

Abstract
When an action potential invades a presynaptic terminal it evokes large, brief Ca2+ signals that trigger vesicle fusion within milliseconds that is followed by a small residual Ca2+ (Cares) signal. At many synapses Cares produces synaptic facilitation that lasts up to hundreds of milliseconds and, although less common, Cares can also evoke asynchronous release (AR) that persists for tens of milliseconds. The properties of facilitation and AR are very different, which suggests that they are mediated by distinct mechanisms. However, recently it has been shown that the slow calcium sensor synaptotagmin 7 (Syt7) mediates facilitation at many synapses where AR does not occur, and conversely Syt7 can mediate AR without mediating facilitation. Here we study cerebellar granule cell synapses onto stellate cells and Purkinje cells in mice of both sexes to assess the role of Syt7 in these phenomena at the same synapse. This is of particular interest at granule cell synapses where AR is much more calcium dependent and shorter-lived than facilitation. We find that Syt7 can mediate these two processes despite their divergent properties. In Syt7 knock-out animals, facilitation and AR are smaller and shorter lived than in wild-type animals, even though the initial probability of release and Cares signals are unchanged. Although there are short-lived Syt7-independent mechanisms that mediate facilitation and AR in Syt7 KO animals, we find that at granule cell synapses AR and facilitation are both mediated primarily by Syt7.
SIGNIFICANCE STATEMENT At synapses made by cerebellar granule cells, presynaptic activity elevates calcium for tens of milliseconds, which in turn evokes both asynchronous release (AR) and synaptic facilitation. AR is more calcium sensitive and shorter-lived than facilitation at these synapses, suggesting that they are mediated by different mechanisms. However, we find that the slow calcium sensor synaptotagmin 7 mediates both of these phenomena. Small, rapidly decaying components of AR and facilitation are present in Syt7 KO animals, indicating that additional mechanisms can contribute to both AR and facilitation at these synapses.
Keywords: asynchronous release, calcium, facilitation, short-term plasticity
Introduction
Activation of presynaptic cells opens voltage gated Ca2+ channels within presynaptic boutons leading to large, fast, highly localized Ca2+ signals that trigger rapid vesicle fusion. Ca2+ then binds to Ca2+-binding proteins and diffuses, thereby collapsing spatial gradients and giving rise to a small residual Ca2+ (Cares) signal that persists for tens of milliseconds and controls neurotransmitter release in multiple ways. Cares can elevate the probability of release (PR) to produce synaptic facilitation of synchronous release for hundreds of milliseconds. Facilitation is present at synapses throughout the brain, and is most prominent at synapses with low initial PR. Cares can also trigger asynchronous release (AR) that lasts for tens of milliseconds following single stimuli (Atluri and Regehr, 1998; Iremonger and Bains, 2007). AR is not apparent at most synapses, but when observed it is most prominent following bursts of presynaptic activity or during sustained activation (Atluri and Regehr, 1998; Lu and Trussell, 2000; Hefft and Jonas, 2005; Iremonger and Bains, 2007; Best and Regehr, 2009; Labrakakis et al., 2009; Peters et al., 2010).
It has been hypothesized that facilitation and AR involve specialized Ca2+ sensors, and that the same sensor might mediate both phenomena (Rahamimoff and Yaari, 1973; Zucker and Regehr, 2002; Jackman and Regehr, 2017). However, many synapses facilitate in the absence of AR, whereas some synapses with AR do not facilitate (Hefft and Jonas, 2005; Labrakakis et al., 2009; Peters et al., 2010). Further, when both phenomena are present at the same synapses, their time course and Ca2+ dependence can be different (Atluri and Regehr, 1998; but see Molgó and Van der Kloot, 1991). These observations made it seem unlikely that the same Ca2+ sensor mediates both AR and facilitation.
Despite the differences between the two processes, recent studies indicate that synaptotagmin 7 (Syt7) can contribute to either facilitation or AR (Wen et al., 2010; Bacaj et al., 2013; Jackman et al., 2014; Luo et al., 2015; Luo and Südhof, 2017; Turecek et al., 2017). The properties of Syt7 make it a strong candidate for both processes because it is the Synaptotagmin isoform with the highest affinity for Ca2+ and the slowest kinetics (Sugita et al., 2002; Brandt et al., 2012), and it is widely expressed (Mittelsteadt et al., 2009). Syt7 was initially implicated in AR at the zebrafish neuromuscular junction, where sustained presynaptic activation led to Syt7-dependent AR (Wen et al., 2010). Initial studies of cultured mammalian synapses found that eliminating Syt7 did not alter synaptic responses (Maximov et al., 2008), but it was subsequently found that in the absence of Syt1 AR is prominent and Syt7-dependent (Bacaj et al., 2013). At the young calyx of Held, sustained high-frequency stimulation led to slow currents that were attributed to AR, and that were strongly reduced in Syt7 KOs (Luo and Südhof, 2017). However, facilitation is absent or very weak at synapses where Syt7 has been implicated in AR. In contrast, at three types of hippocampal synapses and thalamocortical synapses, paired-pulse facilitation is prominent in wild-types, but eliminated in Syt7 KOs (Jackman et al., 2016). At Purkinje cell (PC) synapses and vestibular synapses, Syt7-mediated facilitation is present but it is masked by depression (Turecek et al., 2017). At synapses where Syt7 has been implicated in facilitation, AR is not apparent. Thus, Syt7 mediates facilitation without producing AR at some synapses, but mediates AR without generating facilitation at others.
Here we determine the role of Syt7 in transmission at synapses made by cerebellar granule cells (grCs) onto stellate cells (SCs), and PCs. These synapses are of particular interest because they have large facilitation, AR is prominent after single stimuli, and AR is shorter-lived and more Ca2+ dependent than facilitation (Atluri and Regehr, 1998). We find that Syt7 elimination strongly attenuates both AR and facilitation and decreases their duration, indicating that both processes are mediated primarily by Syt7. We also conclude that additional mechanisms mediate the rapid AR and facilitation remaining in Syt7 KO mice.
Materials and Methods
Animals.
Syt7 KO mice and wild-type littermates (Chakrabarti et al., 2003) of either sex were used. All mice were handled in accordance with NIH guidelines and protocols approved by Harvard Medical Area Standing Committee on Animals. Experiments and analysis shown in Figures 1, 3 and 5 were performed blind. Experiments in Figures 2, 4, 6–8 were initially performed blind but blinding was abandoned because the genotype could be reliably determined from physiology alone.
Figure 1.
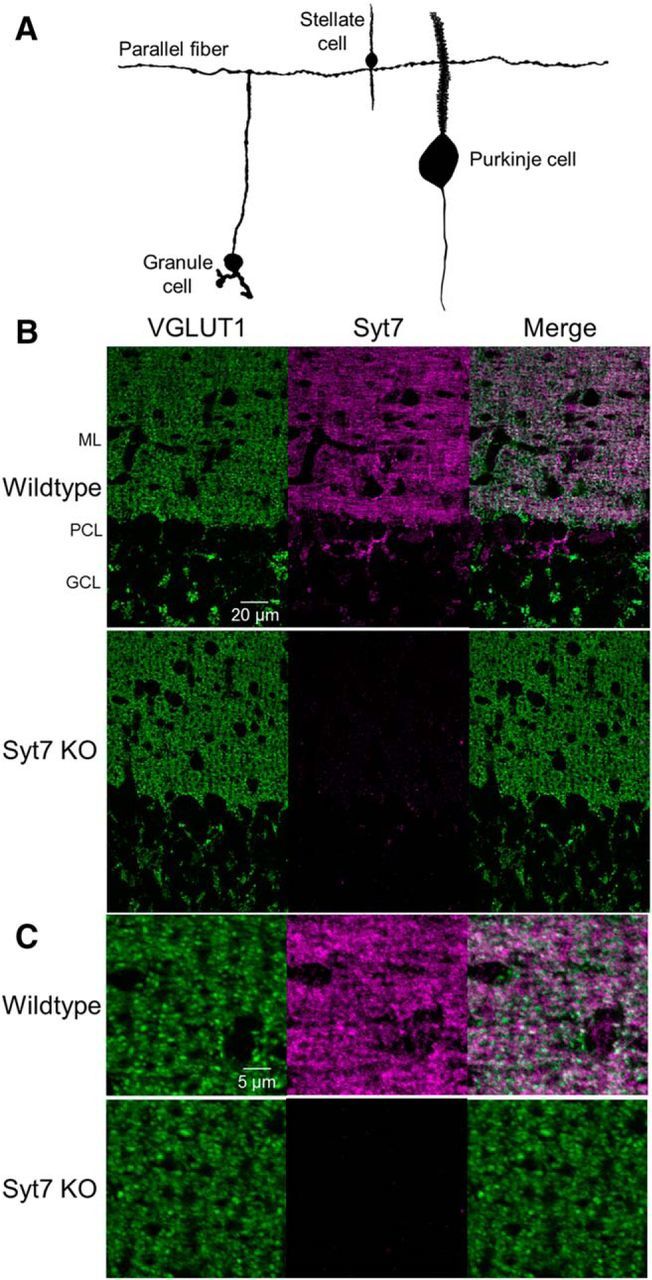
Immunohistochemical localization of Syt7 in the cerebellar cortex. A, Parallel fibers originate from granule cells and extend laterally in transverse sections, forming synapses onto stellate and PCs in the cerebellar cortex (vermal lobule VI). The SC and PC dendrites are oriented perpendicular to the surface of the slice. B, Transverse sections of cerebellar cortex immunolabeled for the presynaptic marker VGLUT1 (green) and Syt7 (magenta) in a wild-type (top) and Syt7 KO (bottom). ML, Molecular layer; PCL, Purkinje cell layer; GCL, granule cell layer. C, Expanded view of the molecular layer for the images in B.
