

Abstract
Decay accelerating factor (DAF or CD55) is a cell associated C3 and C5 convertase regulator originally described in terms of protection of self-cells from systemic complement but now known to modulate adaptive T cell responses. It is expressed on all cell types. We investigated whether nonenzymatic glycation could impair its function and potentially be relevant to complications of diabetes mellitus and other conditions that result in nonenzymatic glycation including cancer, Alzheimer s disease, and aging. ’ Immunoblots of affinity-purified DAF from erythrocytes of patients with diabetes showed pentosidine, glyoxal-AGEs, carboxymethyllysine, and argpyrimidine. HPLC/MS analyses of glucose modified DAF localized the sites of AGE modifications to K125 adjacent to K126, K127 at the junction of CCPs2-3 and spatially near R96, and R100, all identified as being critical for DAF’s function. Functional analyses of glucose or ribose treated DAF protein showed profound loss of its regulatory activity. The data argue that de-regulated activation of systemic complement and de-regulated activation of T cells and leukocytes could result from non-enzymatic glycation of DAF.
Keywords: DAF, Glycation, Diabetes, Complement, AGE
1. Introduction
Decay accelerating factor (DAF or CD55) is a cell-associated regulatory protein initially characterized as a control protein that circumvents activation of autologous complement on self- cell surfaces (Medof et al., 1984). It recently was shown to be a key modulator of T cell activation (Heeger et al., 2005; Strainic et al., 2008). It functions by rapidly dissociating C3 and C5 convertases that assemble on self-cells thereby blocking C3a/C5a generation. In the context of serum complement activation, it not only shields against self-cell injury but also prevents amplification of C3b deposition (that enables interaction with C3b-bearing leukocytes) and C5b deposition [which triggers assembly of the membrane attack complex (MAC)] (Medof et al., 1996). In the context of adaptive T effector cell responses, DAF suppresses the endogenous generation of C3a/C5a by interacting with dendritic cells (DCs) and T cells. This latter function inhibits autocrine signaling through C3a and C5a receptors (C3ar1/C5ar1) in both partners needed for the costimulation and viability of T cells (Strainic et al., 2008; Lalli et al., 2008) as well as for the activation of DCs, macrophages and other antigen presenting cells (APCs) (Liu et al., 2008; Strainic et al., 2008).
In diabetes mellitus hyperglycemia leads to nonenzymatic modification of proteins by glucose and by glucose-derived reactants such as methylglyoxal and glyoxal (reviewed in Monnier et al., 1996). Similar modifications can occur in other conditions including cancer, and Alzheimer’s disease, as well as in aging. These modifications, termed advanced glycation end products (AGEs), have been implicated in the development of the chronic complications of diabetes, i.e., nephropathy, neuropathy, and retinopathy. The AGEs can form extracellularly or intracellularly via oxidation reaction of sugars, dicarbonyls or ascorbate oxidation products with protein amines. Among reported AGEs are N−ε-carboxymethyl lysine (CML) (Ahmed et al., 1986), pentosidine (Sell and Monnier, 1989) and argpyrimidine (Shipanova et al., 1997).
If DAF were to be chemically modified in any of the above conditions so as to lose functionality, several deleterious processes might result. Autologous C3b and its immediate (factor I) cleavage product, iC3b, bound to self-cells could serve as ligands for C3b and iC3b receptors (CR1 and CR3) on leukocytes, (polymorphonuclear cells, monocytes and macrophages) potentially inducing the release of proteolytic enzymes and inflammatory mediators from these cells (reviewed in van Lookeren Campagne et al., 2007). The local generation of autologous C3a and C5a anaphylatoxins that accompanies C3 cleavage by dysregulated C3 convertases could enhance recruitment of these leukocytes as well as cause vasodilation and aggregation of the incoming cells (reviewed in Liszewski and Atkinson, 2015). In addition, local C3a/C5a generation by interacting DCs and T cells could be potentiated and consequent C3ar1/C5ar1 signaling augmented thereby favoring the generation of Th1/Th17 cells (Liu et al., 2008) rather than the generation of Foxp3+ T regulatory cells (Tregs) (Strainic et al., 2013). This would increase the production of inflammatory cytokines by DCs, macrophages, and other APCs (Strainic et al., 2008; Liu et al., 2008).
DAF is composed of 4 tandem ∼60 amino acid long homologous repeats termed short consensus repeats (SCRs) or complement control repeats (CCPs). The four CCPs are attached on a long heavily O-glycosylated stalk which suspends them at an appropriate distance above the cell membrane (Medof et al., 1987; Stafford et al., 1988; Kuttner-Kondo et al., 1996). Structure function analyses of the four CCPs have shown that DAF’s regulatory activity resides principally in residues at and near the junction of CCP2 and CCP3 with some contribution from residues in CCP4 (Kuttner-Kondo et al., 2001, 2007). NMR analyses and crystallography have shown that CCP4 is located in proximity to the active site residues in CCPs2-3 (Kuttner-Kondo et al., 2001, 2007; Uhrinova et al., 2002, 2003; Harris et al., 2005; Lukacik et al., 2004).
Herein, we report that DAF proteins isolated from erythrocytes of diabetic patients contain AGEs. Functional and structural analyses of DAF showed that glucose (and ribose) incubation inactivated it by chemical modification of its critical active site residues. That DAF glycation could enhance complement activation as well as promote T effector responses in diabetes and other conditions argues that this process could contribute to the development of complications.
