

Abstract
Objective
The purpose of this narrative literature review is to discuss the literature regarding the potential role that cytokines play in degenerative disk disease.
Methods
The inclusion criteria were studies that used inflammatory mediators in advancing disk disease processes. Research studies were limited to the last 3 decades that had free full-text available online in English. Exclusion criteria were review articles and articles pertaining to temporomandibular joints and other joints of the body other than the intervertebral disk. The following databases were searched: PubMed, EBSCOhost, and Google Scholar through March 13, 2017.
Results
A total of 82 studies were included in this review. The papers were reviewed for complex mechanisms behind the degenerative cascade, emphasizing the role of proinflammatory cytokines, which may be instrumental in processes of inflammation, neurologic pain, and disk degeneration. Interleukin-1β and tumor necrosis factor α were among the more notable cytokines involved in this cascade. Because monocyte chemoattractant protein-1 stimulates and activates macrophages in the event of infiltration, additional proinflammatory cytokines are released to act on molecules to promote blood and nerve ingrowth, resulting in pain signaling and tissue degradation. Excessive inflammation and/or tissue damage initiates a pathologic imbalance between anabolic and catabolic processes.
Conclusions
This literature review describes how inflammatory and biochemical changes may trigger disk degeneration. Proinflammatory cytokines stimulate microvascular blood and nerve ingrowth, resulting in pain signaling and tissue degradation. This may sensitize a person to chemical and/or mechanical stimuli, contributing to severe low back pain.
Key Indexing Terms: Cytokines, Intervertebral Disk, Intervertebral Disk Degeneration
Introduction
Intervertebral disk (IVD) degeneration (IDD) is an irreversible and progressive process with disabling potential as a result of pain. It is implicated to play a clinical role in the presentation of low back pain (LBP) and lumbar radiculopathy secondary to IVD displacement.1 Age, mechanical pressures, genetics, inflammatory and biochemical changes can all act as triggering agents in this condition.1, 2, 3, 4
Degenerative disorder of the disk is characterized generally as enhanced matrix degradation, angiogenesis/neovascularization, nerve ingrowth, and increased expression of catabolic cytokines, resulting in anatomic and biochemical disk changes.1, 2, 5, 6, 7 The condition may include disk desiccation, decreased disk height, disk bulging, annular fissures, osteophytes, or end plate sclerosis.8 The disk’s nucleus pulposus (NP) is filled with proteoglycans as the surrounding annulus fibrosis (AF) is collagenous. The aged disk has less water, supposedly as a result of loss of negatively charged proteoglycans, consequently affecting one’s ability to bear loads.2, 9 The disk is largely avascular and aneural besides the outer third of the annulus. However, degeneration accompanies the growth of blood vessels and nerves into the inner layers of annulus and nucleus.5, 6, 7
It is now well established that in chronic inflammation the disk experiences extracellular matrix (ECM) loss and degradation. Because the inflammatory cascade involves neovascularization and nerve ingrowth, macrophages are seen as the most populous of any inflammatory cell.10, 11, 12, 13, 14 Once macrophages are activated, they secrete cytokines and disk-degenerative compounds and may even spread to nearby noninjured disk tissue.10, 13 Nonresolved neuroinflammation may play a key role in the development of chronic progressive pain.15 The purpose of this narrative literature review is to discuss the literature regarding the potential role that cytokines play in degenerative disk disease.
Methods
This was a narrative overview. A search was performed using electronic databases: PubMed, Google Scholar, and EBSCOhost generated articles relevant to cytokines and their role in degeneration. Studies were limited to those published in the last 3 decades that had free full-text available online in English. The final selection of studies ranged from 1994 through March 13, 2017. Search terms included “intervertebral disc degeneration,” “cytokines,” and “inflammatory mediators.” EBSCOhost generated 6423 articles, PubMed generated 4519, and Google Scholar generated about 29500. Articles were subcategorized into human participants and animal subjects and methods were further categorized into in vitro and in vivo studies. The reference lists from the included articles were considered as well. Exclusion criteria included articles pertaining to temporomandibular joints and other joints of the body other than the IVD. Eighty-two articles were finally included in the review.
Results
IVD Degeneration
Healthy ECM undergoes a normal cycle of tissue degradation and synthesis. Nutritional compromise, repetitive loading, or natural aging may lead to a disk wearing down as normal matrix turnover loses its remodeling capability, and structural proteins degrade in excess.1 In turn, the disk loses hydration and its structural integrity. Disk degeneration likely involves biochemical and mechanical cell stimulation that activates this matrix destructive pathway.2 Fig 1 provides a representation of the disk-degenerative cascade.
Fig 1.
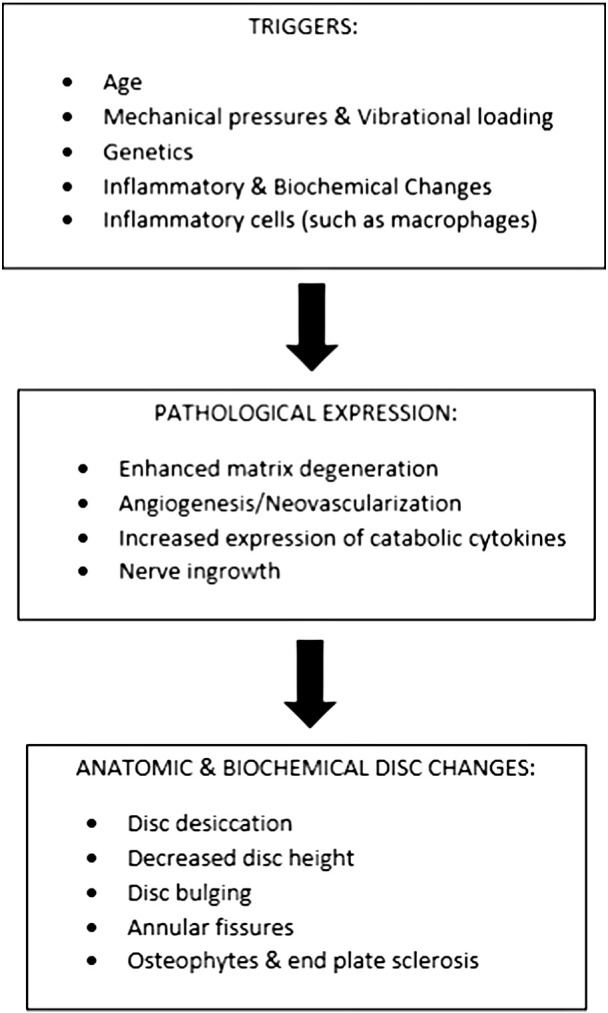
Degenerative cascade.
