

Abstract
Activation-induced cytidine deminase (AID) is crucial for controlling the immunoglobulin (Ig) diversification processes of somatic hypermutation (SHM) and class switch recombination (CSR). AID initiates these processes by deamination of cytosine, ultimately resulting in mutations or double strand DNA breaks needed for SHM and CSR. Levels of AID control mutation rates, and off-target non-Ig gene mutations can contribute to lymphomagenesis. Therefore, factors that control AID levels in the nucleus can regulate SHM and CSR, and may contribute to disease. We previously showed that transcription factor YY1 can regulate the level of AID in the nucleus and Ig CSR. Therefore, we hypothesized that conditional knock-out of YY1 would lead to reduction in AID localization at the Ig locus, and reduced AID-mediated mutations. Using mice that overexpress AID (IgκAID yy1f/f) or that express normal AID levels (yy1f/f), we found that conditional knock-out of YY1 results in reduced AID nuclear levels, reduced localization of AID to the Sμ switch region, and reduced AID-mediated mutations. We find that the mechanism of YY1 control of AID nuclear accumulation is likely due to YY1-AID physical interaction which blocks AID ubiquitination.
Keywords: YY1, AID, Somatic Hypermutation, Class Switch Recombination
Introduction
Formation of functional immunoglobulin (Ig) genes in the B cell lineage requires the somatic rearrangement of variable (V), diversity (D), and joining (J) segments [1]. After antigen stimulation, further Ig gene diversification occurs largely within germinal centers where both Ig light (L) and heavy (H) chain genes undergo somatic hypermutation (SHM) to produce Ig molecules with increased affinity for antigen. Additionally, the IgH locus undergoes class switch recombination (CSR) to place the rearranged VDJ segment adjacent to one of 8–10 distinct constant (C) region segments to produce various Ig isotypes that provide distinct effector functions [2–4].
CSR is a complex process that involves approximately 50 different proteins [5–9]. CSR is induced by various cytokines that activate transcription within switch regions upstream of each IgH C region with the exception of the Cδ region. This transcriptional process enables access of activation-induced cytidine deaminase (AID) that deaminates cytosine to uracil. Action of DNA repair processes (mismatch repair and base excision repair) results in mutations and double strand DNA breaks within the switch regions which are subsequently joined by ligation between the Cμ switch break and the corresponding activated and broken switch region to generate an IgH gene with the same VDJ segment linked to a new C region.
Key for initiating the process of CSR is the mutagenic activity of AID. AID knockout mice, and patients with autosomal recessive AID mutations, generate only low affinity antibodies of IgM isotype and thus suffer from a severe immunodeficiency known as hyper IgM syndrome (HIGM2) [10]. Conversely, overexpression of AID can cause mutations leading to cancer. AID function must be tightly regulated to avoid deleterious mutagenic activity at off-target non-Ig genes. AID catalyzed cytidine deamination is believed to be involved in generation of lymphomagenic mutations and chromosome translocations, and overexpression of AID in transgenic animals leads to T cell lymphomas and tumors in lung epithelium [11–14]. AID expression is also implicated in a growing list of cancers apart from B cell leukemias and lymphomas. AID is aberrantly expressed in numerous solid tumors such as colitis-associated colorectal cancer, hepatocellular carcinoma, gastric cancer, pancreatic cancer, lung cancer and cholangiocarcinoma [15–20].
AID expression levels directly correlate with the frequency of AID-dependent DNA remodeling events and incidence of c-myc/IgH translocations [12, 21–24]. Therefore, limiting AID levels in the nucleus protects the B cell genome from mistargeted mutations and this is regulated by multiple mechanisms. AID is expressed at very low levels in naïve B cells, but is dramatically up-regulated in activated B cells [25]. Most of the AID protein is retained in the cytoplasm with only a small fraction translocating into the nucleus to mediate CSR and SHM [26–29]. AID is actively exported from the nucleus by a CRM1-exportin-dependent mechanism to regulate nuclear AID levels [27–31], and AID stability is greatly reduced in the nucleus by polyubiquitination and consequent degradation in nuclear proteasomes [30]. A number of factors regulate AID stability including eEF1A, REG-γ, and Hsp90 [31–34]. Factors that impact AID nuclear stability and accumulation would have profound impact on AID function in CSR and SHM, and may also contribute to lymphomagenesis if misregulated.
Previously, we showed that transcription factor YY1 controls IgH CSR [35]. Using a conditional ex vivo knock-out system we found that ablation of YY1 in primary splenic B cells results in a large drop in CSR. Loss of YY1 did not impact transcription of switch region sequences or splenic B cell proliferation needed for CSR. Instead, we found that YY1 physically interacts with AID and regulates its nuclear accumulation, apparently by controlling AID stability [35]. As AID is required for CSR, we proposed that YY1 may control CSR, at least in part, by regulating the amount of nuclear AID. We hypothesized here that conditional knock-out of YY1 will reduce localization of AID to the Sμ switch region DNA sequence, and will reduce AID mutagenesis. Our results support this hypothesis and demonstrate that YY1 likely controls AID nuclear stability by regulating AID ubiquitination. Therefore, YY1 regulation of AID protein stability in the nucleus impacts AID mutagenesis and this may relate to lymphomagenesis.
Results
YY1 affects AID mutation frequency
Deletion of YY1 results in dramatic reduction of Ig CSR [35]. YY1 loss does not impact Ig switch region transcripts or cell proliferation. Instead, we found that loss of YY1 reduces the level of nuclear AID, and overexpression of YY1 increases AID by increasing its nuclear half life [35]. The ability of YY1 to control AID stability suggested that it might control AID mutagenic activity. To investigate this, we measured AID mutagenic activity initially using IgκAID transgenic mice that overexpress AID, resulting in high levels of mutation in the IgH mu switch region sequence (Sμ) and elevated CSR [36]. We crossed these mice onto a yy1f/f background so that YY1 could be deleted ex vivo by addition of recombinant TAT-CRE protein [35]. We reasoned that loss of YY1 would result in reduced nuclear AID and reduced CSR, similar to our previous work [35]. Indeed, treatment of IgκAID yy1f/f splenic B cells with recombinant TAT-CRE resulted in loss of YY1, reduced nuclear AID, and reduced CSR (Supporting Information Fig. 1A–D). Thus, we set out to determine if loss of YY1 would result in reduced AID mutagenic activity.