2. Materials and methods
2.1. Proteins and antibodies
DAF protein was purified from extracts of erythrocyte (Ehu) stroma as detailed in (Medof et al., 1986). Murine anti-DAF monoclonal antibodies (mAb) IIH6 and IA10 were prepared as described (Kinoshita et al., 1985). Rabbit polyclonal anti-GXL-AGE antibodies (Sady et al., 2000), glyoxal-modified bovine serum albumin (GXL-BSA), and murine anti-argpyrimidine mAb were similarly prepared as described (Oya et al., 1999). Rabbit polyclonal anti-pentosidine antibodies and pentosidine-modified bovine serum albumin (pentosidine-BSA) were kind gifts of Dr. V. Monnier (Case Western Reserve University, Cleveland, OH).
2.2. Patient samples
Blood samples were obtained from de-identified patients with both Type 1 and Type 2 diabetes. All had longstanding diabetes, were being followed at University Hospitals of Cleveland, and were provided without selection regarding blood glucose or HbA1c levels or any other clinical information.
2.3. Preparation of human erythrocyte (Ehu) extracts
Ehu obtained from diabetic patients and control subjects were lysed in 30 vol of 0.01 M phosphate buffer, pH 7.4, and stroma extracted with 1% Nonidet P-40 in phosphate-buffered saline (PBS) containing 1 mM phenylmethylsulfonylfluoride (Sigma Chemical Co., St. Louis, MO), 0.5 mM EDTA, 0.025% sodium azide, 60 g/ml soybean trypsin inhibitor (Sigma Chemical Co.), 20 g/ml leupeptin, and 20 g/ml aprotinin (Boehringer Mannheim, Indianapolis, IN). After centrifugation at 18,000 × g for 15 min, stromal extracts were stored at −70°.
2.4. Isolation of DAF proteins from Ehu extracts of diabetic patients
Extracts obtained as described above were mixed with 20 l (30% packed) of anti-DAF mAb IA10-conjugated Sepharose 4B beads (Oya et al., 1999) and the suspensions rotated overnight at 4 °C. The beads then were spun down in a microcentrifuge and washed three times with 50 l of 1 M Tris, 0.5 M NaCl, pH 7.4 containing 0.1% 3-[(3-Cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate (CHAPS). Bound DAF protein was eluted by incubating the beads at 20 °C for 3 min with 30 l of 0.05 M Tris, 0.05 M triethylamine, pH 11.2 containing 0.1% CHAPS. After removal of the beads, DAF containing eluate was neutralized with 8.6 l of glycine-saturated 0.5 M Tris, pH 6.0 containing 0.1% CHAPS. In certain cases, eluted fractions from samples that contained low amounts of DAF were pooled and concentrated in Amicon micron 7M-10 centrifugal filters (Millipore, Inc., Bedford, MA).
2.5. Immunoblotting
Immuno-isolated DAF was loaded on non-reduced 7.5% SDS-PAGE gels. Following electrophoresis, the gels were electro-transferred to Immobilon-P membranes using a Bio-Rad Mini Trans-Blot cell (Biorad, Inc., Hercules, CA). The presence and position of the recovered DAF protein were documented in control lanes by probing blots with IIH6 anti-DAF mAb (specific for DAF CCP4). Other lanes of the immunoblot were probed for argpyrimidine with an anti-argpyrimidine mAb, for glyoxal AGEs, and for pentosidine with the respective rabbit polyclonal antibodies described above. Bound antibody on the membranes was detected with horseradish peroxidase (HRP)-labeled sheep anti-mouse or goat anti-rabbit IgG using the enhanced chemiluminescence (ECL) kit (Amersham, Arlington, Hts., IL).
2.6. Glycation of DAF in vitro
Purified Ehu DAF (5 g) was incubated at 20 °C for 10 days without or with 0.5 M glucose or for 7 days without or with 0.5 M ribose in 200 l of 0.1 M phosphate buffer. The 0.5 M concentration of the sugars although not physiologic was used by us as by others (Takahashi et al., 2017; Iannuzzi et al., 2016) to accelerate the nonenzymatic glycation process which proceeds at a slow pace in vivo. Incubations were carried out in 145 mM NaCl, pH 7.4, 0.1% NP40 in a 1.5 ml Eppendorf tube with gentle rocking. To prevent glycoxidation from occurring, the incubation mixtures were blanketed with nitrogen and the tubes kept sealed. Following incubation, the alternatively treated Ehu DAF proteins were dialyzed against isoionic glucose veronal buffer (GVB++) consisting of 145 mM NaCl, 3.12 mM barbital, 1.82 mM sodium barbital, 1.0 mM MgCl2, 0.15 mM CaCl2 (pH 7.4) to which 0.1% gelatin was added.
2.7. Hemolytic assays
The Ehu DAF proteins (2 g/ml in GVB++ containing 0.005% NP40) were incubated for 30 min at 30 °C with 108 antibody sensitized guinea pig erythrocytes (EgpA) in a total volume of 100 l as described in Medof et al. (1984). Following washing, the EgpA bearing the treated or untreated Ehu DAF proteins (107 cells) were then incubated for 60 min at 37 °C with a 1:20 dilution of normal human serum. Cells were pelleted, hemoglobin color measured at 512 nM, and percent hemolysis determined. The in vivo AGE modified DAF proteins immuno-isolated from the diabetic patients were eluted in SDS and present in insufficient amounts for functional assays.
