

Abstract
Multiple lines of evidence indicate that a reduction in the expression and function of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) is associated with neurodegeneration in diseases such as Huntington's disease (HD). Polymorphisms in the PGC-1α gene modify HD progression and PGC-1α expression is reduced in striatal medium spiny neurons (MSNs) of HD patients and mouse models. However, neither the MSN-specific function of PGC-1α nor the contribution of PGC-1α deficiency to motor dysfunction is known. We identified novel, PGC-1α-dependent transcripts involved in RNA processing, signal transduction, and neuronal morphology and confirmed reductions in these transcripts in male and female mice lacking PGC-1α specifically in MSNs, indicating a cell-autonomous effect in this population. MSN-specific PGC-1α deletion caused reductions in previously identified neuronal and metabolic PGC-1α-dependent genes without causing striatal vacuolizations. Interestingly, these mice exhibited a hypoactivity with age, similar to several HD animal models. However, these newly identified PGC-1α-dependent genes were upregulated with disease severity and age in knock-in HD mouse models independent of changes in PGC-1α transcript, contrary to what would be predicted from a loss-of-function etiological mechanism. These data indicate that PGC-1α is necessary for MSN transcriptional homeostasis and function with age and that, whereas PGC-1α loss in MSNs does not replicate an HD-like phenocopy, its downstream genes are altered in a repeat-length and age-dependent fashion. Understanding the additive effects of PGC-1α gene functional variation and mutant huntingtin on transcription in this cell type may provide insight into the selective vulnerability of MSNs in HD.
SIGNIFICANCE STATEMENT Reductions in peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α)-mediated transcription have been implicated in the pathogenesis of Huntington's disease (HD). We show that, although PGC-1α-dependent transcription is necessary to maintain medium spiny neuron (MSN) function with age, its loss is insufficient to cause striatal atrophy in mice. We also highlight a set of genes that can serve as proxies for PGC-1α functional activity in the striatum for target engagement studies. Furthermore, we demonstrate that PGC-1α-dependent genes are upregulated in a dose- and age-dependent fashion in HD mouse models, contrary to what would be predicted from a loss-of-function etiological mechanism. However, given this role for PGC-1α in MSN transcriptional homeostasis, it is important to consider how genetic variation in PGC-1α could contribute to mutant-huntingtin-induced cell death and disease progression.
Keywords: cellular specificity, Huntington's disease, medium spiny neuron, pgc-1alpha, striatum, transcription
Introduction
Reductions in the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) are thought to be a key factor in the progression of multiple neurological disorders, including Huntington's disease (HD). Polymorphisms in the ge ne encoding PGC-1α (Taherzadeh-Fard et al., 2009; Weydt et al., 2009, 2014; Ramos et al., 2012) and polymorphisms in PGC-1α-interacting transcription factors and downstream genes are genetic modifiers of disease progression (Taherzadeh-Fard et al., 2011). These data indicate that dysfunction in PGC-1α-dependent transcription may contribute to disease pathophysiology. In fact, several reports indicate a reduction in PGC-1α expression in the caudate of HD patients (Cui et al., 2006; Weydt et al., 2006) and in HD mouse models (Cui et al., 2006; Weydt et al., 2006; Chaturvedi et al., 2010). Interestingly, PGC-1α expression is dramatically reduced specifically in striatal medium spiny neurons (MSNs) relative to other striatal populations in an HD model (Cui et al., 2006).
MSNs are the most affected neuronal population in HD (Ehrlich, 2012) and mutant huntingtin (mHtt)-mediated impairment in PGC-1α-dependent transcription in this population could contribute to their vulnerability. Animals lacking full-length or C-terminal expression of PGC-1α have vacuolizations throughout the brain, with severity being highest in the striatum (Lin et al., 2004; Leone et al., 2005; Lucas et al., 2012; Szalardy et al., 2013). This phenotype mirrors that seen in genetic mouse models of HD subjected to energy deprivation (Chaturvedi et al., 2010). In addition, PGC-1α-null mice have severe motor deficits, including impaired motor coordination and rearing ability, resting tremor, and hindlimb clasping (Lin et al., 2004; Lucas et al., 2012, 2014b). mHtt is able to repress PGC-1α directly (Cui et al., 2006) and complete loss of PGC-1α in an HD mouse model exacerbates motor coordination deficits, neuronal degeneration, and lesion size in response to mitochondrial inhibition (Cui et al., 2006). Further, extrasynaptic glutamate receptor stimulation can impair CREB-PGC-1α-mediated signaling to augment mHtt-induced cell death (Okamoto et al., 2009). Cultured neurons from the PGC-1α-null striatum exhibit a dramatically reduced neuritic length (Lin et al., 2004) and a loss of PGC-1α dramatically affects the expression of MSN markers (Lucas et al., 2012). These data suggest that PGC-1α plays a significant role in maintaining MSN function, yet knowledge of the downstream genes regulated in this cell type and the behavioral impact of PGC-1α deletion specifically from MSNs have not been determined.
