. 2 A) took variably sized steps averaging 32.2 ± 16.5 nm, including many short forward steps (Fig. 2 C, blue and dark blue) and more frequent backward steps (27%) compared to the LZIPMX parallel myosin X (recordings in Fig. S4, average forward step size 35.4 ± 16.8 nm (SD), 7% backward steps (Fig. 2 D, dark green). The distribution of forward step sizes of APCCMX in single filaments was well fit by two Gaussian components centered at 20 and 40 nm (Fig. 2 C; Table 1).
Figure 2.

Representative trajectories for APCCMX on single actin filaments, (A), and fascin-bundled actin, (B). More frequent backward steps are observed on single filaments. Black data points are calculated positions of the molecule along the filament at each 50 ms frame, and the red trace is a fit by a step-fitting algorithm (Materials and Methods). (C) shows the step size histograms of APCCMX on single actin filaments (dark blue) and fascin-actin bundles (light blue) at 500 nM MgATP; n = 421 and n = 443 steps. (D) shows the step size histogram for LZIPMX on single actin filaments (dark green) and fascin-bundled actin (light green); n = 393, n = 451. Curves are best fits to double Gaussian distributions (single forward and backward stepping peaks). The histogram of APCCMX on single filaments was best fit by a triple Gaussian distribution (two peaks for forward stepping; the individual peaks are shown as blue traces). The short (20 nm) steps account for ∼36% of the total number of forward steps. (E) shows box plots of forward step size at 500 nm [MgATP], showing that APCCMX steps are shorter on single filaments than on bundles or LZIPMX on either single filaments or bundles. (F) gives the fraction of backward steps. Error bars represent mean ± SE, ∗∗ represents p < 0.01, and N.S. represents not significantly different.
Table 1.
Summary of Experimentally Measured and Fitted Parameters for Myosin X Constructs on Single and Fascin-Bundled Actin Filaments
| Construct | APCCMX Single | APCCMX Bundles | LZIP Single | MVCCMX Single |
|---|---|---|---|---|
| Forward step size (nm) ± HWHM | 20.3 ± 9 | 38 ± 13 | 38.2 ± 15 | 39 ± 22 |
| 40.3 ± 16 | ||||
| Backward step size (nm) ± HWHM | 30.2 ± 15 | 30 ± 15 | 26.8 ± 13 | 29.1 ± 19 |
| Backward steps (%) mean ± SE | 26.6 ± 6.3 | 14.7 ± 7.3 | 7 ± 5 | 8.9 ± 3.5 |
| Velocity (nm/s) mean ± SE | 32.9 ± 2 | 39.0 ± 1.2 | 29.0 ± 1.6 | 36.9 ± 2.8 |
| Dwell time (s) ± SD | 0.63 ± 0.7 | 0.74 ± 0.7 | 1.9 ± 1.5 | 2.0 ± 1.9 |
| VMax (nm/s) mean ± SE | N/A | 399 ± 12 | 381 ± 12 | 389 ± 12 |
| VMax (nm/s) fitted | N/A | 344 | 389 | 393 |
| k1 (μM·s)−1 | N/A | 2.8 | 3.9 | 5.0 |
| k2 (s−1) | N/A | 15.3 | 12.5 | 13.0 |
| KM (μM) | N/A | 5.4 | 3.2 | 2.6 |
Forward and backward step sizes are the results of fits to the distributions shown in Fig. 2 measured at 500 nM MgATP. Velocities are the means ± SE of the net displacement/run time for more than 100 trajectories for each construct at 500 nM MgATP. Dwell times (means ± SD) at 500 nM [MgATP] represent >393 dwells for each case. The corresponding distributions are plotted in Fig. 4, A and B. HWHM, halfwidth at half-maximum; N/A, not available.
On fascin-actin bundles, the forward step sizes of APCCMX (Fig. 2 C, cyan) and LZIPMX (Fig. 2 D, light green) were similar—38.0 ± 13 and 39.0 ± 22 nm, respectively—but the APCCMX was still more likely to step backward (15%, Fig. 2 F) than the parallel myosin X (6%, Fig. 2 F). The backward stepping frequency of APCCMX on actin bundles was significantly reduced compared to that on single filaments, whereas LZIPMX exhibited similar step sizes and percentages of backward stepping on single and bundled actin filaments. The average forward step size of APCCMX was larger on actin bundles compared to single filaments because of the reduction of the number of short steps observed on single filaments. For LZIPMX, step size remained the same on single and bundled filaments.
The stepping characteristics of LZIPMX are similar to those of myosin V (29). Myosin X dimerized via the myosin V coiled coil, MVCCMX, exhibited similar stepping characteristics to the LZIPMX (Fig. S5). The differences between APCCMX and the parallel dimerized myosin X constructs suggest that the polarity of dimerization may play an important role in regulating the step size and directionality of myosin X.
APCCMX is weakly processive on single filaments
At 500 nM MgATP run-lengths for APCCMX were shorter (342 ± 24 nm, mean ± SE, Fig. 3 A, blue) on single filaments than on fascin-actin bundles (663 ± 43 nm; p < 0.001, Fig. 3 A, cyan). LZIPMX exhibited processivity on both single (run lengths 714 ± 33 nm, Fig. 3 A, dark green) and bundled actin filaments (739 ± 46 nm, Fig. 3 A, light green). On fascin-actin bundles, run lengths of APCCMX were similar to those of LZIPMX.
Figure 3.

