

Abstract
Stroke has become a more common disease worldwide. Despite great efforts to develop treatment, little is known about ischemic stroke. Cerebral ischemia activates multiple cascades of cell type-specific pathomechanisms. Ischemic brain injury consists of a complex series of cellular reactions in various cell types within the central nervous system (CNS) including platelets, endothelial cells, astrocytes, neutrophils, microglia/macrophages, and neurons. Diverse cellular changes after ischemic injury are likely to induce cell death and tissue damage in the brain. Since cells in the brain exhibit different functional roles at distinct time points after injury (acute/subacute/chronic phases), it is difficult to pinpoint genuine roles of cell types after brain injury. Many experimental studies have shown the association of apoptosis signal-regulating kinase 1 (ASK1) with cellular pathomechanisms after cerebral ischemia. Blockade of ASK1, by either pharmacological or genetic manipulation, leads to reduced ischemic brain injury and subsequent neuroprotective effects. In this review, we present the cell type-specific pathophysiology of the early phase of ischemic stroke, the role of ASK1 suggested by preclinical studies, and the potential use of ASK suppression, either by pharmacologic or genetic suppression, as a promising therapeutic option for ischemic stroke recovery.
1. Introduction
Ischemic stroke is a heterogeneous neurologic disorder characterized by sudden onset and multiple environmental risk factors [1, 2]. Ischemic stroke develops as a result of complex pathomechanisms induced by a critical reduction in cerebral blood flow (CBF) caused by either sudden or gradual occlusion of cerebral arteries [3, 4]. The brain requires large amounts of oxygen and glucose from the blood for energy metabolism; thus, blockage of blood circulation causes neurologic deficits [3–5]. The epicenter of a stroke, the area of the brain with crucially impaired blood flow, is referred to as the “infarct core,” and the neighboring area is referred to as the “ischemic penumbra” or salvageable area [3–5]. Ischemia-related pathologic reactions can last for days to weeks in these areas of the brain [4]. The main pathologic changes involved in ischemic stroke are energy depletion, calcium overload, excessive reactive oxygen species (ROS) generation, inflammatory signals, and ion imbalance, all of which can lead to cell death [3, 5, 6]. These changes are severely detrimental to neuronal, glial, and endothelial cell function [5] and lead to platelet activation, reactive gliosis, immune cell activation, and neuronal cell death in the ischemic brain [3, 5, 7]. Over the last decades, various stroke models have been designed in an effort to find new therapies for stroke [8]. However, therapeutic candidates from preclinical studies have failed to translate into effective therapies [8]. In this review, we will address the underlying pathophysiology of ischemic stroke briefly, focusing on cell type-specific mechanisms generated from preclinical ischemic stroke models. Additionally, we discuss apoptosis signal-regulating kinase 1 (ASK1) as a potential therapeutic target based on preclinical testing.
2. ASK1
ASK1 is a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family, which activates mitogen-activated protein kinase kinase (MAP2K: MKK4/MKK7, MKK3/MKK6) and leads to the subsequent activation of mitogen-activated protein kinase (MAPK) as part of a signaling cascade [9–12]. ASK1 is endogenously expressed in various cell types [13]. It is comprised of 1375 amino acids in humans and 1379 in mice, and it contains a serine/threonine kinase domain in the middle region [14]. The phosphorylation of threonine residues (Thr 838 in human and Thr 845 in mouse) is important for ASK1 activation [10, 15]. In normal conditions, ASK1 is a homooligomer, which binds to another ASK1 via its C-terminal coiled-coil domain. The N-terminal coiled-coil domain of ASK1 binds to thioredoxin (Trx), which suppresses ASK1 kinase activity [9, 15]. Under oxidative stress conditions, oxidized Trx is separated from ASK1, and unbound ASK1 is activated by phosphorylation [11, 16]. Calcium influx and oxidative stress can elicit phosphorylation of the ASK1 Thr residue [10, 15]. In addition, tumor necrosis factor receptor-associated factor 2 (TRAF2) and TRAF6 act as positive regulators of ASK1 after hydrogen peroxide (H2O2) injury [10]. Negative regulators of ASK1, 14-3-3 proteins, block activation of ASK1 in the steady state by binding to the C-terminal of ASK1 after Ser 966 phosphorylation [9, 10, 15]. However, oxidative stress promotes dephosphorylation of ASK1 at Ser 966 and leads to detachment of 14-3-3, which results in activation of ASK1 [10]. ASK1 is activated not only from oxidative stress but also from endoplasmic reticulum stress and bacterial infection [9, 11, 14]. Both Fas death receptor and tumor necrosis factor (TNF) also activate ASK1 [17, 18]. ASK1 is known as an early responder to ROS after cerebral ischemia [19]. After exposure to various stimuli, activated ASK1 initiates multiple signaling cascades, including c-Jun N-terminal kinase (JNK) and p38, and governs cellular mechanisms, including cell death, growth, and differentiation (Figure 1) [10, 14, 20]. Although ASK1 has previously been known to be mainly involved in apoptotic cell death [21], recent research has identified other functions of ASK1, such as its association with thrombosis, brain edema, inflammatory responses, and reactive gliosis after cerebral ischemia [19, 22–24].
Figure 1.
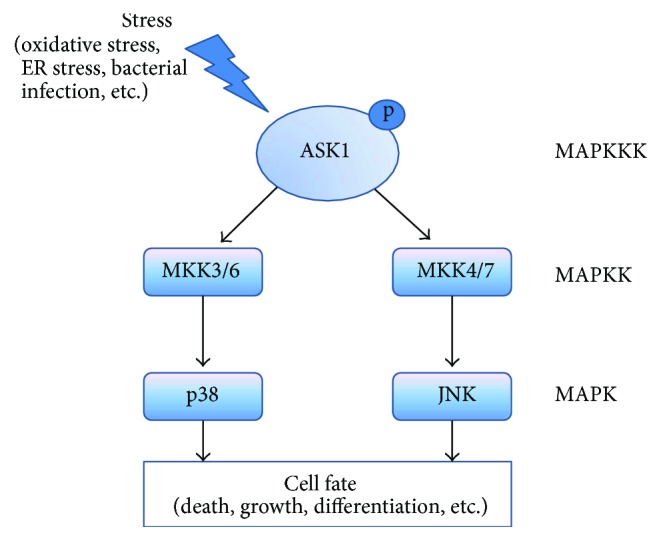
Involvement of ASK1 in the cellular mechanism. After various stresses, the activated form of ASK1 activates MAPKK (MKK3/6 and MKK4/7), thereby activating MAPK including JNK and p38. The ASK1 signaling pathway governs cellular fate such as apoptosis, growth, and differentiation.