To evaluate the contribution of MSN-specific PGC-1α deletion to the behavioral phenotype seen in the PGC-1α-null line and/or an HD-like phenotype, PGC-1α was deleted from all MSNs using cre-lox technology. Here, we show that, whereas no ambulatory changes are seen at 6 months of age, conditional knock-out animals exhibit hypoactivity at 18 months of age. Although a loss of PGC-1α specifically in MSNs does not result in overt cell loss, it does lead to a significant reduction in several PGC-1α-dependent transcripts, including the synchronous release protein complexin 1 (Cplx1), the axonal stabilizing protein neurofilament heavy chain (Nefh), and a novel set of striatal transcripts involved in transcriptional processing, calcium transport, and metabolism. These genes were measured in the HDQ knock-in mouse lines to determine whether this transcriptional profile was similarly reduced in the striatum of this HD model. Although the PGC-1α transcript was unaffected, several of its downstream targets were upregulated, particularly in homozygous HDQ mice with increased CAG repeat length at 6 months and heterozygous HDQ mice at 17 months of age. Together, these data highlight the importance of PGC-1α in maintaining and modulating striatal function and provide insight into how transcriptional dysregulation in this circuit can give rise to aging-associated ambulatory deficits. Further, these data suggest that a reduction in PGC-1α mRNA expression or function is not a key feature of the knock-in mouse models, but rather that upregulation of certain PGC-1α-dependent genes occurs as part of the disease process in MSNs.
Materials and Methods
Animals.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham. Animals lacking the full-length form of PGC-1α (Lin et al., 2004) (gift from Jiandie Lin, University of Michigan) were used for array experiments. Conditional deletion of PGC-1α was achieved by crossing mice with loxP sites flanking exons 3–5 of the PPARGC1A gene (Lin et al., 2004) (gift from Bruce Spiegelman, Dana-Farber Cancer Institute) with those expressing Cre-recombinase driven by the RGS9L promoter to specifically target both direct and indirect pathway MSNs in the striatum (Dang et al., 2006). Females that were heterozygous for both Cre-recombinase and the floxed allele for PGC-1α were bred with males that were heterozygous for the PGC-1α allele and lacking Cre-recombinase. Littermates expressing Cre-recombinase alone were used as controls. Both males and females generated from this breeding scheme (RGS9LCre: PGC-1α+/+, RGS9LCre: PGC-1αfl/fl) were tested at 6 and 18 months of age unless otherwise noted. As an indicator of Cre-mediated recombination, the RGS9LCre line was crossed to the ROSA mT/mG (JAX007576) line to generate progeny in which green fluorescence indicates efficient recombination. All mice were maintained on a C57BL/6J genetic background and housed 2–5 in a cage at 26 ± 2°C room temperature with food and water ad libitum. cDNA from multiple HD knock-in mouse lines for transcriptional studies was isolated as described previously (Kumar et al., 2016). These animals are heterozygous or homozygous for CAG repeats within the huntingtin locus and compared with age-matched controls. The repeat lengths used for HD knockin lines between 5 and 7 months of age are as follows: HDQ250/+, 310/+, 50/50, 100/100, 150/150 (144–164 repeats), and 200/200 (194–211 repeats) compared with control (HDQ7/7). In the HDQ310/+, previously published as HDQ300/+ (Kumar et al., 2016), age of onset is later than that of the HDQ150/150 and earlier than the HDQ200/200 as determined by weight loss, rotarod performance, and activity levels, indicating their placement on the phenotypic severity scale between the two homozygous genotypes. HDQ310/+ mice exhibit motor deficits by 8 months that worsen with age; by 35 weeks of age, HDQ310/+ mice exhibit reductions in dopamine receptor expression; and, by 70 weeks, these mice exhibit aggregates and a significant reduction in brain weight compared with controls. These animals also exhibit an inability to breed, preventing homozygous HDQ310/310 offspring (Kumar et al., 2016). Due to symptom severity, the repeat length for CAG repeats beyond 300 is restricted to the 315/+ mice at 17 months of age and was used as a comparison for transcriptional changes seen in the 18-month-old conditional knock-out line for PGC-1α.
Behavior.
Mice were evaluated, as described previously (Lucas et al., 2012), for the presence of hindlimb clasping by suspending the mouse by the tail for 15 s (clasping of hindlimbs together or forelimb to hindlimb). In addition, mice were observed in a holding cage for signs of tremor.
Open field (n = 8–12/genotype) was assessed as described previously (Lucas et al., 2012). For assessment of baseline activity, animals were placed in a square apparatus (27.9 cm2) consisting of 48 infrared beams (Med Associates) in a dark room for 60 min. Data were collected with Open Field Activity Software (Med Associates) in 1 min intervals over the test period. To evaluate responsivity to alterations in dopaminergic tone, animals were treated via intraperitoneal injection with 5 mg/kg d-amphetamine and immediately tested in the open field as described above.
Oligonucleotide array.
The striatum was dissected from PGC-1α WT and null mice (n = 4/genotype) at postnatal day 30 (P30) (an age before the appearance of vacuolizations to minimize the likelihood of compensatory transcriptional changes) and RNA was isolated as described below. Gene expression analysis was performed using the Mouse Ref-8 BeadChip and iScan system from Illumina. Total RNA was converted to cDNA by reverse transcription, followed by second-strand synthesis, to generate double-stranded cDNA. After purification, the cDNA was converted to biotin-labeled cRNA, hybridized to a Mouse Ref-8 BeadChip and stained with strepavidin-Cy3 for visualization. The Mouse Ref-8 BeadChips contain sequences representing ∼46,000 curated and putative genes and ESTs. Quality standards for hybridization, labeling, staining, background signal, and basal level of housekeeping gene expression for each chip were verified. After scanning the probe array, the resulting image was analyzed using GenomeStudio software (Illumina). Gene lists were created using GeneSpring. Data were analyzed through the use of Ingenuity Pathways Analysis (Ingenuity Systems). The WEB-based Gene SeT AnaLysis Toolkit was used to determine both transcription factor enrichment sequences for genes downregulated by microarray and for gene ontology; hypergeometric analysis was used, followed by Benajamini-Hochberg multiple test adjustment and significance was set at p < 0.01.