2.8. Peptide analysis by liquid chromatography/mass spectroscopy (LC/MS)
DAF treated with glucose or with PBS control for 40 days was in-gel digested with trypsin for peptide analysis by LC/MS as follows: samples were denatured and reduced by boiling in 1X SDS-PAGE sample buffer. After cooling to room temperature, cysteine residues were alkyla ted by ad ding 4-vinyl-pyridine to a final concentration of 0.5% and leaving at room temperature for 15 min. Samples were subjected to SDS-PAGE, stained with Coomassie blue R-250, the protein band excised with a razor blade for in-gel digestion with modified sequencing grade trypsin (Promega). The digest was removed and the gel slice washed with 0.5% acetic acid, 60% acetonitrile to increase peptide recovery. For HPLC analysis, acetonitrile was removed from the combined sample and the combined sample washed by drying under vacuum. The digest was redissolved in 0.5% acetic acid and injected onto a C18 ODS-AQ reversed-phase HPLC column from YMC (2 mm × 10 cm, 5 m particles), using 0.5% acetic acid as solvent A and 0.5% acetic acid in 80% acetonitrile as solvent B. At a flow rate of 75 ul/min, the column was washed with 5% solvent B isocratically for 5 min, then peptides were eluted with a linear gradient from 5% to 60% solvent B over 60 min an d from 60% to 95% solvent B over 10 min. Peptides were eluted directly from the HPLC into the micro-spray ionization source of an LCQ Classic ion trap mass spectrometer (ThermoFinnigan), which had been optimized to detect standard peptides in 0.5% acetic acid, 35% acetonitrile at a flow rate of 75 ul/min. The scan function of the mass spectrometer was set to collect full MS scan from m/z of 400 to 2000 and to collect an MS/MS spectrum for the most abundant ion whenever its intensity exceeded a threshold value, in a data-dependent manner.
2.9. Identication of critical residues for DAF s activity
The 3D picture of DAF’s CCPs 23 utilized the crystal structure of DAF CCPs 1–4 (Lukacik et al., 2004). PDB entry 1OJV. The figure was created with the Schrodinger Small-molecule Drug Discovery Software Suite, release 2015–3.
3. Results
3.1. Identication of AGEs on DAF expressed on erythrocytes of diabetic patients
To establish whether DAF is subject to glycation in vivo and is modified by AGEs analogous to findings reported for other proteins (Monnier et al., 1996), we first affinity purified DAF protein from erythrocytes of diabetic patients with long-standing disease and healthy controls, and probed immunoblots of the immuno-isolates with antibodies against different AGEs. In contrast to DAF recovered from erythrocytes of nondiabetic control subjects, which showed reactivity with anti DAF mAb (Fig. 1A) but no reactivity with the anti-AGE mAbs, DAF on erythrocytes of diabetic patients reacted with anti-glyoxal-AGEs (Fig. 1B), anti-pentosidine (Fig. 1C) and anti-argpyrimidine (Fig. 1 D) Abs. All three AGEs were present on DAF from individual diabetic patients (Fig. 1B and C, lane 4, and D, lane 2) as well as from pooled DAF isolates (Fig. 1B and C, lane 3). In some samples, the DAF immuno-isolate exhibited faint high Mr (~140 kDa) bands (not shown) in addition to the normal Mr (70 kDa) DAF band. The high molecular weight band approximated the size of “DAF-2,” a previously identified but uncharacterized dimeric form of DAF (Takahashi et al., 2017; Iannuzzi et al., 2016). Precise quantitation of erythrocyte DAF levels in the diabetic samples was not done because the AGE modifications of DAF could impair the binding of anti-DAF Abs. Erythrocyte DAF isolated from diabetic erythrocytes showed no evidence of a CML modification (data not shown).
Fig. 1.
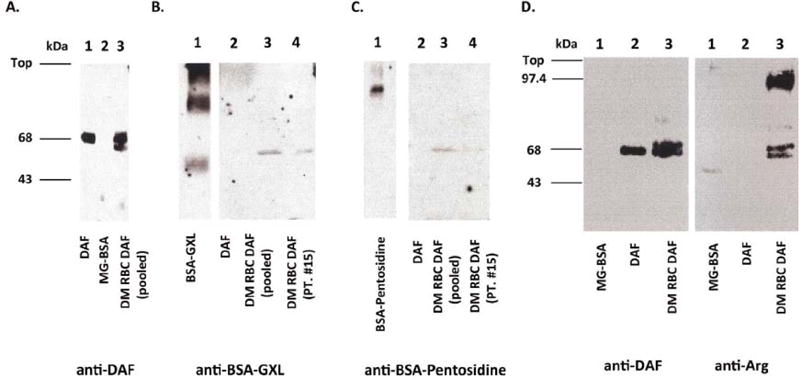
Western blots of Ehu DAF from diabetic (DM) patients to detect AGE products. Immunoblots using affinity purified DAF protein from erythrocytes of a pool (n = 3) of diabetic patients (DM RBC DAF (pooled)) or nondiabetic patients (DAF) developed with (A) IIH6 anti-DAF mAb, (B) rabbit anti-GXL (glyoxal) polyclonal antibody, (C) rabbit anti-pentosidine polyclonal antibody, and (D) anti-argpyrimidine (anti-Arg) mAb. Immunoprecipitated DAF from erythrocytes from a representative individual diabetic patient likewise showed immunoreactivity with anti- GXL and anti-pentoside. In each panel, the lane on the left serves as control.
