

Abstract
Protein disulfide isomerase A1 (PDIA1) is an endoplasmic reticulum (ER)-localized thiol-disulfide oxidoreductase that is an important folding catalyst for secretory pathway proteins. PDIA1 contains two active-site domains (a and a′), each containing a Cys-Gly-His-Cys (CGHC) active-site motif. The two active-site domains share 37% sequence identity, and function independently to perform disulfide-bond reduction, oxidation and isomerization. Numerous inhibitors for PDIA1 have been reported, yet the selectivity of these inhibitors toward the a and a′ sites are poorly characterized. Here, we identify a potent and selective PDIA1 inhibitor, KSC-34, with 30-fold selectivity for the a site over the a′ site. KSC-34 displays time-dependent inhibition of PDIA1 reductase activity in vitro with a kinact/KI = 9.66 × 103 M−1s−1, and is selective for PDIA1 over other members of the PDI family, and other cellular cysteine-containing proteins. We provide the first cellular characterization of an a-site selective PDIA1 inhibitor and demonstrate that KSC-34 has minimal sustained effects on the cellular unfolded protein response (UPR), indicating that a-site inhibition does not induce global protein-folding-associated ER stress. KSC-34 treatment significantly decreases secretion of a destabilized, amyloidogenic antibody light chain, thereby minimizing pathogenic amyloidogenic extracellular proteins that rely on high PDIA1 activity for proper folding and secretion. Given the poor understanding of the contribution of each PDIA1 active site to the (patho)physiological functions of PDIA1, site-selective inhibitors like KSC-34 provide useful tools for delineating the pathological role and therapeutic potential of PDIA1.
INTRODUCTION
Proper folding of nascent proteins in the cell is accelerated by chaperones that serve to stabilize protein domains and overall structure. One such family of protein-folding catalysts comprise dithiol-disulfide oxidoreductases of the protein disulfide isomerase (PDI) family. The PDI family is primarily located in the endoplasmic reticulum (ER), and includes 21 proteins with at least one thioredoxin-like domain. PDIs typically contain two different types of thioredoxin-like domains, catalytic (a) and non-catalytic (b)1, 2. The catalytic a domains of the PDIs are responsible for catalyzing the oxidation, reduction, and isomerization of disulfide bonds in nascent proteins. The non-catalytic b domains are rigid spacers between the catalytic domains that are involved in recognizing and recruiting unfolded protein substrates1.
Protein disulfide isomerase A1 (PDIA1) was the first PDI family member to be discovered3, 4, 5, 6. PDIA1 is a 57 kDa oxidoreductase and molecular chaperone that localizes in the lumen of the ER, and accounts for roughly 0.8% of total cellular protein7. PDIA1 catalyzes the oxidation, reduction, and isomerization of disulfide bonds between cysteine residues on its protein substrates, and is organized into four globular domains, a, b, b′, and a′, as well as a highly acidic C-terminal extension with a KDEL ER-retention sequence (Figure 1A)3, 4. The a and a′ domains functionally resemble thioredoxin, and contain redox catalytic Cys-Gly-His-Cys (CGHC) active-site motifs. It is known that the a and a′ domains operate independently of each other because mutation of either active-site cysteine eliminated 50% of the catalytic activity of PDIA1 in vitro, while mutations in both active sites completely abolished activity8. The non-catalytic domains, b and b′, are primarily for spacing and substrate recruitment. The b′ domain is the major substrate binding site of PDIA1, containing a large hydrophobic cavity between helices α1 and α3 to interact with unfolded protein substrates1, 9.
Figure 1.

Domain organization of PDIA1 and chemical structures of PDIA1 inhibitors. (A) PDIA1 comprises two active site a-type domains that contain a CGHC active site motif, together with two b-domains implicated in substrate recognition and binding. (B) Chemical structure of RB-11-ca, a previously reported a-site inhibitor of PDIA1. RB-11-ca contains a central triazine scaffold, a chloroacetamide reactive group for covalent cysteine modification, an alkyne bioorthogonal handle for CuAAC, and an octylamine diversity element. (C) Predicted binding pose of RB-11-ca in a domain active site of PDIA1 by the covalent docking algorithm from Schrodinger, Inc. (D) Structures of second-generation PDIA1 inhibitors obtained by varying the diversity element of RB-11-ca.
The dysregulation of PDIA1 activity has been implicated in a variety of diseases, including cancer3, 10–12, cardiovascular13–15, and neurodegenerative16–19 diseases. PDIA1 is retained in the ER through a C-terminal KDEL retention signal sequence, however, the presence of PDIA1 in the extracellular milieu is well documented3, 20, 21. Extracellular PDIA1 is involved in many biological processes such as platelet activation, thrombus formation, and viral infection. For example, PDIA1 can catalyze the reduction of structural disulfides on gp120, which results in a major conformational change, allowing the HIV virus to interact with the cell surface20.