FISH.
FISH was performed using the RNAscope Multiplex Fluorescent assay (Advanced Cell Diagnostics) according to the manufacturer's instructions. Mice at 3 months of age (n = 2/genotype) were briefly anesthetized with isoflurane and decapitated and brains were removed and frozen with powdered dry ice for sectioning on a cryostat. Then, 20 μm sections were collected on SuperFrost Plus slides (Thermo Fisher Scientific) and immediately refrozen. Samples were fixed in 4% prechilled paraformaldehyde followed by dehydration in ethanol and pretreatment in protease IV (Advanced Cell Diagnostics). Probes were custom designed to recognize exons 3–5 of PGC-1α (Advanced Cell Diagnostics). Colocalization studies were performed using probes for DARPP32 (Advanced Cell Diagnostics). Tissues were treated with a mixture of probes for 2 h at 40°C followed by fluorescent amplification and mounting with Prolong gold antifade mounting medium containing DAPI (Thermo Fisher Scientific). Images were captured with a Nikon A1+ confocal microscope.
Transcript analyses.
Quantitative real-time PCR was conducted as described previously (Lucas et al., 2012). Mice were anesthetized with isoflurane and decapitated and brains were rapidly removed and dissected. Tissue pieces were flash frozen on dry ice and stored at −80°C until use, at which time they were incubated in RNAlater-ICE (Ambion) according to the manufacturer's instructions. Tissue was homogenized in TRIzol using a Tissue-Tearor homogenizer (Biospec) and RNA was isolated using the TRIzol/choloform-isopropanol method following the manufacturer's instructions (Invitrogen). RNA concentration and purity were determined using a NanoDrop 2000 (Thermo Fisher Scientific). Equivalent amounts of RNA (1 μg) were treated with DNase I (Promega) at 37°C for 30 min and DNase Stop solution at 65°C for 15 min. RNA was reverse-transcribed using the High-Capacity cDNA Archive Kit (Applied Biosystems). Transcripts were measured using mouse-specific primers from Applied Biosystems and JumpStart Taq Readymix (Sigma-Aldrich) using a protocol with an initial ramp (2 min, 50°C; 10 min, 95°C) and 40 subsequent cycles (15 s, 95°C; 1 min, 60°C). Relative concentration of transcript was calculated compared with a standard curve generated from pooled cDNA samples and then diluted (1:5, 1:10, 1:20, 1:40; calibrator method). These values were normalized to β-actin and expressed as ratio to control samples ± SEM (n = 8–10/genotype for conditional knock-out studies; 1–3 animals/sample, n = 9–12 samples/genotype for 6-month HD knock-in studies, n = 13–16/genotype for 17 month HDQ 310/+ studies). The following primer/probe sets were used: Actb (Mm00607939_s1), PGC-1α (Mm00447183_m1), PV (Mm00443100_m1), Got2 (Mm00494703_m1), Ctsa (Mm00447194_g1), Mrps31 (Mm01222255_m1), Nfyc (Mm04204600_g1), Arntl (Mm00500222_m1), Wdr77 (Mm01296590_g1), Trib3 (Mm00454879_m1), Lamc1 (Mm00711821_g1), Atp2b3 (Mm00623637_m1), Rap1gap (Mm01181215_m1), Sbsn (Mm00552057_m1), prodynorphin (Pdyn; Mm00457572_m1), tachykinin 1 (Tac1; Mm01166996_m1), Drd1a (Mm01353211_m1), preproenkephalin (Penk1; Mm01212875_m1), dopamine receptor D2 (Drd2; Mm00438541_m1), Gad1 (Mm00725661_s1), calbindin 1 (Calb1; Mm00486645_m1), opioid receptor, mu 1 (Oprm; Mm01188089_m1), synaptotagmin 2 (Syt2; Mm00436864_m1), Cplx1 (Mm00514378_m1), Nefh (Mm01191456_m1), Nceh1 (Mm00626772_m1), Ak1 (Mm00445475_m1), Inpp5j (Mm00552486_m1), Pdha1 (Mm00468675_m1), St8sia1 (Mm00456915_m1), Stac2 (Mm00524631_m1), Atp50 (Mm01611862_g1), Atp5a1 (Mm00431960_m1), Idh3a (Mm00499674_m1), and Phyh (Mm00477734_m1). PGC-1α splice variants were measured as described previously (Rowe et al., 2012). Relative expression of transcript was calculated using the 2−ΔΔCt method using the mean of two housekeeping genes, 36B4 and HPRT. Sequences for the primers are as follows: 36B4 forward (5′GGAGCCAGCGAGGCCACACTGCTG3′), 36B4 reverse (5′CTGGCCACGTTGCGGACACCCTCC3′), HPRT forward (5′GTTAAGCAGTACAGCCCCAAA3′), HPRT reverse (5′AGGGCATATCCAACAACAAACTT3′), NT-PGC-1α forward (5′TGCCATTGTTAAGACCGAG3′), NT-PGC-1α reverse (5′GGTCACTGGAAGATATGGC3′), alternative PGC-1α forward (5′CCACCAGAATGAGTGACATGGA3′), and alternative PGC-1α reverse (5′GTTCAGCAAGATCTGGGCAAA3′) (Chinsomboon et al., 2009; Thom et al., 2014).
Immunohistochemistry.