3.2. Inhibition of DAF activity by nonenzymatic glycation
To determine if the hyperglycemic state can lead to alterations in DAF that would impair its regulatory activity, we performed in vitro studies in which we incubated purified Ehu DAF protein with 0.5 M glucose, 0.5 M ribose, or with buffer alone for 7 or 10 d. Following this, we assessed the complement regulatory function of the treated proteins by incubating them with EgpA and increasing concentrations of human complement (Medof et al., 1984; Walter et al., 1992) and developing lysis as described in Methods. Unexpectedly, these exeriments (Fig. 2) showed that the DAF incubation with ribose abrogated > 99% of DAF’s complement regulatory activity, thus resulting in lysis of DAF treated EgpA approaching that of untreated EgpA alone. Incubation with glucose, which glycates at a slower rate, resulted in 50 percent inactivation at the 10 d time point (not shown).
Fig. 2.
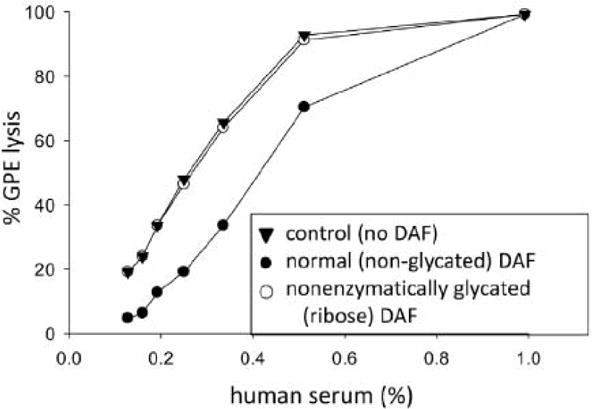
Inactivation of DAF’s function by nonenzymatic glycation. Purified Ehu DAF (20 g/ml) was incubated with either 0.5 M ribose (open circle) or buffer (control; closed circle) as described in Methods. Following incubation of this modified DAF protein with antibody-sensitized guinea pig erythrocytes (EgpA), the erythrocytes were incubated with increasing percentages of human serum (containing complement), and lysis quantitated. Whereas hemolysis of EgpA, was reduced by native non-glycated DAF, hemolysis of EgpA treated with glycated DAF was nearly identical to that of untreated EgpA control not exposed to DAF (closed triangles).
3.3. Localization of the glycated sites in the DAF molecule
To determine the mechanism of the glucose inactivation of DAF’s function, we examined sequential amino acids within DAF for evidence of glycation. To do this, we employed a DAF sample incubated with 0.5 M glucose for a longer time (40 d). We digested the protein with trypsin and performed tryptic peptide analysis by LC/MS. The peptides were compared to those from buffer treated DAF protein. Past studies of albumin documented that this approach allows identification of the glycated peptides (Iberg and Fluckiger, 1986). In the case of non-glycated control DAF, peptides covering most of the backbone from CCP1 through 4 were identified. They are listed in Fig. 3. With glucose-treated DAF, three peptides (CCP2:117–125; CCP3:137–161 and CCP3:162-187) were not recovered. The first two of these peptides contained K125 and K161, which have been documented as being critical in DAF’s function. In addition to the loss of these peptides, other peptides with masses corresponding to glucose adducted peptides were detectable by comparative analysis of glycated peptides from glycated albumin. One of these peptides contains K125 and is adjacent to K126,127 and spatially near R96 and R100 (Fig. 4). That glycation of the K125 residue and possible alterations in the region 76–100 would diminish DAF’s function are in accordance with structure/function studies of DAF (Kuttner-Kondo et al., 2007, 2001) performed by alanine substitution mutagenesis. These analyses showed that the three positively charged K125–127 residues [located between CCPs 2 and 3] and two positively charged R95 and R100 [located near the C-terminus of CCP 2] are essential for DAF’s function (see Introduction and Discussion).
Fig. 3.