Cumulative distributions of run-length for APCCMX (blue) and LZIPMX (green) on single (dark colors) and fascin-bundled (light colors) actin filaments at 500 nM [MgATP]. More than 100 runs were analyzed in each case. Color key indicates constructs and conditions for both (A) and (B). (B) shows run-length versus [MgATP] for APCCMX on single (blue) and bundled (cyan) f-actin, LZIPMX (green), and MVCCMX (gray) constructs on single filaments. Data points represent experimentally measured dwell times mean ± SE. Solid lines represent fits to the kinetic model described later.
Run lengths decreased for myosin X with increasing [MgATP] from 500 nM to 500 μM, independent of construct and dimerization orientation (Fig. 3 B). Above 5 μM MgATP processive runs of APCCMX on single filaments were rarely observed, consistent with APCCMX being nonprocessive on single filaments at physiological [MgATP]. On bundles, APCCMX was nearly as processive as parallel constructs on single filaments.
Parallel and antiparallel myosin X exhibit similar velocities
The dwell time of APCCMX between steps at 500 nM MgATP was found to be shorter than LZIPMX, indicating that the dimerization mode alters the stepping rate of the motor (Fig. 4, A and B; note different abscissa scales). Bundling of actin filaments did not have a significant effect on the dwell times for either APCCMX or LZIPMX (compare light and dark distributions in Fig. 4, A and B). Faster stepping of APCCMX compensates for the substantial increase in backward stepping compared to LZIPMX, leading to similar net velocities of APCCMX and LZIPMX on single actin filaments at 500 nM [MgATP] (Fig. 4 C).
Figure 4.

Stepping rates and velocities of myosin X constructs. Dwell time distributions at 500 nM MgATP for antiparallel (A) and LZIPMX (B) on single actin filaments (dark colors) and fascin-actin bundles (light colors) are given. Note the expanded time axis in (A). Distributions include >393 dwells each. Dashed lines are fits to P(t) = A · exp(−t/τ), where the respective decay times are 0.45, 0.70, 1.9, and 2.4 s for APCCMX on single actin and bundles, and LZIPMX on single actin and bundles, respectively. (C) shows box plots of velocity at 500 nM [MgATP], with symbols as in Fig. 2E. (D) gives velocity versus ATP concentration (means with error bars representing mean ± SE). Curves in (D) are fits by a mathematic model (described later). ∗∗ represents p < 0.01 and N.S. represents not significantly different.
The velocity of APCCMX on fascin-actin bundles (39.0 ± 1.2 nm/s) was faster than on single actin filaments (32.9 ± 2 nm/s, Fig. 4 C). The velocities of LZIPMX on single (29 ± 1.6 nm/s) and bundled (26.5 ± 1.7 nm/s) actin filaments were not different from each other or from APCCMX on single filaments. At 2 mM [MgATP], differences between the velocities of APCCMX and LZIPMX were not significant. The maximal observed velocities for the constructs (Fig. S6; Table 1) were not found to be significantly different.
Velocity followed a hyperbolic dependence on [MgATP], saturating between 350 and 400 nm/s for all processive constructs (Fig. 4 D). The simplest reaction scheme for saturating dependence of velocity on [MgATP] contains two reaction steps in series, leading to the mean dwell time versus [MgATP] given by
| (1) |
where k1 (M−1s−1) is the second order rate constant for MgATP binding to a nucleotide-free myosin head and k2 (s−1) is the rate of an ATP-independent step, probably ADP release. In the case for which significant backward steps are evident, the velocity is given by
| (2) |
where <s> is the mean step size, fBS is the fraction of backward steps, and KM = k2/k1 is the [MgATP] at half-maximal velocity.
Vmax and KM (Table 1) were obtained (Fig. S7) by fitting Eq. 2 to the velocity versus [MgATP] data using mean step size (<s>) and fraction of backward steps (fBS) obtained from the stepping analysis (Fig. 2). Although Vmax is not different between the processive constructs, a higher KM value was found for APCCMX on fascin-bundled actin. The value of KM on bundles was higher for APCCMX—5.4 μM, compared to the parallel myosin X values 3.2 and 2.6 μM—which is attributed to a smaller k1 and larger k2 of APCCMX compared to parallel myosins (Table 1). APCCMX was not processive enough at ATP concentrations above 5 μM on single filaments to obtain Vmax or KM by fitting Eq. 2.
Processive stepping dynamics of myosin X constructs with parallel (LZIPMX and MVCCMX) and antiparallel (APCCMX) dimerizing CC regions indicate that the antiparallel arrangement reduces processivity on single actin filaments. This result suggests that bundle selectivity of myosin X may partly be accomplished by the structural dynamics of the tail. On its own, the more frequent backward stepping of APCCMX on single filaments (Fig. 2, C and F) would cause a ∼30% reduction in the velocity of APCCMX compared to LZIPMX, but the shorter dwell times (Fig. 4, A and B) compensate for the decreased directional persistence, leading to similar velocities of APCCMX and LZIPMX on single filaments at 500 nM MgATP (Fig. 4 C).