Immunofluorescence was performed as described previously (Lucas et al., 2014a). Animals were anesthetized with isoflurane and perfused intracardially with cold PBS and 4% paraformaldehyde (PFA) in PBS. Brains were removed, postfixed in 4% PFA for 24–72 h, and cryoprotected in graded sucrose (5–20%). Next, brains were embedded in 2:1 20% sucrose and Tissue-Tek optimal cutting temperature compound (Sakura Finetek) and stored at −80°C. Tissue was sectioned at 30 μm and mounted onto charged slides (Thermo Fisher Scientific). After drying overnight, slides were stored at −80°C. For the first day of immunohistochemistry, slides were washed in PBS before blocking for 1 h with 10% serum from the host of the secondary antibody in PBS. When antigen retrieval was necessary, slides were incubated in citrate buffer (10 mm citric acid, pH 4.0) at 37°C for 10 min and at room temperature for 20 min. Slides were then incubated with the primary antibody in 3% BSA and 0.3% Triton X-100 (Sigma-Aldrich) in PBS at 4°C. The following primary antibodies were used: Nefh (Abcam;), PV (Swant), and tyrosine hydroxylase (TH; Millipore).
The following day, slides were rinsed and incubated with the corresponding fluorescence-conjugated secondary antibodies (from Jackson ImmunoResearch, Invitrogen, or Thermo Fisher Scientific) for confocal microscopy or IRDye secondary antibodies (LI-COR Biosciences) for quantification experiments for 1 h at room temperature in 5% serum, 3% BSA, and 0.3% Triton X-100 in PBS. When necessary, autofluorescence eliminator reagent was used per the manufacturer's instructions (Millipore).
Sections were coverslipped using Prolong Antifade Gold with DAPI (Invitrogen) and stored at 4°C. Images were captured with a Leica TCS-SP5 laser scanning confocal microscope. All confocal settings, including laser intensity, gain, offset, and zoom, were held constant across all groups for a given experiment. For quantification, images were collected using the ODYSSEY CLx system (LI-COR Biosciences) and intensity and area measurements were obtained using Image Studio Lite Version 4.0.21 (LI-COR Biosciences).
For confocal microscopy, three to four animals/genotype and three to four sections/animal were imaged. For for quantification of infrared immunofluorescence, three to four animals/genotype were used. Every tenth slide was taken per animal for sampling throughout the striatum, resulting in eight to 12 sections/animal. All microscopy images were imported into Adobe Photoshop CS3 for adjustments to contrast, sharpness, and brightness.
Statistical analyses.
Under an a priori hypothesis that transcriptional, immunohistochemistry, and behavioral data in the RGS9LCre: PGC-1αfl/fl would replicate findings from the PGC-1α-null animals, one-tailed Student's t tests were used unless otherwise noted. For the remaining transcriptional studies, data were analyzed using a one-way ANOVA followed by Tukey's multiple comparisons test or Kruskal–Wallis test for unequal variance, followed by Dunn's post hoc test, Student's two-tailed t test, or Mann–Whitney U test. All HDQ data are represented as fold control [WT (HDQ7/7)] and normalized to β-actin. For ambulatory distance, data were analyzed using repeated-measures ANOVA. Area analyses using LI-COR Odyssey software were performed using a two-way ANOVA and two-tailed t tests.
Results
Unbiased identification of PGC-1α-dependent transcripts in the striatum
Although a number of PGC-1α-dependent genes have been identified in various cell types and tissues by us and others, we initially sought to identify PGC-1α-dependent genes within the striatum using unbiased methods. RNA was isolated from striatal homogenate of P30 PGC-1α+/+ and PGC-1α−/− mice. Microarray analysis from these samples revealed 659 genes that were significantly downregulated (p < 0.05) and 429 genes that were upregulated (p > 0.05) in the PGC-1α-null striatum compared with control (GSE100510). Using the online WebGestalt database, gene ontology analysis for biological function revealed that downregulated sets of genes play a role in metabolism, gene expression and RNA processing (Fig. 1A). To limit our transcript analysis to genes that could be potential direct targets of PGC-1α-containing transcriptional complexes, we cross-referenced this striatal array dataset to previously published microarray data (GSE100341) from PGC-1α-overexpressing neuroblastoma cells in which 1067 transcripts were upregulated >2-fold (Lucas et al., 2014a). This comparison revealed an overlap in 11 genes involved in neurite growth, metabolism, and transcription (Fig. 1B) that are distinct from those identified to overlap with PGC-1α and parvalbumin (PV) expression patterns in the cortex.
Figure 1.

Identification of novel PGC-1α-dependent gene programs in the striatum. A, Gene ontology analyses using microarray data from the PGC-1α-null striatum identified a number of gene programs that were significantly downregulated in biological, molecular function categories, where were grouped into distinct cellular components. Hypergeometric analysis was used, followed by Benajamini-Hochberg multiple test adjustment. B, Previously published microarray data of genes upregulated by PGC-1α overexpression in vitro (Lucas et al., 2014a) were cross-referenced with those significantly downregulated in the striatum in vivo to result in a subset of putative PGC-1α-dependent transcripts in this region.