3. Platelets and ASK1
Ischemic stroke is linked to vascular occlusion due to a thrombus or emboli in the brain [25]. Platelets are necessary for thrombosis and thromboembolism formation [26]. Platelet-induced thrombosis is associated with platelet adhesion, activation, and aggregation [27]. Normally, homeostasis is maintained by coagulation, fibrinolysis, and platelet function [28]. When homeostasis breaks down, platelets are involved in thrombus formation through a complex process [27]. For initial adhesion, platelets make connections between platelet surface receptors (glycoprotein (GP) Ib-V-IX complex or integrin αIIbβ3 and α2β1) and adhesive substrates (von Willebrand factor (vWF) and collagen) on an exposed endothelial extracellular matrix (ECM) [29–31]. The platelet GP IIb/IIIa surface receptor mediates platelet aggregation through platelet-platelet interactions with extracellular fibrinogen and vWF [25, 27, 29]. These binding events trigger platelet activation and intracellular signaling pathways, which induce the production of thrombin and promote the release of activating factors (granule contents) (adenosine diphosphate (ADP), epinephrine, and thromboxane A2 (TXA2)) [27]. These factors can increase GP IIb/IIIa and calcium levels [32, 33]. Upregulated calcium can induce an increase in phospholipase A2(PLA2) [34]. A previous study proved the expression of p38, extracellular signal-regulated kinase 2 (ERK2), and JNK1 in platelets, which can be activated by thrombin, collagen, vWF, and ADP [35]. ASK1, expressed in human and murine platelets, has recently been found to be rapidly activated by different platelet agonists (ADP, convulxin, and thrombin) [22]. Genetic depletion of ASK1 leads to defects in platelet aggregation, impaired integrin αIIbβ3 activation, and reduced TXA2 generation [22]. In Ask1−/− platelets, inhibition of cytoplasmic phospholipase A2 (cPLA2), an important enzyme in the generation of TXA2, led to a reduction in thrombin-, collagen-, and convulxin-induced TXA2 production [22]. Impaired platelet functions, caused by ASK1 depletion in platelets, result in thrombosis deterioration, which eventually becomes protective against arterial thrombosis [22]. Hence, ASK1 serves as an important factor in thrombosis, and its activation is associated with an increased risk factor for ischemic stroke.
4. Endothelial Cells and ASK1
The blood-brain barrier (BBB) consists of a highly specialized endothelial structure and maintains brain homeostasis by controlling para- and transcellular transport between blood and the extracellular space [36]. Brain interendothelial junction complex contains adherence junctions (vascular endothelial cadherin, β-catenin), gap junctions (connexins), and tight junctions (claudins, occludins, and junctional adhesion molecules (JAM)), which are important for constructing barriers and cell-cell communication [36–39]. However, pathological conditions, such as ischemic stroke, can change BBB permeability and interactions between endothelial cells [36, 37]. A damaged BBB allows blood to enter the parenchyma and causes brain damage [40], followed by cerebral edema and vasomotor/hemodynamic dysfunctions [40].
Metalloproteinases (MMPs) and proteolytic enzymes can affect BBB integrity by degrading neurovascular substrates [37, 39]. Deterioration of the BBB caused by MMP-mediated tight junction degradation is mainly associated with MMP-9 [37]. BBB breakdown permits water into the extracellular compartments and results in vasogenic edema in the injured brain [41]. MMPs also contribute to edema formation [42]. Several reports have concluded that the excessive activation of MMP-2 and MMP-9 in the ischemic brain and cerebral endothelium leads to cellular damage [39, 43]. By inhibiting MMPs, vascular hyper permeability can be reduced by impeding degradation of gap junction proteins and rearrangement of tight junction proteins [44]. In this context, MMP-9 knockout contributes to preventing BBB disruption and enlargement of brain lesion [45].
Vascular endothelial growth factor (VEGF), a vascular permeability factor, is involved in the formation of vascular leakages and vasogenic edema [44]. VEGF has permeabilizing effects on the endothelium through MMP-9-induced reduction of tight junction proteins (zo-1, occludin) [44]. Previous studies have shown that administration of recombinant human VEGF165 exacerbates the breakdown of BBB integrity, which can be prevented by inhibiting VEGF at an early stage of ischemic stroke [46, 47]. A relationship between increased activation of MMP-9- and VEGF-induced BBB leakage had also been identified [48].
Although ASK1 is necessary for angiogenesis and the recovery of blood flow by direct expression of VEGF and monocyte chemoattractant protein-1 (MCP-1) after unilateral hindlimb ischemia, ASK1 and VEGF play an important role in vascular permeability in cerebral ischemia [24, 49]. Inhibition of ASK1 reduces ischemia-induced edema formation and the expression of VEGF and aquaporin-1 (water channel protein) [24]. In addition, ASK1 silencing by siRNA decreases gene levels of Mmp3, Vegf-a, Vegf-c, and Aquaporin 12 and 18 [24]. ASK1 inhibition reduces MMP-9 activity in both mice and endothelial cell cultures. These results suggest a role for ASK1 in suppressing neuronal cell death [50], based on its profound effect on BBB permeability and brain edema formation after ischemic stroke.
5. Immune Cells and ASK1
After cerebral ischemia, circulating blood leukocytes migrate across disrupted vessel walls into the cerebral parenchyma [40]. During this influx of immune cells, adhesion molecules, such as vascular adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), E-selectin, and P-selectin, promote the transendothelial recruitment of immune cells [51–53]. Infiltrating immune cells accumulate in the brain lesion and trigger the release of inflammatory cytokines, which further promote tissue damage [40]. Peripheral blood cells are involved in a variety of functions, from cell death to cell recovery, depending on the time course of the ischemic stroke [54].
Neutrophils are subpopulations of leukocytes, which exacerbate neuronal damage by participating in the early stages of ischemic stroke [40, 55]. Transmigrated neutrophils possess neurotoxic properties and produce cytokines, protease, chemokines, and ROS [56, 57]. Neutrophils also release neurotoxic-related neutrophil extracellular traps (NETs), composed of proteases and decondensed DNA [56]. Moreover, oxygen free radicals and proteolytic enzymes are also released from penetrated neutrophil [58]. Several studies have proven that the inhibition of either neutrophil accumulation or adhesion can diminish ischemic brain injury [55, 58]. Prevention of neutrophil infiltration toward the ischemic lesion has beneficial effects on the ischemic brain [59]. Inhibition of neutrophils by anti-neutrophil antibody (RP3) efficiently reduces the extent of brain infarction and the cerebral water content [60].