Inflammation may create irreversible biomechanical and structural changes16 because inflammatory stimuli have the ability to increase both hydraulic permeability and cell size of the NP. Even after removing the stimuli, these changes may persist, suggesting that the inflammatory response may irreversibly disrupt the disk’s volume.
The IVD has no nerve or blood supply except for the outer third of the annulus. However, with IDD, microvascular blood vessels and nerve ingrowth may extend into the nucleus.17 Chronically degenerated disks display increased vascularization,18 and such blood vessels are needed to supply activated phagocytes that further release proinflammatory mediators. Lumbar disks of cadavers in their 30s tend to be avascular, whereas those in their 40s and beyond begin to exhibit a new blood supply.19 Vascular granulation tissue also tends to form along a torn annular fissure, releasing inflammatory mediators such as histamine, heparin, tumor necrosis factor α (TNF-α), proteases, interleukins (ILs), and chemoattractants.8
The cascade of IDD involves macrophage infiltration with subsequent cytokine secretions.8 The largest amounts of cytokines are found in the AF, suggesting this to be the predominant site of the degenerative inflammatory response.18 Although most of the precursor to the cytokine TNF-α is found in the annulus, activated TNF-α is able to reach the nucleus via diffusion. Chronic degenerated disk tissues may have greater amounts of cytokines.
Overall, disk degeneration involves a cascade of events involving cytokines and the upregulation of vascular and nerve growth to areas which previously were largely not innervated by a blood or nerve supply.8 This may have implications in subsequent matrix degradation and pain processing mechanisms. Not all cytokines are stimulated by the same mechanism.20 Fig 2 illustrates the complex interrelations of individual cytokines and their influence on the degenerative process.
Fig 2.

Cytokine interrelationships, indicating stimulating or regulating relationships. ADAM10, a disintegrin and metalloproteinase domain 10; BDF, brain-derived neurotrophic factor; CCL, C-C motif ligand; CNS, central nervous system; COX, cyclooxygenase; Csf1, colony stimulating factor 1; DR5, death receptor 5; ECM, extracellular matrix; FasL, Fas ligand; IFN, interferon; IGF, insulin-like growth factor; IL, interleukin; MCP, monocyte chemoattractant protein; MMP, metalloproteinase; NGF, nerve growth factor; NO, nitric oxide; PGE, prostaglandin E; PLA2, phospholipase A2; RANTES, regulated upon activation, normal T cell expressed and secreted; TGF, transforming growth factor; TIMP, tissue inhibitor of metalloproteinase; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand; TSLP, thymic stromal lymphopoietin; VEGF, vascular endothelial growth factor.
IVD Displacement
Often the degenerative process manifests into IVD displacement,1 where a disk ruptures through the posterior longitudinal ligament, exposing NP cells to the vascular system of the epidural space.5 The chemoattractive properties of the nucleus draw peripheral blood mononuclear cells to infiltrate the border of the extruded disk in an autoimmune fashion.1, 5 These monocytes may consequently differentiate into macrophages.21 They also contain inflammatory mediators contributing to inflammation (involving granulation tissue) and disk neovascularization, both leading to phagocytosis and resorption of the disk, serving a beneficial role in removing nerve compression.5, 6
Inflammation involves the formation of granulation tissue consisting of macrophages and T-lymphocytes.1 Inflammatory cytokines are then produced and consequently induce the expression of matrix-degrading proteins. Although granulation tissue is usually associated with chronic inflammation,22 disk displacement may also produce such granulation tissue, consisting of small round cells, fibroblasts, and neovascularization.7 All patients with disk displacement may have some degree of disk degeneration,23 and both conditions have been reported to have substantial macrophage infiltration.24
Macrophages
Most extruded disk tissue naturally decreases in size and sometimes disappears completely. Macrophages are instrumental in this resorption process, leading to the presence of any inflammatory cell.10, 11, 12, 13, 14 They most likely invade disk tissue from new blood vessels formed at the disk’s periphery and may spread to nearby noninjured disk tissues.10, 13 Once activated, macrophages secrete proinflammatory cytokines and disk-degenerative enzymes.5, 12, 25 Macrophages have also been reported to produce chemotactic cytokines as well, recruiting further monocytes to the area.26 Cytokines may stimulate the release of monocyte chemoattractant protein-1 (MCP-1), upregulating additional macrophage infiltration.25, 26, 27, 28 Monocyte chemoattractant protein-1 is involved with initiating inflammation and is suggested to play an important role in the early resorption process of the nucleus.26, 29
Tumor Necrosis Factor α
The inflammatory cytokine TNF-α stimulates the release of vascular endothelial growth factor (VEGF), which is involved in new blood vessel formation. Such new vascularization brings about macrophage infiltration, in turn, generating further cytokines and disk-degrading enzymes, leading to disk degeneration and resorption.27, 28, 30 Tumor necrosis factor-α shows up early after a disk herniates.31 Its levels might then fluctuate throughout the inflammatory process. Disks may produce TNF-α and IL-1β on the first day after IVD displacement, stimulating the production of MCP-1 2 days later.32 Monocyte chemoattractant protein-1 then recruits and activates macrophages, which further stimulate MCP-1 and trigger phagocytosis, the release of matrix metalloproteases (MMPs), and the degradation of ECM. Stimulating the release of TNF-α from macrophages has accelerated herniated disk resorption.27