Identification of PGC-1α-dependent transcripts in striatal MSNs
The biological function of PGC-1α in MSNs is relatively unknown. Although we have previously identified transcriptional changes in the striatum from whole-body PGC-1α-null mice (Lucas et al., 2012), it is not clear whether these changes are occurring in MSN populations or other striatal cell types that express PGC-1α, such as PV-positive interneurons (Lucas et al., 2010, 2014a; Bartley et al., 2015). To determine its role in this population, we generated mice lacking PGC-1α specifically in MSNs using mice expressing Cre-recombinase driven by the RGS9L promoter crossed with mice expressing loxP sites flanking exons 3–5 of the PPARGC1A gene; excision of these exons generates a nonfunctional and unstable protein (Lin et al., 2004). We have used a similar approach to delete PGC-1α from all cells of the CNS by crossing the PGC-1α floxed mouse to a mouse expressing cre recombinase driven by the nestin promoter; this causes complete deletion of PGC-1α mRNA (exons 3–5) in multiple brain regions and profound motor impairment by 3 months of age (Lucas et al., 2012). Antibodies to detect PGC-1α adequately in mouse brain are unavailable. However, previously reported use of the RGS9LCre indicates that recombination in these animals occurs as early as P8 in mice (Dang et al., 2006) in D1-positive and D2-positive neuronal populations (Tecuapetla et al., 2014) of the striatum (Rahman et al., 1999). These studies suggest that conditional knock-out of a gene is occurring in MSNs of the direct and indirect pathways. Here, RGS9LCre-positive mice were crossed to the mTmG reporter line, in which Cre-mediated recombination is indicated by a switch from red fluorescence to green. Recombination was restricted to MSNs of the striatum seen in the coronal plane and recombination was visualized in axonal projections to the globus pallidus and substantia nigra in the sagittal plane at 3 months of age (Fig. 2A), supporting previous documentation that recombination occurs MSNs of both the direct and indirect motor pathways.
Figure 2.
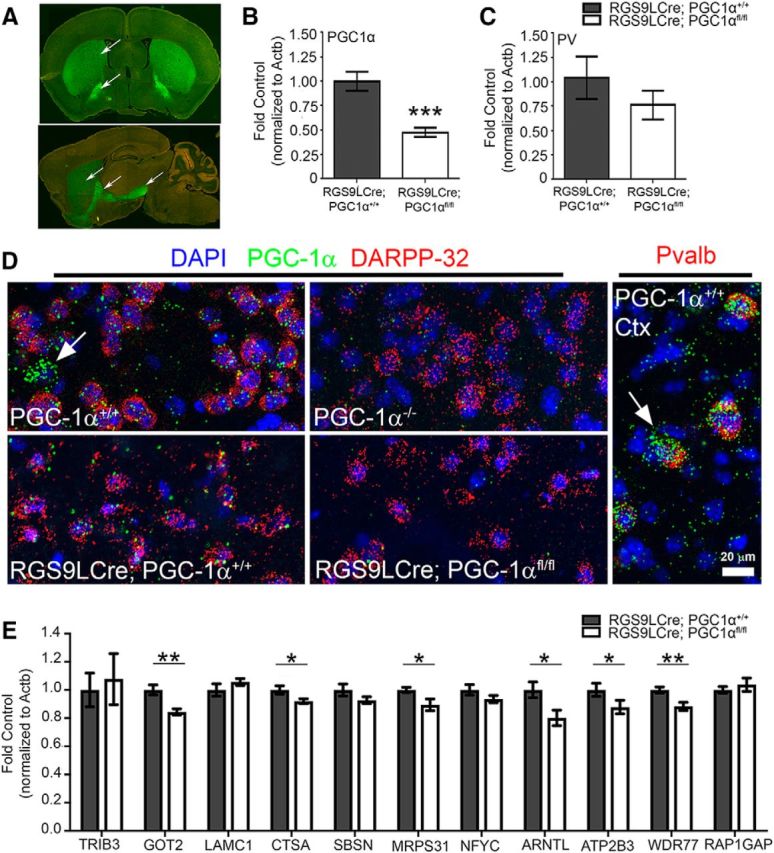
Putative PGC-1α-dependent targets are reduced in conditional knock-outs. A, RGS9L promoter-driven Cre-recombinase activity was validated by crossing the RGS9Lcre line with the mTmG reporter line; green fluorescence indicates recombination in direct and indirect MSN populations (arrows). B, RGS9LCre:PGC-1αfl/fl striatum exhibited an ∼50% reduction in PGC-1α transcript compared with RGS9LCre:PGC-1α+/+ mice. C, Expression of the PGC-1α-dependent gene PV was not significantly changed. D, PGC-1α expression (green) was reduced in both the null and RGS9LCre:PGC-1αfl/fl in DARPP-32-positive neurons (red) in the striatum compared with controls, as shown by FISH. E, Loss of PGC-1α in striatal MSNs significantly reduced the expression Got2, Ctsa, Mrps31, Arntl, Atp2b3, and Wdr77. n = 8–10/genotype. *p < 0.05; **p < 0.01; ***p < 0.001; one-tailed t tests based on previously published a priori hypotheses. Data are presented as mean ± SEM.
To confirm reductions in PGC-1α transcript, primers recognizing exons 5–6 were used to measure PGC-1α knock-down in the RGS9LCre: PGC-1αfl/fl striatum at 18 months. PGC-1α transcript was reduced by 52% (t(16) = 4.48, p = 0.0002, unpaired t test; Fig. 2B). Because green fluorescence was distributed homogenously throughout the dorsal striatum and nucleus accumbens, we predict that residual expression of PGC-1α may be due to other cell types (i.e., interneurons) that express high levels of this coactivator relative to MSNs, such as PV-positive interneurons (Cowell et al., 2007), which do not undergo recombination in the RGS9LCre line. This interpretation is consistent with the absence of changes in PV expression (Fig. 2C); PV transcript remained unaffected in the RGS9LCre: PGC-1αfl/fl striatum (t(16) = 1.01, p = 0.16, unpaired t test), confirming that recombination was not taking place in PV-positive interneurons (PV-INs) in this model because PV is the most robust readout of PGC-1α activity in this cell type in the forebrain. To confirm that PGC-1α knock-down was occurring selectively in MSNs, we developed a custom probe to detect PGC-1α exons 3–5 using FISH (Fig. 2D). Signal for PGC-1α was reduced almost entirely in dorsolateral striatum of whole-body knock-out mice and concentrated highly in WT PV-INs (arrows), demonstrating specificity of the probe. Signal was reduced selectively in DARPP-32-positive neurons in the RGS9LCre: PGC-1αfl/fl striatum.