Brain microglia, representative immune cells of the brain, contribute to the immune systems in the CNS through defense mechanisms such as phagocytosis [40, 61]. After acute cerebral ischemia, microglia are activated in response to the influx of immune cells and become indistinguishable from macrophages [62–64]. Microglia are activated via the Toll-like receptor (TLR) pathway in response to cellular damage after cerebral ischemia and release cytotoxic and cytoprotective substances [40, 61]. Infiltrated blood-borne macrophages in infarcted brain tissue are key modulators of the immune system [63]. Although microglial activation leads to tissue injury during early stages of cerebral ischemia, microglia/macrophages participate in tissue recovery during the late course of ischemia [65]. Astrocytes, fibroblasts, and endothelial cells, as well as resident microglia and peripheral macrophage, are involved in the production of inflammatory cytokines such as interleukin-1β (IL-1β), transforming necrosis factor-α (TNF-α), and transforming growth factor-β (TGF-β) [58, 61, 62, 64, 66]. Although microglia and macrophages are associated with brain plasticity and recovery at later stages of cerebral ischemia, several studies have reported that microglia and macrophages induce neuronal injury through a TLR-4-dependent manner and trigger the proinflammatory mediator in the acute stages of ischemic stroke [67–69]. Pharmacological inhibition of microglia showed protective effects in cerebral ischemia by inhibiting a microglia-derived inflammatory mediator. Suppression of activated microglia by minocycline led to reduced brain infarction, improved neurological deficits, and diminished BBB leakage [70, 71]. Moreover, macrophage-derived angiopoietin-like protein 2 knockout contributed to reduced brain injury [67].
ASK1 is closely related to the immune system and is required in inflammatory responses [14, 72]. It has been reported that TLR4 activates ASK1 to initiate the MAPK pathway and thereby express inflammatory-related genes [73]. MAPK also mediates expression of a variety of inflammatory genes such as cell surface adhesion molecules, chemokines, and cytokines [74]. ASK1 is linked to ventilation-induced cytokine production, neutrophil infiltration, and cell death in the lung [75]. The TLR/ASK1/p38 pathway is important in chemokine production and in triggering neurotoxicity in multiple sclerosis [76]. ASK1 contributes to production of TNF-α and inducible nitric oxide synthase (iNOS) in primary microglia cell culture [77]. However, ASK1 knockout in vivo and in vitro models diminished lipopolysaccharide- (LPS-) induced upregulation of IL-6, IL-1β, and TNF-α and diminished LPS-exaggerated injury [9, 14, 72]. LPS-induced ASK1/p38 signals and cytokine production in the RAW264.7 macrophage cell line are attenuated by antioxidants [14]. From previous studies on ischemic injury, it is known that ASK1 silencing by siRNA reduces infiltrated macrophages/resident microglia in brain regions such as the striatum, cortex, and hippocampus, and ASK1 silencing downregulates proinflammatory cytokines such as IL-6, IL-1β, and TNF-α in the ipsilateral hemisphere at late stages of cerebral ischemia. In the RAW264.7 macrophage cell line and BV2 microglia cell line, ASK1 inhibition diminishes the release of proinflammatory mediators [23]. From a genetic perspective, microarray analysis shows that ASK1 silencing decreased the gene levels of Il1b, Il6, Cxcl2, Cxcl1, and Ccl2 [19]. Considering the relationship between ASK1 and the immune response, ASK1 could be an important regulator of the inflammatory response after ischemic stroke.
6. Astrocytes and ASK1
The key roles of astrocytes in the neuronal system are involved in the maintenance of brain physiology and neuronal support, both structurally and metabolically, through neurotransmitter regulation (glutamate uptake/release), ion buffering, scavenging free radicals, enhancing BBB integrity, and regulating water transports [78–81]. However, astrocytes become hyperactivated in response to ischemic stress and extend their processes, changing morphology with the expression of glial fibrillary acidic protein (GFAP) [7, 78]. Astrocytes migrate toward the injury site and thus accumulate and produce a glial scar [7, 19, 80]. It has been reported that astrocytes produce and release either trophic factors (brain-derived neurotrophic factor (BDNF), fibroblast growth factor-2, and nerve growth factor (NGF)) or inflammatory cytokines (IL-6, IL-1β, TNF-α, and interferon-gamma (IFNγ)) [79, 82]. Trophic factors play crucial roles in neuronal survival and protection, while inflammatory mediators contribute to brain injury [18, 63, 83, 84]. Therefore, it is known that astrocytes play dual roles in the immune system [78]. Although several studies have provided evidence of the relationship between reactive astrocytes and neurogenesis, previous reports have suggested that reactive astrocytes block neuronal regeneration [19, 81, 85–87].
ASK1 is present in astrocytes and is strongly expressed after cerebral ischemia. Readily identified reactive astrocytes in ischemic lesions form a glial scar in the chronic phase of ischemic stroke, which delays extension of neurite and functional recovery [19]. However, siRNA targeting ASK1 reduced reactive astrocyte marker GFAP in both in vivo and in vitro studies, decreased glial scar formation, and promoted neuronal plasticity and functional performance [19]. Moreover, ASK1 deletion suppressed mitochondrial complex I inhibitor 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine- (MPTP-) induced astrocyte activation and protected against degeneration of dopaminergic neurons [88]. In addition, p38, a molecule downstream of ASK1, is also associated with reactive astrogliosis, and a conditional GFAP/p38 knockout reduced astrogliosis [89]. Therefore, several lines of evidence show that ASK1 may play a major role in reactive astrocytes and glial scar formation after ischemic stroke.
7. Neurons and ASK1
After ischemic injury, neurons are harmed by excitotoxicity, acidotoxicity, MMP, nitric oxide (NO), ion imbalances, and free radicals, which results in neuronal death and cerebral damage [90–92]. Inhibition of ATP synthesis in the mitochondria after ischemia depolarizes neuronal plasma membranes [92]. Additionally, the intracellular influx of excess calcium overloads via nonselective cation channels and calcium channels depolarizes neurons [90]. Membrane depolarization induces the release of the excitatory neurotransmitter glutamate, and the increase in glutamate concentration can activate the N-methyl-D-aspartate (NMDA) and α-amino3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors [92, 93]. Calcium-permeable NMDA receptors induce further membrane depolarization, which aggravates calcium overload [92]. Ion imbalances cause excessive ROS in the intracellular system [94]. Increased NO production and free radicals can enhance BBB leakage and contribute to apoptotic signaling cascades [91].