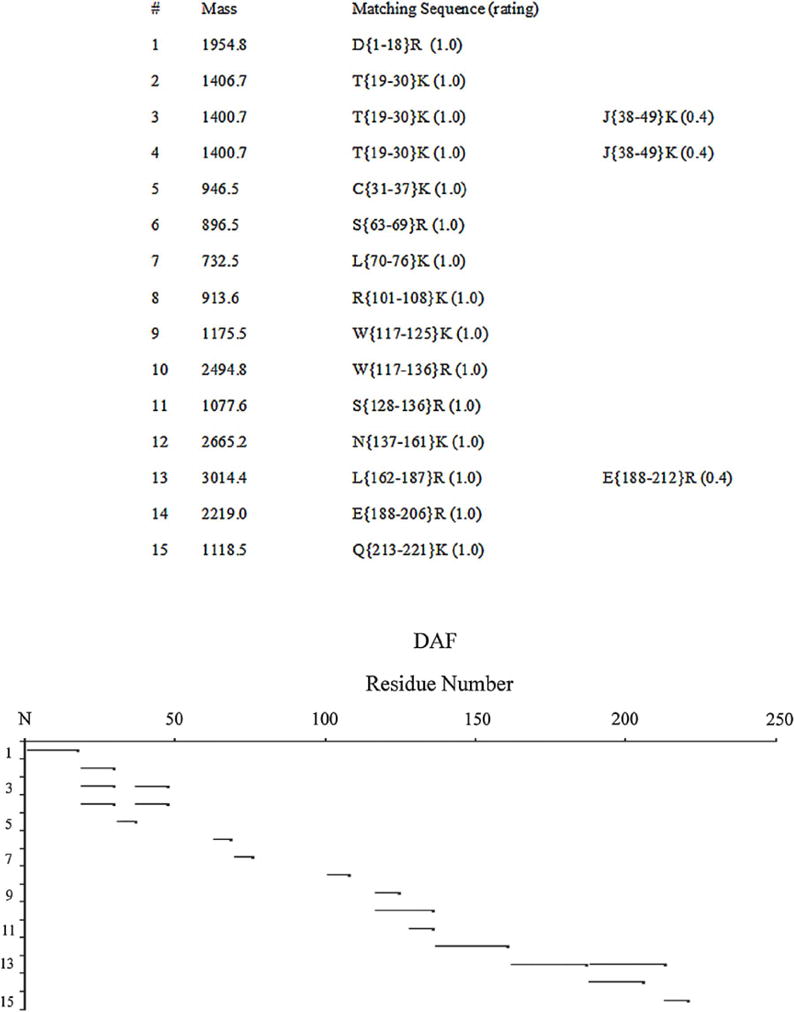
LC/MS analysis of non-enzymatically glycated (glucose-treated) DAF protein. Schematic representation of peptides recovered from LC/MS analysis of glucose treated DAF protein. Peptide 117–125 was not observed with glycated DAF.
Fig. 4.
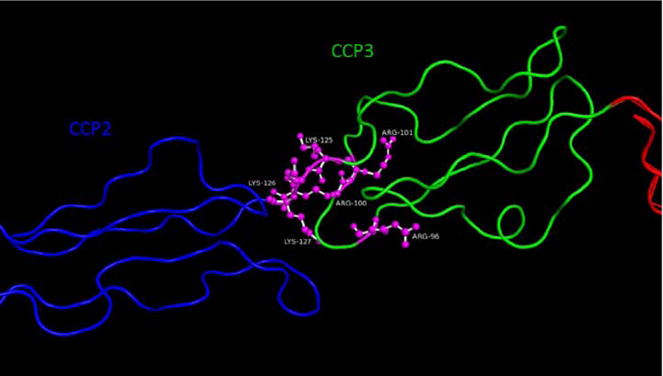
Crystal structure of DAF CCP’s 2 and 3, PDB ID 1OJV, shown in backbone representation with lysine and arginine residues at the interface between CCP’s 2 and 3 shown in ball-and-stick representation. Site specific alanine mutagenesis identified (Oya et al., 1999; Lukacik et al., 2004) identified the amino acids important for DAF function. present between CCPs 2 and 3 (Lukacik et al., 2004; Kuttner-Kondo et al., 2001; Brodbeck et al., 1996).
3.4. Mapping the altered amino acids on the crystal structure of DAF
To precisely localize whether, and if so how, the glycated sites in DAF are clustered and relate to DAF’s active site region localized between CCPs 2 and 3, we placed the glycated amino acids on the crystal structure DAF. Remarkably, all precisely localized in DAF’s active site region which mediates C3/C5 convertase decay acceleration of systemic complement and suppresses local C3a/C5a production and consequent C3ar1/C5ar1 signaling which drives T effector cell activation (Fig. 4).
4. Discussion
DAF was originally characterized as a cell surface, i.e. “intrinsic” complement regulatory protein that protects self- tissues from injury that could be induced by autologous complement activation on their surfaces (Medof et al., 1984). In this context, it acts on systemic complement and functions as an inhibitor of the central C3/C5 amplification convertases of the complement cascade. More recent studies unexpectedly found that DAF also plays a pivotal role in regulating adaptive T cell responses and in modulating monocyte/macrophage/DC function (Strainic et al., 2008; Liu et al., 2008). In this context, DAF acts on complement that is endogenously produced by immune cells themselves and functions to inhibit local C3a/C5a generation and consequently suppress autocrine C3ar1/C5ar1 signaling.
Herein, we report that non-enzymatic glycation impairs the biological activity of DAF. Potentially relevant to the complications of long standing hyperglycemia in diabetes as well as other disorders connected with nonenzymatic glycation, we show that DAF isolated from erythrocytes is modified by several types of AGEs. These findings suggest that in diabetes and potentially other conditions, DAF dependent protection of self-tissues from uptake of autologous C3b/C5b, as well as physiological control of inflammatory Th1/Th17 T cell responses and mononuclear cell activation could be attenuated and contribute to the development of complications. It could contribute in other ways since the systemic complement system is an important component of multiple inflammatory processes implicated in the development of long-term complications of diabetes (Tang and Kern, 2011; Lim and Tesch, 2012; Navarro-Gonzalez et al., 2011).
When we investigated the specific modifications of DAF that occur in vivo in diabetic patients, we found that glyoxal-AGE, pentosidine, and argpyrimidine were present in DAF expressed on circulating erythrocytes. These modifications are produced by reaction of aldehydes and ketones with amino groups of lysine and arginine. Long-lived extracellular proteins such as skin collagen (Vishwanath et al., 1986; Genuth et al., 2015; Monnier et al., 2013) and eye lens protein (Perry et al., 1987) and retina have been shown to be targets of these carbohydrate-mediated reactions but nonenzymatic glycation occurs also on short-lived proteins (Bidasee etal., 2003). The pentosidine modification identified in DAF of diabetic patients is known to induce crosslinking involving the nearby R residues.