Splenic B cells were isolated from six individual IgκAID yy1f/f mice. Cells were either mock treated or treated with recombinant TAT-CRE protein to delete YY1, then cultured in LPS plus IL4 for 4 days. DNA was isolated and then evaluated by Sanger dideoxy sequencing after subcloning of the PCR-amplified IgH Sμ switch region. Mock treated samples showed an average mutation frequency of 58.7 × 10−4, in close agreement with the previously published frequency (57.4 × 10−4) [36] (Fig. 1A). However, the TAT-CRE treated samples showed a significant drop in average mutation frequency to 45.4 × 10−4 (p<0.003) (Fig. 1A). Data from individual mice are shown in Supporting Information Fig. 2, and Supporting Information Table 1. Deletion of YY1 also resulted in a 2.5 fold increase in the number of sequences with no mutations (14.8% vs 5.9%) (Fig. 1B). Thus, loss of YY1 in IgκAID yy1f/f splenic B cells resulted in a drop in mutation frequency at Sμ. Conversely, YY1 deletion had no impact on the mutation frequency at the AID non-target transmembrane activator and CAML interactor (Taci) gene (0.67 × 10−4 for mock and 0.62 × 10−4 for TAT-CRE treated samples) (Fig. 1C and Supporting Information Table 2).
Figure 1.
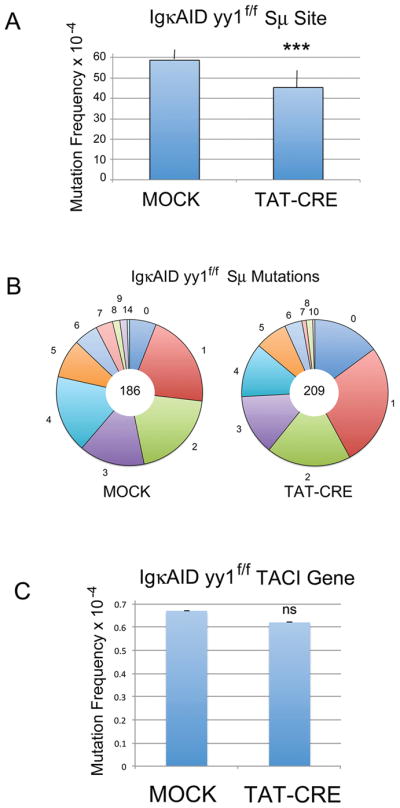
YY1 effect on AID-mediated mutations within the Sμ region in IgκAID yy1f/f mice. (A) Average mutation frequency at the Sμ region from 6 independent IgκAID yy1f/f mice. Isolated splenic B cells were either mock treated, or treated with TAT-CRE to delete the yy1 gene, then induced with LPS plus IL4 in culture for 4 days. DNA was isolated, the Sμ region was amplified by PCR, cloned, and subjected to Sanger sequencing. Error bars indicate the standard deviation from the mean. The three asterisks denote p<0.002 in a two tailed T-test. (B) The frequency of Sμ mutation numbers in a sequence (indicated by the numbers around the periphery of the circle) are shown by the size of the pie slice. The total number of sequenced clones is shown in the middle of each pie. (C) Average mutation frequency at the Taci gene from 6 individual mice detected by Sanger sequencing of mock and TAT-CRE treated samples. The same DNA from 6 independent mice used in (A) above was amplified with primers to the Taci gene promoter and individual clones were sequenced. Error bars represent the standard deviation from the mean. There was no statistical difference at the Taci gene between Mock and TAT-CRE treated samples (ns) in a two tailed T-test.
To test the effect of YY1 on AID mutagenesis in a more physiological setting (non-AID overexpressing mice) we deleted YY1 in splenic B cells from yy1f/f mice that express wildtype levels of AID. As in the previous experiment, we compared mock treated and TAT-CRE treated splenic B cells that were cultured ex vivo for 4 days in LPS and IL4. We analyzed regions known to accumulate AID mutations, i.e., the IgH Sμ region and IgH Jh4 intron sequences. We expected much lower levels of overall AID-mediated mutations compared to IgκAID yy1f/f B cells which overexpress AID [36]. To detect this much lower mutation frequency (up to 10−7) we used ultra-deep next generation sequencing (NGS) yielding very high coverage (0.6 to 1.5 billion total base pairs were sequenced depending on the locus) (Supporting Information Tables 3, 4). Raw sequence data were processed by a deep-single nucleotide variant (deep-SNV) algorithm which identifies single nucleotide variants (SNVs) above the sequencing background as a result of pairwise comparison of mock and TAT-CRE treated samples detecting statistically significant differences in variant allele frequency (VAF) at individual nucleotide positions (for details see Material and Methods). We evaluated DNA mutations in six independent experiments, and observed significantly reduced accumulation of SNVs in IgH Sμ and Jh4 loci after TAT-CRE treatment (p<0.05, Wilcoxon test; Fig. 2A, Supporting Information Table 3). Supporting Information Figs. 3–5 show VAF in mock and TAT-CRE treated samples compared to sequencing background in AID knockout samples.
Figure 2.

YY1 effect on AID mediated mutations and AID genomic localization in ex vivo stimulated splenic B cells from yy1f/f mice. (A) Frequencies of single nucleotide variants (SNVs) in Sμ, Jh4, and Cd83 promoter regions detected by NGS. Splenic B cells isolated from six individual yy1f/f mice were either mock treated or treated with TAT-CRE to delete the yy1 gene. After induction for 4 days with LPS plus IL4, DNA was isolated and DNA sequences at the Sμ, Jh4, and CD83 promoter were subjected to NGS DNA sequencing. The frequencies of single nucleotide varients are shown in mock treated (black columns) or TAT-CRE treated (white columns) mice. Differences between mock and TAT-CRE were calculated using the Wilcoxon test. (B and C) Recruitment of AID to the Sμ region, and YY1 to the Eμ enhancer and rpL30 promoter. Mock and TAT-CRE treated splenic yy1f/f B cells were transduced with retroviral vector pMX-HA-AID, and two days later cells were subjected to ChIP with control anti-IgG or anti-HA antibody (B), or YY1 antibody (C). QPCR was performed with primers that amplify the IgH Sμ region (B) or Eμ enhancer and rpL30 promoter (C). Error bars indicate standard deviation from the mean, and asterisks indicate p<0.001 in a two tailed T-test. (D) Quantitative RT-PCR of IgM germline transcripts in mock and TAT-CRE treated samples. RNA isolated from Mock and TAT-CRE treated samples was evaluated by quantitative RT-PCR with primers that detect the germline IgM transcript. Data are from six independent experiments and error bars show the standard deviation of the mean.