Overexpression of ASK1 promotes apoptotic cell death, and JNK/p38 MAP kinases closely interact with this process [95, 96]. Increased ASK1 levels after cerebral ischemia also induce apoptosis, which leads to neuronal cell death and the development of infarct lesions [21]. Calcium influx activates p38 signals in the ASK1+/+ mice-derived neuron, but these p38 activations are suppressed in the ASK1−/− mice-derived neuron [10]. ASK1 is closely related to Ca2+/calmodulin-dependent protein kinase II (CaMK II), which is activated by calcium influx [10, 97]. It has been reported that CaMKII directly phosphorylates ASK1 at Thr 838 [10, 15, 97]. CaMKII inhibition reduces Ca2+-induced activation of ASK1 [15, 97]. NO activates ASK1, and the nitric oxide synthase (nNOS) inhibitor 7-NI and the NMDA receptor antagonist MK801 reduce ASK1 activity [98]. In addition, an AMPA receptor blocker and a free radical scavenger prevent activation of ASK1 and JNK [99]. These previous studies demonstrated the neuroprotective effects from genetic knockdown or pharmacological inhibition of ASK1 after cerebral ischemia [21, 100, 101]. Neuroprotective drugs show beneficial effects by suppression of ASK1/JNK signals [102]. Based on previous studies, ASK1 may be involved in calcium influx, oxidative stress, neuronal cell death, and cerebral infarctions after ischemic stroke. (Table 1).
Table 1.
Genetic/pharmacological inhibition of ASK1. bEnd.3 cell line (mouse endothelial cells), BV2 cell line (mouse microglia cells), RAW 264.7 cell line (mouse macrophage cells), HAEC cells (human aortic endothelial cells), and MEF cells (mouse embryonic fibroblasts). MPO: myeloperoxidase; ICV: intracerebrovascular injection; IV: intravenous injection; IH: intrahemispheric injection.
| Genetic/pharmacological inhibition of ASK1 | Dosage | Application | Injury or duration | Main outcome | Reference |
|---|---|---|---|---|---|
| ASK1 knockout (KO) | ASK1 KO mice | Optic nerve injury | Eye: apoptotic cell death↓, MCP-1↓, TNF-α↓, iNOS↓ | [77] | |
| MEF cells | H2O2 or TNF treatment | Apoptotic cell death↓ | [96] | ||
| Platelets | Thromboxane A2↓, platelet granule secretion↓ | [22] | |||
| ASK1 KO mice | FeCl3-induced injury | Carotid artery thrombosis↓ | [22] | ||
| Small interfering RNA (siRNA) for ASK1 | 5 μM/mice (ICV) | C57/BL6 mice | 3 days before ischemic/reperfusion (I/R) injury | Brain: brain edema↓, VEGF↓, AQP-1↓ | [24] |
| 5 μM/mice (ICV) | C57/BL6 mice | 3 days before I/R injury | Brain: neuronal cell death↓, MMP-9↓ | [50] | |
| 5 μM/mice (ICV) | C57/BL6 mice | 3 days before I/R injury | Brain: GFAP↓, glial scar formation↓ | [19] | |
| 5 μM/mice (ICV) | C57/BL6 mice | 3 days before I/R injury | Brain: IL-6↓,TNF-α↓, IL-1β↓ | [23] | |
| 5 μM/mice (ICV) | C57/BL6 mice | 3 days before I/R injury | Brain: infarct volume↓ | [21] | |
| Nanoparticles with anti-ASK1 short hairpin RNA (shRNA) | 150 μg/rat (IV) | Spraque-Dawley rats | 2 days before I/R injury | Brain: apoptotic cell death↓, infarct injury↓ | [101] |
| NQDI-1, ASK1 inhibitor | 600 nM | bEnd.3 cell line | 3 hrs before oxygen/glucose deprivation (OGD) | VEGF↓ | [24] |
| 600 nM | bEnd.3 cell line | 1 hr before OGD/6 hrs during OGD | Released MMP-9↓ | [50] | |
| 600 nM | BV2 cell line | 1 hr before OGD/4 hrs during OGD | IL-6↓, TNF-α↓, IL-1β↓, iNOS↓ | [23] | |
| 600 nM | RAW 264.7 cell line | 1 hr before OGD/4 hrs during OGD | IL-6↓, TNF-α↓, IL-1β↓, iNOS↓ | [23] | |
| 250 nmol/pup (IH) | Spraque-Dawley rats | 30 mins before hypoxia-ischemia (HI) injury | Brain: caspase-3↓, infarct volume↓, apoptotic cell death↓ | [100] | |
| AGI-1067, ASK1 inhibitor | 10 μM | HAEC cells | 1 hr before LPS/during LPS treatment | VCAM-1↓, E-selectin↓, IL-6↓, MCP-1↓ | [73] |
| Thioredoxin | 2 μg/g (IV) | C57/BL6 mice | 10 mins before ventilation | Infiltration of neutrophils↓, MPO↓ | [75] |
| MSC2032964A, ASK1 inhibitor | 10 μM | Primary microglia cells | 1 hr before LPS treatment | TNF-α↓, iNOS↓ | [77] |
8. Conclusion
Ischemic stroke is a complex neurologic disorder with limited treatment options, which amplifies the need for drug development. This review focuses on cell type-specific pathomechanisms, mainly targeting platelets, endothelial cells, immune cells, astrocytes, and neurons in preclinical ischemic stroke models. We focus on ASK1 as a major target molecule in the etiology of ischemic stroke. Pharmacologic and genetic inhibition of ASK1 has been shown to provide neuroprotective effects in cerebral ischemia. Therefore, we would like to highlight the importance of ASK1 as a key target in drug development for ischemic stroke.
Acknowledgments
This article was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIP) to Bon-Nyeo Koo (NRF-2017R1A2B4009478) and Eun Jung Kim (NRF-2017R1C1B5017506).