Consistent with the presence of AGEs on DAF from erythrocytes of diabetic patients, in preliminary studies of DAF immuno-isolated from retinas of diabetic patients, we obtained evidence for argpyrimidine modification, the most detectable modification of erythrocyte DAF. In addition to the native DAF band, a higher molecular weight anti-argpyrimidine DAF band was prominent, consistent with intermolecular cross-linking, possibly from other AGEs. As DAF levels on vascular endothelial cells and activated monocytes/macrophages are many times higher than those on erythrocytes (0.5–2 × 104/cell (Asch et al., 1986) vs. 2.5 × 103/cell16) (Kinoshita et al., 1985) and DAF is present on retinal neurons (Fett et al., 2012) which comprise the majority of the retina, it is unlikely that the DAF derived from erythrocytes.
In our in vitro studies, we found that prolonged exposure to glycating agents virtually abolished DAF’s regulatory function. Of relevance to the current findings, similar results have been reported by others for CD59 (Acosta et al., 1996; Qin et al., 2004), another intrinsic cell surface complement regulator which functions later in the complement cascade to inhibit systemic C5b-9-mediated attack. Effects of nonenzymatic glycation on DAF control of CD4+ cell and antigen presenting cell activities, and on C3ar1 and C5ar1 signaling involved in endothelial cell homeostasis have not been investigated.
Comparative LC/MS analyses showed that among the 12 peptides identified, MS peak 588.7 corresponding to DAF peptide 117–125 was present in the buffer-treated sample but was not recoverable from the ribose-treated sample. As indicated (Results), this peptide encompasses K125 of the DAF molecule, which is immediately adjacent to K126 and K127, and, according to the structural characterizations of DAF protein (Uhrinova et al., 2003; Lukacik et al., 2004), is in close proximity to R96 and R100. The peptide(s) encompassing the latter R residues were not recovered from either the ribose-treated or the control sample. Site-specific mutagenesis of DAF (Kuttner-Kondo et al., 2001) showed that K125A substitution decreases DAF’s activity to 32% in the classical pathway and to 62% in the alternative pathway. Relevant to DAF’s function in regulating systemic complement, the K126A and K127A mutations decrease DAF’s activity to 9% and 14% in the classical pathway. Relevant to DAF’s regulation of local complement activation that controls T cell and monocyte/macrophage function (as well as amplifies systemic complement activation), the substitutions decrease DAF’s activity to < 1% and to ~14% in the alternative pathway. While our failure to detect peptides spanning positions 76–100 is consistent with nonenzymatic glycation, it does not prove modification of the peptides by this process. It is noteworthy that R96A substitution totally abolishes DAF’s activity in both pathways and R100A substitution decreases DAF’s activity to 12% in the classical pathway and to 25% in the alternative pathway. According to DAF’s structure (see Fig. 4) (Lukacik et al., 2004) 1) modification of K125 could interfere sterically with K126 and K127, and 2) nearby R96 and R100 (and R101), in the presence of PO4, could catalyze the formation of the AGEs on the K125 residue as well as promote intramolecular crosslinking.
5. Conclusions
Taken together, our results indicate that augmented systemic complement activation that could result from impairment of DAF’s function by nonenzymatic glycation could participate in the local inflammation that has been implicated in diabetes (Lukacik et al., 2004; Tang and Kern, 2011) cancer, aging, and other conditions. Since activated monocytes/macrophages serve as professional antigen presenting cells, and vascular endothelial cells can serve as nonprofessional antigen presenting cells in that they express class II MHC and costimulatory molecules under conditions of inflammation, impairment of DAF’s function by nonenzymatic glycation, in principle, could also promote local C3a/C5a production, and thereby contribute to adaptive immune responses.
Acknowledgments
The authors thank Dr. V. Monnier (Institute of Pathology, Case Western Reserve University) for providing the anti-pentosidine antibody and Dr. Koji Uchida (Nagoya University, Japan) for providing the argpyrimidine antibody. We thank Dr. Jose Halperin for supporting RF for this work and helpful suggestions, Dr. Judith Hettinga and Dr. Nicole Krumrei for establishing the hemolytic assay, Dr. John Rush from the Howard Hughes Medical Institute, Harvard Medical School for performing the LC/MS analyses, Sara Cechner for manuscript preparation, and the CWRU Visual Science Research Center Core Facilities (P30EY11373) for help with Figures.
Abbreviations
- AGEs
advanced glycation end products
- CCP
complement control protein repeat
- DAF
decay accelerating factor
- GPCR
G protein coupled receptor
Footnotes
This work was supported by National Institutes of Health grants EY11288, AI071125, and HL109561, AR067182 (MEM), EY022938, EY024864 and a Merit grant from the Department of Veteran Affairs (TSK), EY023286 (RHN), and EY11373 (Core), DK52855 (RF).