ADAM10 and Fas Ligand
Fas ligand (FasL) is a member of the TNF family. This molecule may protect the disk from autoimmune attack by maintaining the disk’s avascular nature. This ligand binds with Fas located on a target immune cell and induces vascular endothelial cell apoptosis, protecting against progressive disk degeneration.33 In the nucleus, ADAM10 (a disintegrin and metalloproteinase domain 10) may regulate the expression of FasL.34 Because FasL may stimulate the production of proinflammatory cytokines, reducing ADAM10 may reduce the production of inflammatory cytokines. Exposing spinal dorsal nerve roots to the disk nucleus may upregulate FasL levels, which may induce apoptosis to reestablish homeostasis in the dorsal root ganglion.35 However, greater levels of FasL may exist in normal NP cells compared with degenerated ones.36
Interleukins
Interleukin-1β may be the key cytokine responsible for disk degeneration.17 It stimulates angiogenesis and nerve innervations in the human IVD via its upregulating effect on VEGF, nerve growth factor, and brain-derived neurotrophic factor. Interleukin-1β is thought to play a role in the degenerative cascade by inhibiting ECM synthesis, stimulating matrix-degrading enzymes and furthering cytokine production, as well as indirectly speeding up apoptosis rates.17, 37, 38, 39 Interleukin-1 may also increase the gene expression of proinflammatory proteins that can enhance disk degeneration.2 Interleukin-1(α and β) and its inhibitor, IL-1Ra, are in balance in the healthy disk.40 The degenerative disk, however, has elevated IL-1(α and β), whereas IL-1Ra is not changed, resulting in increased cell expression of matrix-degrading enzymes. Interleukin-1Ra may almost completely eliminate matrix-degrading activity.
Tumor necrosis factor-α may also be upregulated in disk degeneration, but its predominant receptor, TNF-R1, is reduced, leading to reduced bioactivity of TNF-α. This suggests that IL-1 may play a larger role in disk degeneration and therefore may serve as a better target for therapeutic intervention.
In addition to being released from the disk itself, IL-1 may also be a product of mechanical, or vibrational, loading or from inflammatory cells such as macrophages.38 The potential inflammation, neurologic pain, and disk degeneration resulting from IL-1β indicates the clinical importance of controlling the levels of this cytokine.
Interleukin-6 and its receptor, IL-6R, may play important parts in acute inflammation. In their protective roles, they may stimulate tissue inhibitor of metalloproteinase (TIMP), which inhibits excessive breakdown of ECM.5, 41 They may also stimulate vascular epithelial growth factor, promoting new blood vessel formation.
Substance P
The neuropeptide substance P can stimulate macrophages to release IL-1α and TNF-α.18 Higher levels of substance P found in chronic disk degeneration could explain the greater levels of these 2 other cytokines. Substance P has upregulated other inflammatory cytokines as well, in both the annulus and nucleus.42
Nitric Oxide
In herniated lumbar disks, nitric oxide (NO) may be produced mainly by the macrophages in granulation tissue surrounding the herniated disk material.7 NO synthesis may be stimulated by TNF-α and IL-1β, lipopolysaccharide, or interferon-γ.24, 7 It has been known to help maintain blood pressure by dilating vessels and help kill foreign invaders in the immune response. Nitric oxide is proinflammatory in the sense of being a vasodilator and increasing vascular leakiness, suppressing proteoglycans, and contributing to neuropathic pain. Paradoxically, it can be anti-inflammatory in its ability to inhibit prostaglandin E2, thromboxane, and IL-6 synthesis.43, 9
Interferon-γ
Interferon-γ is a proinflammatory cytokine produced by multiple cell types that may activate microglia and other cells to upregulate further cytokines and contribute to macrophage recruitment.24, 44
Transforming Growth Factor-β
Although the more notable role of transforming growth factor (TGF)-β is to encourage matrix production and limit ECM degradation, it may serve as a contributing cytokine in the downregulation of leukocyte formation during disk displacement.45 Tumor necrosis factor-α and IL-1 may upregulate this cytokine. Transforming growth factor-β also contributes to fibrosis and angiogenesis, proteoglycan stimulation, and decreased active MMP-2 levels.8 The role of TGF-β1 in ligamentum flavum hypertrophy is discussed later.
Matrix Breakdown and Disk Resorption
Disk resorption results when disk-destroying enzyme activity outweighs matrix protection.5 Matrix metalloproteases are a set of zinc-dependent proteolytic enzymes contributing to the breakdown of disk matrix components, especially collagen and proteoglycans. As described earlier, the NP is filled with proteoglycans, whereas the enclosing annulus fibrosus is collagenous.38 Greater amounts of MMPs and inflammatory mediators are apparent in degenerative disks.2 Herniated lumbar disk tissue also have high levels of MMPs, which degrade core proteoglycans in cartilage. This increased enzyme production may be a natural response to remodel and help resorb the herniated disk.9
Although other enzymes may be involved in degradation of displaced IVDs, MMP-1 and MMP-3 are thought to be 2 major MMPs involved in the process. Nucleus pulposus cells produce MMP-1 and MMP-3, which may explain the reduced herniated disk volume accompanying spontaneous healing (not requiring surgery) as well as reduced pain and neurologic symptoms.5 Macrophages, once activated, generate inflammatory cytokines such as IL-1β and TNF-α, potent MMP inducers.