Figure 3.
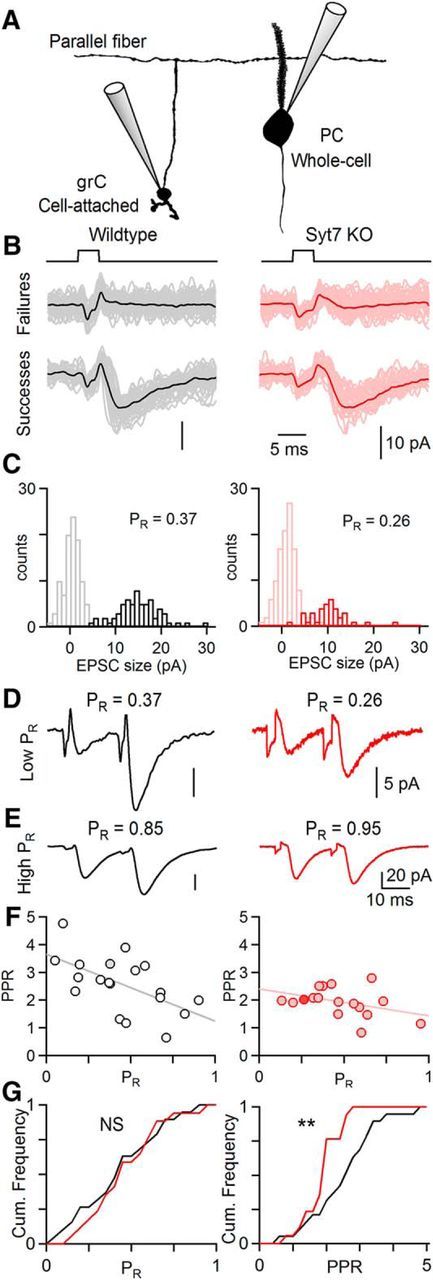
The probability of release at single granule cell to PC synapses is unaltered in Syt7 KO animals. Cell-attached recordings were used to stimulate single grCs and PC responses were measured. A, Experimental configuration. A single grC was stimulated in cell-attached mode and whole-cell responses were recorded in PCs at least 150 μm away in the transverse plane. B, Synaptic currents evoked by a voltage step to a grC cell body (top) were recorded in PCs. Averaged synaptic responses of failed and successful release events for wild-type (left) and Syt7 KOs (right). Voltage steps applied to grCs generated a small stimulus artifact preceding synaptic currents. C, Histogram of response amplitudes for pairs shown in B. PR was calculated as the fraction successes. D, Averaged synaptic currents evoked by two stimuli (20 ms) for cells from wild-type and Syt7 KO animals shown in B and C. E, Example of averaged synaptic currents at grC to PC pairs that had high initial PR. F, All grC to PC pairs plotted by PPR versus PR for wild-types (left; n = 19) and Syt7 KOs (right; n = 17) with linear fits. Filled markers indicate cells shown in B–D. G, Cumulative histograms of PR (left) and PPR (right) for all paired recordings, two-sample K-S test, **p < 0.01.
Figure 5.

Presynaptic Ca2+ signaling is unaltered in Syt7 KO animals. A, Parallel fibers were loaded with the low affinity Ca2+ indicator Mg-Green-AM (Kd = 7 μm for Ca2+), and stimulated. Ca2+-dependent presynaptic fluorescence changes are shown for average wild-type (black) and Syt7 KO animal (red). The half decay time was measured and summarized (right; wild-type: n = 6, Syt7 KO: n = 6). B, Same as in A, but for brief stimulus trains (5 stimuli at 50 Hz). Averages are shown for wild-type and Syt7 KOs (left) with single stimuli overlaid. The ratio between the amplitude of the first and second transient in the train are summarized (right; wild-type: n = 7, Syt7 KO: n = 6). C, Parallel fibers were labeled with the high affinity Ca2+ indicator Fura-2-AM (Kd = 0.2 μm) and responses were evoked by one and two stimuli. Averages are shown for a wild-type (black) and Syt7 KO (red). The ratio of the amplitudes of the responses evoked by two stimuli and one stimulus provides a measure of the magnitude of presynaptic Ca2+ entry, and is summarized (right; wild-type: n = 9, Syt7 KO: n = 10). Traces are normalized to peak values evoked by a single stimulus. Data are mean ± SEM.
Figure 2.

Facilitation is strongly attenuated in Syt7 KO animals at synapses made by granule cells onto PCs and SCs. Paired-pulse facilitation in wild-type and Syt7 KO mice. A, Representative paired-pulse facilitation at the grC to PC synapse in wild-type (top) and Syt7 KO (middle). PPR as a function of interstimulus interval (Δt) is summarized (bottom) for wild-type (n = 9) and Syt7 KOs (n = 10). Fits to single (solid) and double (dashed) exponential fits are shown. B, Same as in A, but at grC to SC synapses for wild-type (n = 6) and Syt7 KO (n = 7). Data are mean ± SEM. For some points error bars are occluded by markers.
Figure 4.
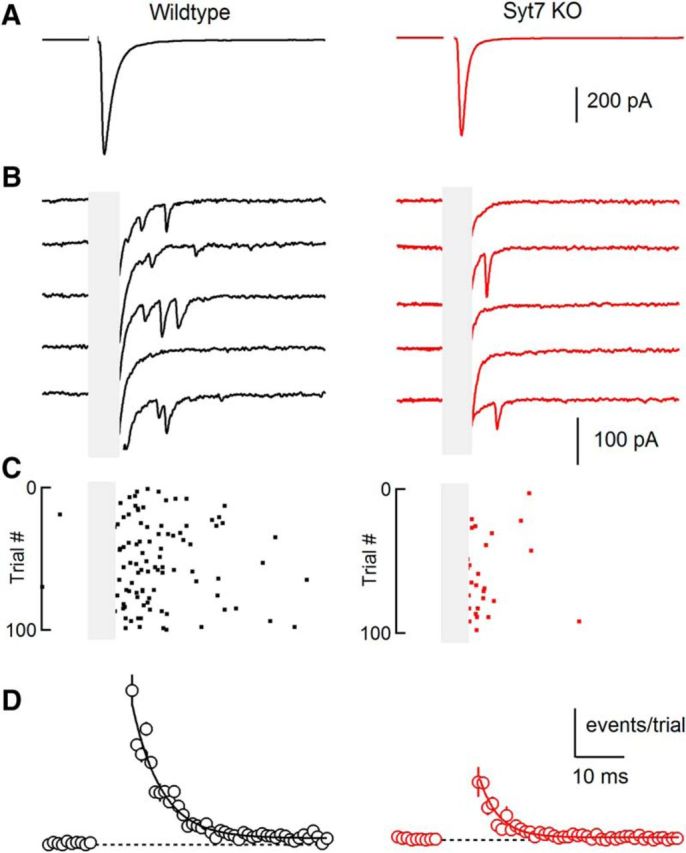
Asynchronous release at the granule cell to SC synapse is mediated primarily by Syt7. A, Extracellular activation of grC parallel fibers with single stimuli evoked synaptic responses in SCs: the average EPSC (100 trials) showed a large prominent synchronous component in both wild-types (left) and Syt7 KOs (right). B, Asynchronous quantal events were apparent in individual trials (stimulus artifact and synchronous EPSC has been blanked). C, Raster plot of quantal events for all trials for cells shown in A and B. D, The time course and amplitude of quantal events evoked by single stimuli summarized across cells for wild-types (left; n = 17) and Syt7 KOs (right; n = 10). Data are mean ± SEM. For some points error bars are occluded by markers.
Figure 6.
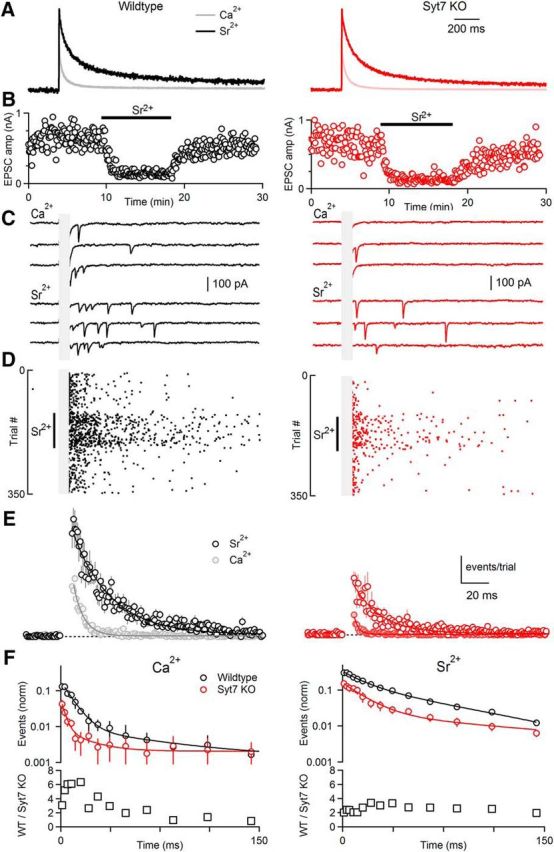
The Sr2+ dependence of asynchronous release in wild-type and Syt7 KO animals. A, Presynaptic divalent levels were measured in the presence of extracellular Ca2+ and extracellular Sr2+ using Mg-Green in wild-type and Syt7 KO animals as described in Materials and Methods. B, Synaptic currents were evoked by single stimuli in the presence of external Ca2+ and Sr2+ and peak EPSC amplitudes are shown as a function of time for representative cells. C, Example traces from cells shown in B in the presence of Ca2+ and Sr2+. D, Raster of quantal events shown during the application and washout of Sr2+ for example cells shown in B and C. E, Summary of delayed events evoked by single stimuli in the presence of Ca2+ and Sr2+ for wild-type (n = 7) and Syt7 KO mice (n = 7). F, Left, The frequency of quantal events in the presence of Ca2+ is shown as a function of time for wild-type (top graph, black) and Syt7 KO animals (top graph, red) in a semilogarithmic plot with logarithmic binning. A point-by-point ratio of the rate of events of wild-type to Syt7 KO animals is plotted in the lower graph. Right, Similar analysis but for events in the presence of Sr2+. Data are mean ± SEM. For some points error bars are occluded by markers.