To determine whether transcripts identified as putative PGC-1α targets (Fig. 1D) were reduced in MSNs lacking PGC-1α, we tested this set in the 18-month RGS9LCre: PGC-1αfl/fl striatum. Got2 (t(16) = 3.45, p = 0.002, unpaired t test), Ctsa (t(16) = 2.15, p = 0.02, unpaired t test), Mrps31 (t(16) = 2.47, p = 0.01, unpaired t test), Arntl (t(16) = 2.49, p = 0.01, unpaired t test), Atp2b3 (t(16) = 1.79, p = 0.046, unpaired t test), and Wdr77 (t(16) = 3.29, p = 0.002, unpaired t test) were significantly reduced in the RGS9LCre: PGC-1αfl/fl striatum. Trib3 (t(16) = 0.37, p = 0.36, unpaired t test), Lamc1 (t(16) = 1.04, p = 0.16, unpaired t test), Sbsn (t(16) = 1.39, p = 0.09, unpaired t test), Nfyc (t(16) = 1.35, p = 0.10, unpaired t test), and Rap1gap (t(16) = 0.75, p = 0.23, unpaired t test) were unaffected by a loss of PGC-1α (Fig. 2E).
Absence of PGC-1α in MSNs does not lead to overt neuronal loss
Our previous studies have reported significant changes in general MSN markers and markers specific to the direct or indirect pathways in the PGC-1α-null striatum at 3 months of age (Lucas et al., 2012). These alterations include significant upregulation of the matrix marker Calb1, the striosome marker Oprm, the direct pathway markers Pdyn and Tac1, and the indirect pathway markers Penk1 and Drd2 (Lucas et al., 2012). To determine whether alterations in MSN markers occurred cell autonomously in the PGC-1α-null striatum, we measured transcript for Gad1 (t(16) = 0.47, p = 0.32, unpaired t test), Calb1 (t(16) = 0.72, p = 0.24, unpaired t test), and Oprm (t(16) = 0.87, p = 0.20, unpaired t test) in the 18-month RGS9LCre: PGC-1αfl/fl striatum, none of which were significantly different. We then measured the direct pathway markers Pdyn (t(16) = 1.19, p = 0.13, unpaired t test), Tac1 (t(16) = 0.41, p = 0.34, unpaired t test), and Drd1a (t(16) = 0.45, p = 0.33, unpaired t test) and the indirect pathway markers Penk1 (t(16) = 1.35, p = 0.10, unpaired t test) and Drd2 (t(16) = 0.83, p = 0.21, unpaired t test); the expression of these transcripts was unaffected in the RGS9LCre: PGC-1αfl/fl striatum (Fig. 3A–C). These data suggest that deletion of PGC-1α in MSNs is not sufficient to cause the transcriptional changes seen in the striatum of PGC-1α-null mice. In addition, a lack of change in MSN markers suggests that PGC-1α deletion within this population does not result in overt cell loss.
Figure 3.

Previously identified PGC-1α-dependent genes are reduced in conditional knock-outs. A–C, Markers of MSNs in either the direct or indirect motor pathways were unaffected by a loss of PGC-1α. D, Among the previously identified nonmetabolic PGC-1α-dependent transcripts, Cplx1 and Nefh were significantly reduced in the RGS9LCre:PGC-1αfl/fl striatum. E, Six of 10 previously identified PGC-1α-dependent metabolic transcripts were reduced in the RGS9LCre:PGC-1αfl/fl striatum. n = 8–10/genotype. *p < 0.05; **p < 0.01; one-tailed t tests based on previously published a priori hypotheses. Data are presented as mean ± SEM.
Reduction in previously identified PGC-1α-dependent transcripts in striatal MSNs
We have shown previously that genes involved in calcium buffering (PV), synchronous neurotransmitter release (Syt2, Cplx1), and axonal structure (Nefh) are significantly reduced in the cortex of both PGC-1α-null mice and mice lacking PGC-1α specifically in PV-INs (Lucas et al., 2014a). These transcripts were tested in the RGS9LCre: PGC-1αfl/fl striatum to determine whether they are similarly regulated by PGC-1α in MSNs. Interestingly, whereas Syt2 transcript was unaffected (t(16) = 0.10, p = 0.46, unpaired t test), that of Cplx1 (t(16) = 1.87, p = 0.04, unpaired t test) and Nefh (t(16) = 1.83, p = 0.04, unpaired t test) were significantly downregulated (Fig. 3D). These data indicate that PGC-1α regulates genes for synchronous neurotransmitter release and axonal integrity in MSNs.