The interaction between peripheral blood mononuclear cells and displaced disk tissue causes an upregulation of MMP-3 and the formation of inflammatory granulation tissue.5 Proinflammatory cytokines (ie, TNF-α, IL-1) stimulate the production of MMP-3, leading to disk degeneration and resorption.18, 27 The role of MMP-3 in cartilage degradation, also occurring in arthritic conditions, serves in the spontaneous healing of displaced tissue and removes nerve compression.5
Both vascular epithelial growth factor and TNF-α upregulate urokinase-type plasminogen activator, which transforms plasminogen into plasmin, a matrix degrading enzyme, which activates certain MMPs.30 Tumor necrosis factor-α may also directly stimulate the intracellular production of latent MMPs, but these proteinases may need extracellular stimulation before being able to degrade substances.
Cell Aging and Programmed Cell Death
Cell senescence represents the period of biological aging where normal cells stop dividing. Cells can reach senescence prematurely via stresses induced by reactive oxygen species, mechanical load, or cytokines, implying that these stressors may prematurely place a cell at growth arrest.46 Apoptosis is a term used to describe programmed cell death, a normal component of cellular biology. Excessive apoptosis may lead to cellular atrophy, whereas inadequate apoptosis may lead to uncontrolled cellular proliferation. Fas-L, TNF-α, and TRAIL (TNF-α-related apoptosis inducing ligand) all exhibit potent cytotoxic activity and induce apoptosis in susceptible cells.47 Death receptor 5 (TRAIL receptor 2) is a receptor involved in apoptosis. It exists in both normal and herniated disks but in greater amounts in the latter as a result of its hypoxic, hypoglycemic, and acidic environment, implying that excessive apoptosis may result from proinflammatory cytokine activity after disk injury.
Implication in Pain Processing
Diskogenic Pain
Mechanical factors, such as disk displacement, may be an initial trigger for acute LBP.43 However, LBP appears to be multifactorial, depending on both mechanical and biochemical mechanisms. Not all degenerative disks lead to painful symptoms. In disk degeneration, pain is believed to result from fissures extending from the nucleus to the outer annulus, exposing nerve endings to enzymes and degradation substances.8 Degenerative disks that cause LBP contain more nociceptive nerve endings in the endplate and nucleus.20 After disk degeneration, nerve growth factor and brain-derived neurotrophic factor may promote nerve ingrowth, contributing to pain.17 Meanwhile, VEGF may induce new blood vessel ingrowth. Injury to the annulus, such as tearing its outer layers, has potential to create diskogenic LBP. Cytokines such as IL-6, produced by cells in the outer annulus, may produce pain at such free nerve endings, contributing to the development of diskogenic pain.
Those with disk degeneration have greater inflammation and vascular penetration compared with those with disk displacement.8 The ingrowth of unmyelinated nerve fibers, which are very sensitive to chemical and/or mechanical stimuli, may explain why these patients tend to have more severe LBP than those with disk displacement. Those with only LBP have greater amounts of sensory nerves. It is unknown exactly why degenerated disks produce more inflammatory mediators. Degenerative disks may release chemotactic substances that aid in the ingrowth of nerves that may produce LBP. Figure 3 illustrates the events resulting in pain and tissue destruction.
Fig 3.

IVD degeneration cascade. ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; ECM, extracellular matrix; IVD, intervertebral disk; MCP, monocyte chemoattractant protein; MMP, metalloproteinase; mRNA, microRNA; NP, nucleus pulposus; PLL, posterior longitudinal ligament; TGF, transforming growth factor.
Radicular Pain
In disk displacement, pain results from the chemical irritation and compression of spinal nerve roots.8, 31 Lumbar disk displacement involves chronic local inflammation.15 A lumbar displacement that creates pain radiating into one’s leg along the distribution of the sciatic nerve is known as sciatica. Although a protruded disk into the spinal canal may compress a nerve, irritate it, and create sciatic pain, the extent of radicular symptoms does not depend exclusively on the size of a herniated disk.12 Merely compressing a noninflamed nerve may result in motor and sensory deficits without pain.48, 49 Mechanical stimulation preceded by exposure to the NP, however, may lead to sciatic pain, reinforcing the notion that a nerve must be inflamed to experience pain.
Exposing spinal dorsal nerve roots to the nucleus may significantly boost a C-fiber response and increase levels of TNF-α, IL-1β, colony stimulating factor 1, and FasL, which are thought to contribute to central sensitization.35 C-fibers have unmyelinated axons and are more sensitive to inflammation than are Aδ fibers. The latter are, however, myelinated and are more sensitive to compression.31 Even small amounts of TNF-α may be adequate to produce radicular pain, either directly by stimulating the nerve root or indirectly via nucleus-induced nerve root injury.45 Applying TNF-α along the sciatic nerve may stimulate more C-fiber firing compared with Aδ firing.31
Inflammatory mediators may directly affect nerve endings that exist in the outer annulus and longitudinal ligaments as well as the nearby dorsal root ganglion and spinal nerve root. In disk displacement, the NP increases its expression of cytokines, MMPs, and other inflammatory mediators.3 The resulting inflammation may promote axonal growth of dorsal root ganglion neurons and cause more nerve fibers to penetrate the disk.
Damaged neurons in peripheral nerve injuries release MCP-1, which activates microglia, the local macrophages of the central nervous system.35 Also, enhanced expression of neurotrophic factors (ie, nerve growth factor and brain-derived neurotrophic factor) in the dorsal root ganglion regulate dorsal horn neurons and may contribute to neuropathic pain.29 In the central nervous system, enhanced colony stimulating factor 1 levels may also activate microglia.