These results indicate that YY1 plays a role in accumulation of mutations in the IgH variable region and switch Sμ sequences. As YY1 knock-out reduces AID nuclear levels [35], we observed dramatically reduced AID binding at the Sμ region (Fig. 2B). Loss of YY1 protein due to TAT-CRE treatment also greatly reduced YY1 binding at the Eμ enhancer and the rpL30 promoter (Fig. 2C). Reduced AID and YY1 binding at the IgH locus was not due to reduced transcription of the locus, as IgM germline transcripts were unchanged by TAT-CRE treatment (Fig. 2D).
Next, we asked whether YY1 affects AID mutagenesis outside of Ig loci. AID exerts its mutagenic activity genome-wide with a strong preference for promoter proximal regions of highly expressed genes associated with stalled RNA polymerase and Spt5 transcriptional pausing factor [37, 38]. We sequenced about 1 kb downstream of the Cd83 promoter, as this genomic region accumulates high levels of AID mutations in wildtype mice in vivo, and in IgAID Ung−/− B cells stimulated ex vivo [14, 38, 39]. Using NGS, we detected a 2.9 fold lower SNV frequency in TAT-CRE treated compared to mock treated samples (Fig. 2A, p<0.05, Wilcoxon test, Supporting Information Table 3). Our sequencing results were highly reproducible as demonstrated by multiple independent sequencing runs yielding similar results using DNA samples from the same mouse (data not shown).
YY1 knock-out does not alter AID mutation spectrum
AID preferentially mutates the RGYW/WRCY hotspot motif (W=A/T, R=A/G, Y=C/T) [14, 40]. Therefore, we evaluated whether mutations we detected in Sμ, Jh4 and Cd83 loci were within RGYW/WRCY hotspots. Indeed, we found increased targeting of AID hotspot motifs in all three loci. RGYW/WRCY motifs were targeted in 63% or 72% of all mutations in mock or TAT-CRE samples in the Ig switch regions, 35% or 17% of mutations in the Jh4 intron, and 78% or 79% in mock and TAT-CRE samples in the Cd83 gene (Table 1). There was a two-fold drop in hotspot mutations at the Jh4 intron, though the reason for this drop is unclear. Even more mutations accumulated in more degenerate motifs GYW/WRC and GNW/WNC also known to be targeted by AID [41] (Table 1). Thus, loss of YY1 did not change AID hotspot preference apart from the two-fold drop detected in Jh4.
Table 1.
Percent Mutations in AID Hotspots. Percent mutations and transitions in G/C nucleotides.
| Locus | Sequencing Technique | Mouse Genotype | Sample | All Mut in RGYW/WRCY (%) | All Mut in GYW/WRC (%) | All Mut in GNW/WNC (%) | Mut in G/C (%) * | Transitions in G/C (%) * |
|---|---|---|---|---|---|---|---|---|
| Sμ | NGS | yy1 f/f | MOCK | 63 | 76 | 82 | 95 | 63 |
| Sμ | NGS | yy1 f/f | TAT-CRE | 72 | 82 | 83 | 92 | 63 |
| Jh4 | NGS | yy1 f/f | MOCK | 35 | 50 | 56 | 68 | 78 |
| Jh4 | NGS | yy1 f/f | TAT-CRE | 17 | 38 | 42 | 53 | 70 |
| Cd83 | NGS | yy1 f/f | MOCK | 78 | 97 | 97 | 98 | 85 |
| Cd83 | NGS | yy1 f/f | TAT-CRE | 79 | 93 | 93 | 91 | 96 |
| Sμ | Sanger | IgκAID yy1f/f | MOCK | 59 | 70 | 83 | 95 | 67 |
| Sμ | Sanger | IgκAID yy1f/f | TAT-CRE | 61 | 74 | 84 | 97 | 70 |
C – Cytosine, G – Guanine, R – purine bases (A or G), Y – pyrimidine bases (C or T), W – A or T, N – any nucleotide
corrected for base composition
G/C content of individual loci: Sμ 46.3%, Jh4 42.8%, Cd83 59.2%
Next, we examined the proportion of G/C vs A/T nucleotides as well as the number of transitions versus transversions in our dataset. Both YY1-containing and YY1-deficient samples showed very high targeting of mutations to G or C nucleotides, and the mutations were skewed towards transitions (Table 1). Overall, our results show that YY1 deficiency resulted in a significant drop of AID mutation frequency in Ig and non-Ig loci, and the mutations that we detected by both Sanger and NGS sequencing displayed a typical AID mutation signature.
YY1 does not affect AID nuclear export
The results presented here are consistent with our previous observations that YY1 influences AID nuclear stability [35]. YY1 could also potentially regulate AID by reducing its nuclear export. AID is actively exported from the nucleus by a Crm1-mediated mechanism that requires the C-terminal AID amino acids 188–198, termed the nuclear export sequence (NES) [27, 28, 29 ]. Deletion of these AID amino acids ablates AID nuclear export, and therefore all AID remains in the nucleus. If the function of YY1 is to reduce AID nuclear export, it should have no impact on nuclear levels of the AIDΔNES mutant. However, transfection of CMV-YY1 with either Flag-AID or Flag-AIDΔNES resulted in increased nuclear accumulation of both proteins (Fig. 3). Thus, YY1-mediated nuclear accumulation of AID is not due to regulation of AID nuclear export.