Kinetic model for myosin processivity
A kinetic model for myosin processivity was developed to discriminate the specific biochemical or mechanical steps within the reaction pathway of the myosin X dimer that may be affected by the polarity of dimerization (Fig. 5). The scheme is an extension of the model described by Elting et al. (35) used to quantify myosin VI kinetics and to describe minimal requirements for processivity. Here, we incorporate backward stepping, which increases the number of accessible states in the mechanochemical cycle from 9 to 15. The available binding sites on actin in a single kinetic cycle are labeled α, β, γ, and δ. The net direction of motility (the plus end of the filament) is to the right. The reaction sequence begins with both heads bound to actin subunits β and γ and ADP on each head (state C1). One head releases ADP, and the molecule enters a state with ADP (D) bound to either the leading head (state A1) or the trailing head (B1). ADP can be released, or ATP (T) can bind to the nucleotide-free head, thereby dissociating it. Hydrolysis of ATP to ADP · Pi occurs in the detached head, and then it can bind in the forward or backward direction, release Pi, and complete a forward or backward step, resulting in state A0, B0, A2, or B2, depending on which head has ADP bound. F0 and F1 are singly actin-bound states that are vulnerable to run termination if ATP binds to the attached head before the detached one rebinds to actin. G is the detached endpoint of a run. After a complete step, corresponding reaction steps continue from states A0, B0, A2, and B2 until both heads detach (state G), ending the processive run. Runs can end from the nucleotide-free state (E) by head dissociation without ATP binding. Adding this possibility was motivated by the relatively high nucleotide-free detachment rate of myosin X (36). ATPase cycles can occur without stepping (futile cycling) when the molecule returns to state A1 or B1 from C1 (two ADPs bound), F0, or F1.
Figure 5.

State diagram of myosin kinetic cycle illustrating the predominantly occupied states of the motor during a mechanochemical cycle that can result in either a forward step (FS), backward step (BS), or a futile cycle for each ATP consumed. Leading and trailing myosin heads are labeled with nucleotide state: O = nucleotide free (rigor), D = ADP bound, and T = ATP or ADP · Pi bound. Similar states of the motor are denoted by letters: A = rigor trailing head, ADP leading head, B = ADP trailing head, rigor leading head, C = ADP bound to both heads, D = ADP bound to attached head and ATP or ADP · Pi bound to detached head, E = both heads in rigor, F = bound head in rigor and ATP or ADP · Pi bound to detached head, and G = both heads detached, the endpoint of a run. Numerical subscripts denote position on actin: 0 = backward, 1 = central, and 2 = forward. Binding sites on actin are labeled from left to right in plus-end direction of travel: α, β, γ, and δ. Arrows denote allowable transitions between states and are color-coded to denote the biochemical process that occurs for each state transition: ATP binding, ADP release, myosin rebinding to actin, or detachment from rigor.
For any given set of rate constants describing the transitions from one state to available states, a Markov matrix (37) specifies the probability of taking a specific reaction branch to the next state. The full set of equations will be published elsewhere. Briefly, the relative occupancy of the 15 states after each transition is given by multiplying a vector describing their occupancy by the Markov transition matrix. The initial occupancy vector has state C1 occupied. After a large number of transitions, G (detached myosin) is the sole occupied state. From the number of transitions into A0, B0, A2, and B2, the average number of forward and backward steps are calculated. The net number of steps (forward steps − backward steps) multiplied by the average forward and backward step sizes (Fig. 2) determines the run length. The sum of the dwell times for each state multiplied by the number of visitations gives the total run time. The velocity is the net run length divided by the run time.
Kinetic differences for ATP binding and ADP dissociation between the leading and trailing heads of the dimer are termed biochemical gating. Preference of the singly attached head to rebind in the forward direction versus backward is a type of mechanical gating. Elting et al. (35) showed that biochemical gating is not essential for two-headed myosin motors to produce processive motility, although the mechanochemical reaction scheme they used to simulate myosin VI did not include allowance of backward steps.
Since our model to describe myosin X incorporates backward steps, asymmetry of the biochemical steps that causes the trailing head to detach more often than the leading one or else mechanical asymmetry that favors head reattachment in the forward direction is necessary to obtain plus-end-directed processivity. Otherwise, symmetry between forward and backward stepping causes the average run length and velocity to be zero (diffusive motion). Three different steps in this model were candidates for biochemical or mechanical asymmetry between leading and trailing heads: 1) ATP binding, 2) hydrolysis byproduct release (thought to be limited by ADP release), and 3) bias for a detached head to rebind to available actin subunits in the forward direction.
To simulate the mechanical characteristics for each of the constructs studied, approximate magnitudes of the biochemical reaction rates were taken from biochemical experiments (36) as listed in Table S1. Fitting the model simultaneously to the velocity, run length, and backward stepping data versus ATP concentration measured in our experiment provides strong constraints to the model parameters and yields estimates of the biochemical transition rates and backward versus forward rebinding for the myosin X constructs on their respective tracks (Table 2). Comparing these rates enables a description of how kinetic alterations between parallel and antiparallel dimerization produce bundle-selective or nonselective processivity. Cross correlations between model parameters, however, prevents determination of values by global optimization to more than two significant figures.
Table 2.
Table of Parameters Derived from the Kinetic Model
| Construct | kADPoff-rear (s−1) | kADPoff-front (s−1) | kATPon-rear (μM−1s−1) | kATPon-front (μM−1s−1) | krebind-for (s−1) | krebind-back (s−1) | krigor-detach (s−1) |
|---|---|---|---|---|---|---|---|
| APCCMX | 21 | 21 | 5.1 | 2.2 | 130 | 110 | 1.5 |
| APCCMX-BUNDLES | 21 | 7 | 4.8 | 1.9 | 160 | 100 | 1.5 |
| LZIPMX | 17.4 | 7.2 | 2.7 | 1.6 | 150 | 35 | 1.45 |
| MVCCMX | 18 | 6.4 | 3.0 | 1.6 | 150 | 45 | 1.55 |
Curves predicted by the fitted model of run length (Fig. 3 B), velocity (Fig. 4 D), and proportion of backward stepping (Fig. S8), all versus [MgATP], capture the main features of the measured ATP-dependence of these parameters. The model predicts that the proportion of backwards stepping may depend on the MgATP concentration (Fig. S8).