Epidural fat encloses the nerve root and can secrete adipokines. In patients with sciatica, TNF-α levels are significantly higher in epidural fat.50 Ex vivo results predict epidural fat itself to produce proinflammatory cytokines, such as TNF-α.
Other chemical irritants besides cytokines may play a role in pain and inflammation, including immunoglobulins (eg, immunoglobulin G), hydrogen, NO and enzymes (eg, phospholipase A2 and MMPs), fibroblast growth factor, leukotrienes, thromboxanes, and prostaglandins.48 The activation of cyclooxygenase-2, whose arachidonic cascade creates prostaglandin E2, leads to the ectopic firing of nerve roots, with consequent pain and inflammation.27, 31
Chronic Pain
Acute pain relief may be due to neural decompression, whereas persistent pain progression could be representative of an unresolved neuroinflammatory process.15 Granulation and inflammatory tissue proliferates as the duration of radicular pain increases.41 The longer the nucleus has contact with the dorsal root ganglion, the greater the potential for developing chronic pain becomes.48 Differing levels of inflammatory responses may account for the degree and length of disability with those experiencing radiculopathy.38 The longer duration the nucleus physically interacts with its surrounding environment, the more inflammatory mediators are produced.31, 20 Once a chronic pain mechanism is triggered, removing the source may not end the pain cycle. Thus, a longer inflammatory process may lead to pain chronification. Currently, it is unknown what the specific timeframe is for a cycle to become chronic.
Contributing and Complicating Factors
The main function of the IVD is to bear and distribute load. In normal loading, the nucleus experiences hydrostatic and shear stress, whereas the annulus experiences tensile strain. Fluid movement within the disk creates shear stress, whereas spinal movement creates compression, tension, and shear stress.2 Aging and degeneration can create adverse strain where the extracellular matrix integrity is compromised and mechanical forces are altered. Excessive loading creates a catabolic shift within disk tissue where protease activity increases.2 High mechanical strain during degeneration could accelerate degeneration and promote neuronal infiltration, thereby contributing to diskogenic pain.4 Excessive mechanical loading can trigger inflammatory and cytokine responses, potentially contributing to spinal disk degeneration and LBP. Asymmetric disk loading can upregulate the production of cytokine mediators, lowering the threshold response to mechanical load. Exposing disks to IL-1β may render AF cells more vulnerable to injury from excessive load.
Damage, especially to the outer annulus, may predispose the disk to prolapse of the nucleus.3 The onset of disk damage seems to be the cleavage of the junction where the endplates of the disk and vertebra meet. Degenerative changes may be more pronounced in displaced disks than with spondylolytic disks.3
Hypertrophied ligamentum flavum, which covers the posterior lateral portion of the lumbar spinal canal, can lead to lumbar spine stenosis, encroaching the canal and compressing nerve roots, or cauda equina syndrome.51 Transforming growth factor-β1 may be responsible for pathologic tissue fibrosis in diseased states. Fibroblasts stimulate a higher expression of TGF-β1, which may contribute to the hypertrophy of ligamentum flavum in lumbar spinal stenosis.51 Serum elevations of TGF-β1, TIMP-1, and TIMP-2 may all be involved with ligamentum flavum hypertrophy.52 TIMPs may also play a role in fibrotic diseases by inhibiting MMP action, impairing matrix degradation. Inflammatory granulation tissue may also break down the collagenous component of the posterior longitudinal ligament, contributing to structural weakness.53
Discussion
There is a complex interworking among proinflammatory mediators, collectively participating in the pathologic expressions behind LBP. Inflammation, blood and nerve ingrowth, tissue breakdown, and pain processing all have cytokine involvement. The secretion of cytokines and disk degenerative enzymes may result in disk resorption, degeneration, and neuropathic pain.
A nerve may need to be inflamed to experience pain, because nerve compression alone may simply result in sensory and motor deficits. Coupled with nerve sensitization, some cytokines are implicated in generating painful IDD. Cytokine secretion and expression may rely on independent pathways from one another. By removing or inhibiting certain influential players in a cascade, consequent cytokine production may be impeded.
Limitations
This was a narrative literature review; therefore, conclusions cannot necessarily be made about cause and effect. It is possible that some highly relevant studies were missed. To identify a relationship, additional studies are needed. Further research may focus on the clinical context of reducing LBP and attenuating disk degeneration by means of controlling cytokines. Because much of the research is limited to the laboratory setting, these findings may not translate to caring for a patient who is in need of effective treatment.
Conclusion
Cytokines may have an important role in the biochemical mechanisms related to IDD.
Funding Sources and Conflicts of Interest
No funding sources or conflicts of interest were reported for this study.
Contributorship Information
Concept development (provided idea for the research): C.M.D.
Design (planned the methods to generate the results): C.M.D.
Supervision (provided oversight, responsible for organization and implementation, writing of the manuscript): C.M.D.
Data collection/processing (responsible for experiments, patient management, organization, or reporting data): C.M.D.
Analysis/interpretation (responsible for statistical analysis, evaluation, and presentation of the results): C.M.D.
Literature search (performed the literature search): C.M.D.
Writing (responsible for writing a substantive part of the manuscript): C.M.D.
Critical review (revised manuscript for intellectual content, this does not relate to spelling and grammar checking): C.M.D.
Practical Applications
-
•
This paper addresses the consequences from directly and indirectly manipulating individual and groups of inflammatory mediators, external factors that may influence cytokine expression and inflammation, and complicating factors that may affect the condition.
-
•
The implications of this review may stimulate further research and provide in-depth, updated, and collective information on the basis with how inflammation is involved with degenerative disk disease and how disruption to normal disk cell physiologic function via the inflammatory cascade may advance the stages of disease progression.