Abbreviations
- CNS:
Central nervous system
- ASK1:
Apoptosis signal-regulating kinase 1
- CBF:
Cerebral blood flow
- ROS:
Reactive oxygen species
- MAPKKK:
Mitogen-activated protein kinase kinase kinase
- MAP2K:
Mitogen-activated protein kinase kinase
- MAPK:
Mitogen-activated protein kinase
- Trx:
Thioredoxin
- H2O2:
Hydrogen peroxide
- JNK:
c-Jun N-terminal kinase
- vWF:
von Willebrand factor
- ADP:
Adenosine diphosphate
- TXA2:
Thromboxane A2
- PLA2:
Phospholipase A2
- ERK2:
Extracellular signal-regulated kinase 2
- cPLA2:
Cytoplasmic phospholipase A2
- BBB:
Blood-brain barrier
- MMPs:
Metalloproteinases
- VEGF:
Vascular endothelial growth factor
- MCP-1:
Monocyte chemoattractant protein-1
- VCAM-1:
Vascular adhesion molecule-1
- ICAM-1:
Intercellular adhesion molecule-1
- NETs:
Neutrophil extracellular traps
- TLR:
Toll-like receptor
- IL-1β:
Interleukin-1β
- TNF-α:
Transforming necrosis factor-α
- TGF-β:
Transforming growth factor-β
- iNOS:
Inducible nitric oxide synthase
- LPS:
Lipopolysaccharide
- GFAP:
Glial fibrillary acidic protein
- BDNF:
Brain-derived neurotrophic factor
- NGF:
Nerve growth factor
- IFNγ:
Interferon-gamma
- NO:
Nitric oxide
- NMDA:
N-Methyl-D-aspartate
- AMPA:
α-Amino3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CaMK II:
Ca2+/calmodulin-dependent protein kinase II
- nNOS:
Nitric oxide synthase.
Conflicts of Interest
The authors declare that they have no competing interests.
Authors' Contributions
So Yeong Cheon, Eun Jung Kim, Jeong Min Kim, and Bon-Nyeo Koo participated in writing the manuscript, and all authors approved the final version. So Yeong Cheon and Eun Jung Kim contributed equally to this work.
References
- 1.Kleinschnitz C., Fluri F., Schuhmann M. Animal models of ischemic stroke and their application in clinical research. Drug Design, Development and Therapy. 2015;9:3445–3454. doi: 10.2147/dddt.s56071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bi B. L., Wang H. J., Bian H., Tian Z. T. Identification of therapeutic targets of ischemic stroke with DNA microarray. European Review for Medical and Pharmacological Sciences. 2015;19(21):4012–4019. [PubMed] [Google Scholar]
- 3.Durukan A., Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacology, Biochemistry, and Behavior. 2007;87(1):179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 4.Dirnagl U., Iadecola C., Moskowitz M. A. Pathobiology of ischaemic stroke: an integrated view. Trends in Neurosciences. 1999;22(9):391–397. doi: 10.1016/S0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 5.Khoshnam S. E., Winlow W., Farzaneh M., Farbood Y., Moghaddam H. F. Pathogenic mechanisms following ischemic stroke. Neurological Sciences. 2017;38(7):1167–1186. doi: 10.1007/s10072-017-2938-1. [DOI] [PubMed] [Google Scholar]
- 6.Mehta S. L., Manhas N., Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Research Reviews. 2007;54(1):34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Pekny M., Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50(4):427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 8.O'Collins V. E., Macleod M. R., Donnan G. A., Horky L. L., van der Worp B. H., Howells D. W. 1,026 experimental treatments in acute stroke. Annals of Neurology. 2006;59(3):467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- 9.Matsukawa J., Matsuzawa A., Takeda K., Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. The Journal of Biochemistry. 2004;136(3):261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- 10.Shiizaki S., Naguro I., Ichijo H. Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Advances in Biological Regulation. 2013;53(1):135–144. doi: 10.1016/j.jbior.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Hayakawa R., Hayakawa T., Takeda K., Ichijo H. Therapeutic targets in the ASK1-dependent stress signaling pathways. Proceedings of the Japan Academy, Series B. 2012;88(8):434–453. doi: 10.2183/pjab.88.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekine Y., Takeda K., Ichijo H. The ASK1-MAP kinase signaling in ER stress and neurodegenerative diseases. Current Molecular Medicine. 2006;6(1):87–97. doi: 10.2174/156652406775574541. [DOI] [PubMed] [Google Scholar]
- 13.Matsuzawa A., Nishitoh H., Tobiume K., Takeda K., Ichijo H. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxidants & Redox Signaling. 2002;4(3):415–425. doi: 10.1089/15230860260196218. [DOI] [PubMed] [Google Scholar]
- 14.Hayakawa T., Matsuzawa A., Noguchi T., Takeda K., Ichijo H. The ASK1-MAP kinase pathways in immune and stress responses. Microbes and Infection. 2006;8(4):1098–1107. doi: 10.1016/j.micinf.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Kawarazaki Y., Ichijo H., Naguro I. Apoptosis signal-regulating kinase 1 as a therapeutic target. Expert Opinion on Therapeutic Targets. 2014;18(6):651–664. doi: 10.1517/14728222.2014.896903. [DOI] [PubMed] [Google Scholar]
- 16.Matsuzawa A., Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochimica et Biophysica Acta (BBA) - General Subjects. 2008;1780(11):1325–1336. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Chang H. Y., Nishitoh H., Yang X., Ichijo H., Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998;281(5384):1860–1863. doi: 10.1126/science.281.5384.1860. [DOI] [PubMed] [Google Scholar]
- 18.Yagi T., Jikihara I., Fukumura M., et al. Rescue of ischemic brain injury by adenoviral gene transfer of glial cell line-derived neurotrophic factor after transient global ischemia in gerbils. Brain Research. 2000;885(2):273–282. doi: 10.1016/S0006-8993(00)02956-5. [DOI] [PubMed] [Google Scholar]
- 19.Cheon S. Y., Cho K. J., Song J., Kim G. W. Knockdown of apoptosis signal-regulating kinase 1 affects ischaemia-induced astrocyte activation and glial scar formation. European Journal of Neuroscience. 2016;43(7):912–922. doi: 10.1111/ejn.13175. [DOI] [PubMed] [Google Scholar]
- 20.Nagai H., Noguchi T., Takeda K., Ichijo H. Pathophysiological roles of ASK1-MAP kinase signaling pathways. BMB Reports. 2007;40(1):1–6. doi: 10.5483/bmbrep.2007.40.1.001. [DOI] [PubMed] [Google Scholar]