APCCMX exhibits reduced gating on single actin filaments
Rates of the kinetic model fitted to the data for the parallel LZIPMX and MVCCMX indicate that ADP release and a bias to rebind in the forward direction are the primary mechanisms of gating in myosin X. The overall modeled rates are compatible with the magnitude of rates measured in solution (36) and suggest that the specific mechanisms of gating are reduced rates for ADP dissociation and ATP binding to the leading head (instead of accelerating kinetics of the trailing head) and low backward rebinding rate. Moderately slower binding of MgATP to the leading head compared to the trailing head enables the simulation to capture the peak in the average run length between 0.5 and 2.5 μM [MgATP] (Fig. 3 B).
Because of the high proportion of backward stepping observed for APCCMX, bias toward forward rebinding is much less prevalent in APCCMX compared to the parallel constructs, leading to the ratio of forward to backward binding rates of 130 s−1/110 s−1. APCCMX rebinds backward slightly less often on fascin-actin bundles (forward/backward binding rates 160 s−1/100 s−1). The parallel constructs exhibited much stronger bias for forward binding (150 s−1/∼40 s−1 (Table 2)). The decreased bias toward forward rebinding in APCCMX may result from the antiparallel dimerization of myosin X introducing more mechanical compliance between the heads than the parallel coiled coils, thereby enabling binding to backward actin subunits. The reduced step size of the APCCMX on single filaments suggests frequent binding to actin subunits close to the nonstepping head. Some of these sites might be blocked by the fascin-bundling of actin, resulting in the increased forward binding bias of APCCMX on fascin bundles.
The simulation suggests that APCCMX on fascin-actin bundles exhibits similar ADP release gating to LZIPMX. No significant difference in the gating of ATP binding was observed between the APCCMX on single filaments or bundles, whereas the rate of ADP release and ATP binding at the leading head appeared to be less for the parallel constructs compared to APCCMX. Gating of ADP release was eliminated in APCCMX on single filaments for the model to simulate the observed loss of processivity at high [ATP]. On the other hand, APCCMX on fascin bundles exhibited similar ADP release gating to the parallel constructs (LZIPMX and MVCCMX).
Run-terminating detachment from the doubly actin-bound rigor state (E) was introduced into the model to avoid run-length becoming infinite as [MgATP] approaches zero. The apparent value of the rigor detachment rate (1.5 s−1) was not found to vary between fits of the model to the various myosin X constructs. Its magnitude is of the same order as the 1 s−1 rate measured for myosin X subfragment-1 heads in solution (36) and is much faster than the corresponding rate for myosin V (38) or myosin II (39).
MC simulations match experimental distributions
The rates extracted from the analytical equations describing the kinetic model rely on the average values of run length, run time, and backward stepping versus [ATP]. Probability distributions of these parameters were simulated using the fitted kinetic rates in MC simulations (Fig. 6). The simulated probability distributions agree well with the corresponding experimentally observed distributions of dwell time, velocity, and run length (Figs. 3 and 4).
Figure 6.
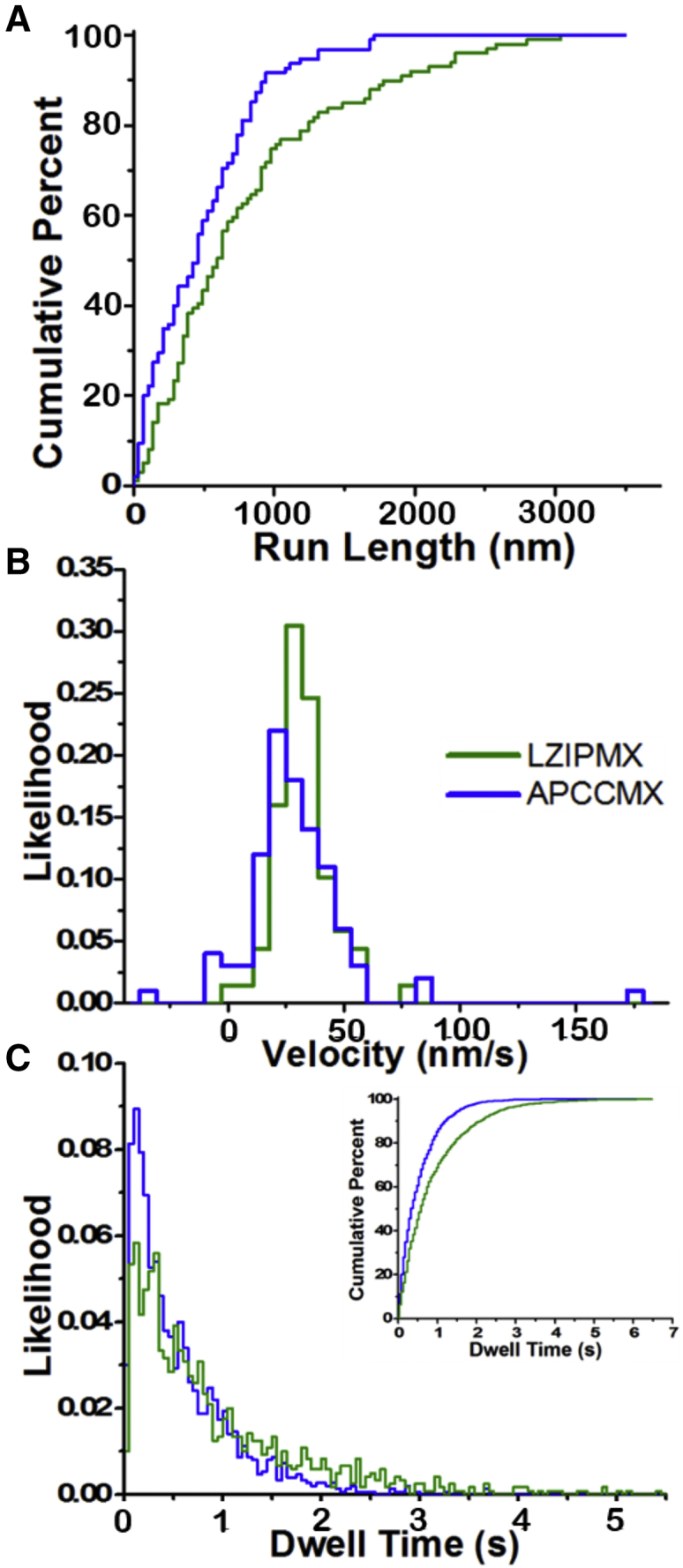