Figure 8.

Facilitation and asynchronous release under physiological conditions in wild-type and Syt7 KO animals. Experiments were performed at 35°C in 1.5 mm external Ca2+. A, Paired-pulse facilitation in representative PCs for wild-type and Syt7 KO animals. B, Summarized paired-pulse facilitation curves for wild-types (n = 5) and Syt7 KOs (n = 5). Single (solid) and double (dashed) exponential fits are shown. Asynchronous release was examined in wild-type and Syt7 KO animals (C–F). C, Average evoked current by single stimuli in a representative wild-type SC. D, Single stimuli evoked delayed events during single trials for cells shown in C. Synchronous component of the EPSC and stimulus artifact are blanked. E, Raster plot of quantal events. F, The time course and amplitude of quantal events evoked by single stimuli summarized across cells in wild-type (n = 9) and Syt7 KOs (n = 8). Data are mean ± SEM. For some points error bars are occluded by markers.
Slice preparation.
P16–P23 animals of both sexes were anesthetized with ketamine/xylazine and transcardially perfused with solution containing the following (in mm): 110 choline Cl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 0.5 CaCl2, 7 MgCl2, 3.1 Na pyruvate, 11.6 Na ascorbate, 0.002 (R)-CPP, 0.005 NBQX, oxygenated with 95% O2/5% CO2, kept at 4°C. The cerebellum was removed and horizontal/transverse slices (250 μm) were made using a Leica 1200S vibratome in choline ACSF maintained at 4°C. Slices were then transferred to a holding chamber with solution containing the following (in mm): 127 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2, 1 MgCl2, and allowed to recover at 35°C for at least 30 min before cooling to room temperature.
Electrophysiology.
Experiments in Figures 2–7 were performed at room temperature and experiments in Figure 8 were performed at 35°C. Unless otherwise stated, the composition of ACSF used for recording was the same as for slice incubation. Whole-cell voltage-clamp recordings were made from PCs or molecular layer interneurons in the outer third of the molecular layer (SCs) in vermal lobules V and VI. Borosilicate electrodes (1–1.5 MΩ for PCs, 1.5–2.5 MΩ for SCs) were filled with the following (in mm): 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, pH 7.3 with CsOH. A recording electrode (1.5–2.5 MΩ) filled with ACSF was placed several hundred micrometers away in the molecular layer to stimulate parallel fibers (0.2 ms pulses, 5–40 μA). For PCs, the tip of the stimulation electrode was placed within 10–50 μm of the PC layer to activate parallel fibers synapses close to the PC body. For SCs, the electrode was placed in alignment with the SC body and at a depth to prevent activation of off-beam fibers (Carter and Regehr, 2000). For all experiments measuring facilitation, the average initial EPSC amplitude was kept between 100 and 250 pA to minimize the activation of off-beam fibers and to maintain voltage-clamp. PCs were held at −60 mV, with series resistance (1–10 MΩ) compensated up to 80% and whole-cell capacitance compensated only for the cell body (5 pF). SCs were held at −70 mV and series resistance and whole-cell capacitance were left uncompensated. Experiments were performed in the presence of bicuculine (20 μm) or SR-95531 (5 μm) to block GABAARs. Experiments measuring mEPSCs were performed in SCs in the presence of 0.5 μm TTX and performed in slices unperturbed by stimulation electrodes.
Figure 7.

Facilitation in wild-type and Syt7 KO animals is dependent on residual Ca2+. A, Paired-pulse facilitation for a representative PC shown before (control; gray) and after bath application of EGTA-AM (20 μm for 15 min; bottom, black). B, Same as in A, but for a Syt7 KO. C, Summary of the effect of EGTA on paired-pulse facilitation in wild-type grC to PC synapses (control: n = 17; EGTA: n = 8). D, Same as in C, but for Syt7 KOs (control: n = 11; EGTA: n = 9). Data are mean ± SEM. For some points error bars are occluded by markers.
In paired recordings between grCs and PCs, cell-attached recordings were obtained from grCs using electrodes filled with the following (in mm): 135 K-gluconate, 20 KCl, 2 MgCl2, 10 HEPES, 0.1 EGTA pH 7.3 using KOH. When searching for grCs, brief infrequent (0.1 Hz) pulses of positive pressure were applied in the grC layer. Regions of the grC layer in which puffs elicited synaptic currents were then targeted for single-cell recordings. grCs were at least 150 μm away from the recorded PC to avoid ascending branch synapses which are reported to have different properties than parallel fiber synapses (Sims and Hartell, 2005). grCs were held in cell-attached mode with a holding potential of −60 mV and 3–5 ms voltage steps of 50–200 mV were pulsed to elicit action currents as previously described (Schmidt et al., 2013). For grC to PC pairs, PCs were recorded using leaded glass electrodes (1–1.5 MΩ) filled with the following (in mm): 110 Cs2SO4, 10 HEPES, 10 EGTA, 4 CaCl2, 1.5 MgCl2, 5.5 MgSO4, 4 Na2-ATP, 0.1 D600, and 5 QX-314, pH 7.3 with CsOH. For these experiments, PCs were held at −70 mV and series resistance and whole-cell capacitance were left uncompensated.
For experiments measuring the effect of EGTA-AM, 20 μm EGTA-AM was washed into the bath for 15 min. Data were collected from multiple cells within the same slice following wash-in. Thus, data shown in Figure 7 are from some cells in which recordings were maintained before, during and after wash-ins, as well as cells in which recordings were made following wash-in. Experiments measuring AR in Sr2+ were performed as previously described (Xu-Friedman and Regehr, 2000). Briefly, control ACSF contained the following (in mm): 2 CaCl2 and 1 MgCl2, which was then replaced with ACSF containing 2 EGTA, 4 SrCl2, and 1 MgCl2. EGTA binds Ca2+ with high affinity (Kd of ∼100 nm), Sr2+ with moderate affinity (Kd ∼30 μm) and Mg2+ with low affinity (Kd ∼15 mm), resulting in estimated free divalent levels of 2 Sr2+, 1 Mg2+, and very low free Ca2+. All data shown in Figure 6 are wash-ins.
Ca2+ transients were measured from either Mg-Green AM or Fura-2 AM (240 μm) loaded into parallel fibers as previously described (Regehr and Tank, 1991; Regehr and Atluri, 1995; Atluri and Regehr, 1996; Brenowitz and Regehr, 2014). Briefly, indicator was loaded into a pipette (8–15 μm diameter), which was lowered into the molecular layer under positive pressure for 3 min. Fast green (1%) was included to allow visualization under bright field. A suction pipette (15–20 μm diameter) was positioned near the outflow of the labeling pipette to confine the area of indicator loading. Slices were imaged using a 60× objective and custom-built photodiode at least 1 h following loading, with imaging sites >300 μm from the loading site. A stimulus electrode was placed >400 μm from the imaging site to excite parallel fibers. Mg-Green and Fura-2 were excited by a tungsten and xenon lamp, respectively.
Sr2+levels were determined as described previously (Xu-Friedman and Regehr, 1999, 2000). Briefly, fluorescence transients in Sr2+were normalized to transients in Ca2+ at the same imaging site and converted to relative concentrations by adjusting for the binding affinity of Mg-Green for Ca2+versus Sr2+(multiplying Sr2+ transients by 5.5 = KD-Sr/KD-Ca).
Analysis.
Recordings were collected using a MultiClamp 700B (Molecular Devices) sampled at 20 kHz and filtered at 2 kHz. All data were collected in Igor Pro (WaveMetrics). All analysis was performed using custom-written scripts in MATLAB (MathWorks). Paired-pulse facilitation was measured from averages of at least 10 trials per interstimulus interval. For short interstimulus intervals in PCs, EPSCs were elicited during the decay of the initial EPSC. In these cases, the initial EPSC from long duration interstimulus intervals was subtracted. All data in figures are summarized as mean ± SEM across individual cells. In some cases, error bars are occluded by markers. Number of experiments are presented in figure legends as number of cells, except for Ca2+ imaging experiments where the number of slices are indicated. In text, fit coefficients are provided ± SD, and all other values are summarized as mean ± SEM.
To quantify AR, events were detected for each trial. Histograms of asynchronous release versus time were generated by summing the events across all trials, and dividing by the number of trials. The peak amplitude of the synchronous component (within 9 ms following stimulation) was used as a measure of the number of synapses stimulated. Histograms of asynchronous release versus time for each cell were therefore divided by the peak amplitude of the synchronous EPSC, measured from the averaged trace of all trials for that cell.
In paired recordings, only trials in which action potentials were evoked in the grC were included for analysis. The amplitude of the evoked current in PCs for each trial was measured as the average of 2 ms around the peak current following stimulation. At least 120 acceptable trials were collected per cell, and up to 600 trials were collected for cells with low PR. Trials in which spontaneous release obscured the detection of postsynaptic events were discarded. A double Gaussian fit was applied to histograms of EPSC amplitudes for each cell and used to estimate the number of successes and failures. For all cells, release events were also detected manually by visual inspection, which resulted in good agreement with Gaussian fits. In rare cases, histograms of evoked responses contained two peaks, and were discarded. To determine the paired-pulse ratio (PPR), all trials were averaged and the PPR was measured from the averaged trace as the peak amplitude of the second EPSC divided by the peak amplitude of the first. The potency was determined by averaging all trials in which there was a successful release event on the first stimulus and measuring the peak amplitude of the first EPSC.