Figure 3.

Effects of YY1 on AID nuclear export. HEK293T cells were transfected with plasmids expressing either CMV-FlagAID or CMV-FlagAIDΔNES (which deletes the AID nuclear export sequence causing loss of AID nuclear export), along with 6 or 18μg of pCDNA3 vector expressing YY1 (CMV-YY1). Cells were harvested 2 days later and 20 or 40μg of nuclear extract protein was probed on western blots with anti-Flag, anti-YY1, or anti-TFIIB. Representative results from three independent experiments are shown.
YY1 controls AID ubiquitination
As YY1 can control AID stability, we sought to determine whether YY1 impacted AID ubiquitination. We transfected Flag-AID into HEK293T cells in either the presence or absence of YY1 expression vector, and inhibited proteasomal degradation with MG-132. Nuclear extracts were isolated, denatured to dissociate interacting proteins, and immunoprecipitated with anti-Flag antibody, then blotted with anti-ubiquitin antibody. AID was clearly ubiquitinated as evidenced by the distinct bands larger than unmodified AID (arrows in Fig. 4A). The high molecular weight signal also suggested that AID could be polyubiquitinated (Fig. 4A, marked by an asterisk). Overexpression of YY1 caused a significant drop in the level of AID ubiquitination (Fig. 4A). Data from 9 independent experiments are shown in Supporting Information Fig. 6A. YY1 can physically interact with AID through sequences contained within YY1 amino acids 1–200 [35]. Transfection of a plasmid expressing YY1 sequences 1–200 also resulted in a significant drop in AID ubiquitination (Fig. 4B). Data from 7 independent experiments are show in Supporting Information Fig 6B. To explore whether YY1 inhibition of AID ubiquitination is likely due to YY1-AID physical interaction, we tested YY1 deletion mutant, YY1 1–200Δ16–80 which greatly reduces YY1-AID interaction [35]. This mutant was six times less efficient, on average, at reducing AID ubiquitination compared to YY1 1–200 (Fig. 4C). Results from 6 independent experiments are shown in Supporting Information Fig. 6C. Thus, we conclude that YY1 can reduce AID ubiquitination, and this block likely requires YY1-AID physical interaction.
Figure 4.

Effects of YY1 on AID ubiquitination. HEK293T cells were transfected with CMV-FlagAID plus either GALYY1 or various YY1 mutants. Two days after transfection cells were treated with MG132 to inhibit proteosomal degradation, nuclear extracts were prepared, heated to dissociate protein interactions, immunoprecipitated with anti-Flag, then subjected to western blot with anti-ubiquitin antibody. Blots were subsequently stripped and probed with anti-Flag to indicate the level of AID. Expression of various GAL-YY1 fusion proteins was detected by western blot with anti-GAL4 antibody. (A) Experiments with wild type YY1 fused to the GAL4 DNA binding domain. Arrows show ubiquitination bands higher than the size of AID, and the asterisk denotes possible AID polyubiquitination. (B) Experiments with GALYY1 compared to the GALYY1 1–200 mutant containing YY1 amino acids 1–200. (C) Experiments with GALYYY1 1–200 compared to GALYY1 1–200Δ16–80. In all cases the top panel shows signals with anti-ubiquitin antibody, middle panel the amount of immunoprecipitated Flag-AID, and the bottom panel, the western signal with anti-GAL4 antibody. Quantification of 9, 7, and 6 independent experiments is shown in the right panel of each figure, respectively. The densitometric signal in each lane with the anti-ubiquitin antibody was normalized to the amount of AID in the same sample observed with FLAG antibody. Error bars show standard deviation from the mean. Two asterisks denote p<0.02 and a single asterisk denotes p<0.05 in a two tailed T-test.
Discussion
In this work, we have demonstrated that YY1 can regulate the accumulation of AID-mediated mutations, apparently by controlling AID ubiquitination, subsequent stability, and binding to the Sμ region. As AID is directly responsible for development of diffuse large B cell lymphoma [11], these results may relate to lymphomagenesis. Although YY1 could impact AID stability by indirect mechanisms such as regulating expression of genes that control AID stability, we favor a direct physical interaction model. We previously showed that in the nucleus, YY1 physically interacts with AID and can regulate AID nuclear half life [35]. We propose YY1-AID interaction blocks AID ubiquitination, resulting in AID stabilization. Conversely, loss of YY1 results in reduced AID stability, nuclear accumulation, Sμ DNA binding, and AID-mediated mutation.
Recently, it was shown that activation of Parp1 by DNA damage results in reduced proteasomal degradation of AID, and increased nuclear accumulation [42]. YY1 does not regulate expression of Parp1, but our previous RNA transcript data in LPS plus IL4 activated splenic B cells show that YY1 positively regulates Parp2 expression [43]. Whether YY1 and Parp1 function by the same mechanism to regulate AID nuclear levels is not clear. It is currently not known whether the Parp1-mediated stabilization of AID is due to direct physical interaction with AID or to indirect mechanisms. On the contrary, REG-γ can regulate nuclear AID accumulation apparently by direct physical interaction [33]. REG-γ is implicated in ubiquitin-independent degradation and its physical association with AID leads to accelerated proteasomal degradation of AID [33]. It will be interesting to determine if YY1 competes with REG-γ for interaction with AID, thus enabling AID stabilization and binding to the Sμ region. Within the cytoplasm, AID stability also can be regulated by physical interaction with eIF1A and Hsp90 [31, 34, 44]. These factors could also augment AID function by increasing overall AID accumulation.
The AID mutation distribution we observed here showed some differences compared with vivo data reported in wildtype mice where frequency of mutations at A/T and G/C bases were similar (reviewed in [45]). However, our results showing the preference for G/C mutations and transitions pointed to AID activity. In addition, preferred G/C targeting in ex vivo culture systems was previously reported by others [46, 47] and might be caused by altered expression of BER or MMR repair enzymes and/or error-prone polymerases (particularly DNA polymerase eta known for targeting WA motifs) which contribute to mutations in A/T bases during somatic hypermutation.