Distributions of run length, velocity, and dwell time at 500 nM MgATP calculated from an MC simulation using the rates (Table 2 from the analytical (Markov) model) for LZIPMX (green) and APCCMX (blue) on single filaments. (A) Cumulative distribution of run lengths simulated for LZIPMX (dark green) and APCCMX (dark blue). (B) Histogram of run velocity simulated for LZIPMX (dark green) and APCCMX (dark blue). (C) Histogram of average dwell times simulated for LZIPMX (dark green) and APCCMX (dark blue). The inset in (C) shows the cumulative distribution of dwell times. The simulation was repeated for n = 100 runs for each construct.
The distributions of 100 simulated run-lengths for APCCMX and LZIPMX are approximately single exponential distributions with mean run lengths of 460 ± 40 and 830 ± 85 nm for APCCMX and LZIPMX, respectively (Fig. 6 A), similar to the experimentally measured values of 342 ± 24 and 714 ± 33 nm, respectively, on single actin filaments.
Simulated velocity distributions are approximately Gaussian, with APCCMX and LZIPMX exhibiting similar mean velocities of 29 ± 2.3 and 31 ± 1.8 nm/s, respectively (Fig. 6 B), similar to the corresponding measured values of 29.0 ± 1.6 and 32.9 ± 2.0 nm/s, respectively, at 500 nM [ATP]. The distribution of velocities is wider for APCCMX compared to LZIPMX. Because of backward stepping, APCCMX exhibits some runs with negative velocity in the MC simulation, predicting that a fraction of molecules make short runs in the direction of actin’s pointed end.
MC simulations of dwell time distributions also approximate the experimental data, showing that APCCMX steps at roughly double the rate of the LZIPMX on single actin filaments exhibiting average dwell times of 0.56 ± 0.01 and 0.91 ± 0.03 s, respectively, and for over 1197 dwells in each case (Fig. 6 C). The distributions are expected to have rising and falling components versus dwell time because of the two sequential transitions (ATP-dependent, kATPon-rear, and ATP-independent, kADPoff-rear) required for each forward step. The main component is an exponential falling phase, but a very brief rising phase is apparent in the first bin of both distributions (Fig. 6 C) because of the ATP-independent transition at ∼20 s−1. The rising phase is observable in the simulations in which there is a 1 ms time step but cannot be captured in the experimental dwell-time distributions (Fig. 4, A and B), which are limited mainly by recording frame rate (50 ms). MC-simulated dwell times are slightly (15–30%) less than experimentally measured dwell times, which can result from two factors: short dwells were missed by the 50 ms camera frame rate in the experiments, and/or some motors exhibited relatively long stalls (Figs. 2, A and B and S4).
Discussion
Myosin X is the only isoform in the myosin superfamily whose heavy chains are thought to associate in an antiparallel coiled coil and to translocate on actin bundles more effectively than on single filaments. Whether these two unusual features are related has been the subject of several past studies without resolution. Our results indicate that antiparallel dimerization of myosin X (APCCMX) reduces processivity on single actin filaments compared to artificially parallel dimers formed by a leucine zipper insert (LZIPMX) or the myosin V coiled coil (MVCCMX), whereas all three of the myosins studied here are strongly processive on bundled actin filaments. Thus, antiparallel orientation of the dimerization motif does seem to confer bundle selectivity on myosin X.