Experimental design and statistical analysis.
Facilitation curves and AR were averaged across cells and fit with a single exponential and offset of the form: PPR = (c + Ae−(t−t0)/τ) or release = (c + Ae−(t−t0)/τ) For facilitation curves, the offset was set to 1, and t0 was the smallest interstimulus interval (10 ms for 25°C, 5 ms for 35°C). For AR, t0 was taken as the time when individual events could first be detected following synchronous release (9 ms following stimulation for experiments at 25°C, 5 ms for experiments at 35°C). In all cases data were well-fit by single exponentials (R2 > 0.95). Once single exponential fits were obtained, permutation tests were used to determine whether fit coefficients between wild-type and Syt7 KO were significantly different (Quinn and Keough, 2002). Wild-type and Syt7 KO data were randomly assigned to two groups and the averages were fit with the above equations. The residuals, or the differences between the fit coefficients between the two groups (A1–A2; τ1–τ2), were collected. This process was repeated 10,000 times to generate a distribution of residuals. The difference between observed, unpermutated, wild-type, and Syt7 KO data were then compared with the generated distribution. A p value was obtained as the number of generated residuals greater than the observed difference, with p < 0.01 considered significant.
For imaging experiments, results were compared using two-tailed unpaired Student's t tests. For paired grC to PC recordings, the distributions of PR, PPR, and potency were compared between wild-types and Syt7 KOs using a two-sample Kolmogorov–Smirnov (K–S) test. The distributions of mEPSC amplitudes and mEPSC frequencies were compared between wild-types and Syt7 KOs using two-sample K–S tests.
Immunohistochemistry.
P21 female mice were anesthetized with ketamine/xylazine (100/10 mg/kg) and perfused transcardially with PBS followed by 4% paraformaldehyde (PFA) in PBS. The brain was removed and kept in PFA overnight. Transverse sections of the cerebellum (50 μm) were permeabilized using 0.2% Triton X-100 in PBS for 10 min and blocked with 4% normal goat serum in 0.1% Triton X-100 for 1 h at room temperature. Slices were incubated overnight at 4°C with primary Syt7 antibodies (mouse anti-Syt7 targeting the C2A domain, UC Davis/NIH NeuroMab Facility, clone N275/14; RRID:AB_11030371; 1 μg/ml, 1:100). Following overnight incubation with Syt7 antibodies VGLUT1 primary antibodies (guinea pig anti-VGLUT1; Synaptic Systems 135304; 1 μg/ml, 1:1600) were applied alone for 2 h at room temperature to reduce background. Slices were then incubated with secondary antibodies for 2 h at room temperature (anti-guinea pig-AlexaFluor488, Abcam, ab150185; anti-mouse-AlexaFluor647, Abcam, ab150115). All tissue was stained and processed in parallel. z-Stacks of lobule VI along the vermis were collected using an Olympus Fluoview1000 confocal microscope. Images were collected using the same settings, and processed identically in ImageJ.
Results
Syt7 is present in the molecular layer of the cerebellum
We used immunohistochemistry to determine whether Syt7 is expressed in parallel fibers of the cerebellum. In transverse sections of the cerebellum, grCs give rise to parallel fibers that traverse the slice, whereas PC and SC dendrites are oriented orthogonally into the slice (Fig. 1A). Because of the preservation of parallel fibers, this orientation was used for both anatomy and physiology experiments. grCs form glutamatergic synapses onto both PCs and SCs in the molecular layer of the cerebellum and express VGLUT1. In wild-type animals Syt7 labeling was prominent in the molecular layer where VGLUT1-expressing grC synapses are enriched (Fig. 1B,C). Syt7 labeling was weak in KO animals, whereas the expression of VGLUT1 in grC synapses was comparable.
Facilitation and asynchronous release are strongly attenuated in Syt7 KO animals
We examined synaptic facilitation by stimulating with pairs of stimuli separated by different interstimulus intervals. The resulting synaptic currents evoked for different interstimulus intervals are shown for the grC to PC synapse for wild-type animals (Fig. 2A, top) and for Syt7 KO animals (Fig. 2A, middle) and the average paired-pulse facilitation curves for many cells are shown (Fig. 2A, bottom). The facilitation curves were fit to the equation (1 + Ae−(t−t0)/τ), where A is the amplitude of facilitation and τ is the time constant of facilitation. The magnitude and time constant of facilitation were reduced, respectively, from 2.12 ± 0.04 to 1.02 ± 0.06 and from 85 ± 5 to 46 ± 7 ms in Syt7 KO mice. Similar experiments were performed at the grC to SC synapse (Fig. 2B), and the magnitude and time constant of facilitation were reduced, respectively, from 1.52 ± 0.07 to 0.67 ± 0.08 and from 132 ± 20 to 35 ± 10 ms in Syt7 KO mice. The decreases in amplitudes and time constants of facilitation were statistically significant (p < 0.01, permutation test; see Materials and Methods). These results indicate that Syt7 mediates most of the facilitation at these synapses, but there is also a Syt7-independent mechanism that mediates shorter-lived facilitation in Syt7 KO mice.
The observation that PPR decays more rapidly in Syt7 KO animals than in wild-type animals raises the question as to whether facilitation is normally mediated by two processes with different time courses in wild-type animals:
and for Syt7 KO animals the slow component is selectively eliminated and facilitation is mediated only by the fast component:
A fit to Equation 1 for the average PPR in grC to PC synapses in wild-type animals yielded constants {Afast, τfast, Afast, τslow} of {1.12, 47 ms, 1.04, 147 ms}, and a for a fit to Equation 2 for PPR in Syt7 KO animals {Afast, tfast} are {1.02, 47 ms}. The similarity of Afast and τfast for wild-type and Syt7 KO would seem to suggest that PPR is a consequence of a fast and a slow component in wild-type animals, and that the slow component is eliminated selectively in Syt7 KO animals. However, the double exponential fit is essentially indistinguishable from the single exponential fit (Fig. 2A) and both are good fits to the data (R2 >0.95) making it unclear whether a double exponential fit is more appropriate. Moreover, the four free parameters of the double exponential fits cannot be precisely estimated because there are only seven points. Thus, we conclude that the faster decay of facilitation in Syt7 KO animals is compatible with either an acceleration of a single exponential decay, or with the selective elimination of a slow component of facilitation.
Synaptic facilitation can be reduced either as a direct effect on the mechanism of facilitation, or indirectly by increasing the PR. Previous studies indicated that the initial PR is not altered at the CA3 to CA1 synapse in Syt7 KO animals (Jackman et al., 2016). At that synapse, PR was assessed with input/output curves and with the use-dependent block of NMDARs by MK801. These approaches are more difficult to apply at grC synapses. Input/output curves are highly sensitive to slice orientation for grC parallel fibers, and use-dependent blockade by MK-801 is impractical because at this age PCs do not express NMDARs and the NMDAR component in SCs is extremely small. Most grCs form a single contact onto SCs and PCs, allowing a measure of release from single putative release sites. Given the extremely low connection probability between grCs and SCs, we therefore determined whether initial PR was altered by determining the properties of single grC to PC connections (Fig. 3). We stimulated single grCs using brief voltage steps in a cell-attached configuration that also allowed us to detect the presynaptic action potential. We targeted grCs at least 150 μm lateral from the PC (Fig. 3A), which provide parallel fiber inputs to PCs that consist of single contacts. grC stimulation generated artifacts in the PC that were followed by synaptic currents in some trials, and by failures in others (Fig. 3B,C). Successes and failures were evident as two distinct distributions in the histogram of the response amplitudes (Fig. 3C), and the fraction of trials in which release was detected was used as a measure of PR. There was considerable variability in the magnitude of PR, but there was no significant difference between the initial PR between wild-type and Syt7 KOs (Fig. 3G, left; wild-type: 0.44 ± 0.06, n = 19; Syt7 KO: 0.48 ± 0.05 n = 17; p = 0.80, K–S test). There was also no difference in the potency, or the amplitude of successful events, between wild-types and Syt7 KOs (wild-type: 16.9 ± 2.8 pA, n = 19; Syt7 KO: 14.4 ± 2.4 pA, n = 17; p = 0.32, K–S test). This suggests that differences in facilitation are not a secondary consequence of alterations in the initial PR at grC to PC synapses.
We also examined facilitation at synapses between individual grCs and PCs to provide additional insight into the mechanism of facilitation. Average synaptic responses evoked by pairs of stimuli are shown for synapses with a low initial PR (Fig. 3D) and high PR (Fig. 3E) for both wild-type and Syt7 KO animals. For wild-type animals, facilitation was variable but was generally larger at synapses with low initial PR, as illustrated by plotting PPR as a function of PR and fitting to a line, with PPR = (1 + 2.65) − 2.41 × PR (Fig. 3F, left; R2 = 0.34). The more prominent facilitation at low initial PR synapses is consistent with more severe vesicle depletion at high PR synapses (Zucker and Regehr, 2002). Facilitation in Syt7 KO animals is also variable and dependent on PR, but it is less steeply dependent upon PR than for wild-type animals, with PPR = (1 + 1.39) − 0.97 × PR (Fig. 3F, right; R2 = 0.16). Facilitation (PPR-1) was significantly reduced in Syt7 KOs (Fig. 3G, right; wild-type: 1.58 ± 0.23, n = 19; Syt7 KO: 0.93 ± 0.12, n = 17; p < 0.01 K–S test).