We previously demonstrated that YY1 deletion in B cells ex vivo dramatically reduces class switch recombination (CSR) [35] and we proposed that loss of CSR is caused at least in part by reduced nuclear AID levels. However, we recently found that YY1 also controls other functions that impact CSR. Specifically, we found that YY1 conditional knock-out ablates the long-distance 220kb DNA loop between the Eμ and 3′RR enhancers believed to be necessary for CSR [43]. We further showed that the YY1 C-terminal half which lacks the transcriptional activation domain is sufficient for controlling this loop [43]. These results indicate that YY1 impacts B cell function by numerous mechanisms including control of gene expression, long-distance DNA loops, and AID stability and nuclear function.
SHM and CSR generally occur in germinal centers (GC), and YY1 has been proposed to be a master regulator of the GC-specific transcriptional program [48]. Recently, we and others showed that YY1 plays a critical role in GC development and maintenance. YY1 conditional ablation in mice results in a significant decrease of GC B cells and germinal centers [49, 50, 51]. However, RNA transcript analyses suggest that the impact of YY1 on GC development is not a stage-specific B cell effect, but a more general effect [43, 50]. YY1 knock-out impacts numerous cellular processes including mitochondrial function. In addition, YY1 conditional deletion at multiple B cell stages halts further development suggesting that a more basic cellular function common to all cells is being impacted [50].
Our results here clearly indicate that YY1 can impact AID-mediated mutation frequencies. The mechanism of this control likely involves regulation of AID nuclear stability and concomitant AID DNA binding and mutagenesis. The consequences of this regulation on development of DLBCL and other lymphomas will require additional studies.
Materials and methods
Mice
YY1f/f mice described in Liu H et al. [52] were a gift from Yang Shi (Harvard). IgκAID mice described in Robbiani et al. [36] and AID knock-out mice were provided by Michel Nussenzweig (Rockefeller University). We crossed IgκAID and yy1f/f mice to generate IgκAID yy1f/f mice on a C57BL/6 background. Male and female animals between 8 and 12 weeks of age were used for experiments. All animal studies were performed in compliance with the U.S. Department of Health and Human Services guidelines and were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
YY1 deletion by TAT-CRE treatment in activated splenic B cells and measurement of CSR
Isolation of splenic B cells from either IgκAIDyy1f/f or yy1f/f mice, deletion of YY1 by recombinant TAT-CRE treatment, LPS plus IL4 treatment, and measurement of CSR to IgG1, were performed as previously described [35]. Briefly, follicular B cells were purified from mouse spleen with anti-CD23-biotin (eBioscience) and streptavidin microbeads (MACS, Miltenyi Biotec), and conditional YY1 knock-out was performed ex vivo using TAT-CRE enzyme purified from bacteria. Cells were activated ex vivo with 10 μg/ml LPS (Sigma) plus 20 ng/ml IL-4 (Peprotech). Splenic B cells were stained with PE anti-mouse IgG1 (BD Pharmingen) and 7-amino-actinomycin D (Invitrogen) and isotype switching was measured by flow cytometry (dead cells stained with 7AAD were excluded from analysis). Flow cytometry was performed on a FACS Canto II machine, and data was analyzed using FlowJo software.
Mutational analysis
DNA samples (200 ng) from individual IgκAID yy1f/f mice either mock treated or treated with recombinant TAT-CRE protein were amplified in 24 cycles of PCR using Q5® High-Fidelity DNA polymerase (NEB). The IgH switch sequence preceding the Sμ core region was amplified with primers Sm.F: GACCCAGGCTAAGAAGGCAATC and Sm.R: GCGGCCCGGCTCATTCCAGTTCATTACAG yielding a 542 bp product [36]. The Taci gene sequence was amplified with primers: GTCAGGTCAGACAACTCAGGAAGG and GTTTGCCACCCACATCAAGC. Amplified products were A-tailed and cloned into the pGEM-T vector (Promega) for Sanger DNA sequencing. The Cd83 locus was amplified with MusgCd83.F2 CTCCTCCGACTGGGGAGT and MusgCd83.R2 CAATGTTGGAGTCTGAGGGCT yielding a 1020 bp product. The Jh4 intron sequence was amplified in a nested PCR with MusVhJ558.F 5′-GGAATTCGCCTGACATCTGAGGACTCTGC-3′ and MusJh4intron.R 5′-CTGGACTTTCGGTTTGGTG-3′ in the first round (14 cycles) and MusJh4intron.NF 5′-GGTCAAGGAACCTCAGTCA-3′ and MusJh4intron.NR 5′-TCTCTAGACAGCAACTAC-3′ in the second round of PCR (21 cycles) yielding a 581 bp product. PCR products were purified using the Agencourt AMPure XP kit (Beckman Coulter). Sequencing libraries of DNA from either mock or TAT-CRE treated splenic B cells from yy1f/f mice, were prepared using the Nextera XT DNA Sample Preparation kit (Illumina) and then sequenced using MiSeq Reagent kit v2 (300 cycles) on an Miseq instrument (Illumina) according to the manufacturer’s recommendations.
NGS data analysis
Sequencing reads were mapped onto reference genome GRCm38 using the BWA-MEM algorithm [53]. For SAM to BAM conversion and sorting and indexing of BAM files SAMtools was used [53]. For detecting low-level mutations, quantitative variant caller deep-SNV from the R/Bioconductor package repository was applied. The beta-binomial model was used to discriminate low-level single nucleotide variants (SNVs) from sequencing errors at each of the loci. Paired mock and TAT-CRE samples were directly compared and positions differing in variant allele frequency (VAF) with statistical significance p<0.05 were scored. Obtained p-values were adjusted by the Benjamini-Hochberg correction.
Analysis of Sanger sequencing data
Sanger sequences in the ab1 format were imported into CLC bio (https://www.qiagenbioinformatics.com/). After mapping onto the reference genome GRCm38, variant calling was performed. Only good quality sequences and variants, devoid of clones and mixed traces, were used for mutational pattern analyses.