Studying processive motility by single molecule fluorescence techniques requires dilution of the protein into the nM range in order to reduce fluorescence background intensity relative to the individual labeled motors. Homodimerization of myosin X heavy chains, though, exhibits relatively weak affinity (0.6 μM), leading investigators to adopt various strategies to obtain dimers sufficiently stable for in vitro motility and processivity assays. Nagy et al. (18) fused the GNC4 leucine zipper sequence into the native CC domain, yielding a construct that takes short steps (18 nm) on both single actin filaments and fascin-actin bundles and shows much longer run lengths on bundles. This dimerization domain was later shown to be antiparallel (21). Sun et al. (23) replaced the native sequence with the stable coiled coil of myosin V and obtained robust processivity on both bundles and single filaments, the difference from Nagy et al.’s study seemingly being caused by slightly different insertion points. Sun et al. (23) also obtained transient dimerization of an almost-native sequence by utilizing actin decoration in the absence of ATP to bring monomers adjacent to each other and then adding ATP to observe motility of these proximity-mediated dimers. This approach followed from cargo- and actin-assisted dimerization experiments in myosin VI (40). The run lengths of these myosin X transient dimers on single actin filaments at a low (10 μM) MgATP concentration was approximately half of that observed with the forced parallel constructs, either because of dissociation of the myosin monomers or bundle selectivity. Bao et al. (41) added a leucine zipper closely following the native coiled coil to generate a processive myosin X with 36 nm steps and double the processivity on bundled actin relative to single filaments. Sato et al. (42) utilized full-length myosin X stabilized with a leucine zipper at the C-terminus. This construct exhibited a wide distribution in step sizes and a meandering path on fascin-bundled actin, with nearly 50% of steps directed onto adjacent actin filaments. The effectiveness of the leucine zipper at the end of the full-length construct in rigidly stabilizing antiparallel dimerization, however, was not clear. Ropars et al. (22) introduced an elegant approach by using a leucine zipper placed 19 residues beyond the native coiled coil, which structural studies suggested would stabilize the native antiparallel dimerization orientation. This myosin X construct exhibited a wide distribution of step sizes and a significant number of backward steps on both single actin filaments and bundles. It exhibited increased processivity on actin bundles with double the velocity and run length relative to single filaments. The velocity of motility on fascin bundled actin (630 nm/s) was consistent with that measured on filopodia in cells. Ropars et al. suggested that filament switching in a bundle and large steps (up to 54 nm) optimize myosin X for motility on fascin-actin bundles.
Our approach to obtain a stable antiparallel myosin X species was to extend the native dimerization sequence by about half its length by adding two heptads of the native coiled-coil (14 residues) that are critical for antiparallel CC formation to reinforce dimerization by improving the affinity. According to single molecule FRET experiments with short, labeled dimerization sequences (Fig. S2), this approach was successful, although some heterogeneity of the interprobe distances was observed in both the parallel and antiparallel designs. On fascin-bundled actin filaments, we found antiparallel dimerized myosin X (APCCMX) to be strongly processive, exhibiting run lengths similar to the parallel constructs. On single filaments, APCCMX takes shorter average forward steps (averaging 32 nm, composed of 20 and 40 nm steps) and a larger number (27%) of backward steps than on bundles of filaments (38 nm forward step size with 7% backward), consistent with a loss of biochemical gating in APCCMX associated with the shorter step size. Dwell times between steps at 500 nM ATP are shorter for APCCMX than for the parallel constructs, leading to comparable velocities between the three constructs on single actin filaments. Neither parallel myosin X species exhibited appreciable selectivity for bundles versus single actin filaments.
These findings may help explain the discrepancies among the groups studying the processivity and bundle selectivity of myosin X in vitro (18, 23, 24), as the dimerization domains and orientations of the constructs were different. As mentioned, recent works have also reported that antiparallel myosin X takes variable step sizes, including larger ones, on single actin filaments and also many backward steps (22, 42). The work here supports these findings and advances understanding by analysis of a two-headed kinetic model that elucidates the specific alterations of gating mechanisms causing differences in processivity and bundle selectivity. Results from this modeling suggest that APCCMX exhibits reduced gating of ADP release on single actin filaments compared to the parallel constructs, resulting in increased backward stepping that strongly impacts its net velocity.
Antiparallel dimerization extends the distance between the lever arms of myosin X by 5–7 nm and presumably reduces rigidity by insertion of an additional hinge in between the two heads. Compared to parallel myosins, the shorter steps of APCCMX on single filaments are consistent with this higher flexibility enabling reattachment of a detached head to a wider target zone of actin subunits, including ones on the side of the filament (e.g., seven or eight subunits along the filament) as well as straight ahead (11, 13, and 15 subunits). A shorter span for APCCMX should have several effects that follow from each other: 1) it should reduce intermolecular force between two actin-bound heads, thereby 2) reducing gating (slowing of ADP release from the leading head), 3) reducing the dwell time between steps, and 4) diminishing coordination that promotes forward stepping and processive runs. Reduced gating of ADP release on single actin filaments compared to the parallel constructs, as suggested by the kinetic model (Table 1), is responsible for a reduction in velocity and run length with increasing [MgATP] due to an increased number of backward steps.
APCCMX exhibits increased gating of ADP release (lower dissociation rate of ADP from the front actin-bound head) on fascin-bundled actin, leading to robust processive behavior at high ATP concentrations. Fascin bundles, which present an array of appropriately spaced binding sites, may enable antiparallel myosin X to attain a more regular stride length (e.g., 36 nm) and improve processivity by blocking the subunits on the sides of actin. Flexibility increased by the extra hinge would facilitate shifting to neighboring filaments or even adopting a “waddling” type of motility on two adjacent filaments. We showed earlier that myosin X follows a left-handed helical path on unobstructed filaments and bundles (23), and sideways motions and short steps were detected in Sato et al. (42) and Ropars et al. (22), all implying filament switching. These considerations suggest that antiparallel dimerized myosins are not optimized for motility on single actin filaments because of the availability of actin subunit binding sites that disrupt coordination between the heads. The larger and more consistent strides on actin bundles enable the molecule to effectively gate its biochemical reaction rates and maintain processivity. These features are appropriate for myosin X’s role in establishing and maintaining actin bundles, facilitating production of filopodia, and targeting cargoes to filopodial tips along the bundled actin.