Facilitation at individual synapses showed the same trend as the facilitation evoked by extracellular stimulation of many parallel fibers (Fig. 2; 2.12 for wild-type animals and 1.02 for Syt7 KO animals for an interstimulus interval of 20 ms), but facilitation for individual sites was slightly smaller than extracellular stimulation. This may reflect the contribution of individual synapses that have a very low initial PR (<0.1). In paired recordings, such synapses are expected to be difficult to identify, and have large facilitation, particularly in wild-type animals. We found several such synapses in which postsynaptic currents could be detected only by evoking a brief burst in a grC, but events following the first stimulus were too infrequent to be quantified to measure PR. As PR could not be measured, these cells were not included in the analysis. It is therefore expected that difficulty in detecting very low PR synapses could have increased average PR and reduced the average PPR of paired recordings.
We also examined the role of Syt7 in AR by comparing AR in wild-type and Syt7 KO animals. Previous studies have shown that the properties of AR at grC to SC and grC to PC synapses are similar (Atluri and Regehr, 1998; Xu-Friedman and Regehr, 1999, 2000), but that it is much easier to quantify AR in SCs because quantal events are large, rapid and readily detected and there is much less noise and spontaneous activity compared with PCs. We therefore studied asynchronous release at grC to SC synapses. AR at this synapse consists of a prominent fast component and a much smaller slow component (Atluri and Regehr, 1998). Here we focus on the fast component of AR. In wild-type animals a single stimulus evoked an average EPSC with rapid kinetics (Fig. 4A). When individual trials were examined on an expanded vertical axis individual quantal events were apparent (Fig. 4B) that could be detected and quantified (Fig. 4C). The high release rates associated with synchronous release made it impractical to detect quantal events closely time-locked to stimulation (Fig. 4B,C, gray region). The AR in wild-type animals (Fig. 4D) had a time constant of 6.2 ± 0.4 ms. In Syt7 KO animals, the average EPSC (Fig. 4A) was qualitatively similar to that observed in wild-type animals. However, as shown in individual trials (Fig. 4B, C) AR was strongly attenuated in Syt7 KOs. A summary of AR in KOs indicates that compared with wild-type animals the magnitude of AR is reduced to ∼44% (p < 0.01, permutation test) and the time constant of decay following a single stimulus significantly decreased from 6.2 ± 0.1 ms for wild-type animals to 4.0 ± 0.1 ms for Syt7 KO animals (p < 0.01, permutation test). On average the total number of asynchronous events was reduced to 31% in Syt7 KOs. The reduction in asynchronous events could not be accounted for by changes in quantal size because we found no differences in mEPSC amplitude in SCs, which receive their glutamatergic inputs exclusively from parallel fibers (wild-type: 66.9 ± 2.2 pA, n = 18; Syt7 KO: 61.8 ± 3.4 pA, n = 8; K–S test, p = 0.49). We also did not observe differences in mEPSC frequency (wild-type: 0.43 ± 0.12 Hz, n = 18; Syt7 KO: 0.30 ± 0.05 Hz, n = 8; K–S test, p = 0.35). Thus, just as for synaptic facilitation, AR at the grC to SC synapse is primarily mediated by Syt7.
Presynaptic Ca2+ signaling in granule cells is unaltered in Syt7 KO animals
Facilitation and AR at the grC to PC and SC synapses is Ca2+ dependent (Atluri and Regehr, 1996), and it is possible that changes in presynaptic Ca2+ could alter facilitation and AR in Syt7 KO animals. We compared presynaptic Ca2+ signals in wild-type and Syt7 KO animals by using the low affinity fluorescent Ca2+ indicator Mg green (Fig. 5A), as has been used extensively to characterize presynaptic Ca2+ signals in cerebellar parallel fibers (Regehr and Tank, 1991; Sabatini and Regehr, 1995; Dittman and Regehr, 1997; Xu-Friedman and Regehr, 1999). This approach relies on labeling many parallel fibers with a small amount of indicator and recording Ca2+-dependent fluorescence transients from many presynaptic boutons. This has been a useful approach to characterize presynaptic Ca2+ entry because low levels of indicator can be used to avoid significant perturbation of presynaptic Ca2+ signals (Sabatini and Regehr, 1995; Dittman and Regehr, 1997). We have also previously shown that low affinity Ca2+ indicators provide a good measure of the time course of presynaptic Ca2+ signals (Regehr and Atluri, 1995; Kreitzer et al., 2000). We found that the time courses of Ca2+ signals evoked by single stimuli were similar in wild-type and Syt7 KO animals (wild-type: t1/2 = 18.1 ± 1.4 ms, n = 6; Syt7 KO: t1/2 = 16.4 ± 0.4 ms, n = 6; unpaired two-tailed Student's t test, t(10) = 1.12, p = 0.29). We used trains of five stimuli at 50 Hz to determine whether there could be changes in Ca2+ signals between stimuli (Fig. 5B), but found that ratio of the first and second stimuli were similar in wild-type and Syt7 KO animals (wild-type: ΔF2/ΔF1 = 1.06 ± 0.01, n = 7; Syt7 KO: ΔF2/ΔF1 = 1.06 ± 0.1, n = 6; unpaired two-tailed Student's t test, t(10) = −0.43, p = 0.68).
It is difficult to use low affinity Ca2+ indicators to quantify the magnitude of presynaptic Ca2+ signals because fluorescence changes are proportional to both the number of activated fibers, and to the size of the presynaptic Ca2+ signal in each fiber (Regehr and Atluri, 1995). However, high affinity Ca2+ indicators such as fura-2 can be used to estimate the amplitude of presynaptic Ca2+ entry (Fig. 5C). This approach relies on the observation that Ca2+ entry for each of two closely spaced stimuli is similar (Regehr and Atluri, 1995; Kreitzer et al., 2000), but the second stimulus produces a smaller increase in fura-2 fluorescence (Sabatini and Regehr, 1995; Chen and Regehr, 1997). This is because the first stimulus produces such large Ca2+ signals that most of the fura-2 binds Ca2+, and there is a less unbound fura-2 present for the second stimulus. If the presynaptic Ca2+ entry evoked by a single pulse is increased, then less fura-2 is available to respond to the second stimulus and ΔF2/ΔF1 decreases (Sabatini and Regehr, 1995). We found, however, that ΔF2/ΔF1 was not different in wild-type and Syt7 KO animals (wild-type: ΔF2/F1 = 1.40 ± 0.02, n = 9; Syt7 KO: ΔF2/F1 = 1.38 ± 0.02, n = 10; unpaired two-tailed Student's t test, t(17) = 0.77, p = 0.45). We also used a related approach and measured the maximal change in fluorescence (Fmax) and determined ΔF1/ΔFmax, which provides another measure of the magnitude of presynaptic Ca2+ signals (Maravall et al., 2000). There was also no significant change in ΔF1/ΔFmax (wild-type: ΔF1/Fmax = 0.32 ± 0.02, n = 9; Syt7 KO: ΔF1/Fmax = 0.32 ± 0.01, n = 10; unpaired two-tailed Student's t test, t(17) = −0.05 p = 0.96). Thus, we conclude that differences in facilitation and AR in Syt7 KO animals and wild-type animals are not a consequence of alterations in presynaptic Ca2+ signaling.
The Sr2+ dependence of asynchronous release
AR is often studied by replacing extracellular Ca2+ with Sr2+, which increases the amplitude and time course of AR while attenuating the amplitude of synchronous release (Dodge et al., 1969; Zengel and Magleby, 1980; Goda and Stevens, 1994; Searl and Silinsky, 2002; Bekkers, 2003; Shin et al., 2003; Calakos et al., 2004; Zhao et al., 2006; H. Yang and Xu-Friedman, 2010; Babai et al., 2014). We therefore examined the properties of AR in wild-type and Syt7 KO animals in the presence of Sr2+. Before determining the properties of AR in the presence of Sr2+ we measured presynaptic Sr2+ signals in wild-type and Syt7 KO animals using an established approach (Xu-Friedman and Regehr, 1999, 2000; see Materials and Methods). Presynaptic Sr2+ signals are larger and longer lasting than presynaptic Ca2+ signals in wild-type animals (Fig. 6A), which is a consequence of less-effective presynaptic buffering and extrusion of Sr2+ relative to Ca2+ (Xu-Friedman and Regehr, 1999, 2000). Presynaptic Sr2+ signals were the same in wild-type and Syt7 KO animals (Fig. 6A), indicating that any differences in AR occurs downstream of Sr2+ signaling. Changing from external Ca2+ to Sr2+ reversibly attenuated the synchronous EPSC amplitude, and increased the magnitude and duration of AR in both wild-type and Syt7 KO animals (Fig. 6B). AR was more prominent in the presence of Sr2+ than in the presence of Ca2+ (Fig. 6C–E). A comparison of the AR evoked in the presence of Sr2+ in wild-type and Syt7 KO animals revealed an ∼2- to 3-fold reduction in AR in Syt7 KO mice (Fig. 6F, right, squares; Table 1). These findings establish that just as for AR mediated by Ca2+, most AR mediated by Sr2+ relies on Syt7, but a component of AR remains even when Syt7 is eliminated.
Table 1.