- 21.Kim H. W., Cho K. J., Lee S. K., Kim G. W. Apoptosis signal-regulating kinase 1 (Ask1) targeted small interfering RNA on ischemic neuronal cell death. Brain Research. 2011;1412:73–78. doi: 10.1016/j.brainres.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 22.Naik M. U., Patel P., Derstine R., et al. Ask1 regulates murine platelet granule secretion, thromboxane A2 generation, and thrombus formation. Blood. 2017;129(9):1197–1209. doi: 10.1182/blood-2016-07-729780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheon S. Y., Kim E. J., Kim J. M., Kam E. H., Ko B. W., Koo B. N. Regulation of microglia and macrophage polarization via apoptosis signal-regulating kinase 1 silencing after ischemic/hypoxic injury. Frontiers in Molecular Neuroscience. 2017;10:p. 261. doi: 10.3389/fnmol.2017.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song J., Cheon S. Y., Lee W. T., Park K. A., Lee J. E. The effect of ASK1 on vascular permeability and edema formation in cerebral ischemia. Brain Research. 2015;1595:143–155. doi: 10.1016/j.brainres.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 25.Murray V., Norrving B., Sandercock P. A. G., Terént A., Wardlaw J. M., Wester P. The molecular basis of thrombolysis and its clinical application in stroke. Journal of Internal Medicine. 2010;267(2):191–208. doi: 10.1111/j.1365-2796.2009.02205.x. [DOI] [PubMed] [Google Scholar]
- 26.Kinsella J. A., Tobin W. O., Hamilton G., McCabe D. J. H. Platelet activation, function, and reactivity in atherosclerotic carotid artery stenosis: a systematic review of the literature. International Journal of Stroke. 2013;8(6):451–464. doi: 10.1111/j.1747-4949.2012.00866.x. [DOI] [PubMed] [Google Scholar]
- 27.Franchi F., Angiolillo D. J. Novel antiplatelet agents in acute coronary syndrome. Nature Reviews Cardiology. 2015;12(1):30–47. doi: 10.1038/nrcardio.2014.156. [DOI] [PubMed] [Google Scholar]
- 28.Kim J. H., Lim K. M., Gwak H. S. New anticoagulants for the prevention and treatment of venous thromboembolism. Biomolecules & Therapeutics. 2017;25(5):461–470. doi: 10.4062/biomolther.2016.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoll G., Kleinschnitz C., Nieswandt B. Molecular mechanisms of thrombus formation in ischemic stroke: novel insights and targets for treatment. Blood. 2008;112(9):3555–3562. doi: 10.1182/blood-2008-04-144758. [DOI] [PubMed] [Google Scholar]
- 30.Kulkarni S., Dopheide S. M., Yap C. L., et al. A revised model of platelet aggregation. The Journal of Clinical Investigation. 2000;105(6):783–791. doi: 10.1172/JCI7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennett J. S., Kolodziej M. A. Disorders of platelet function. Disease-a-Month. 1992;38(8):577–631. [PubMed] [Google Scholar]
- 32.Wilson J. M., Ferguson J. J., III Platelet-endothelial interactions in atherothrombotic disease: therapeutic implications. Clinical Cardiology. 1999;22(11):687–698. doi: 10.1002/clc.4960221103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin J., Quinton T. M., Zhang J., Rittenhouse S. E., Kunapuli S. P. Adenosine diphosphate (ADP)-induced thromboxane A2 generation in human platelets requires coordinated signaling through integrin αIIbβ3 and ADP receptors. Blood. 2002;99(1):193–198. doi: 10.1182/blood.V99.1.193. [DOI] [PubMed] [Google Scholar]
- 34.Church D. J., Braconi S., Vallotton M. B., Lang U. Protein kinase C-mediated phospholipase A2 activation, platelet-activating factor generation and prostacyclin release in spontaneously beating rat cardiomyocytes. Biochemical Journal. 1993;290(2):477–482. doi: 10.1042/bj2900477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adam F., Kauskot A., Rosa J. P., Bryckaert M. Mitogen-activated protein kinases in hemostasis and thrombosis. Journal of Thrombosis and Haemostasis. 2008;6(12):2007–2016. doi: 10.1111/j.1538-7836.2008.03169.x. [DOI] [PubMed] [Google Scholar]
- 36.Schoknecht K., David Y., Heinemann U. The blood-brain barrier—gatekeeper to neuronal homeostasis: clinical implications in the setting of stroke. Seminars in Cell & Developmental Biology. 2015;38:35–42. doi: 10.1016/j.semcdb.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 37.Posada-Duque R. A., Barreto G. E., Cardona-Gomez G. P. Protection after stroke: cellular effectors of neurovascular unit integrity. Frontiers in Cellular Neuroscience. 2014;8:p. 231. doi: 10.3389/fncel.2014.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallez Y., Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2008;1778(3):794–809. doi: 10.1016/j.bbamem.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Amtul Z., Hepburn J. D. Protein markers of cerebrovascular disruption of neurovascular unit: immunohistochemical and imaging approaches. Reviews in the Neurosciences. 2014;25(4):481–507. doi: 10.1515/revneuro-2013-0041. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q., Tang X., Yenari M. The inflammatory response in stroke. Journal of Neuroimmunology. 2007;184(1-2):53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nag S., Manias J. L., Stewart D. J. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathologica. 2009;118(2):197–217. doi: 10.1007/s00401-009-0541-0. [DOI] [PubMed] [Google Scholar]
- 42.Jianliu K., Rosenberg G. Matrix metalloproteinases and free radicals in cerebral ischemia. Free Radical Biology & Medicine. 2005;39(1):71–80. doi: 10.1016/j.freeradbiomed.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 43.Lee S. R., Lo E. H. Induction of caspase-mediated cell death by matrix metalloproteinases in cerebral endothelial cells after hypoxia-reoxygenation. Journal of Cerebral Blood Flow & Metabolism. 2004;24(7):720–727. doi: 10.1097/01.WCB.0000122747.72175.47. [DOI] [PubMed] [Google Scholar]
- 44.Bauer A. T., Burgers H. F., Rabie T., Marti H. H. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. Journal of Cerebral Blood Flow & Metabolism. 2010;30(4):837–848. doi: 10.1038/jcbfm.2009.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asahi M., Wang X., Mori T., et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. The Journal of Neuroscience. 2001;21(19):7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Z. G., Zhang L., Jiang Q., et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. The Journal of Clinical Investigation. 2000;106(7):829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H. T., Zhang P., Gao Y., et al. Early VEGF inhibition attenuates blood-brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Molecular Medicine Reports. 2017;15(1):57–64. doi: 10.3892/mmr.2016.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valable S., Montaner J., Bellail A., et al. VEGF-induced BBB permeability is associated with an MMP-9 activity increase in cerebral ischemia: both effects decreased by Ang-1. Journal of Cerebral Blood Flow & Metabolism. 2005;25(11):1491–1504. doi: 10.1038/sj.jcbfm.9600148. [DOI] [PubMed] [Google Scholar]