Acknowledgments
We thank Drs. Russell J. Composto and Jody A. Dantzig and Mr. Jeffrey Hallock for useful comments. We thank Dr. Dan Safer for fascin and Ms. Sanjula Wickramasinghe for help with the custom peptides.
The work was supported by National Institutes of Health grants R01-GM086352 and R35-GM118139 to Y.E.G. and T32-HL007954 to M.A.C., University of Texas STARS PLUS award to M.I., and National Science Foundation grant DMR32802 to the Nano/Bio Interface Center at the University of Pennsylvania.
Editor: David Warshaw.
Footnotes
Supporting Materials and Methods, eight figures, and one table are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(18)30200-5.
Supporting Citations
References (43, 44, 45) appear in the Supporting Material.
Supporting Material
References
- 1.Courson D.S., Cheney R.E. Myosin-X and disease. Exp. Cell Res. 2015;334:10–15. doi: 10.1016/j.yexcr.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sousa A.D., Cheney R.E. Myosin-X: a molecular motor at the cell’s fingertips. Trends Cell Biol. 2005;15:533–539. doi: 10.1016/j.tcb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Sandquist J.C., Larson M.E., Hine K.J. Myosin-10 independently influences mitotic spindle structure and mitotic progression. Cytoskeleton (Hoboken) 2016;73:351–364. doi: 10.1002/cm.21311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwano S., Satou A., Toyoshima F. PCTK1 regulates integrin-dependent spindle orientation via protein kinase A regulatory subunit KAP0 and myosin X. Mol. Cell. Biol. 2015;35:1197–1208. doi: 10.1128/MCB.01017-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohil A.B., Robertson B.W., Cheney R.E. Myosin-X is a molecular motor that functions in filopodia formation. Proc. Natl. Acad. Sci. USA. 2006;103:12411–12416. doi: 10.1073/pnas.0602443103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg J.S., Cheney R.E. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat. Cell Biol. 2002;4:246–250. doi: 10.1038/ncb762. [DOI] [PubMed] [Google Scholar]
- 7.Tokuo H., Mabuchi K., Ikebe M. The motor activity of myosin-X promotes actin fiber convergence at the cell periphery to initiate filopodia formation. J. Cell Biol. 2007;179:229–238. doi: 10.1083/jcb.200703178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe T.M., Tokuo H., Ikebe M. Myosin-X induces filopodia by multiple elongation mechanism. J. Biol. Chem. 2010;285:19605–19614. doi: 10.1074/jbc.M109.093864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He K., Sakai T., Ikebe M. Myosin X is recruited to nascent focal adhesions at the leading edge and induces multi-cycle filopodial elongation. Sci. Rep. 2017;7:13685. doi: 10.1038/s41598-017-06147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H., Berg J.S., Strömblad S. Myosin-X provides a motor-based link between integrins and the cytoskeleton. Nat. Cell Biol. 2004;6:523–531. doi: 10.1038/ncb1136. [DOI] [PubMed] [Google Scholar]
- 11.Umeki N., Jung H.S., Ikebe M. Phospholipid-dependent regulation of the motor activity of myosin X. Nat. Struct. Mol. Biol. 2011;18:783–788. doi: 10.1038/nsmb.2065. [DOI] [PubMed] [Google Scholar]
- 12.Arjonen A., Kaukonen R., Ivaska J. Filopodia and adhesion in cancer cell motility. Cell Adhes. Migr. 2011;5:421–430. doi: 10.4161/cam.5.5.17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao R., Chen J., Zhu X. Elevated expression of myosin X in tumours contributes to breast cancer aggressiveness and metastasis. Br. J. Cancer. 2014;111:539–550. doi: 10.1038/bjc.2014.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kureishy N., Sapountzi V., Adams J.C. Fascins, and their roles in cell structure and function. BioEssays. 2002;24:350–361. doi: 10.1002/bies.10070. [DOI] [PubMed] [Google Scholar]
- 15.Vignjevic D., Kojima S., Borisy G.G. Role of fascin in filopodial protrusion. J. Cell Biol. 2006;174:863–875. doi: 10.1083/jcb.200603013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikawa R., Sakamoto T., Kohama K. Polarized actin bundles formed by human fascin-1: their sliding and disassembly on myosin II and myosin V in vitro. J. Neurochem. 2003;87:676–685. doi: 10.1046/j.1471-4159.2003.02058.x. [DOI] [PubMed] [Google Scholar]
- 17.Tokuo H., Ikebe M. Myosin X transports Mena/VASP to the tip of filopodia. Biochem. Biophys. Res. Commun. 2004;319:214–220. doi: 10.1016/j.bbrc.2004.04.167. [DOI] [PubMed] [Google Scholar]
- 18.Nagy S., Ricca B.L., Rock R.S. A myosin motor that selects bundled actin for motility. Proc. Natl. Acad. Sci. USA. 2008;105:9616–9620. doi: 10.1073/pnas.0802592105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagy S., Rock R.S. Structured post-IQ domain governs selectivity of myosin X for fascin-actin bundles. J. Biol. Chem. 2010;285:26608–26617. doi: 10.1074/jbc.M110.104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ricca B.L., Rock R.S. The stepping pattern of myosin X is adapted for processive motility on bundled actin. Biophys. J. 2010;99:1818–1826. doi: 10.1016/j.bpj.2010.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vavra K.C., Xia Y., Rock R.S. Competition between coiled-coil structures and the impact on myosin-10 bundle selection. Biophys. J. 2016;110:2517–2527. doi: 10.1016/j.bpj.2016.