Mutational pattern analysis
Only regions with coverage higher than 10,000 reads/position (based on NGS) were further analyzed (genomic coordinates in Supporting Information Table 4). Regions of interest were extracted from the mouse reference genome by bedtools2 [54]. Sequences representing genes coded on the reverse strand (Supporting Information Table 4) were converted into the reverse complement by http://www.bioinformatics.org/sms/rev_comp.html. AID motifs were localized in given regions of interest by http://www.bioinformatics.org/sms2/dna_pattern.html. Detected variants were annotated for their presence and position in these motifs by an in-house script.
Transient expression assays, ubiquitination analysis, and western blots
HEK293T cells were transfected by Lipofectamine 2000 (Invitrogen) according to the manufacturer’s procedures. For nuclear export experiments, 6μg CMV-FlagAID or CMV-FlagΔNES [29] plasmids were co-transfected with 6 μg or 18 μg of CMVYY1. Nuclear extracts were prepared as previously described [55] two days after transfection and then immunoblotted with anti-Flag (M2, Sigma), anti-YY1 (H414, Santa Cruz Biotechnology), and anti-TFIIB (Santa Cruz Biotechnology) antibodies. For ubiquitination experiments, plasmids CMV-FlagAID, GAL-YY1, GAL-YY1 1–200, and GAL-YY1 1–200Δ16–80 were previously described [35, 47, 56, 57]. For ubiquitination analysis, HEK293T cells were transfected with 6μg CMV-FlagAID and either 6 or 18μg of each YY1-expressing construct. Two days after transfection, MG132 was added for 5 hrs, and nuclear fractions were prepared as described [55]. SDS was added to final concentration 1%, samples were heated to 95°C for 10 minutes, diluted 10 fold with immunoprecipitation buffer and 0.5% NP-40 before overnight immunoprecipitation with anti-Flag antibody. Western blot was performed with anti-ubiquitin antibody conjugated to HRP (FK2H) and after stripping, reprobed with anti-Flag to indicate the level of AID. Western blots were performed with the following antibodies: anti-Flag (M2, Sigma), anti-YY1 (H414, Santa Cruz), anti-TFIIB (Santa Cruz), anti-ubiquitin FK2H (Enzo Life Sciences), and anti-GAL4 (sc577; Santa Cruz). For western blots of splenic B cell extracts, the following antibodies were used: Anti-AID (L7E7, Cell Signaling Technologies), anti-YY1 (H414, Santa Cruz), and anti-TBP (Cell Signaling Technologies).
Chromatin Immunoprecipitation and RNA transcript analyses
CD23+ cells were isolated, mock treated or treated with TAT-CRE protein to delete YY1, and cultured in RPMI media in the presence of LPS and IL4. After 24 hours, both the mock and TAT-CRE treated cells were transduced with pMX-HA or pMX-HA-AID retrovirus (a gift from M. Nussenzweig, Rockefeller University). At 72 hours, cells were harvested, cross-linked with formaldehyde and ChIP assays were performed as described earlier [58, 59] with modifications. Chromatin was sonicated using a Covaris AFA Focused-ultrasonicator and 100 ug of chromatin was taken for each immunoprecipitation (IP) with anti-YY1 (C-20X, Santa Cruz) or anti-HA (ab9110, Abcam), antibodies. Purified DNA was taken for qPCR with primers designed in the Sμ region where Robbiani et al. [36] found the maximum number of mutations (F1: GCTGAGCAAAATTAAGGGAACAA; R1 TCAGAGAAGCCCACCCATCT and F2 GGTGGGCTTCTCTGAGTGCTTCTA; R2 GCTCATTCCAGTTCATTACAGTCT). Efficiency of YY1 deletion by TAT-CRE treatment was verified by monitoring YY1 binding at the Eμ enhancer (EμF: GGAATGGGAGTGAGGCTCTCTC, EμR: GGACTTTCGGTTTGGTGG) and rpL30 promoter sites (rpL30 F: AGCAACCAACTACCGCAGACTACT and rpL30 R: ATCCAGAGCGTCAAACACCAGCTA). The data was plotted as percentage of input and rabbit IgG (2729S, Cell Signaling Tech.) was used as the negative control. The data represents the average of three replicate experiments with qPCRs being performed twice in triplicates. To quantitate IgM germline transcripts, RNA was isolated from spleens of six individual animals that had been either mock treated or TAT-CRE treated, and QPCR was performed using primers 5′-CTCTGGCCCTGCTTATTGTTG-3′ and 5′-GAAGACATTTGGGAAGGACTGACT-3′. Values were normalized to the mock treated sample and error bars show the standard deviation of the mean.
Supplementary Material
Acknowledgments
We thank Yang Shi (Harvard) for yy1f/f mice, and Michel Nussenzweig (Rockefeller) for IgκAID and AID−/− mice as well as for retrovirus pMX and pMXAID plasmids. This work was supported by NIH R01 grants AI079002 and GM111384, and DoD grant W81XWH-14-1-0171 to MLA. This work was also supported by project No. 3SGA5792 financed by the SoMoPro II Programme that has acquired a financial grant from the People Programme (Marie Curie Action) of the Seventh Framework Programme of EU according to the REA Grant Agreement No. 291782 and was further co-financed by the South-Moravian Region (KZ). This publication reflects only the author’s views and the Union is not liable for any use that may be made of the information contained therein. Part of the work was carried out with the support of Genomics Core Facility of CEITEC – Central European Institute of Technology under CEITEC – open access project, ID number LM2011020, funded by the Ministry of Education, Youth and Sports of the Czech Republic under the activity “Projects of major infrastructures for research, development and innovations” (BT). This research also has been financially supported by the Ministry of Education, Youth and Sports of the Czech Republic under the project CEITEC 2020 (LQ1601) and by the project of Faculty of Medicine, Masaryk University MUNI/A/1106/2016 (NT). The authors have no conflict of interest.
Abbreviations
- CSR
Class Switch Recombination
- Ig
immunoglobulin
- YY1
Yin Yang1
- AID
activation-induced cytidine deaminase
Footnotes
Animal Studies Permission
Animal studies were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania (Protocol 803080).
Conflict of Interest Statement
None of the authors have a financial conflict of interest pertaining to any of these studies.