- 49.Izumi Y., Kim-Mitsuyama S., Yoshiyama M., et al. Important role of apoptosis signal-regulating kinase 1 in ischemia-induced angiogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(9):1877–1883. doi: 10.1161/01.ATV.0000174801.76234.bd. [DOI] [PubMed] [Google Scholar]
- 50.Cheon S. Y., Cho K. J., Kim S. Y., Kam E. H., Lee J. E., Koo B. N. Blockade of apoptosis signal-regulating kinase 1 attenuates matrix metalloproteinase 9 activity in brain endothelial cells and the subsequent apoptosis in neurons after ischemic injury. Frontiers in Cellular Neuroscience. 2016;10:p. 213. doi: 10.3389/fncel.2016.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindsberg P. J., Carpen O., Paetau A., Karjalainen-Lindsberg M. L., Kaste M. Endothelial ICAM-1 expression associated with inflammatory cell response in human ischemic stroke. Circulation. 1996;94(5):939–945. doi: 10.1161/01.CIR.94.5.939. [DOI] [PubMed] [Google Scholar]
- 52.Zhang R., Chopp M., Zhang Z., Jiang N., Powers C. The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Research. 1998;785(2):207–214. doi: 10.1016/S0006-8993(97)01343-7. [DOI] [PubMed] [Google Scholar]
- 53.Jander S., Pohl J., Gillen C., Schroeter M., Stoll G. Vascular cell adhesion molecule-1 mRNA is expressed in immune-mediated and ischemic injury of the rat nervous system. Journal of Neuroimmunology. 1996;70(1):75–80. doi: 10.1016/S0165-5728(96)00109-9. [DOI] [PubMed] [Google Scholar]
- 54.Zheng Z., Yenari M. A. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurological Research. 2004;26(8):884–892. doi: 10.1179/016164104X2357. [DOI] [PubMed] [Google Scholar]
- 55.Hallenbeck J. M. Significance of the inflammatory response in brain ischemia. Mechanisms of Secondary Brain Damage in Cerebral Ischemia and Trauma. 1996;66:27–31. doi: 10.1007/978-3-7091-9465-2_5. [DOI] [PubMed] [Google Scholar]
- 56.Allen C., Thornton P., Denes A., et al. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. The Journal of Immunology. 2012;189(1):381–392. doi: 10.4049/jimmunol.1200409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wright H. L., Moots R. J., Bucknall R. C., Edwards S. W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology. 2010;49(9):1618–1631. doi: 10.1093/rheumatology/keq045. [DOI] [PubMed] [Google Scholar]
- 58.Huang J., Upadhyay U. M., Tamargo R. J. Inflammation in stroke and focal cerebral ischemia. Surgical Neurology. 2006;66(3):232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 59.Jiang N., Moyle M., Soule H. R., Rote W. E., Chopp M. Neutrophil inhibitory factor is neuroprotective after focal ischemia in rats. Annals of Neurology. 1995;38(6):935–942. doi: 10.1002/ana.410380615. [DOI] [PubMed] [Google Scholar]
- 60.Matsuo Y., Onodera H., Shiga Y., et al. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994;25(7):1469–1475. doi: 10.1161/01.STR.25.7.1469. [DOI] [PubMed] [Google Scholar]
- 61.Brea D., Sobrino T., Ramos-Cabrer P., Castillo J. Inflammatory and neuroimmunomodulatory changes in acute cerebral ischemia. Cerebrovascular Diseases. 2009;27(1):48–64. doi: 10.1159/000200441. [DOI] [PubMed] [Google Scholar]
- 62.Kawabori M., Yenari M. Inflammatory responses in brain ischemia. Current Medicinal Chemistry. 2015;22(10):1258–1277. doi: 10.2174/0929867322666150209154036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vogelgesang A., Becker K. J., Dressel A. Immunological consequences of ischemic stroke. Acta Neurologica Scandinavica. 2014;129(1):1–12. doi: 10.1111/ane.12165. [DOI] [PubMed] [Google Scholar]
- 64.Denes A., Thornton P., Rothwell N. J., Allan S. M. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain, Behavior, and Immunity. 2010;24(5):708–723. doi: 10.1016/j.bbi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 65.Amantea D., Nappi G., Bernardi G., Bagetta G., Corasaniti M. T. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. The FEBS Journal. 2009;276(1):13–26. doi: 10.1111/j.1742-4658.2008.06766.x. [DOI] [PubMed] [Google Scholar]
- 66.Iadecola C., Anrather J. The immunology of stroke: from mechanisms to translation. Nature Medicine. 2011;17(7):796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amadatsu T., Morinaga J., Kawano T., et al. Macrophage-derived angiopoietin-like protein 2 exacerbates brain damage by accelerating acute inflammation after ischemia-reperfusion. PLoS One. 2016;11(11, article e0166285) doi: 10.1371/journal.pone.0166285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lehnardt S., Massillon L., Follett P., et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Amantea D., Micieli G., Tassorelli C., et al. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Frontiers in Neuroscience. 2015;9:p. 147. doi: 10.3389/fnins.2015.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou R., Yang Z., Tang X., Tan Y., Wu X., Liu F. Propofol protects against focal cerebral ischemia via inhibition of microglia-mediated proinflammatory cytokines in a rat model of experimental stroke. PLoS One. 2013;8(12, article e82729) doi: 10.1371/journal.pone.0082729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yenari M. A., Xu L., Tang X. N., Qiao Y., Giffard R. G. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37(4):1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- 72.Takeda K., Noguchi T., Naguro I., Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annual Review of Pharmacology and Toxicology. 2008;48(1):199–225. doi: 10.1146/annurev.pharmtox.48.113006.094606. [DOI] [PubMed] [Google Scholar]
- 73.Zheng S., Long L., Li Y., et al. A novel ASK inhibitor AGI-1067 inhibits TLR-4-mediated activation of ASK1 by preventing dissociation of thioredoxin from ASK1. Cardiovascular Pharmacology: Open Access. 2015;4(1) doi: 10.4172/2329-6607.1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berk B. C., Abe J.-I., Min W., Surapisitchat J., Yan C. Endothelial atheroprotective and anti-inflammatory mechanisms. Annals of the New York Academy of Sciences. 2001;947(1):93–111. doi: 10.1111/j.1749-6632.2001.tb03932.x. [DOI] [PubMed] [Google Scholar]
- 75.Li L. F., Liao S. K., Lee C. H., Tsai Y. H., Huang C. C., Quinn D. A. Ventilation-induced neutrophil infiltration and apoptosis depend on apoptosis signal-regulated kinase 1 pathway. Critical Care Medicine. 2005;33(9):1913–1921. doi: 10.1097/01.CCM.0000178179.48050.BC. [DOI] [PubMed] [Google Scholar]