The demonstrated role of increased PDIA1 activity in various diseases has spurred the development of a variety of small-molecule inhibitors targeting PDIA1. Juniferdin, discovered in a high-throughput screen of natural products, inhibits extracellular PDIA1-mediated disulfide-bond reduction of gp120 and inhibits HIV entry into host cells20. Quercetin-3-rutinoside binds to the b′x region of extracellular PDIA1 to induce a compact conformation, and inhibit thrombus formation22. T8, and derivative JP04–042, sensitizes cancer cells to sub-toxic concentrations of etoposide through inhibition of PDIA1 by reversible binding23. Another reversible inhibitor of PDIA1, LOC14, was identified from a high-throughput screen of ~10,000 compounds, and induces an oxidized conformation that is neuroprotective in PC12 cells expressing mutant huntingtin protein24. PACMA31 is an irreversible PDIA1 inhibitor that reacts through a propynoic acid carbamoyl methyl amide moiety, and suppresses tumor growth in a mouse xenograft model of ovarian cancer25. S-CW3554 possesses a 2-chloropropionamide reactive group, and is cytotoxic to various multiple myeloma cell lines26. 16F16, an irreversible inhibitor that reacts with PDIA1 via a chloroacetamide electrophile, prevents neuronal cell death in a cell-based model of Huntington’s disease19. Lastly, P1, an irreversible inhibitor, which reacts via a vinyl-sulfone electrophile, displays anti-proliferative effects in a variety of cancers27.
In 2013 we reported RB-11-ca (Figure 1B), a cell-permeable, irreversible inhibitor of PDIA128. RB-11-ca is a tri-functionalized triazine-based covalent inhibitor that contains an alkyne-handle for reporter-tag conjugation via copper-catalyzed azide-alkyne cycloaddition (CuAAC), an octylamine diversity element for binding specificity, and an electrophilic chloroacetamide for irreversible modification of proximal cysteine residues. PDIA1 was identified as the primary target of RB-11-ca after CuAAC-mediated conjugation of a biotin group, avidin enrichment, and subsequent target identification via LC/LC-MS/MS. The specific cysteine residue targeted by RB-11-ca was determined using cysteine-to-alanine mutants of each cysteine residue within the CXXC motifs of the two a domains. RB-11-ca was confirmed to bind to C53, the N-terminal cysteine in the a domain of PDIA128. RB-11-ca was therefore unique in that it was the first characterized a-domain selective inhibitor for PDIA1.
Herein, we report on KSC-34, an optimized PDIA1 inhibitor, which demonstrates a 30-fold selectivity for the a domain over the a′ domain. KSC-34 displays high selectivity for PDIA1 in complex proteomes with minimal engagement of other members of the PDI family. Given the critical role of PDIA1 in normal protein homeostasis, PDIA1 inhibitors that shut down both active sites demonstrate high cytotoxicity. By selectively inhibiting a single active-site domain of PDIA1, we hypothesized that basal PDIA1 activity could be maintained to minimize cytotoxicity, whilst selectively inhibiting pathogenic functions that are dependent on elevated PDIA1 activity. We demonstrate that a-site selective PDIA1 inhibition shows negligible toxicity, and minimal induction of sustained cellular protein-folding stress as demonstrated by low activation of the unfolded protein response (UPR). PDIA1 inhibition by KSC-34 has the potential to display targeted effects on the secretion of pathogenic proteins that utilize PDIA1 for folding and secretion. In particular, antibody light chains contain a disulfide bond within the hydrophobic core of the protein, and the folding of antibody light chains has been shown to involve PDIA1 activity29. Dysregulated light-chain secretion from a clonal expansion of plasma cells can lead to immunoglobulin light-chain amyloidosis30. Here we demonstrate that KSC-34 treatment decreases secretion of a destabilized, amyloidogenic antibody light chain at non-toxic concentrations. Together, we identify a potent and selective PDIA1 inhibitor that uniquely targets the a active-site domain of PDIA1 with the ability to affect the secretion of pathogenic proteins whose folding involves PDIA1 activity.
MATERIALS/EXPERIMENTAL DETAILS
See the Supporting Information for details related to materials, synthesis and structural characterization of all compounds, and methods for in-gel fluorescence, mass-spectrometry proteomics, UPR target gene analysis and FT-ALLC secretion.
RESULTS AND DISCUSSION
Generation of a targeted library of RB-11-ca analogs
In previous published studies, we identified a cysteine-reactive triazine-based compound, RB-11-ca, as an a-site selective covalent modifier of PDIA128. In order to further optimize the selectivity and potency of RB-11-ca, we sought to develop a second-generation library of triazine-based covalent inhibitors for PDIA1. To gain deeper insight into the binding mode of our lead compound, RB-11-ca (Figure 1B) within the a domain of PDIA1, we embarked on some preliminary docking studies. Only one high-resolution crystal structure of full-length PDIA1 is available31, and this structure was utilized to predict a binding model for RB-11-ca using the Covalent Dock workflow from Schrodinger, Inc (New York, NY) (Figure 1C)32. In the predicted binding pose, RB-11-ca is covalently bound to the N-terminal cysteine (C53) of the CGHC active-site motif within the a domain, and the octylamine diversity element interacts with a large hydrophobic pocket at the domain periphery. Based on this predicted binding mode of RB-11-ca, we hypothesized that variations to the octylamine diversity element could likely enhance the predicted interactions with the observed hydrophobic pocket, and serve to improve the potency of the compound. Therefore, a second-generation library of 15 compounds (Figure 1D) was synthesized according to previous methods with minor modifications (Scheme S1)28. These second-generation compounds contain diversity elements that possess hydrocarbons of varying lengths, cyclic, branched, and benzyl-functionalized hydrocarbons.