Fit parameters for average paired-pulse facilitation and asynchronous release
| Figure | Expt | Cell type | Conditions | Genotype | A | τ, ms | c |
|---|---|---|---|---|---|---|---|
| 2 | PPR | PC | 2 Ca, 25°C | Wild-type | 2.12 ± 0.04 | 85 ± 5 | 1 |
| 2 | PPR | PC | 2 Ca, 25°C | Syt7 KO | 1.02 ± 0.06 | 46 ± 7 | 1 |
| 2 | PPR | SC | 2 Ca, 25°C | Wild-type | 1.52 ± 0.07 | 132 ± 20 | 1 |
| 2 | PPR | SC | 2 Ca, 25°C | Syt7 KO | 0.67 ± 0.08 | 35 ± 10 | 1 |
| 4 | AR | SC | 2 Ca, 25°C | Wild-type | 0.122 ± 0.005 | 6 ± 1 | 0.005 ± 0.002 |
| 4 | AR | SC | 2 Ca, 25°C | Syt7 KO | 0.054 ± 0.003 | 4 ± 1 | 0.002 ± 0.001 |
| 6 | AR | SC | 2 Sr, 25°C + Sr | Wild-type | 0.272 ± 0.003 | 28 ± 1 | 0.006 ± 0.001 |
| 6 | AR | SC | 2 Sr, 25°C + Sr | Syt7 KO | 0.131 ± 0.003 | 20 ± 1 | 0.004 ± 0.003 |
| 7 | PPR | PC | 2 Ca, 25°C + EGTA | Wild-type | 1.21 ± 0.08 | 49 ± 9 | 1 |
| 7 | PPR | PC | 2 Ca, 25°C + EGTA | Syt7 KO | 0.51 ± 0.09 | 22 ± 9 | 1 |
| 8B | PPR | PC | 1.5 Ca, 35°C | Wild-type | 1.78 ± 0.06 | 64 ± 6 | 1 |
| 8B | PPR | PC | 1.5 Ca, 35°C | Syt7 KO | 0.866 ± 0.033 | 17 ± 2 | 1 |
| 8F | AR | SC | 1.5 Ca, 35°C | Wild-type | 0.090 ± 0.002 | 4 ± 1 | 0.001 ± 0.001 |
| 8F | AR | SC | 1.5 Ca, 35°C | Syt7 KO | 0.033 ± 0.001 | 3 ± 1 | 0.002 ± 0.001 |
The PPR and AR evoked by single stimuli were fit with equations of the form PPR = (c + Ae−(t − t0)/τ) or release = (c + Ae−(t − t0)/τ). For facilitation curves, c was set to 1, and t0 was the smallest interstimulus interval (10 ms for 25°C, 5 ms for 35°C). For AR, t0 was taken as the time when individual events could first be detected following synchronous release (9 ms following stimulation for experiments at 25°C, 5 ms for experiments at 35°C).
Expt, Experiment.
The dependence of facilitation on residual Ca2+
It has previously been shown that the slow Ca2+ chelator EGTA can be used to accelerate the decay of residual Ca2+ in grC presynaptic boutons (Atluri and Regehr, 1996). This in turn decreased the amplitude and accelerated the decay of facilitation and AR, and established that these phenomena depend upon residual Ca2+. Facilitation was not completely eliminated by even the highest concentration of EGTA-AM used. This suggested that this component of facilitation might be driven by high local Ca2+ rather than residual Ca2+, or that it involved a different mechanism. We therefore assessed whether the remaining facilitation in Syt7 KO mice is also sensitive to EGTA using bath application of EGTA-AM in wild-type and Syt7 KO animals. We used a concentration of EGTA-AM (20 μm) that accelerates the decay but does not affect the peak of Ca2+ transients in parallel fibers (Atluri and Regehr, 1996). In wild-types we found after exposure to EGTA-AM the magnitude of facilitation was reduced to 57% of control (Control: A = 2.12 ± 0.04; EGTA: A = 1.21 ± 0.08; p < 0.01, permutation test) and time constant of decay was reduced to 57% of control (Control: τ = 85 ± 5 ms; EGTA: τ = 49 ± 9 ms; p < 0.01, permutation test; Fig. 7A,B). In Syt7 KOs EGTA-AM reduced the magnitude of facilitation to 50% of control (Control: A = 1.02 ± 0.06; EGTA: A = 0.51 ± 0.09; p < 0.01, permutation test) and the time constant of decay to 48% of control (Control: τ = 46 ± 7 ms; EGTA: τ = 22 ± 9 ms; p < 0.01, permutation test; Fig. 7C,D). These findings suggest that the component of facilitation that remains in Syt7 KO animals has approximately the same Ca2+ dependence as the facilitation present in wild-type animals. We did not examine the effect of EGTA on AR in Syt7 KO animals because AR is exceedingly small and the decay is very rapid.
Facilitation and asynchronous release in physiological conditions
Our studies thus far have been performed at room temperature in the presence of 2 mm external Ca2+, which are the conditions of most previous studies of facilitation and AR at these synapses. It is known that presynaptic waveform, presynaptic Ca2+signaling, and neurotransmitter release are highly dependent on temperature and external Ca2+ levels. This raises the possibility that the contribution of Syt7 to synaptic transmission, AR, and facilitation could be different under physiological conditions. We therefore extended our studies to more physiological conditions (35°C, and 1.5 mm external Ca2+). Under these conditions in wild-type animals, facilitation at the grC to PC synapse was prominent and decayed slightly faster than at room temperature (τ = 63 ms). In Syt7 KO animals the amplitude of facilitation was significantly smaller (wild-type: A = 1.78 ± 0.06; Syt7 KO: A = 0.87 ± 0.03, p < 0.01, permutation test) and faster (wild-type: τ = 64 ± 6 ms; Syt7 KO: τ = 17 ± 2 ms; p < 0.01, permutation test; Fig. 8A,B). The accelerated decay of facilitation in Syt7 KO animals is particularly obvious under these experimental conditions. A fit to Equation 1 for PPR in wild-type animals yielded constants {Afast, τfast, Aslow, τslow} of {0.44, 15 ms, 1.40, 85 ms}, and a for a fit to Equation 2 for PPR in Syt7 KO animals {Afast, τfast} are {0.87, 17 ms}. It is difficult to precisely estimate the parameters of the double exponential fit, and as for room temperature experiments the faster decay of facilitation in Syt7 KO animals is compatible with either an acceleration of a single exponential decay or with the selective elimination of a slow component of facilitation.
We also studied AR under these experimental conditions at the grC to SC synapse. In wild-type animals, AR was less prominent and decayed more rapidly than in room temperature experiments (Fig. 8C–F). As at room temperature and 2 mm external Ca2+, AR was dominated by a rapid component that could be well described by a single exponential fit. Under near physiological conditions, AR was reduced to 30% in Syt7 KOs (p < 0.01, permutation test). Differences in time course of AR were not statistically significant (wild-type: τ = 3.8 ± 0.1 ms; Syt7 KO: τ = 2.8 ± 0.1 ms; p = 0.50, permutation test), although there was a trend toward an accelerated decay. These studies indicate that the qualitative contribution of Syt7 to AR and facilitation are the same at 35°C in 1.5 mm external Ca2+ as at 24°C in 2 mm external Ca2+. We find, however, that under physiological conditions AR is less prominent, and the Syt7-independent component of facilitation is very short-lived.
Discussion
Our primary finding is that Syt7 can mediate both AR and facilitation at the same synapses, even though AR is much shorter-lived and much more Ca2+ dependent than facilitation. Although most facilitation and AR at grC synapses is reliant on Syt7, they are also partially mediated by a Syt7-independent mechanism.
Syt7 mediates both facilitation and asynchronous release
Previous studies suggested that Cares produces both facilitation and AR, but it was not known whether the same Ca2+ sensor mediates both phenomena (Zucker and Lara-Estrella, 1983; Van der Kloot and Molgó, 1993), The observations that AR decays much more rapidly than facilitation, and that reducing extracellular Ca2+ strongly attenuates AR but increases facilitation, suggested that the mechanisms underlying these phenomena must differ (Atluri and Regehr, 1998). Nonetheless, here we show that despite these differences, Syt7 mediates both phenomena at the same synapses.
A recent model can qualitatively account for some aspects of the role of Syt7 in these two phenomena (Jackman and Regehr, 2017). According to this model, Syt1 and Syt7 bind to phospholipids in the presence of Ca2+, reducing the energy barrier for vesicle fusion. For low-frequency stimulation high local Ca2+ drives rapid synchronous release mediated entirely by Syt1 and does not activate Syt7 due to its high affinity and slow kinetics. Syt7 is instead activated after several milliseconds and this activation persists for tens of milliseconds. If both Syt1 and Syt7 are activated, as occurs for closely spaced stimuli, they lead to a slightly larger reduction in the energy barrier than for Syt1 alone, which leads to facilitation. AR could be produced by modest Syt1 activation for many milliseconds after an action potential acting in concert with Syt7 to produce AR.
This scheme provides a framework that accounts qualitatively for the roles of Syt7 in AR and facilitation, but it is highly speculative. Moreover, there are many unknown features that could also contribute to how Syt7 mediates facilitation and AR at different synapses. The precise location of Syt7 is important, but is not known. Syt7 is thought to be present on plasma membranes (Sugita et al., 2001), but its location relative to docked vesicles is unknown, and whether it might also be present at low levels in vesicles is difficult to rule out. The absolute level of Syt7 expression could also affect its function, and it is possible that AR and facilitation are differentially sensitive to Syt7 levels. Syt7 is alternatively spliced (Sugita et al., 2001; Fukuda et al., 2002), and it is possible that the relative expression of different splice variants could determine the extent of facilitation or AR. The manner in which Syt7 and Syt1 interact to produce facilitation is not known. Finally, the extent of interactions between Syt7 and other proteins has not been characterized. Further studies will be required to distinguish between these possibilities, and to determine how Syt7 can play diverse roles across many different synapses.