- 76.Guo X., Harada C., Namekata K., et al. Regulation of the severity of neuroinflammation and demyelination by TLR‐ASK1‐p38 pathway. EMBO Molecular Medicine. 2010;2(12):504–515. doi: 10.1002/emmm.201000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Katome T., Namekata K., Guo X., et al. Inhibition of ASK1-p38 pathway prevents neural cell death following optic nerve injury. Cell Death & Differentiation. 2013;20(2):270–280. doi: 10.1038/cdd.2012.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Becerra-Calixto A., Cardona-Gomez G. P. The role of astrocytes in neuroprotection after brain stroke: potential in cell therapy. Frontiers in Molecular Neuroscience. 2017;10:p. 88. doi: 10.3389/fnmol.2017.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takuma K., Baba A., Matsuda T. Astrocyte apoptosis: implications for neuroprotection. Progress in Neurobiology. 2004;72(2):111–127. doi: 10.1016/j.pneurobio.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 80.Dienel G. A., Hertz L. Astrocytic contributions to bioenergetics of cerebral ischemia. Glia. 2005;50(4):362–388. doi: 10.1002/glia.20157. [DOI] [PubMed] [Google Scholar]
- 81.Chen Y., Swanson R. A. Astrocytes and brain injury. Journal of Cerebral Blood Flow & Metabolism. 2003;23(2):137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- 82.Lau L. T., Yu A. C. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. Journal of Neurotrauma. 2001;18(3):351–359. doi: 10.1089/08977150151071035. [DOI] [PubMed] [Google Scholar]
- 83.del Zoppo G., Ginis I., Hallenbeck J. M., Iadecola C., Wang X., Feuerstein G. Z. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathology. 2000;10(1):95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saito A., Tominaga T., Chan P. H. Neuroprotective role of neurotrophins: relationship between nerve growth factor and apoptotic cell survival pathway after cerebral ischemia. Current Atherosclerosis Reports. 2005;7(4):268–273. doi: 10.1007/s11883-005-0018-0. [DOI] [PubMed] [Google Scholar]
- 85.Tiveron M. C., Barboni E., Pliego Rivero F. B., et al. Selective inhibition of neurite outgrowth on mature astrocytes by Thy-1 glycoprotein. Nature. 1992;355(6362):745–748. doi: 10.1038/355745a0. [DOI] [PubMed] [Google Scholar]
- 86.Duan C. L., Liu C. W., Shen S. W., et al. Striatal astrocytes transdifferentiate into functional mature neurons following ischemic brain injury. Glia. 2015;63(9):1660–1670. doi: 10.1002/glia.22837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Song H., Stevens C. F., Gage F. H. Astroglia induce neurogenesis from adult neural stem cells. Nature. 2002;417(6884):39–44. doi: 10.1038/417039a. [DOI] [PubMed] [Google Scholar]
- 88.Lee K. W., Zhao X., Im J. Y., et al. Apoptosis signal-regulating kinase 1 mediates MPTP toxicity and regulates glial activation. PLoS One. 2012;7(1, article e29935) doi: 10.1371/journal.pone.0029935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roy Choudhury G., Ryou M. G., Poteet E., et al. Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Research. 2014;1551:45–58. doi: 10.1016/j.brainres.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tuttolomondo A., Di Sciacca R., Di Raimondo D., et al. Neuron protection as a therapeutic target in acute ischemic stroke. Current Topics in Medicinal Chemistry. 2009;9(14):1317–1334. doi: 10.2174/156802609789869646. [DOI] [PubMed] [Google Scholar]
- 91.Guo M. F., Yu J. Z., Ma C. G. Mechanisms related to neuron injury and death in cerebral hypoxic ischaemia. Folia Neuropathologica. 2011;49(2):78–87. [PubMed] [Google Scholar]
- 92.Doyle K. P., Simon R. P., Stenzel-Poore M. P. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55(3):310–318. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Song M., Yu S. P. Ionic regulation of cell volume changes and cell death after ischemic stroke. Translational Stroke Research. 2014;5(1):17–27. doi: 10.1007/s12975-013-0314-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Birben E., Sahiner U. M., Sackesen C., Erzurum S., Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organization Journal. 2012;5(1):9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ichijo H., Nishida E., Irie K., et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 96.Tobiume K., Matsuzawa A., Takahashi T., et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Reports. 2001;2(3):222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takeda K., Matsuzawa A., Nishitoh H., et al. Involvement of ASK1 in Ca2+‐induced p38 MAP kinase activation. EMBO Reports. 2004;5(2):161–166. doi: 10.1038/sj.embor.7400072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu D. H., Yuan F. G., Hu S. Q., et al. Endogenous nitric oxide induces activation of apoptosis signal-regulating kinase 1 via S-nitrosylation in rat hippocampus during cerebral ischemia-reperfusion. Neuroscience. 2013;229:36–48. doi: 10.1016/j.neuroscience.2012.10.055. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Q., Zhang G., Meng F., Tian H. Biphasic activation of apoptosis signal-regulating kinase 1-stress-activated protein kinase 1-c-Jun N-terminal protein kinase pathway is selectively mediated by Ca2+-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors involving oxidative stress following brain ischemia in rat hippocampus. Neuroscience Letters. 2003;337(1):51–55. doi: 10.1016/s0304-3940(02)01295-8. [DOI] [PubMed] [Google Scholar]
- 100.Hao H., Li S., Tang H., et al. NQDI-1, an inhibitor of ASK1 attenuates acute perinatal hypoxic-ischemic cerebral injury by modulating cell death. Molecular Medicine Reports. 2016;13(6):4585–4592. doi: 10.3892/mmr.2016.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.An S., Kuang Y., Shen T., et al. Brain-targeting delivery for RNAi neuroprotection against cerebral ischemia reperfusion injury. Biomaterials. 2013;34(35):8949–8959. doi: 10.1016/j.biomaterials.2013.07.060. [DOI] [PubMed] [Google Scholar]
- 102.Wang Q., Zhang Q. G., Wu D. N., Yin X. H., Zhang G. Y. Neuroprotection of selenite against ischemic brain injury through negatively regulating early activation of ASK1/JNK cascade via activation of PI3K/AKT pathway. Acta Pharmacologica Sinica. 2007;28(1):19–27. doi: 10.1111/j.1745-7254.2007.00469.x. [DOI] [PubMed] [Google Scholar]