Identification of KSC-34 as a potent and selective covalent modifier of PDIA1
To streamline the selection of an optimized PDIA1 inhibitor, we first screened for compounds with improved covalent modification of PDIA1 using a gel-based fluorescence screening platform. Given that the extent of covalent modification of active-site cysteines directly correlate with PDIA1 inhibition, screening for improved covalent modifiers acts as a proxy for increased PDIA1 inhibition. In the first screening step, covalent modification of purified, recombinant PDIA1 was evaluated to identify compounds that demonstrated increased covalent modification of PDIA1 relative to RB-11-ca. Briefly, PDIA1 (50 μg/mL) was incubated with RB-11-ca and second-generation library members (5 μM) for 1 hour. After incubation, covalent modification of PDIA1 was monitored by appending a tetramethylrhodamine (TAMRA) fluorophore to the alkyne group of each compound using CuAAC 33, 34, followed by in-gel fluorescence to quantify the amount of compound-bound PDIA1 (Figure 2A). Seven library members (KSC-4, 7, 10, 11, 24, 26 and 34) that possessed equal or greater potency compared to RB-11-ca were advanced to the next round of selection.
Figure 2.

Characterization of PDIA1 second-generation library members (A) Library members (5 μM) were incubated with purified recombinant PDIA1 (50 μg/mL) in PBS, and protein labeling by each compound was evaluated after CuAAC-mediated incorporation of a TAMRA fluorophore, SDS-PAGE, and in-gel fluorescence (complete gel images provided in Figure S1). Library members with equal or greater potency than RB-11-ca progressed to the next step of the assay. (B) In-gel fluorescence studies of library members (5 μM) incubated with MCF-7 cell lysate (2 mg/mL) to assess potency and selectivity for endogenous PDIA1 in a complex proteome (complete gel images provided in Figure S3). (C) To assess cell permeability as well as selectivity of the library, probes (5 μM) were incubated with MCF-7 cells for 3 hours at 37 °C. Following cell lysis and in-gel fluorescence, KSC-34 was found to be ~8-fold more potent than the lead compound, RB-11-ca, in whole cells against PDIA1 (left panel). Coomassie gel (right panel is provided to show normalized protein abundance). Fluorescence intensity quantified by Image J (NIH, Bethesda, MD). All data are normalized to RB-11-ca and error bars represent standard deviation (SD) for n=3 experiments.
In the second screening step, library members were assessed against purified, recombinant PDIA1 (50 μg/mL) in the background of MCF-7 cell lysates (1 mg/mL) to evaluate whether other cellular proteins interfere with the ability of each compound to covalently modify PDIA1 (Figure S2). Four library members (KSC-7, 24, 26, and 34) possessed equal or greater potency to RB-11-ca, and thus were advanced to the next stage. In the third screening step, library members were evaluated in MCF-7 lysates (2 mg/mL) for their ability to covalently modify endogenous PDIA1 (Figure 2B). This analysis afforded three library members (KSC-24, 26 and 34) that had equal or greater potency toward PDIA1 compared to RB-11-ca. Of these, the two most potent library members, KSC-24 and KSC-34, were advanced to the final stage.
In the fourth and final step of this multilayered assay, the cell permeability and ability of the compounds to engage with ER-localized PDIA1 directly in living cells was assessed. MCF-7 cells were treated with compound for 3 hours at 37 °C, followed by in-gel fluorescence analysis (Figure 2C). KSC-34, which contains a (4-phenylbutyl)methylamine diversity element, was found to covalently modify PDIA1 directly in living cells with approximately 8-fold increased potency compared to RB-11-ca. The presence of a single robust fluorescent band at ~60 kD demonstrates the high potency and selectivity of KSC-34 for covalent modification of PDIA1 within the background of other highly abundant cysteine-containing proteins.
KSC-34 demonstrates time-dependent inhibition of PDIA1 in vitro
To confirm that improved covalent modification of PDIA1 by KSC-34 correlated with increased inhibitory potency, we utilized an in vitro insulin turbidity assay that monitors PDI reductase activity35. The reduction of disulfide bonds in insulin results in aggregation of the insulin B chain causing a measurable increase in turbidity. Due to the covalent nature of RB-11-ca and KSC-34, PDIA1-induced insulin aggregation was monitored upon treatment with each inhibitor at varying concentrations and pre-incubation times. We also compared RB-11-ca and KSC-34 to the commercially available PDIA1 inhibitor, 16F1619, which also contains a chloroacetamide electrophile for covalent modification of PDIA1, similar to our triazine-based compounds. KSC-34 inhibited PDIA1 in a concentration and time-dependent manner, with a kinact/KI = 9.66 × 103 M−1 s−1 (Figure 3A), which is ~3-fold more potent than RB-11-ca (kinact/KI = 3.35 × 103 M−1 s−1), and 38-fold more potent than the commercial PDIA1 inhibitor 16F16 (kinact/KI = 2.52 × 102 M−1 s−1).
Figure 3.