References
- 1.Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Ann Rev Immunol. 2006;24:541–570. doi: 10.1146/annurev.immunol.23.021704.115830. [DOI] [PubMed] [Google Scholar]
- 2.Honjo T, Kinoshita K, Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Ann Rev Immunol. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 3.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 4.Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 5.Stavnezer J, Schrader CE. IgH chain class switch recombination: mechanism and regulation. J Immunol. 2014;93:5370–5378. doi: 10.4049/jimmunol.1401849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaidyanathan B, Yen WF, Pucella JN, Chaudhuri J. AIDing chromatin and transcription-coupled orchestration of immunoglobulin class-switch recombination. Front Immunol. 2014:5. doi: 10.3389/fimmu.2014.00120. http://dx.doi.org/10.3389/fimmu.2014.00120. [DOI] [PMC free article] [PubMed]
- 7.Matthews AJ, Zhang S, DiMenna LJ, Chaudhuri J. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv Immunol. 2014;122:1–57. doi: 10.1016/B978-0-12-800267-4.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vuong BQ, Chaudhuri J. Combinatorial mechanisms regulating AID-dependent DNA deamination: Interacting proteins and post-translational modifications. Semin Immunol. 2012;24:264–272. doi: 10.1016/j.smim.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol. 2012;12:517–531. doi: 10.1038/nri3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanai O, Catalan N, Forveille M, Dufourcq-Lagelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 11.Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HCI, Nussenzweig MC, Dalla-Favera R. AID is required for germinal center-derived lymphomagenesis. Nature Genetics. 2008;40:108–112. doi: 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- 12.Takizawa M, Tolarova H, Li Z, Dubois W, Lim S, Callen E, Franco S, Mosaico M, Feigenbaum L, Alt FW, Nussenzweig A, Potter M, Casellas R. AID expression levels determine the extent of cMyc oncogenic tranlocations and the incidence of B cell tumor development. J Exp Med. 2008;205:1949–1957. doi: 10.1084/jem.20081007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okazaki IM, Kotani A, Honjo T. Role of AID in tumorigensis. Adv Immunol. 2007;94:245–273. doi: 10.1016/S0065-2776(06)94008-5. [DOI] [PubMed] [Google Scholar]
- 14.Liu M, Schatz DG. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. 2009;30:173–181. doi: 10.1016/j.it.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Endo Y, Marusawa H, Kou T, Nakase H, Fujii S, Fujimori T, Kinoshita K, Honjo T, Chiba T. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008;135:889–898. 898.e881–883. doi: 10.1053/j.gastro.2008.06.091. [DOI] [PubMed] [Google Scholar]
- 16.Endo Y, Marusawa H, Kinoshita K, Morisawa T, Sakurai T, Okazaki IM, Watashi K, Shimotohno K, Honjo T, Chiba T. Expression of activation-induced cytidine deaminase in human hepatocytes via NF-kappaB signaling. Oncogene. 2007;26:5587–5595. doi: 10.1038/sj.onc.1210344. [DOI] [PubMed] [Google Scholar]
- 17.Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, Azuma T, Okazaki IM, Honjo T, Chiba T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–476. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 18.Sawai Y, Kodama Y, Shimizu T, Ota Y, Maruno T, Eso Y, Kurita A, Shiokawa M, Tsuji Y, Uza N, Matsumoto Y, Masui T, Uemoto S, Marusawa H, Chiba T. Activation-Induced Cytidine Deaminase Contributes to Pancreatic Tumorigenesis by Inducing Tumor-Related Gene Mutations. Cancer Res. 2015;75:3292–3301. doi: 10.1158/0008-5472.CAN-14-3028. [DOI] [PubMed] [Google Scholar]
- 19.Morisawa T, Marusawa H, Ueda Y, Iwai A, Okazaki IM, Honjo T, Chiba T. Organ-specific profiles of genetic changes in cancers caused by activation-induced cytidine deaminase expression. Int J Cancer. 2008;123:2735–2740. doi: 10.1002/ijc.23853. [DOI] [PubMed] [Google Scholar]
- 20.Komori J, Marusawa H, Machimoto T, Endo Y, Kinoshita K, Kou T, Haga H, Ikai I, Uemoto S, Chiba T. Activation-induced cytidine deaminase links bile duct inflammation to human cholangiocarcinoma. Hepatology. 2008;47:888–896. doi: 10.1002/hep.22125. [DOI] [PubMed] [Google Scholar]
- 21.Sernandez IV, de Yebenes VG, Dorsett Y, Ramiro AR. Haploinsufficiency of activation-induced deaminase for antibody diversification and chromosome translocations both in vitro an in vivo. PLoS One. 2008;3:e3927. doi: 10.1371/journal.pone.0003927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Yebenes VG, Belver L, Pisana DG, Gonzalez S, Villasante A, Croce C, He L, Ramiro AR. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med. 2008;205:2199–2206. doi: 10.1084/jem.20080579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, Di Virgilio M, San-Martin BR, Heidkamp G, Schwickert TA, Eisenreich T, Rajewsky K, Nussenzweig MC. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity. 2008;28:630–638. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity. 2008;28:621–629. doi: 10.1016/j.immuni.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 26.Rada C, Jarvis JM, Milstein C. AID-GFP chimeric protein increases hypermutation of Ig genes with no evidence of nuclear localization. Proc Natl Acad Sci USA. 2002;99:7003–7008. doi: 10.1073/pnas.092160999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito S, Nagaoka H, Shinkura R, Begum N, Muramatsu M, Nakata M, Honjo T. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc Natl Acad Sci USA. 2004;101:1975–1980. doi: 10.1073/pnas.0307335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brar SS, Watson M, Diaz M. Activation-induced cytosine deaminase (AID) is actively exported out of the nucleus but retained by the induction of DNA breaks. J Biol Chem. 2004;279:26395–26401. doi: 10.1074/jbc.M403503200. [DOI] [PubMed] [Google Scholar]