Alt-text: Image 1
References
- 1.Tsarouhas A, Soufla G, Katonis P, Pasku D, Vakis A, Spandidos DA. Transcript levels of major MMPs and ADAMTS-4 in relation to the clinicopathological profile of patients with lumbar disc herniation. Eur Spine J. 2011;20(5):781–790. doi: 10.1007/s00586-010-1573-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elfervig MK, Minchew JT, Francke E, Tsuzaki M, Banes AJ. IL-1beta sensitizes intervertebral disc annulus cells to fluid-induced shear stress. J Cell Biochem. 2001;82(2):290–298. doi: 10.1002/jcb.1153. [DOI] [PubMed] [Google Scholar]
- 3.Kokubo Y, Uchida K, Kobayashi S. Herniated and spondylotic intervertebral discs of the human cervical spine: histological and immunohistological findings in 500 en bloc surgical samples. Laboratory investigation. J Neurosurg Spine. 2008;9(3):285–295. doi: 10.3171/SPI/2008/9/9/285. [DOI] [PubMed] [Google Scholar]
- 4.Gawri R, Rosenzweig DH, Krock E. High mechanical strain of primary intervertebral disc cells promotes secretion of inflammatory factors associated with disc degeneration and pain. Arthritis Res Ther. 2014;16(1):R21. doi: 10.1186/ar4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doita M, Kanatani T, Ozaki T, Matsui N, Kurosaka M, Yoshiya S. Influence of macrophage infiltration of herniated disc tissue on the production of matrix metalloproteinases leading to disc resorption. Spine (Phila Pa 1976) 2001;26(14):1522–1527. doi: 10.1097/00007632-200107150-00004. [DOI] [PubMed] [Google Scholar]
- 6.Doita M, Kanatani T, Harada T, Mizuno K. Immunohistologic study of the ruptured intervertebral disc of the lumbar spine. Spine (Phila Pa 1976) 1996;21(2):235–241. doi: 10.1097/00007632-199601150-00015. [DOI] [PubMed] [Google Scholar]
- 7.Hashizume H, Kawakami M, Nishi H, Tamaki T. Histochemical demonstration of nitric oxide in herniated lumbar discs. A clinical and animal model study. Spine (Phila Pa 1976) 1997;22(10):1080–1084. doi: 10.1097/00007632-199705150-00005. [DOI] [PubMed] [Google Scholar]
- 8.Lee S, Moon CS, Sul D. Comparison of growth factor and cytokine expression in patients with degenerated disc disease and herniated nucleus pulposus. Clin Biochem. 2009;42(15):1504–1511. doi: 10.1016/j.clinbiochem.2009.06.017. [DOI] [PubMed] [Google Scholar]
- 9.Kang JD, Georgescu HI, McIntyre-Larkin L, Stefanovic-Racic M, Donaldson WF, Evans CH. Herniated lumbar intervertebral discs spontaneously produce matrix metalloproteinases, nitric oxide, interleukin-6, and prostaglandin E2. Spine (Phila Pa 1976) 1996;21(3):271–277. doi: 10.1097/00007632-199602010-00003. [DOI] [PubMed] [Google Scholar]
- 10.Habtemariam A, Virri J, Grönblad M, Holm S, Kaigle A, Karaharju E. Inflammatory cells in full-thickness anulus injury in pigs. An experimental disc herniation animal model. Spine (Phila Pa 1976) 1998;23(5):524–529. doi: 10.1097/00007632-199803010-00002. [DOI] [PubMed] [Google Scholar]
- 11.Kanerva A, Kommonen B, Grönblad M. Inflammatory cells in experimental intervertebral disc injury. Spine (Phila Pa 1976) 1997;22(23):2711–2715. doi: 10.1097/00007632-199712010-00002. [DOI] [PubMed] [Google Scholar]
- 12.Sen O, Aydin MV, Bagdatoglu C. Can E-selectin be a reliable marker of inflammation in lumbar disc disease? Neurosurg Rev. 2005;28(3):214–217. doi: 10.1007/s10143-005-0388-3. [DOI] [PubMed] [Google Scholar]
- 13.Grönblad M, Virri J, Tolonen J. A controlled immunohistochemical study of inflammatory cells in disc herniation tissue. Spine (Phila Pa 1976) 1994;19(24):2744–2751. doi: 10.1097/00007632-199412150-00002. [DOI] [PubMed] [Google Scholar]
- 14.Virri J, Grönblad M, Seitsalo S, Habtemariam A, Kääpä E, Karaharju E. Comparison of the prevalence of inflammatory cells in subtypes of disc herniations and associations with straight leg raising. Spine (Phila Pa 1976) 2001;26(21):2311–2315. doi: 10.1097/00007632-200111010-00004. [DOI] [PubMed] [Google Scholar]
- 15.Andrade P, Hoogland G, Garcia MA, Steinbusch HW, Daemen MA, Visser-Vandewalle V. Elevated IL-1β and IL-6 levels in lumbar herniated discs in patients with sciatic pain. Eur Spine J. 2013;22(4):714–720. doi: 10.1007/s00586-012-2502-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maidhof R, Jacobsen T, Papatheodorou A, Chahine NO. Inflammation induces irreversible biophysical changes in isolated nucleus pulposus cells. PloS One. 2014;9(6) doi: 10.1371/journal.pone.0099621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JM, Song JY, Baek M. Interleukin-1β induces angiogenesis and innervation in human intervertebral disc degeneration. J Orthop Res. 2011;29(2):265–269. doi: 10.1002/jor.21210. [DOI] [PubMed] [Google Scholar]
- 18.Schroeder M, Viezens L, Schaefer C. Chemokine profile of disc degeneration with acute or chronic pain. J Neurosurg Spine. 2013;18(5):496–503. doi: 10.3171/2013.1.SPINE12483. [DOI] [PubMed] [Google Scholar]