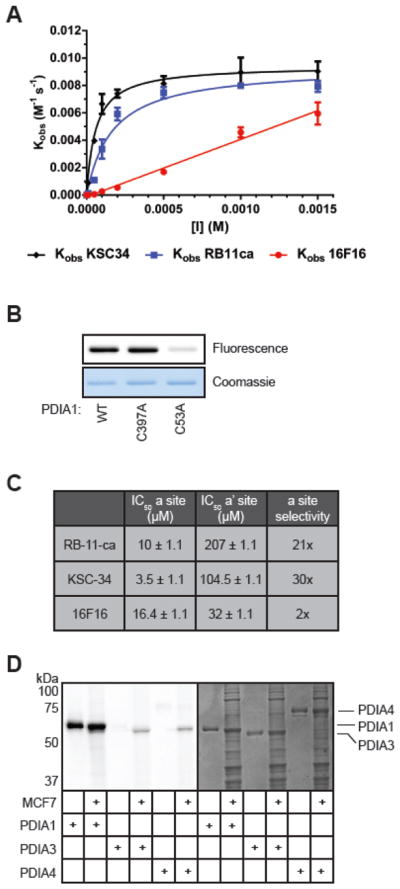
Evaluation of potency and selectivity of KSC-34. (A) Concentration and time-dependent inhibition of PDIA1 by KSC-34, RB-11-ca, and 16F16. PDIA1 reductase activity was measured using an insulin reduction assay in a 100 μL reaction volume containing 0.5 μM PDIA1, 0.16 mM insulin, and 1 mM DTT in assay buffer. Turbidity of the insulin solution was measured over time at various concentrations and pre-incubation times with KSC-34. Error bars represent SD from n=2 experiments. (B) Labeling of WT, C397A and C53A by KSC-34. Each recombinant PDIA1 protein (50 μg/ml) was incubated with KSC-34 (5 μM) for 1 hour, and subjected to CuAAC with TAMRA-azide prior to in-gel fluorescence. (C) Active-site selectivity for KSC-34, RB-11-ca, and 16F16. KSC-34 was found to have a 30-fold selectivity for the a domain over the a′ domain, compared to RB-11-ca which exhibited a 21-fold selectivity and 16F16 which exhibited 2-fold selectivity. Error is calculated by PRISM as +/− SEM from n=3 experiments. (D) PDI isoform selectivity. KSC-34 was found to only covalently modify PDIA1, over PDIA3 and PDIA4, two closely related family members. Fluorescent bands in cell lysate lanes for PDIA3 and PDIA4 samples represent endogenous PDIA1 protein.
KSC-34 is a highly selective covalent modifier of PDIA1
Upon confirmation of the ability of KSC-34 to covalently modify and inhibit PDIA1 activity, we sought to determine the selectivity of KSC-34 for the a and a′ active sites of PDIA1. Given that our first-generation compound RB-11-ca was a-site selective, we expected KSC-34 to bind in a similar fashion. Site-selectivity was assessed by recombinantly expressing wild-type PDIA1 (WT) and cysteine-to-alanine mutants of each of the two active-site nucleophilic residues (C53A and C397A). Each recombinant PDIA1 protein (50 μg/mL) was incubated with KSC-34 (5 μM) for 1 hour, and subjected to CuAAC prior to in-gel fluorescence. The absence of fluorescence in the C53A sample indicates that the binding of KSC-34 to PDIA1 is reliant on the presence of C53. Therefore, KSC-34 maintains selectivity for C53 in the a domain of PDIA1 with minimal binding to C397 in the a′ domain. To further quantify the selectivity for the a and a′ domains, IC50 values were obtained for binding to each active site (Figure 3C). To attain IC50 values, PDIA1 C53A and C397A (50 μg/mL), in the background of MCF-7 lysates (1 mg/mL), were treated with increasing concentrations of KSC-34, RB-11-ca and 16F16. Following incubation, samples were then treated with chloroacetamide-rhodamine (CA-Rh) (8 μM, Figure S4), to append a fluorophore to residual, unmodified active-site cysteines in each PDIA1 mutant. Loss in CA-Rh labeling of each active-site mutant is indicative of inhibitor binding, and informs on the affinity of each compound to the a and a′ domains. KSC-34 demonstrated a 30-fold selectivity for the a domain over the a′ domain, with RB-11-ca and 16F16 displaying 21-fold and 2-fold selectivity, respectively (Figure 3B). KSC-34 is therefore the most selective and potent a-site inhibitor of PDIA1 reported to date.
Owing to structural similarity amongst PDI family members, we next assessed the selectivity of KSC-34 for PDIA1 relative to two other PDI family members, PDIA3 and PDIA4, which display 34% and 36% similarity to PDIA1, respectively. Briefly, PDIA1, PDIA3 and PDIA4 (50 μg/mL) were incubated in the presence or absence of MCF-7 lysates (1 mg/mL), and subjected to CuAAC, and in-gel fluorescence (Figure 3D). As indicated by the absence of fluorescence in the PDIA3 and PDIA4 samples, KSC-34 is selective for PDIA1 over other PDI family members. The band observed in the cell-lysate samples for PDIA3 and PDIA4 is the endogenous PDIA1 in the MCF-7 cell lysates, further confirming the selectivity of KSC-34 in that the presence of a large excess of PDIA3 and PDIA4 does not affect the targeting of PDIA1 by KSC-34.
To more comprehensively assess the selectivity of KSC-34 across other cellular PDIs, as well as any proteins bearing highly reactive cysteine residues, the protein targets of KSC-34 were globally investigated. To achieve this, MCF-7 cells were incubated with KSC-34 (5 μM) or DMSO. Upon cell lysis, KSC-34-labeled proteins were then appended to biotin-azide using CuAAC, enriched on streptavidin beads, subjected to on-bead tryptic digestion, and subsequent LC/LC-MS/MS analysis. Spectral counts (number of fragmentation spectra) generated for each protein in the KSC-34-treated samples were compared to the DMSO control (Figure 4, Table S2). In KSC-34-treated samples, an average of ~2000 spectral counts were matched to PDIA1 (with an average of ~5 spectral counts in the DMSO control). The second most enriched protein was prostaglandin E synthase 2 (PTGES2), which was identified with an average of ~120 spectral counts in the KSC-34-treated sample, significantly lower than that for PDIA1. PDIA6 was the only other PDI that was enriched by KSC-34, albeit with low spectral counts of ~20. KSC-34 is therefore highly selective for PDIA1 over other PDIs, as well as cellular proteins bearing reactive cysteines that could potentially be modified by the chloroacetamide electrophile on KSC-34.