Next, we tested whether previously identified PGC-1α-dependent metabolic transcripts in the cortex were affected in the striatum (Lucas et al., 2014a). Nceh1 (t(16) = 3.30, p = 0.002, unpaired t test), Ak1 (t(16) = 2.24, p = 0.02, unpaired t test), St8sia1 (t(16) = 2.66, p = 0.009, unpaired t test), Stac2 (t(16) = 1.94, p = 0.04, unpaired t test), Idh3a (t(16) = 3.33, p = 0.002, unpaired t test), and Phyh (t(16) = 2.21, p = 0.021, unpaired t test) were significantly reduced in the conditional knock-out striatum, whereas Inpp5j (t(16) = 0.71, p = 0.24, unpaired t test), Pdha1 (t(16) = 1.71, p = 0.05, unpaired t test), Atp5o (t(16) = 1.63, p = 0.06, unpaired t test), and Atp5a1 (t(16) = 0.80; p = 0.22, unpaired t test) were unchanged (Fig. 3E).
Loss of PGC-1α in MSNs does not affect striatal volume
To determine whether knock-down of PGC-1α in MSNs causes striatal vacuolizations observed in PGC-1α-null mice and/or a reduction in striatal volume, we stained sections with hematoxylin and eosin. No vacuolizations are observed in the RGS9LCre: PGC-1αfl/fl striatum by hematoxylin and eosin staining (Fig. 4A). To determine whether a loss of PGC-1α in MSNs affects overall striatal volume, sections sampled throughout the entirety of the region of both RGS9LCre: PGC-1αfl/fl and RGS9LCre: PGC-1α+/+ mice were labeled for TH and NEFH immunoreactivity using infrared immunofluorescence quantification. Representative sections for each genotype for quantification are shown in Figure 4A. There were no differences in striatal area (t(6) = 1.82, p = 0.12, unpaired t test; Fig. 4B), hemispheric area (t(6) = 1.82, p = 0.12, unpaired t test; Fig. 4C), or the ratio of the two (t(6) = 1.03, p = 0.34, unpaired t test; Fig. 4D) between genotypes. Immunoreactivity for TH was not different in the RGS9LCre: PGC-1αfl/fl striatum (U = 3, p = 0.20, Mann–Whitney; data not shown).
Figure 4.

Nefh immunoreactivity is reduced in the conditional knock-out striatum. Shown is hematoxylin and eosin staining of RGS9LCre:PGC-1α+/+ and RGS9LCre:PGC-1αfl/fl striatum. The LI-COR Odyssey was used for volumetric analyses and to measure Nefh immunoreactivity (IR) intensity. A, Representative images for each genotype throughout the striatum are shown. B–D, No differences in striatal or hemispheric area nor ratio between the two were found in the RGS9LCre:PGC-1αfl/fl striatum compared with control. E, Quantification of Nefh-IR using the LI-COR Odyssey shows significant reduction in intensity. F, Although Nefh-IR is present striosomes of the RGS9LCre:PGC-1αfl/fl striatum (arrowheads), there is a loss of the lighter Nefh-IR in the striatal matrix (arrows), as visualized by confocal microscopy. n = 2 sections/slide, 12–14 slides/animal and 3–4 animals/genotype in A–E; n = 3–4 animals/genotype and 3–4 sections/animal were imaged in F. All settings were held constant across genotypes for microscopy images. *p < 0.05, two-way ANOVA and two-tailed t test in B–D; one-tailed t tests based a priori hypotheses in E. Data are presented as mean ± SEM.
Reductions in Nefh immunoreactivity in the striatum
Among the most robust reductions in gene expression was that of the PGC-1α-responsive gene Nefh. To quantify changes in immunoreactivity, Nefh intensity was measured using LI-COR Odyssey software. The reduction in Nefh intensity throughout the striatum was significant (t(5) = 2.22; p = 0.038, unpaired t test; Fig. 4E). Using confocal microscopy, dense Nefh immunoreactivity can be seen in the striatal matrix of control animals (Fig. 4F, white arrows) and, at a higher magnification, specifically in the cell bodies of MSNs. The cell body staining, although apparent in WT striatum, is reduced in the RGS9LCre: PGC-1αfl/fl striatum. Some Nefh expression can still be seen in the RGS9LCre: PGC-1αfl/fl matrix and in striosomes (Fig. 4F, white arrowheads). The remaining patch-like immunoreactivity of Nefh is likely due to projections from cortical pyramidal neurons and local PV-INs.
Absence of PGC-1α in MSNs causes age-related hypoactivity
Both PGC-1α-null animals and animals lacking PGC-1α specifically in neurons exhibit dramatic ambulatory and coordination deficits (Lin et al., 2004; Lucas et al., 2012, 2014b). To determine the contribution of MSN-specific PGC-1α deletion to motor deficits, RGS9LCre:PGC-1αfl/fl mice were tested in the open field at 1, 2, and 3 months of age (data not shown) and 6 and 18 months of age. At 6 months of age and earlier, there was no significant effect of genotype (F(1,18) = 0.15, p = 0.70, ANOVA) or interaction between genotype and time (F(11,198) = 0.40, p = 0.95, ANOVA; (Fig. 5A). By 18 months of age, repeated-measures ANOVA revealed a significant effect of genotype (F(1,17) = 11.86, p = 0.0031, ANOVA), with no interaction between genotype and time (F(11,187) = 0.34, p = 0.98, ANOVA), indicating a hypoactive phenotype in RGS9LCre:PGC-1αfl/fl mice (Fig. 5B).
Figure 5.

Deletion of PGC-1α in MSNs results in age-related hypoactivity. Ambulatory activity of mice lacking PGC-1α specifically in MSNs was assessed in the open field. A, Mice up to the age of 6 months showed no differences between genotypes in ambulatory distance throughout the test session. B, Mice at 18 months of age exhibited hypoactivity throughout the test session. C, D, There was a lack of genotype-dependent effect on d-amphetamine-induced ambulation at 6 months (C) or 18 months (D) of age. n/genotype are indicated on histograms. *p < 0.05, repeated-measures ANOVA. Data are presented as mean ± SEM.