- 19.Ito T, Yamada M, Ikuta F. Histologic evidence of absorption of sequestration-type herniated disc. Spine (Phila Pa 1976) 1996;21(2):230–234. doi: 10.1097/00007632-199601150-00014. [DOI] [PubMed] [Google Scholar]
- 20.Burke JG, Watson RW, McCormack D, Dowling FE, Walsh MG, Fitzpatrick JM. Intervertebral discs which cause low back pain secrete high levels of proinflammatory mediators. J Bone Joint Surg Br. 2002;84(2):196–201. doi: 10.1302/0301-620x.84b2.12511. [DOI] [PubMed] [Google Scholar]
- 21.Kawaguchi S, Yamashita T, Katahira G, Yokozawa H, Torigoe T, Sato N. Chemokine profile of herniated intervertebral discs infiltrated with monocytes and macrophages. Spine (Phila Pa 1976) 2002;27(14):1511–1516. doi: 10.1097/00007632-200207150-00006. [DOI] [PubMed] [Google Scholar]
- 22.Kang JD, Georgescu HI, McIntyre-Larkin L, Stefanovic-Racic M, Evans CH. Herniated cervical intervertebral discs spontaneously produce matrix metalloproteinases, nitric oxide, interleukin-6, and prostaglandin E2. Spine (Phila Pa 1976) 1995;20(22):2373–2378. doi: 10.1097/00007632-199511001-00001. [DOI] [PubMed] [Google Scholar]
- 23.Grad S, Bow C, Karppinen J. Systemic blood plasma CCL5 and CXCL6: potential biomarkers for human lumbar disc degeneration. Eur Cell Mater. 2016;31:1–10. doi: 10.22203/ecm.v031a01. [DOI] [PubMed] [Google Scholar]
- 24.Shamji MF, Setton LA, Jarvis W. Proinflammatory cytokine expression profile in degenerated and herniated human intervertebral disc tissues. Arthritis Rheum. 2010;62(7):1974–1982. doi: 10.1002/art.27444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang KY, Lin RM, Chen WY, Lee CL, Yan JJ, Chang MS. IL-20 may contribute to the pathogenesis of human intervertebral disc herniation. Spine (Phila Pa 1976) 2008;33(19):2034–2040. doi: 10.1097/BRS.0b013e31817eb872. [DOI] [PubMed] [Google Scholar]
- 26.Haro H, Komori H, Okawa A, Murakami S, Muneta T, Shinomiya K. Sequential dynamics of monocyte chemotactic protein-1 expression in herniated nucleus pulposus resorption. J Orthop Res. 1997;15(5):734–741. doi: 10.1002/jor.1100150516. [DOI] [PubMed] [Google Scholar]
- 27.Iwabuchi S, Ito M, Chikanishi T, Azuma Y, Haro H. Role of the tumor necrosis factor-alpha, cyclooxygenase-2, prostaglandin E2, and effect of low-intensity pulsed ultrasound in an in vitro herniated disc resorption model. J Orthop Res. 2008;26(9):1274–1278. doi: 10.1002/jor.20525. [DOI] [PubMed] [Google Scholar]
- 28.Haro H, Crawford HC, Fingleton B, Shinomiya K, Spengler DM, Matrisian LM. Matrix metalloproteinase-7-dependent release of tumor necrosis factor-alpha in a model of herniated disc resorption. J Clin Invest. 2000;105(2):143–150. doi: 10.1172/JCI7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SJ, Park SM, Cho YW. Changes in expression of mRNA for interleukin-8 and effects of interleukin-8 receptor inhibitor in the spinal dorsal horn in a rat model of lumbar disc herniation. Spine (Phila Pa 1976) 2011;36(25):2139–2146. doi: 10.1097/BRS.0b013e31821945a3. [DOI] [PubMed] [Google Scholar]
- 30.Kato T, Haro H, Komori H, Shinomiya K. Sequential dynamics of inflammatory cytokine, angiogenesis inducing factor and matrix degrading enzymes during spontaneous resorption of the herniated disc. J Orthop Res. 2004;22(4):895–900. doi: 10.1016/j.orthres.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Takada T, Nishida K, Maeno K. Intervertebral disc and macrophage interaction induces mechanical hyperalgesia and cytokine production in a herniated disc model in rats. Arthritis Rheum. 2012;64(8):2601–2610. doi: 10.1002/art.34456. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida M, Nakamura T, Sei A, Kikuchi T, Takagi K, Matsukawa A. Intervertebral disc cells produce tumor necrosis factor alpha, interleukin-1beta, and monocyte chemoattractant protein-1 immediately after herniation: an experimental study using a new hernia model. Spine (Phila Pa 1976) 2005;30(1):55–61. doi: 10.1097/01.brs.0000149194.17891.bf. [DOI] [PubMed] [Google Scholar]
- 33.Liu ZH, Sun Z, Wang HQ. FasL expression on human nucleus pulposus cells contributes to the immune privilege of intervertebral disc by interacting with immunocytes. Int J Med Sci. 2013;10(8):1053–1060. doi: 10.7150/ijms.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto J, Maeno K, Takada T. Fas ligand plays an important role for the production of pro-inflammatory cytokines in intervertebral disc nucleus pulposus cells. J Orthop Res. 2013;31(4):608–615. doi: 10.1002/jor.22274. [DOI] [PubMed] [Google Scholar]
- 35.Egeland NG, Moen A, Pedersen LM, Brisby H, Gjerstad J. Spinal nociceptive hyperexcitability induced by experimental disc herniation is associated with enhanced local expression of Csf1 and FasL. Pain. 2013;154(9):1743–1748. doi: 10.1016/j.pain.2013.05.034. [DOI] [PubMed] [Google Scholar]