Figure 4.

Evaluation of proteome-wide selectivity of KSC-34. Mass spectrometry analysis of proteins enriched in KSC-34 (5 μM) and DMSO treated MCF-7 cells. The spectral count difference between KSC-34-treated and DMSO-treated samples are plotted for all proteins identified (Table S2). Error bars indicate SD for n=2 experiments.
Inhibition of PDIA1 has minimal effects on the cellular unfolded protein response
Given the critical role of PDIA1 in disulfide-bond formation on nascent proteins in the ER, complete loss of PDIA1 activity will likely result in the accumulation of unfolded or misfolded proteins in the ER. Misfolded protein accumulation in the ER typically activates a series of complex stress-responsive signaling pathways known as the unfolded protein response (UPR)36–38. UPR activation involves three different pathways, which implicate inositol-requiring 1α (IRE1α), PKR-like ER kinase (PERK), and activating transcription factor-6α (ATF6α)39, 40. Initial consequences of UPR activation include inhibition of translation, and increased ER protein-folding capacity. Chronic and sustained ER stress will eventually trigger cellular apoptosis. We sought to investigate the effects of KSC-34-mediated a-site inhibition of PDIA1 on each of these three arms of the UPR.
First, we identified an optimal concentration range of KSC-34 to utilize in our UPR assays, which would provide maximal PDIA1 target engagement with minimal toxicity. To quantify PDIA1 target occupancy at varying concentrations of KSC-34, we first synthesized a non-alkyne containing analog, KSC-34na, with identical PDIA1 binding properties to KSC-34, but lacking the biorthogonal handle for further functionalization with reporter tags (Figure S5). To determine PDIA1 occupancy in cells, MCF-7 cells were treated with varying concentrations of KSC-34na. Following cell lysis, protein concentrations were normalized, and the lysates were incubated with KSC-34 (5 μM) to covalently modify any residual, non-inhibitor bound a-site C53 on PDIA1. In-gel fluorescence measurements were used to quantify the amount of PDIA1 that was not occupied by KSC-34na at each concentration. These measurements provided an EC50 of 4 μM for KSC-34na labeling of PDIA1 in MCF-7 cells, and demonstrated that complete occupation of C53 of PDIA1 in cells occurs at concentrations less than ~10 μM. To determine toxicity of KSC-34 at these same concentrations, MTT cell-viability assays were performed in MCF-7 cells, generating an EC50 value of 82 μM (Figure S6), which underscores the relatively low toxicity of a-site selective PDIA1 inhibitors in MCF-7 cells.
Upon identifying non-toxic concentrations of KSC-34 for cellular studies, quantitative transcriptional-profiling methods were utilized to monitor activation of known UPR target genes. Briefly, MCF-7 cells were treated with DMSO, thapsigargin (Tg) (5 μM), and varying concentrations of KSC-34 (4–40 μM) for 3 hours at 37 °C. Tg is a sesquiterpene lactone that induces ER stress by inhibiting sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA). Treated cells were then subjected to qPCR analysis for mRNA encoding known downstream targets preferentially regulated by each of the three arms of the UPR, including SEC24D and ERDJ4 (IRE1α-regulated), BIP and HYOU1 (IRE1α and ATF6-regulated), CHOP and GADD34 (PERK-regulated) and, GRP94 (ATF6-regulated). KSC-34 treatment showed no significant activation of the PERK and ATF6 arms of the UPR (Figure 5A). Interestingly, a small but reproducible increase in SEC24D and ERDJ4 mRNA levels was observed (~2-fold) at >20 μM concentrations of KSC-34, suggesting selective activation of the IRE1α arm under these conditions consistent with minor induction of XBP1 splicing upon KSC-34 treatment (Figure S7). Treatment with an IRE1α inhibitor, 4μ8c, confirmed that the observed effects on SEC24D, ERDJ4, BiP, and HYOU1 were mediated directly through IRE1α (Figure 5B and Figure S6). Further characterization determined that IRE1α activation only occurs within a short timeframe (<6 hours), since longer incubation times led to a loss in upregulation of IRE1α-dependent transcripts (Figure S8). Evaluation of other cell lines demonstrated that this effect cell type-dependent, as no significant effects were observed in SKOV-3 and A549 cells (Figure S9). Together, these data suggest that a-site inhibition of PDIA1 by KSC-34 has minimal effects on activation of the PERK and ATF6 arms of the UPR, with some short-lived, and cell line-dependent effects on the IRE1α arm.
Figure 5.

Effects of KSC-34 on cell viability and the unfolded protein response. (A) qPCR analysis of UPR target genes following concentration-dependent treatment of MCF-7 cells with KSC-34 for 3 hours at 37 °C. qPCR data are reported as the mean fold change relative to the corresponding DMSO-treated cells +/− SEM from n=3 biological replicates. (B) qPCR analysis of UPR target genes following co-treatment of MCF-7 cells with KSC-34 (20 μM) and IRE1α inhibitor, 4u8c. qPCR data are reported relative to the corresponding DMSO-treated cells +/− SEM from n=3 biological replicates.