Syt7 and facilitation
This study adds to the list of synapses in which facilitation is mediated primarily by Syt7. At multiple hippocampal synapses and at corticothalamic synapses, paired-pulse plasticity is entirely mediated by Syt7 (Jackman et al., 2016). At PC and vestibular synapses, Syt7 mediates facilitation that is normally masked by depression, but that is revealed during frequency step trains or when PR is reduced (Turecek et al., 2017). However, in contrast to other synapses, some facilitation remains at grC to PC and SC synapses in Syt7 KOs
Previous studies suggest that the presence of Syt7 in presynaptic boutons does not always result in obvious facilitation. In many instances a high initial PR results in partial vesicle depletion that obscures facilitation (Müller et al., 2010; Lu and Trussell, 2016; Turecek et al., 2016; Chen et al., 2017b). This occurs at vestibular and PC synapses that depress in normal external Ca2+, but Syt7-dependent facilitation is revealed in low external Ca2+ when the initial probability is reduced (Turecek et al., 2017). This could explain the apparent lack of facilitation for cultured synapses and the zebrafish neuromuscular junction, but postsynaptic mechanisms such as receptor desensitization could also obscure facilitation. It is more difficult to understand why Syt7 does not mediate facilitation at the calyx of Held even when the initial PR is lowered (Luo and Südhof, 2017). It is possible that Syt7 expression levels are not sufficiently high at the calyx of Held to facilitate release. It will be informative to compare Syt7 expression levels at the calyx of Held with synapses where the role of Syt7 in facilitation is well established.
Syt7 and asynchronous release
It has been shown that Syt7 can mediate the prominent AR observed when fast synaptotagmin isoforms are eliminated (Bacaj et al., 2013), and asynchronous events during prolonged trains at the zebrafish neuromuscular junction (Wen et al., 2010). It has also been observed that at the calyx of Held prolonged high-frequency trains evoke a Syt7-dependent tonic current that has been attributed to AR (Luo and Südhof, 2017). Here we show that a single stimulus can trigger Syt7-dependent AR even in wild-type animals where fast synaptotagmin isoforms are present. We also used Sr2+ to examine AR because it had been proposed that the Ca2+ sensor that mediates AR had a higher sensitivity to Sr2+ than to Ca2+. However, we found that AR triggered by Sr2+ was mediated by both Syt7-dependent and Syt7-independent mechanisms. This suggests that although Sr2+ is a useful tool to produce large and prolonged AR, it is not particularly helpful in identifying the Ca2+ sensor for AR.
AR is not a prominent feature of most synapses, even those containing Syt7. Why does Syt7 evoke AR at grC synapses but not at others? One possibility is that AR occurs, but is obscured for technical reasons. In SCs, synchronous release is rapid, allowing the detection of delayed events several milliseconds after stimulation. In PCs evoked EPSCs and quantal events are much smaller and slower, which can obscure short-lived AR (Diamond and Jahr, 1995; Atluri and Regehr, 1998; Rudolph et al., 2011). Another factor is the Ca2+ signal seen by Syt7. AR is highly Ca2+-sensitive (Atluri and Regehr, 1998). Parallel fiber boutons have large residual Ca2+ signals that could be effective at promoting Syt7-mediated AR (Brenowitz and Regehr, 2007), whereas residual Ca2+ signals elsewhere may not be sufficiently large to promote AR (Scott and Rusakov, 2006; Delvendahl et al., 2015). An additional factor is the presence of fast synaptotagmin isoforms that can prevent AR (Xu et al., 2007; Kochubey and Schneggenburger, 2011; Chen et al., 2017a). It is unclear whether this is a clamping effect in which Syt1/2 directly suppresses AR, or whether it reflects competition between sensors. More generally other presynaptic proteins may suppress AR, just as complexin and Syt1/2 suppress spontaneous vesicle fusion (Xu et al., 2007; X. Yang et al., 2010).
Syt7-independent AR and facilitation
The mechanisms responsible for the short-lived components of AR and facilitation present in Syt7 KO animals are not known. Whether these components are also present in wild-type animals, or are compensatory mechanisms only present in Syt7 KO animals is also not known. There are several candidate mechanisms that could mediate the short-lived Ca2+-dependent facilitation present in Syt7 KO animals (Jackman and Regehr, 2017). The remaining facilitation is much smaller in amplitude and decays ∼3 times more rapidly than facilitation in wild-type animals, and is EGTA-sensitive suggesting it is Ca2+-dependent. The remaining component could be mediated by a Ca2+ sensor with more rapid kinetics than Syt7. Another possibility is Ca2+-dependent facilitation of Ca2+ influx through P/Q-type Ca2+ channels (Borst and Sakmann, 1998; Cuttle et al., 1998). P/Q-type channels account for a large fraction of the Ca2+ channels in grC synapses (Mintz et al., 1995). Modest enhancement of Ca2+ entry (∼20%) would be sufficient to account for the remaining facilitation, but increases in Ca2+ entry could be difficult to detect whether they are confined to those channels that trigger vesicle fusion. The small, transient component of AR remaining in Syt7 KO animals could also be mediated by an additional sensor (Saraswati et al., 2007; Yao et al., 2011; Kaeser and Regehr, 2014; but see Groffen et al., 2010; Pang et al., 2011). Another possibility is that Ca2+ increases in grC presynaptic boutons are sufficient to activate fast synaptotagmins to a small extent and trigger a small amount of AR. Further experiments are needed to determine the mechanisms of Syt7-independent AR and facilitation and to determine whether they are mediated by the same Ca2+ sensor, or whether they are mediated by distinct mechanisms.
Footnotes
This work was supported by Grants from the NIH (R01NS032405 and R35NS097284) and a Nancy Lurie Marks Grant to W.R., the Vision Core, and NINDS P30 Core Center Grant (NS072030) to the Neurobiology Imaging Center at Harvard Medical School, and support for statistical analysis from R. Betensky of Harvard Catalyst. We thank P. Kaeser for comments on the paper, S.L. Jackman for conversations about Syt7, and Y.X. Chu for preliminary experiments that were not included.
The authors declare no competing financial interests.
References
- Atluri PP, Regehr WG (1996) Determinants of the time course of facilitation at the granule cell to purkinje cell synapse. J Neurosci 16:5661–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG (1998) Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci 18:8214–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babai N, Kochubey O, Keller D, Schneggenburger R (2014) An alien divalent ion reveals a major role for Ca2+ buffering in controlling slow transmitter release. J Neurosci 34:12622–12635. 10.1523/JNEUROSCI.1990-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Südhof TC (2013) Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80:947–959. 10.1016/j.neuron.2013.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM. (2003) Convolution of mini distributions for fitting evoked synaptic amplitude histograms. J Neurosci Methods 130:105–114. 10.1016/j.jneumeth.2003.09.018 [DOI] [PubMed] [Google Scholar]
- Best AR, Regehr WG (2009) Inhibitory regulation of electrically coupled neurons in the inferior olive is mediated by asynchronous release of GABA. Neuron 62:555–565. 10.1016/j.neuron.2009.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B (1998) Facilitation of presynaptic calcium currents in the rat brainstem. J Physiol 513:149–155. 10.1111/j.1469-7793.1998.149by.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt DS, Coffman MD, Falke JJ, Knight JD (2012) Hydrophobic contributions to the membrane docking of synaptotagmin 7 C2A domain: mechanistic contrast between isoforms 1 and 7. Biochemistry 51:7654–7664. 10.1021/bi3007115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2007) Reliability and heterogeneity of calcium signaling at single presynaptic boutons of cerebellar granule cells. J Neurosci 27:7888–7898. 10.1523/JNEUROSCI.1064-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2014) Presynaptic calcium measurements using bulk loading of acetoxymethyl indicators. Cold Spring Harb Protoc 2014:750–757. 10.1101/pdb.prot081828 [DOI] [PubMed] [Google Scholar]
- Calakos N, Schoch S, Südhof TC, Malenka RC (2004) Multiple roles for the active zone protein RIM1alpha in late stages of neurotransmitter release. Neuron 42:889–896. 10.1016/j.neuron.2004.05.014 [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG (2000) Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci 20:4423–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS, Andrews NW (2003) Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol 162:543–549. 10.1083/jcb.200305131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG (1997) The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci 17:8687–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Arai I, Satterfield R, Young SM Jr, Jonas P (2017a) Synaptotagmin 2 is the fast Ca2+ sensor at a central inhibitory synapse. Cell Rep 18:723–736. 10.1016/j.celrep.2016.12.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Satterfield R, Young SM Jr, Jonas P (2017b) Triple function of synaptotagmin 7 ensures efficiency of high-frequency transmission at central GABAergic synapses. Cell Rep 21:2082–2089. 10.1016/j.celrep.2017.10.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T (1998) Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol 512:723–729. 10.1111/j.1469-7793.1998.723bd.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvendahl I, Jablonski L, Baade C, Matveev V, Neher E, Hallermann S (2015) Reduced endogenous Ca2+ buffering speeds active zone Ca2+ signaling. Proc Natl Acad Sci U S A 112:E3075–3084. 10.1073/pnas.1508419112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (1995) Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron 15:1097–1107. 10.1016/0896-6273(95)90098-5 [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG (1997) Mechanism and kinetics of heterosynaptic depression at a cerebellar synapse. J Neurosci 17:9048–9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge FA Jr, Miledi R, Rahamimoff R (1969) Strontium and quantal release of transmitter at the neuromuscular junction. J Physiol 200:267–283. 10.1113/jphysiol.1969.sp008692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Ogata Y, Saegusa C, Kanno E, Mikoshiba K (2002) Alternative splicing isoforms of synaptotagmin VII in the mouse, rat and human. Biochem J 365:173–180. 10.1042/bj20011877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Stevens CF (1994) Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A 91:12942–12946. 10.1073/pnas.91.26.12942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M (2010) Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 327:1614–1618. 10.1126/science.1183765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P (2005) Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8:1319–1328. 10.1038/nn1542 [DOI] [PubMed] [Google Scholar]