References
- Acosta JA, Benzaquen LR, Goldstein DJ, Tosteson MT, Halperin JA. The transient pore formed by homologous terminal complement complexes functions as a bidirectional route for the transport of autocrine and paracrine signals across human cell membranes. Mol Med. 1996;2:755–765. [PMC free article] [PubMed] [Google Scholar]
- Ahmed MU, Thorpe SR, Baynes JW. Identification of N epsilon-carboxymethyllysine as a degradation product of fructoselysine in glycated protein. J Biol Chem. 1986;261:4889–4894. [PubMed] [Google Scholar]
- Asch AS, Kinoshita T, Jaffe EA, Nussenzweig V. Decay-accelerating factor is present on cultured human umbilical vein endothelial cells. J Exp Med. 1986;163:221–226. doi: 10.1084/jem.163.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidasee KR, Nallani K, Yu Y, Cocklin RR, Zhang Y, Wang M, Dincer UD, Besch HR., Jr Chronic diabetes increases advanced glycation end products on cardiac ryanodine receptors/calcium-release channels. Diabetes. 2003;52:1825–1836. doi: 10.2337/diabetes.52.7.1825. [DOI] [PubMed] [Google Scholar]
- Brodbeck WG, Liu D, Sperry J, Mold C, Medof ME. Localization of classical and alternative pathway regulatory activity within the decay-accelerating factor. J Immunol. 1996;156:2528–2533. [PubMed] [Google Scholar]
- Fett AL, Hermann MM, Muether PS, Kirchhof B, Fauser S. Immunohistochemical localization of complement regulatory proteins in the human retina. Histol Histopathol. 2012;27:357–364. doi: 10.14670/HH-27.357. [DOI] [PubMed] [Google Scholar]
- Genuth S, Sun W, Cleary P, Gao X, Sell DR, Lachin J, Group, D.E.R. Monnier VM. Skin advanced glycation end products glucosepane and methylglyoxal hydroimidazolone are independently associated with long-term microvascular complication progression of type 1 diabetes. Diabetes. 2015;64:266–278. doi: 10.2337/db14-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CL, Abbott RJ, Smith RA, Morgan BP, Lea SM. Molecular dissection of interactions between components of the alternative pathway of complement and decay accelerating factor (CD55) J Biol Chem. 2005;280:2569–2578. doi: 10.1074/jbc.M410179200. [DOI] [PubMed] [Google Scholar]
- Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, Xu Y, Medof ME. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. 2005;201:1523–1530. doi: 10.1084/jem.20041967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannuzzi C, Borriello M, Carafa V, Altucci L, Vitiello M, Balestrieri ML, Ricci G, Irace G, Sirangelo I. D-ribose-glycation of insulin prevents amyloid aggregation and produces cytotoxic adducts. Biochim Biophys Acta. 2016;1862:93–104. doi: 10.1016/j.bbadis.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Iberg N, Fluckiger R. Nonenzymatic glycosylation of albumin in vivo. Identification of multiple glycosylated sites. J Biol Chem. 1986;261:13542–13545. [PubMed] [Google Scholar]
- Kinoshita T, Medof ME, Silber R, Nussenzweig V. Distribution of decay-accelerating factor in the peripheral blood of normal individuals and patients with paroxysmal nocturnal hemoglobinuria. J Exp Med. 1985;162:75–92. doi: 10.1084/jem.162.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttner-Kondo L, Medof ME, Brodbeck W, Shoham M. Molecular modeling and mechanism of action of human decay-accelerating factor. Protein Eng. 1996;9:1143–1149. doi: 10.1093/protein/9.12.1143. [DOI] [PubMed] [Google Scholar]
- Kuttner-Kondo LA, Mitchell L, Hourcade DE, Medof ME. Characterization of the active sites in decay-accelerating factor. J Immunol. 2001;167:2164–2171. doi: 10.4049/jimmunol.167.4.2164. [DOI] [PubMed] [Google Scholar]
- Kuttner-Kondo L, Hourcade DE, Anderson VE, Muqim N, Mitchell L, Soares DC, Barlow PN, Medof ME. Structure-based mapping of DAF active site residues that accelerate the decay of C3 convertases. J Biol Chem. 2007;282:18552–18562. doi: 10.1074/jbc.M611650200. [DOI] [PubMed] [Google Scholar]
- Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112:1759–1766. doi: 10.1182/blood-2008-04-151068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AK, Tesch GH. Inflammation in diabetic nephropathy. Mediators Inflamm. 2012;2012:146154. doi: 10.1155/2012/146154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszewski MK, Atkinson JP. Complement regulators in human disease: lessons from modern genetics. J Intern Med. 2015;277:294–305. doi: 10.1111/joim.12338. [DOI] [PubMed] [Google Scholar]
- Liu J, Lin F, Strainic MG, An F, Miller RH, Altuntas CZ, Heeger PS, Tuohy VK, Medof ME. IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J Immunol. 2008;180:5882–5889. doi: 10.4049/jimmunol.180.9.5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacik P, Roversi P, White J, Esser D, Smith GP, Billington J, Williams PA, Rudd PM, Wormald MR, Harvey DJ, Crispin MD, Radcliffe CM, Dwek RA, Evans DJ, Morgan BP, Smith RA, Lea SM. Complement regulation at the molecular level: the structure of decay-accelerating factor. Proc Natl Acad Sci U S A. 2004;101:1279–1284. doi: 10.1073/pnas.0307200101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof ME, Kinoshita T, Nussenzweig V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med. 1984;160:1558–1578. doi: 10.1084/jem.160.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof ME, Walter EI, Roberts WL, Haas R, Rosenberry TL. Decay accelerating factor of complement is anchored to cells by a C-terminal glycolipid. Biochemistry. 1986;25:6740–6747. doi: 10.1021/bi00370a003. [DOI] [PubMed] [Google Scholar]