PDIA1 Inhibition by KSC-34 decreases secretion of an amyloidogenic light chain
PDIA1 influences the folding of disulfide-containing secretory proteins including antibody light chains29, 41–43. Therefore, we sought to evaluate the functional consequence of KSC-34-mediated inhibition of PDIA1 in cell-culture models expressing the destabilized, disease-associated antibody light chain ALLC44. We first performed co-immunoprecipitation (co-IP) experiments to determine how KSC-34-dependent inhibition of PDIA1 influences its interaction with flag-tagged ALLC (FTALLC) in HEK293DAX cells38. PDIA1 was enriched in FTALLC IPs in the absence of KSC-34, confirming that PDIA1 interacts with this destabilized light chain in mammalian cells (Figure 6A). The addition of KSC-34 disrupted this interaction, shown by a decrease in the co-isolation of PDIA1 with FTALLC (Figure 6A). However, the closely related ER protein PDIA4 co-purifies with FTALLC in cells treated with or without KSC-34, demonstrating that this compound does not influence the interaction between these proteins. These results show that KSC-34 selectively disrupts the interaction between FTALLC and PDIA1, demonstrating the high selectivity of KSC-34 for PDIA1 under these cellular conditions.
Figure 6.

KSC-34 reduces secretion of destabilized ALLC from mammalian cells. (A) Immunoblot of anti-FLAG IPs of lysates prepared from HEK293DAX cells transiently transfected with FTALLC and pre-treated for 1 h with vehicle or KSC-34 (40 μM). Cells were crosslinked for 30 min with DSP (500 μM) prior to lysis. Mock transfected cells are included as a control. (B) Graph showing secreted FTALLC (grey) and viability (blue) of HEK293Trex cells stably expressing FTALLC pretreated for 4 h with KSC-34 (40 μM). Media was conditioned for 2 h in the presence or absence of KSC-34 (40 μM) prior to quantification of secreted FTALLC by ELISA. Viability was measured following media conditioning by Cell Titre Glo. All data are normalized to vehicle-treated cells. Error bars show SEM for n=3 experiments. ***p<0.005. (C) Representative autoradiogram and quantification of the fraction [35S]-labeled FTALLC secreted from HEK293DAX cells using the experimental paradigm shown. Experiments were performed in the absence or presence of KSC-34 (40 μM) added 1 h prior to labeling and then again throughout the experiment. Fraction secreted was calculated as described in the Supporting Information 44. Error bars show SEM for n=4. ***p<0.005. (D) Graph showing secreted FTALLC (grey) and viability (blue) of HEK293DAX cells transiently expressing FTALLC pretreated for 4 h with KSC-34 (40 μM). HEK293DAX cells stably expressing PDIA1 shRNA are indicated. Media was conditioned for 2 h in the presence or absence of KSC-34 (40 μM) prior to quantification of secreted FTALLC by ELISA. Viability was measured following media conditioning by Cell Titre Glo. All data are normalized to vehicle-treated controls. Error bars show SEM for n=3 experiments. *p<0.05. (E) Graph showing the fraction [35S]-labeled FTALLC secreted at t=4 h from HEK293DAX cells transiently transfected with FTALLC quantified using the same experimental paradigm shown in Fig. 6C. HEK293DAX cells stably expressing PDIA1 shRNA are indicated. Experiments were performed in the absence or presence of KSC-34 (40 μM) added 1 h prior to labeling and then again throughout the experiment. Error bars show SEM for n=4 experiments. *p<0.05, ***p<0.005.
Next, we determined whether KSC-34-dependent inhibition of PDIA1 influences ALLC secretion. We treated HEK293Trex cells stably expressing FTALLC with KSC-34, and monitored secretion by ELISA. Treatment with KSC-34 reduced levels of FTALLC in the conditioned media by 40%, as compared to vehicle (Figure 6B). This treatment did result in a modest 20% reduction in cellular viability, however this decrease is not sufficient to account for the observed changes in secreted FTALLC. To further define the impact of KSC-34 on FTALLC secretion, we monitored the fraction of newly-synthesized FTALLC secreted from HEK293DAX cells treated with or without KSC-34 using [35S] metabolic labeling (Figure 6C). Pretreatment with KSC-34 reduced the fraction of [35S]-labeled FTALLC secreted by 30%, confirming the results observed by ELISA (Figure 6C). Next, we sought to determine the PDIA1-dependence of these effects on LC secretion utilizing HEK293DAX cells stably expressing PDIA1 shRNA. We confirmed PDIA1 knockdown in these cells by qPCR (Figure S10). Depletion of PDIA1 attenuated the KSC-34-dependent reduction in FTALLC secretion measured by ELISA (Figure 6D) and [35S] metabolic labeling (Figure 6E, Figure S10). This shows that KSC-34 decreases destabilized ALLC secretion through a mechanism dependent on PDIA1. These data support the utility of a-site selective PDIA1 inhibitors to selectively affect secretion of amyloidogenic proteins like destabilized antibody light chains.