- Iremonger KJ, Bains JS (2007) Integration of asynchronously released quanta prolongs the postsynaptic spike window. J Neurosci 27:6684–6691. 10.1523/JNEUROSCI.0934-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SL, Regehr WG (2017) The mechanisms and functions of synaptic facilitation. Neuron 94:447–464. 10.1016/j.neuron.2017.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SL, Beneduce BM, Drew IR, Regehr WG (2014) Achieving high-frequency optical control of synaptic transmission. J Neurosci 34:7704–7714. 10.1523/JNEUROSCI.4694-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SL, Turecek J, Belinsky JE, Regehr WG (2016) The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 529:88–91. 10.1038/nature16507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG (2014) Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol 76:333–363. 10.1146/annurev-physiol-021113-170338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochubey O, Schneggenburger R (2011) Synaptotagmin increases the dynamic range of synapses by driving Ca2+-evoked release and by clamping a near-linear remaining Ca2+ sensor. Neuron 69:736–748. 10.1016/j.neuron.2011.01.013 [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Gee KR, Archer EA, Regehr WG (2000) Monitoring presynaptic calcium dynamics in projection fibers by in vivo loading of a novel calcium indicator. Neuron 27:25–32. 10.1016/S0896-6273(00)00006-4 [DOI] [PubMed] [Google Scholar]
- Labrakakis C, Lorenzo LE, Bories C, Ribeiro-da-Silva A, De Koninck Y (2009) Inhibitory coupling between inhibitory interneurons in the spinal cord dorsal horn. Mol Pain 5:24. 10.1186/1744-8069-5-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HW, Trussell LO (2016) Spontaneous activity defines effective convergence ratios in an inhibitory circuit. J Neurosci 36:3268–3280. 10.1523/JNEUROSCI.3499-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Trussell LO (2000) Inhibitory transmission mediated by asynchronous transmitter release. Neuron 26:683–694. 10.1016/S0896-6273(00)81204-0 [DOI] [PubMed] [Google Scholar]
- Luo F, Südhof TC (2017) Synaptotagmin-7-mediated asynchronous release boosts high-fidelity synchronous transmission at a central synapse. Neuron 94:826–839.e823. 10.1016/j.neuron.2017.04.020 [DOI] [PubMed] [Google Scholar]
- Luo F, Bacaj T, Südhof TC (2015) Synaptotagmin-7 is essential for Ca2+-triggered delayed asynchronous release but not for Ca2+-dependent vesicle priming in retinal ribbon synapses. J Neurosci 35:11024–11033. 10.1523/JNEUROSCI.0759-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravall M, Mainen ZF, Sabatini BL, Svoboda K (2000) Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J 78:2655–2667. 10.1016/S0006-3495(00)76809-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Lao Y, Li H, Chen X, Rizo J, Sørensen JB, Südhof TC (2008) Genetic analysis of synaptotagmin-7 function in synaptic vesicle exocytosis. Proc Natl Acad Sci U S A 105:3986–3991. 10.1073/pnas.0712372105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG (1995) Calcium control of transmitter release at a cerebellar synapse. Neuron 15:675–688. 10.1016/0896-6273(95)90155-8 [DOI] [PubMed] [Google Scholar]
- Mittelsteadt T, Seifert G, Alvárez-Barón E, Steinhäuser C, Becker AJ, Schoch S (2009) Differential mRNA expression patterns of the synaptotagmin gene family in the rodent brain. J Comp Neurol 512:514–528. 10.1002/cne.21908 [DOI] [PubMed] [Google Scholar]
- Molgó J, Van der Kloot W (1991) Quantal release and facilitation at frog neuromuscular junctions at about 0 degrees C. J Neurophysiol 65:834–840. 10.1152/jn.1991.65.4.834 [DOI] [PubMed] [Google Scholar]
- Müller M, Goutman JD, Kochubey O, Schneggenburger R (2010) Interaction between facilitation and depression at a large CNS synapse reveals mechanisms of short-term plasticity. J Neurosci 30:2007–2016. 10.1523/JNEUROSCI.4378-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC (2011) Doc2 supports spontaneous synaptic transmission by a Ca2+-independent mechanism. Neuron 70:244–251. 10.1016/j.neuron.2011.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Smith SM, Andresen MC (2010) Primary afferent activation of thermosensitive TRPV1 triggers asynchronous glutamate release at central neurons. Neuron 65:657–669. 10.1016/j.neuron.2010.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn GP, Keough MJ (2002) Experimental design and data analysis for biologists. Cambridge, UK; New York: Cambridge UP. [Google Scholar]
- Rahamimoff R, Yaari Y (1973) Delayed release of transmitter at the frog neuromuscular junction. J Physiol 228:241–257. 10.1113/jphysiol.1973.sp010084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Atluri PP (1995) Calcium transients in cerebellar granule cell presynaptic terminals. Biophys J 68:2156–2170. 10.1016/S0006-3495(95)80398-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Tank DW (1991) Selective fura-2 loading of presynaptic terminals and nerve cell processes by local perfusion in mammalian brain slice. J Neurosci Methods 37:111–119. 10.1016/0165-0270(91)90121-F [DOI] [PubMed] [Google Scholar]
- Rudolph S, Overstreet-Wadiche L, Wadiche JI (2011) Desynchronization of multivesicular release enhances purkinje cell output. Neuron 70:991–1004. 10.1016/j.neuron.2011.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG (1995) Detecting changes in calcium influx which contribute to synaptic modulation in mammalian brain slice. Neuropharmacology 34:1453–1467. 10.1016/0028-3908(95)00129-T [DOI] [PubMed] [Google Scholar]
- Saraswati S, Adolfsen B, Littleton JT (2007) Characterization of the role of the synaptotagmin family as calcium sensors in facilitation and asynchronous neurotransmitter release. Proc Natl Acad Sci U S A 104:14122–14127. 10.1073/pnas.0706711104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Brachtendorf S, Arendt O, Hallermann S, Ishiyama S, Bornschein G, Gall D, Schiffmann SN, Heckmann M, Eilers J (2013) Nanodomain coupling at an excitatory cortical synapse. Curr Biol 23:244–249. 10.1016/j.cub.2012.12.007 [DOI] [PubMed] [Google Scholar]
- Scott R, Rusakov DA (2006) Main determinants of presynaptic Ca2+ dynamics at individual mossy fiber-CA3 pyramidal cell synapses. J Neurosci 26:7071–7081. 10.1523/JNEUROSCI.0946-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searl TJ, Silinsky EM (2002) Evidence for two distinct processes in the final stages of neurotransmitter release as detected by binomial analysis in calcium and strontium solutions. J Physiol 539:693–705. 10.1113/jphysiol.2001.013129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OH, Rhee JS, Tang J, Sugita S, Rosenmund C, Südhof TC (2003) Sr2+ binding to the Ca2+ binding site of the synaptotagmin 1 C2B domain triggers fast exocytosis without stimulating SNARE interactions. Neuron 37:99–108. 10.1016/S0896-6273(02)01145-5 [DOI] [PubMed] [Google Scholar]
- Sims RE, Hartell NA (2005) Differences in transmission properties and susceptibility to long-term depression reveal functional specialization of ascending axon and parallel fiber synapses to purkinje cells. J Neurosci 25:3246–3257. 10.1523/JNEUROSCI.0073-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Han W, Butz S, Liu X, Fernández-Chacón R, Lao Y, Südhof TC (2001) Synaptotagmin VII as a plasma membrane Ca2+ sensor in exocytosis. Neuron 30:459–473. 10.1016/S0896-6273(01)00290-2 [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin OH, Han W, Lao Y, Südhof TC (2002) Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J 21:270–280. 10.1093/emboj/21.3.270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek J, Jackman SL, Regehr WG (2016) Synaptic specializations support frequency-independent purkinje cell output from the cerebellar cortex. Cell Rep 17:3256–3268. 10.1016/j.celrep.2016.11.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek J, Jackman SL, Regehr WG (2017) Synaptotagmin 7 confers frequency invariance onto specialized depressing synapses. Nature 551:503–506. 10.1038/nature24474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Kloot W, Molgó J (1993) Facilitation and delayed release at about 0 degree C at the frog neuromuscular junction: effects of calcium chelators, calcium transport inhibitors, and okadaic acid. J Neurophysiol 69:717–729. 10.1152/jn.1993.69.3.717 [DOI] [PubMed] [Google Scholar]
- Wen H, Linhoff MW, McGinley MJ, Li GL, Corson GM, Mandel G, Brehm P (2010) Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc Natl Acad Sci U S A 107:13906–13911. 10.1073/pnas.1008598107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC (2007) Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54:567–581. 10.1016/j.neuron.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG (1999) Presynaptic strontium dynamics and synaptic transmission. Biophys J 76:2029–2042. 10.1016/S0006-3495(99)77360-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG (2000) Probing fundamental aspects of synaptic transmission with strontium. J Neurosci 20:4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA (2010) Developmental mechanisms for suppressing the effects of delayed release at the endbulb of held. J Neurosci 30:11466–11475. 10.1523/JNEUROSCI.2300-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Südhof TC (2010) Complexin clamps asynchronous release by blocking a secondary Ca2+ sensor via its accessory alpha helix. Neuron 68:907–920. 10.1016/j.neuron.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Gaffaney JD, Kwon SE, Chapman ER (2011) Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell 147:666–677. 10.1016/j.cell.2011.09.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengel JE, Magleby KL (1980) Differential effects of Ba2+, Sr2+, and Ca2+ on stimulation-induced changes in transmitter release at the frog neuromuscular junction. J Gen Physiol 76:175–211. 10.1085/jgp.76.2.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JP, Phillips MA, Constantine-Paton M (2006) Long-term potentiation in the juvenile superior colliculus requires simultaneous activation of NMDA receptors and L-type Ca2+ channels and reflects addition of newly functional synapses. J Neurosci 26:12647–12655. 10.1523/JNEUROSCI.3678-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Lara-Estrella LO (1983) Post-tetanic decay of evoked and spontaneous transmitter release and a residual-calcium model of synaptic facilitation at crayfish neuromuscular junctions. J Gen Physiol 81:355–372. 10.1085/jgp.81.3.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]