- Medof ME, Lublin DM, Holers VM, Ayers DJ, Getty RR, Leykam JF, Atkinson JP, Tykocinski ML. Cloning and characterization of cDNAs encoding the complete sequence of decay-accelerating factor of human complement. Proc Natl Acad Sci U S A. 1987;84:2007–2011. doi: 10.1073/pnas.84.7.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medof ME, Nagarajan S, Tykocinski ML. Cell-surface engineering with GPI-anchored proteins. FASEB J. 1996;10:574–586. doi: 10.1096/fasebj.10.5.8621057. [DOI] [PubMed] [Google Scholar]
- Monnier VM, Nagaraj RH, Portero-Otin M, Glomb M, Elgawish AH, Sell DR, Friedlander MA. Structure of advanced Maillard reaction products and their pathological role. Nephrol Dial Transplant. 1996;11(Suppl. 5):20–26. doi: 10.1093/ndt/11.supp5.20. [DOI] [PubMed] [Google Scholar]
- Monnier VM, Sell DR, Strauch C, Sun W, Lachin JM, Cleary PA, Genuth S, Group, DR The association between skin collagen glucosepane and past progression of microvascular and neuropathic complications in type 1 diabetes. J Diabetes Complicat. 2013;27:141–149. doi: 10.1016/j.jdiacomp.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327–340. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- Oya T, Hattori N, Mizuno Y, Miyata S, Maeda S, Osawa T, Uchida K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J Biol Chem. 1999;274:18492–18502. doi: 10.1074/jbc.274.26.18492. [DOI] [PubMed] [Google Scholar]
- Perry RE, Swamy MS, Abraham EC. Progressive changes in lens crystallin glycation and high-molecular-weight aggregate formation leading to cataract development in streptozotocin-diabetic rats. Exp Eye Res. 1987;44:269–282. doi: 10.1016/s0014-4835(87)80011-8. [DOI] [PubMed] [Google Scholar]
- Qin X, Goldfine A, Krumrei N, Grubissich L, Acosta J, Chorev M, Hays AP, Halperin JA. Glycation inactivation of the complement regulatory protein CD59: a possible role in the pathogenesis of the vascular complications of human diabetes. Diabetes. 2004;53:2653–2661. doi: 10.2337/diabetes.53.10.2653. [DOI] [PubMed] [Google Scholar]
- Sady C, Jiang CL, Chellan P, Madhun Z, Duve Y, Glomb MA, Nagaraj RH. Maillard reactions by alpha-oxoaldehydes: detection of glyoxal-modified proteins. Biochim Biophys Acta. 2000;1481:255–264. doi: 10.1016/s0167-4838(00)00133-3. [DOI] [PubMed] [Google Scholar]
- Sell DR, Monnier VM. Structure elucidation of a senescence cross-link from human extracellular matrix. Implication of pentoses in the aging process. J Biol Chem. 1989;264:21597–21602. [PubMed] [Google Scholar]
- Shipanova IN, Glomb MA, Nagaraj RH. Protein modification by methylglyoxal: chemical nature and synthetic mechanism of a major fluorescent adduct. Arch Biochem Biophys. 1997;344:29–36. doi: 10.1006/abbi.1997.0195. [DOI] [PubMed] [Google Scholar]
- Stafford HA, Tykocinski ML, Lublin DM, Holers VM, Rosse WF, Atkinson JP, Medof ME. Normal polymorphic variations and transcription of the decay accelerating factor gene in paroxysmal nocturnal hemoglobinuria cells. Proc Natl Acad Sci U S A. 1988;85:880–884. doi: 10.1073/pnas.85.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, Medof ME. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14:162–171. doi: 10.1038/ni.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Takabatake Y, Kimura T, Maejima I, Namba T, Yamamoto T, Matsuda J, Minami S, Kaimori JY, Matsui I, Matsusaka T, Niimura F, Yoshimori T, Isaka Y. Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes. 2017;66:1359–1372. doi: 10.2337/db16-0397. [DOI] [PubMed] [Google Scholar]
- Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011;30:343–358. doi: 10.1016/j.preteyeres.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrinova S, Lin F, Uhrin D, Medof ME, Barlow PN. Resonance assignments of the central complement control protein module pair of human decay accelerating factor. J Biomol NMR. 2002;23:167–168. [PubMed] [Google Scholar]
- Uhrinova S, Lin F, Ball G, Bromek K, Uhrin D, Medof ME, Barlow PN. Solution structure of a functionally active fragment of decay-accelerating factor. Proc Natl Acad Sci U S A. 2003;100:4718–4723. doi: 10.1073/pnas.0730844100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lookeren Campagne M, Wiesmann C, Brown EJ. Macrophage complement receptors and pathogen clearance. Cell Microbiol. 2007;9:2095–2102. doi: 10.1111/j.1462-5822.2007.00981.x. [DOI] [PubMed] [Google Scholar]
- Vishwanath V, Frank KE, Elmets CA, Dauchot PJ, Monnier VM. Glycation of skin collagen in type I diabetes mellitus. Correlation with long-term complications. Diabetes. 1986;35:916–921. doi: 10.2337/diab.35.8.916. [DOI] [PubMed] [Google Scholar]
- Walter EI, Ratnoff WD, Long KE, Kazura JW, Medof ME. Effect of glycoinositolphospholipid anchor lipid groups on functional properties of decay-accelerating factor protein in cells. J Biol Chem. 1992;267:1245–1252. [PubMed] [Google Scholar]