In conclusion, we report the discovery and characterization of KSC-34, a potent and selective inhibitor of PDIA1. KSC-34 was identified from a targeted library of compounds generated around an initial lead compound, RB-11-ca. KSC-34 contains a (4-phenylbutyl)methylamine diversity element for optimized binding to the active site of the a domain of PDIA1 with a chloroacetamide electrophile for covalent modification of C53 on PDIA1. Notably, the presence of a bioothogonal alkyne handle on KSC-34 and derivatives, facilitated the rapid screening of these compounds for potency and selectivity both in vitro against purified PDIs, as well as directly in living cells. Importantly, despite the prominent reactivity of the chloroacetamide electrophile on KSC-34, extremely high selectivity is observed for PDIA1 within a complex proteome. The high inhibitory potency of KSC-34 was confirmed in an in vitro insulin-reduction assay, where KSC-34 proved to be approximately 38-fold more potent than the commercially available PDIA1 inhibitor 16F16. Importantly, KSC-34 is unique in that it displays selective binding to the a domain of PDIA1, and is the most site-selective PDIA1 inhibitor reported to date. KSC-34 treatment generally results in minimal sustained activation of the UPR, although some low cell-specific, short-lived effects were observed with the IRE1α arm in MCF-7 cells. Lastly, the ability of KSC-34 to reduce the extracellular pathogenic load of amyloidogenic antibody light-chain is demonstrated, highlighting the potential of site-selective PDIA1 inhibitors to have therapeutic value by selectively downregulating a subset of PDIA1 functions in cells. The development of KSC-34 validates the potential to selectively, and potently pharmacologically modulate individual thioredoxin-like active-site domains within a single member of the PDI family. KSC-34 is a useful tool compound to further interrogate the cellular functions of the a domain of PDIA1, and additionally, to explore the therapeutic value of site-selective PDI inhibitors.
Supplementary Material
Acknowledgments
We thank members of the Weerapana, Shoulders and Wiseman Labs for assistance with experiments and manuscript preparation. We thank Tyler Bechtel and Inchul You from the Weerapana Lab for providing purified recombinant PDIA3 and PDIA4. This work was funded by NIH grant 1R01GM118431-01A1 (E.W.), 1R01DK107604 (R.L.W.), and 1R01AR071443 (M.D.S.).
Footnotes
Notes
The authors declare no competing financial interests.
Supplemental Information includes Supplemental Experimental Procedures, 9 Supplemental Figures (Figures S1–S9), and 2 data tables (Tables S1–S2). This information can be found with this article online.
References
- 1.Kozlov G, Maattanen P, Thomas DY, Gehring K. A structural overview of the PDI family of proteins. FEBS J. 2010;277:3924–3936. doi: 10.1111/j.1742-4658.2010.07793.x. [DOI] [PubMed] [Google Scholar]
- 2.Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta. 2008;1783:535–548. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Xu S, Sankar S, Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today. 2014;19:222–240. doi: 10.1016/j.drudis.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 4.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 5.Goldberger RF, Epstein CJ, Anfinsen CB. Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J Biol Chem. 1963;238:628–635. [PubMed] [Google Scholar]
- 6.Venetianer P, Straub FB. The enzymic reactivation of reduced ribonuclease. Biochim Biophys Acta. 1963;67:166–168. doi: 10.1016/0006-3002(63)91812-2. [DOI] [PubMed] [Google Scholar]
- 7.Freedman RB. Native Disulfide Band Formation in Protein-Biosynthesis - Evidence for the Role of Protein Disulfide Isomerase. Trends Biochem Sci. 1984;9:438–441. [Google Scholar]
- 8.Vuori K, Myllyla R, Pihlajaniemi T, Kivirikko KI. Expression and site-directed mutagenesis of human protein disulfide isomerase in Escherichia coli. This multifunctional polypeptide has two independently acting catalytic sites for the isomerase activity. J Biol Chem. 1992;267:7211–7214. [PubMed] [Google Scholar]
- 9.Klappa P, Ruddock LW, Darby NJ, Freedman RB. The b′ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 1998;17:927–935. doi: 10.1093/emboj/17.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Network TCGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shai R, Shi T, Kremen TJ, Horvath S, Liau LM, Cloughesy TF, Mischel PS, Nelson SF. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene. 2003;22:4918–4923. doi: 10.1038/sj.onc.1206753. [DOI] [PubMed] [Google Scholar]
- 12.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 13.Shibata E, Ejima K, Nanri H, Toki N, Koyama C, Ikeda M, Kashimura M. Enhanced protein levels of protein thiol/disulphide oxidoreductases in placentae from pre-eclamptic subjects. Placenta. 2001;22:566–572. doi: 10.1053/plac.2001.0693. [DOI] [PubMed] [Google Scholar]
- 14.Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U, Frede S, Toietta G, Di Rocco G, Bussani R, Silvestri F, Piro M, Liuzzo G, Biasucci LM, Mellone P, Feroce F, Capogrossi M, Baldi F, Fandrey J, Ehrmann M, Crea F, Abbate A, Baldi A. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol. 2007;50:1029–1037. doi: 10.1016/j.jacc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Laurindo FR, Fernandes DC, Amanso AM, Lopes LR, Santos CX. Novel role of protein disulfide isomerase in the regulation of NADPH oxidase activity: pathophysiological implications in vascular diseases. Antioxid Redox Signal. 2008;10:1101–1113. doi: 10.1089/ars.2007.2011. [DOI] [PubMed] [Google Scholar]