- 36.Sun Z, Wan ZY, Guo YS. FasL on human nucleus pulposus cells prevents angiogenesis in the disc by inducing Fas-mediated apoptosis of vascular endothelial cells. Int J Clin Exp Pathol. 2013;6(11):2376–2385. [PMC free article] [PubMed] [Google Scholar]
- 37.Kepler CK, Markova DZ, Dibra F. Expression and relationship of proinflammatory chemokine RANTES/CCL5 and cytokine IL-1β in painful human intervertebral discs. Spine (Phila Pa 1976) 2013;38(11):873–880. doi: 10.1097/BRS.0b013e318285ae08. [DOI] [PubMed] [Google Scholar]
- 38.Kang JD, Stefanovic-Racic M, McIntyre LA, Georgescu HI, Evans CH. Toward a biochemical understanding of human intervertebral disc degeneration and herniation. Contributions of nitric oxide, interleukins, prostaglandin E2, and matrix metalloproteinases. Spine (Phila Pa 1976) 1997;22(10):1065–1073. doi: 10.1097/00007632-199705150-00003. [DOI] [PubMed] [Google Scholar]
- 39.Le Maitre CL, Hoyland JA, Freemont AJ. Catabolic cytokine expression in degenerate and herniated human intervertebral discs: IL-1beta and TNFalpha expression profile. Arthritis Res Ther. 2007;9(4):R77. doi: 10.1186/ar2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoyland JA, Le Maitre C, Freemont AJ. Investigation of the role of IL-1 and TNF in matrix degradation in the intervertebral disc. Rheumatology (Oxford) 2008;47(6):809–814. doi: 10.1093/rheumatology/ken056. [DOI] [PubMed] [Google Scholar]
- 41.Specchia N, Pagnotta A, Toesca A, Greco F. Cytokines and growth factors in the protruded intervertebral disc of the lumbar spine. Eur Spine J. 2002;11(12):145–151. doi: 10.1007/s00586-001-0361-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kepler CK, Markova DZ, Hilibrand AS. Substance P stimulates production of inflammatory cytokines in human disc cells. Spine (Phila Pa 1976) 2013;38(21):E1291–E1299. doi: 10.1097/BRS.0b013e3182a42bc2. [DOI] [PubMed] [Google Scholar]
- 43.Pozzuoli A, Valvason C, Bernardi D. YKL-40 in human lumbar herniated disc and its relationships with nitric oxide and cyclooxygenase-2. Clin Exp Rheumatol. 2007;25(3):453–456. [PubMed] [Google Scholar]
- 44.Cuéllar JM, Borges PM, Cuéllar VG, Yoo A, Scuderi GJ, Yeomans DC. Cytokine expression in the epidural space: a model of noncompressive disc herniation-induced inflammation. Spine (Phila Pa 1976) 2013;38(1):17–23. doi: 10.1097/BRS.0b013e3182604baa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahn SH, Cho YW, Ahn MW, Jang SH, Sohn YK, Kim HS. mRNA expression of cytokines and chemokines in herniated lumbar intervertebral discs. Spine (Phila Pa 1976) 2002;27(9):911–917. doi: 10.1097/00007632-200205010-00005. [DOI] [PubMed] [Google Scholar]
- 46.Zhao CQ, Wang LM, Jiang LS, Dai LY. The cell biology of intervertebral disc aging and degeneration. Ageing Res Rev. 2007;6(3):247–261. doi: 10.1016/j.arr.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 47.Chen B, Ma B, Yang S, Xing X, Gu R, Hu Y. DR5 and DcR2 are expressed in human lumbar intervertebral discs. Spine (Phila Pa 1976) 2009;34(19):E677–E681. doi: 10.1097/BRS.0b013e3181b4d4ee. [DOI] [PubMed] [Google Scholar]
- 48.de Souza Grava AL, Ferrari LF, Defino HL. Cytokine inhibition and time-related influence of inflammatory stimuli on the hyperalgesia induced by the nucleus pulposus. Eur Spine J. 2012;21(3):537–545. doi: 10.1007/s00586-011-2027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olmarker K, Nutu M, Størkson R. Changes in spontaneous behavior in rats exposed to experimental disc herniation are blocked by selective TNF-alpha inhibition. Spine (Phila Pa 1976) 2003;28(15):1635–1641. doi: 10.1097/01.BRS.0000083162.35476.FF. discussion 1642. [DOI] [PubMed] [Google Scholar]
- 50.Genevay S, Finckh A, Payer M. Elevated levels of tumor necrosis factor-alpha in periradicular fat tissue in patients with radiculopathy from herniated disc. Spine (Phila Pa 1976) 2008;33(19):2041–2046. doi: 10.1097/BRS.0b013e318183bb86. [DOI] [PubMed] [Google Scholar]
- 51.Park JB, Chang H, Lee JK. Quantitative analysis of transforming growth factor-beta 1 in ligamentum flavum of lumbar spinal stenosis and disc herniation. Spine (Phila Pa 1976) 2001;26(21):E492–E495. doi: 10.1097/00007632-200111010-00007. [DOI] [PubMed] [Google Scholar]
- 52.Kim HJ, Park JB, Won HY, Chang H. Serum Levels of TGF-beta1, TIMP-1 and TIMP-2 in Patients with Lumbar Spinal Stenosis and Disc Herniation. Asian Spine J. 2007;1(1):8–11. doi: 10.4184/asj.2007.1.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsui Y, Maeda M, Nakagami W, Iwata H. The involvement of matrix metalloproteinases and inflammation in lumbar disc herniation. Spine (Phila Pa 1976) 1998;23(8):863–868. doi: 10.1097/00007632-199804150-00005. discussion 868-869. [DOI] [PubMed] [Google Scholar]