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ropars V., Yang Z., Houdusse A. The myosin X motor is optimized for movement on actin bundles. Nat. Commun. 2016;7:12456. doi: 10.1038/ncomms12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Y., Sato O., Goldman Y.E. Single-molecule stepping and structural dynamics of myosin X. Nat. Struct. Mol. Biol. 2010;17:485–491. doi: 10.1038/nsmb.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takagi Y., Farrow R.E., Molloy J.E. Myosin-10 produces its power-stroke in two phases and moves processively along a single actin filament under low load. Proc. Natl. Acad. Sci. USA. 2014;111:E1833–E1842. doi: 10.1073/pnas.1320122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Q., Ye F., Zhang M. Antiparallel coiled-coil-mediated dimerization of myosin X. Proc. Natl. Acad. Sci. USA. 2012;109:17388–17393. doi: 10.1073/pnas.1208642109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pardee J.D., Spudich J.A. Purification of muscle actin. Methods Cell Biol. 1982;24:271–289. doi: 10.1016/s0091-679x(08)60661-5. [DOI] [PubMed] [Google Scholar]
- 27.Ono S., Yamakita Y., Matsumura F. Identification of an actin binding region and a protein kinase C phosphorylation site on human fascin. J. Biol. Chem. 1997;272:2527–2533. doi: 10.1074/jbc.272.4.2527. [DOI] [PubMed] [Google Scholar]
- 28.Sato O., Komatsu S., Ikebe M. Human myosin VIIa is a very slow processive motor protein on various cellular actin structures. J. Biol. Chem. 2017;292:10950–10960. doi: 10.1074/jbc.M116.765966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forkey J.N., Quinlan M.E., Goldman Y.E. Three-dimensional structural dynamics of myosin V by single-molecule fluorescence polarization. Nature. 2003;422:399–404. doi: 10.1038/nature01529. [DOI] [PubMed] [Google Scholar]
- 30.Meeusen R.L., Cande W.Z. N-ethylmaleimide-modified heavy meromyosin. A probe for actomyosin interactions. J. Cell Biol. 1979;82:57–65. doi: 10.1083/jcb.82.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattheyses A.L., Shaw K., Axelrod D. Effective elimination of laser interference fringing in fluorescence microscopy by spinning azimuthal incidence angle. Microsc. Res. Tech. 2006;69:642–647. doi: 10.1002/jemt.20334. [DOI] [PubMed] [Google Scholar]
- 32.Ruhnow F., Zwicker D., Diez S. Tracking single particles and elongated filaments with nanometer precision. Biophys. J. 2011;100:2820–2828. doi: 10.1016/j.bpj.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerssemakers J.W., Munteanu E.L., Dogterom M. Assembly dynamics of microtubules at molecular resolution. Nature. 2006;442:709–712. doi: 10.1038/nature04928. [DOI] [PubMed] [Google Scholar]
- 34.Woody M.S., Lewis J.H., Ostap E.M. MEMLET: an easy-to-use tool for data fitting and model comparison using maximum-likelihood estimation. Biophys. J. 2016;111:273–282. doi: 10.1016/j.bpj.2016.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elting M.W., Bryant Z., Spudich J.A. Detailed tuning of structure and intramolecular communication are dispensable for processive motion of myosin VI. Biophys. J. 2011;100:430–439. doi: 10.1016/j.bpj.2010.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Homma K., Ikebe M. Myosin X is a high duty ratio motor. J. Biol. Chem. 2005;280:29381–29391. doi: 10.1074/jbc.M504779200. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh P., Ghosh S., Das S.K. A markov model based analysis of stochastic biochemical systems. Proceedings of LSS Computational Systems Bioinformatics Conference (CSB). 2007;6:121–132. [PubMed] [Google Scholar]
- 38.De La Cruz E.M., Wells A.L., Sweeney H.L. The kinetic mechanism of myosin V. Proc. Natl. Acad. Sci. USA. 1999;96:13726–13731. doi: 10.1073/pnas.96.24.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor E.W. Kinetic studies on the association and dissociation of myosin subfragment 1 and actin. J. Biol. Chem. 1991;266:294–302. [PubMed] [Google Scholar]
- 40.Park H., Ramamurthy B., Sweeney H.L. Full-length myosin VI dimerizes and moves processively along actin filaments upon monomer clustering. Mol. Cell. 2006;21:331–336. doi: 10.1016/j.molcel.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 41.Bao J., Huck D., Sakamoto T. Actin structure-dependent stepping of myosin 5a and 10 during processive movement. PLoS One. 2013;8:e74936. doi: 10.1371/journal.pone.0074936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato O., Jung H.S., Ikebe M. Activated full-length myosin-X moves processively on filopodia with large steps toward diverse two-dimensional directions. Sci. Rep. 2017;7:44237–44249. doi: 10.1038/srep44237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jamiolkowski R.M., Chen C., Cooperman B.S., Goldman Y.E. tRNA fluctuations observed on stalled ribosomes are suppressed during ongoing protein synthesis. Biophys. Journ. 2017;113:2326–2335. doi: 10.1016/j.bpj.2017.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roy R., Hohng S., Ha T. A practical guide to single molecule FRET. Nat. Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen C., Stevens B., Kaur J., Cabral D., Liu H., Wang Y., Zhang H., Rosenblum G., Smilansky Z., Goldman Y.E., Cooperman B.S. Single-molecule fluorescence measurements of ribosomal translocation dynamics. Mol. Cell. 2011;42:367–377. doi: 10.1016/j.molcel.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.