- 16.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 17.Unterberger U, Hoftberger R, Gelpi E, Flicker H, Budka H, Voigtlander T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol. 2006;65:348–357. doi: 10.1097/01.jnen.0000218445.30535.6f. [DOI] [PubMed] [Google Scholar]
- 18.Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 19.Hoffstrom BG, Kaplan A, Letso R, Schmid RS, Turmel GJ, Lo DC, Stockwell BR. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol. 2010;6:900–906. doi: 10.1038/nchembio.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan MM, Simizu S, Lai NS, Kawatani M, Shimizu T, Osada H. Discovery of a small molecule PDI inhibitor that inhibits reduction of HIV-1 envelope glycoprotein gp120. ACS Chem Biol. 2011;6:245–251. doi: 10.1021/cb100387r. [DOI] [PubMed] [Google Scholar]
- 21.Jiang XM, Fitzgerald M, Grant CM, Hogg PJ. Redox control of exofacial protein thiols/disulfides by protein disulfide isomerase. J Biol Chem. 1999;274:2416–2423. doi: 10.1074/jbc.274.4.2416. [DOI] [PubMed] [Google Scholar]
- 22.Lin L, Gopal S, Sharda A, Passam F, Bowley SR, Stopa J, Xue G, Yuan C, Furie BC, Flaumenhaft R, Huang M, Furie B. Quercetin-3-rutinoside Inhibits Protein Disulfide Isomerase by Binding to Its b′x Domain. J Biol Chem. 2015;290:23543–23552. doi: 10.1074/jbc.M115.666180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eirich J, Braig S, Schyschka L, Servatius P, Hoffmann J, Hecht S, Fulda S, Zahler S, Antes I, Kazmaier U, Sieber SA, Vollmar AM. A small molecule inhibits protein disulfide isomerase and triggers the chemosensitization of cancer cells. Angew Chem Int Ed Engl. 2014;53:12960–12965. doi: 10.1002/anie.201406577. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan A, Gaschler MM, Dunn DE, Colligan R, Brown LM, Palmer AG, 3rd, Lo DC, Stockwell BR. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc Natl Acad Sci U S A. 2015;112:E2245–2252. doi: 10.1073/pnas.1500439112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, Zandi E, Petasis NA, Neamati N. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc Natl Acad Sci U S A. 2012;109:16348–16353. doi: 10.1073/pnas.1205226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allimuthu D, Adams DJ. 2-Chloropropionamide As a Low-Reactivity Electrophile for Irreversible Small-Molecule Probe Identification. ACS Chem Biol. 2017;12:2124–2131. doi: 10.1021/acschembio.7b00424. [DOI] [PubMed] [Google Scholar]
- 27.Ge J, Zhang CJ, Li L, Chong LM, Wu X, Hao P, Sze SK, Yao SQ. Small molecule probe suitable for in situ profiling and inhibition of protein disulfide isomerase. ACS Chem Biol. 2013;8:2577–2585. doi: 10.1021/cb4002602. [DOI] [PubMed] [Google Scholar]
- 28.Banerjee R, Pace NJ, Brown DR, Weerapana E. 1,3,5-Triazine as a modular scaffold for covalent inhibitors with streamlined target identification. J Am Chem Soc. 2013;135:2497–2500. doi: 10.1021/ja400427e. [DOI] [PubMed] [Google Scholar]
- 29.Mayer M, Kies U, Kammermeier R, Buchner J. BiP and PDI cooperate in the oxidative folding of antibodies in vitro. J Biol Chem. 2000;275:29421–29425. doi: 10.1074/jbc.M002655200. [DOI] [PubMed] [Google Scholar]
- 30.Gertz MA. Immunoglobulin light chain amyloidosis: 2016 update on diagnosis, prognosis, and treatment. Am J Hematol. 2016;91:947–956. doi: 10.1002/ajh.24433. [DOI] [PubMed] [Google Scholar]
- 31.Wang C, Li W, Ren J, Fang J, Ke H, Gong W, Feng W, Wang CC. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid Redox Signal. 2013;19:36–45. doi: 10.1089/ars.2012.4630. [DOI] [PubMed] [Google Scholar]
- 32.Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, Harder E. Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J Chem Inf Model. 2014;54:1932–1940. doi: 10.1021/ci500118s. [DOI] [PubMed] [Google Scholar]
- 33.Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 34.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 35.Holmgren A. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J Biol Chem. 1979;254:9627–9632. [PubMed] [Google Scholar]
- 36.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 37.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 38.Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, 3rd, Su AI, Kelly JW, Wiseman RL. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013;3:1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cawley K, Deegan S, Samali A, Gupta S. Assays for detecting the unfolded protein response. Methods Enzymol. 2011;490:31–51. doi: 10.1016/B978-0-12-385114-7.00002-7. [DOI] [PubMed] [Google Scholar]
- 40.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 41.Borth N, Mattanovich D, Kunert R, Katinger H. Effect of increased expression of protein disulfide isomerase and heavy chain binding protein on antibody secretion in a recombinant CHO cell line. Biotechnol Prog. 2005;21:106–111. doi: 10.1021/bp0498241. [DOI] [PubMed] [Google Scholar]
- 42.Lilie H, McLaughlin S, Freedman R, Buchner J. Influence of protein disulfide isomerase (PDI) on antibody folding in vitro. J Biol Chem. 1994;269:14290–14296. [PubMed] [Google Scholar]
- 43.Roth RA, Pierce SB. In vivo cross-linking of protein disulfide isomerase to immunoglobulins. Biochemistry. 1987;26:4179–4182. doi: 10.1021/bi00388a001. [DOI] [PubMed] [Google Scholar]
- 44.Cooley CB, Ryno LM, Plate L, Morgan GJ, Hulleman JD, Kelly JW, Wiseman RL. Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc Natl Acad Sci U S A. 2014;111:13046–13051. doi: 10.1073/pnas.1406050111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.