- 29.McBride KM, Barreto V, Ramiro AR, Stavropoulos P, Nussenzweig MC. Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J Exp Med. 2004;199:1235–1244. doi: 10.1084/jem.20040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aoufouchi S, Faili A, Zober C, D’Orlando O, Weller S, Weill JC, Reynaud CA. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med. 2008;205:1357–1368. doi: 10.1084/jem.20070950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patenaude AM, Orthwein A, Hu Y, Campo VA, Kavli B, Buschiazzo A, Di Noia JM. Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nature Struct Molec Biol. 2009;16:517–527. doi: 10.1038/nsmb.1598. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol Cell. 2007;26:843–852. doi: 10.1016/j.molcel.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uchimura Y, Barton LF, Rada C, Neuberger MS. REG-γ associates with and modulates the abundance of nuclear activation-induced deaminase. J Exp Med. 2011;208:2385–2391. doi: 10.1084/jem.20110856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orthwein A, Patenaude AM, Affar EB, Lamarre A, Young JC, Di Noia JM. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med. 2010;207:2751–2765. doi: 10.1084/jem.20101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaprazna K, Atchison ML. YY1 controls immunoglobulin class switch recombination and nuclear activation-induced deaminase levels. Mol Cell Biol. 2012;32:1542–1554. doi: 10.1128/MCB.05989-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR, Ried T, Nussenzweig A, Nussenzweig MC. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, San-Martin BR, Barreto V, Nieland TJ, Root DE, Casellas R, Nussenzweig MC. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12:62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 40.Hackney JA, Misaghi S, Senger K, Garris C, Sun Y, Lorenzo MN, Zarrin AA. DNA targets of AID evolutionary link between antibody somatic hypermutation and class switch recombination. Adv Immunol. 2009;101:163–189. doi: 10.1016/S0065-2776(08)01005-5. [DOI] [PubMed] [Google Scholar]
- 41.Rogozin IB, Diaz M. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J Immunol. 2004;172:3382–3384. doi: 10.4049/jimmunol.172.6.3382. [DOI] [PubMed] [Google Scholar]
- 42.Tepper S, Jeschke J, Bottcher K, Schmidt A, Davari K, Muller P, Kremmer E, Hemmerich P, Pfeil I, Jungnickel B. PARP activation promotes nuclear AID accumulation in lymphoma cells. Oncotarget. 2016;7:13197–13208. doi: 10.18632/oncotarget.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehra P, Gerasimova T, Basu A, Jha V, Banerjee A, Sindhava V, Gray F, Berry CT, Sen R, Atchison ML. YY1 controls Em-3′RR DNA loop formation and immunoglobulin heavy chain class switch recombination. Blood Adv. 2016;1:15–20. doi: 10.1182/bloodadvances.2016000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montamat-Sicotte D, Litzler LC, Abreu C, Safavi S, Zahn A, Orthwein A, Muschen M, Oppezzo P, Munoz DP, Di Noia JM. HSP90 inhibitors decrease AID levels and activity in mice and in human cells. Eur J Immunol. 2015;45:2365–2376. doi: 10.1002/eji.201545462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steele EJ. Mechanism of somatic hypermutation: critical analysis of strand biased mutation signatures at A:T and G:C base pairs. Mol Immunol. 2009;46:305–320. doi: 10.1016/j.molimm.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 46.Nagaoka H, Muramatsu M, Yamamura N, Kinoshita K, Honjo T. Activation-induced deaminase (AID)-directed hypermutation in the immunoglobulin Smu region: implication of AID involvement in a common step of class switch recombination and somatic hypermutation. J Exp Med. 2002;195:529–534. doi: 10.1084/jem.20012144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barreto VM, Reina San-Martin BR, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Molec Cell. 2003;12:501–508. doi: 10.1016/s1097-2765(03)00309-5. [DOI] [PubMed] [Google Scholar]
- 48.Green MR, Monti S, Dalla-Favera R, Pasqualucci L, Walsh NC, Schmidt-Supprian M, Kutok JL, Rodig SJ, Neuberg DS, Rajewsky K, Golub TR, Alt FW, Shipp MA, Manis JP. Signatures of murine B-cell development implicate Yy1 as a regulator of the germinal center-specific program. Proc Natl Acad Sci U S A. 2011;108:2873–2878. doi: 10.1073/pnas.1019537108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banerjee A, Sindhava V, Vuyyuru R, Jha V, Hodawadekar S, Manser T, Atchison ML. YY1 is required for germinal center B cell development. PLoS One. 2016;11:e0155311. doi: 10.1371/journal.pone.0155311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kleiman E, Jia H, Loguercio S, Su AI, Feeney AJ. YY1 plays an essential role at all stages of B-cell differentiation. Proc Natl Acad Sci U S A. 2016;113:E3911–3920. doi: 10.1073/pnas.1606297113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trabucco SE, Gerstein RM, Zhang H. YY1 Regulates the Germinal Center Reaction by Inhibiting Apoptosis. J Immunol. 2016;197:1699–1707. doi: 10.4049/jimmunol.1600721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu H, Schmidt-Supprain M, Shi Y, Hobeika E, Barteneva N, Jumaa H, Pelanda R, Reth M, Skok J, Rajewsky K, Shi Y. Yin Yang 1 is a critical regulator of B-cell development. Genes Dev. 2007;21:1179–1189. doi: 10.1101/gad.1529307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts”, prepared from small number of cells. Nuc Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bushmeyer SM, Atchison ML. Identification of YY1 sequences necessary for association with the nuclear matrix and for transcriptional repression functions. J Cell Biochem. 1998;68:484–499. [PubMed] [Google Scholar]
- 57.Park K, Atchison ML. Isolation of a candidate repressor/activator, NF-E1 (YY-1, d), that binds to the immunoglobulin k 3′ enhancer and the immunoglobulin heavy-chain mE1 site. Proc Natl Acad Sci USA. 1991;88:9804–9808. doi: 10.1073/pnas.88.21.9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilkinson FH, Park K, Atchison ML. Polycomb recruitment to DNA in vivo by the YY1 REPO domain. Proc Natl Acad Sci USA. 2006;103:19296–19301. doi: 10.1073/pnas.0603564103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Basu A, Atchison ML. CtBP levels control intergenic transcripts, PHO/YY1 DNA binding, and PcG recruitment to DNA. J Cell Biochem. 2010;109:478–486. doi: 10.1002/jcb.22487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.