To determine whether this hypoactivity was due to reduced sensitivity to dopamine in MSNs, we evaluated whether RGS9LCre:PGC-1αfl/fl mice were capable of responding to the dopamine transporter blocker d-amphetamine. Repeated-measures ANOVA did not reveal a significant effect of genotype (F(1,18) = 0.79, p = 0.39, ANOVA) or any interaction between genotype and time (F(11,198) = 0.59, p = 0.84, ANOVA) at 6 months of age (Fig. 5C). Further, there was no effect of genotype (F(1,17) = 2.50, p = 0.13, ANOVA) or interaction between genotype and time (F(11,187) = 0.46, p = 0.92, ANOVA) at 18 months (Fig. 5D). These data demonstrate that the hypoactivity is not due to a decreased ability of MSNs to respond to dopaminergic input and this is consistent with a lack of alteration in DRD1a and DRD2 mRNA expression in these mice.
PGC-1α-dependent pathways in HDQ mice
To explore whether PGC-1α and mHtt converge on similar transcriptional pathways in HD, we measured the levels of our newly identified MSN-specific PGC-1α-dependent genes in the striatum of knock-in models of HD. We measured Drd2 transcript in both the homozygous and heterozygous HDQ mouse lines at 6 and 15–17 months of age to determine whether we were able to replicate previous findings showing that its expression is downregulated (Kumar et al., 2016). One-way ANOVA revealed a significant difference among the HD knock-in lines for Drd2 transcript levels at 6 months (F(6,64) = 1.02, p < 0.0001, ANOVA). Drd2 was significantly reduced in the 150/150 (p = 0.0038, Tukey's multiple comparisons) and 200/200 striata (p = 0.0033, Tukey's multiple comparisons) compared with WT (Fig. 6A). Further, Drd2 was significantly downregulated in the heterozygous HDQ200/+ (t(13) = 2.53, p = 0.025, unpaired t test; Fig. 6C) and HDQ310/+ (U = 26, p = 0.0003, Mann–Whitney; Fig. 6E) at 15–17 months of age. With regard to total PGC-1α transcript levels, no differences were detected in the HDQ homozygous HDQ knock-ins (H = 18.36, p = 0.0054, Kruskal–Wallis test; p > 0.9999, Dunn's multiple comparisons), HDQ250/+ (H = 18.36, p = 0.0054, Kruskal–Wallis test; p = 0.16, Dunn's multiple comparisons) or HDQ310/+ or heterozygous mice (H = 18.36, p = 0.0054, Kruskal–Wallis test; p = 0.16, Dunn's multiple comparisons) at 6 months of age (Fig. 6B). PGC-1α transcript was unchanged at 15–17 months for the HDQ200/+ (t(13) = 0.10, p = 0.92, unpaired t test; Fig. 6D) or HDQ310/+ (t(27) = 0.34, p = 0.74, unpaired t test) mice compared with control (Fig. 6F). We also used primers that recognize the N-terminal (NT) splice variant (to measure total NT levels) and the alternately spliced variant of PGC-1α to determine whether there are changes in variants of PGC-1α in both the homozygous and heterozygous knock-in genotypes at 6 months. The level of total NT-PGC-1α or the alternatively spliced PGC-1α variants in the homozygous knock-in lines did not differ compared with WT (p = 0.28 and p = 0.45, respectively; Tukey's multiple comparisons) and there were no differences detected among the heterozygous genotypes for total NT (F(2,29) = 0.13, p = 0.88; one-way ANOVA) or alternatively spliced (F(2,29) = 0.29, p = 0.75; one-way ANOVA) PGC-1α variants compared with WT.
Figure 6.

PGC-1α expression is unaffected in the HDQ striatum. A, Among the HDQ knock-in mouse lines, Drd2 transcript is significantly reduced in the HDQ150/150 and 200/200 striata compared with WT at 6 months. B, PGC-1α transcript was unaffected at this time. Drd2 was significantly reduced in the HDQ200/+ (C) and HDQ310/+ (E) striatum at 15 and 17 months compared with WT, respectively, whereas PGC-1α transcript was unchanged (D,F). n = 1–3 animals/sample, n = 9–12 samples/genotype in A and B; n = 7–9/genotype in C and D; n = 13–16/genotype in E and F. *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA followed by Tukey's multiple comparisons or Kruskal–Wallis followed by Dunn's test, Student's t test, or Mann–Whitney U test. Data are presented as mean ± SEM.
HD knock-in mouse models exhibit late-onset motor phenotypes, with longer CAG repeat lengths causing symptom presentation at younger ages (Menalled, 2005; Rising et al., 2011). We assessed the expression of PGC-1α-dependent genes before the onset of behavioral abnormalities (i.e., HDQ150/150; Heng et al., 2007; Kumar et al., 2016) and after symptom onset (i.e., HDQ310/+; Kumar et al., 2016) to provide insight into the role that PGC-1α-dependent gene deficiency may play in the progression of HD pathology. Transcripts that were reduced in the RGS9LCre:PGC-1αfl/fl striatum were tested in the HDQ lines at 6 months of age. Among transcripts identified through the array (Fig. 7A–F), Wdr77 transcript was increased in the HDQ200/200 striatum compared with control (H = 11.63, p = 0.02, Kruskal–Wallis; p = 0.04, Dunn's multiple comparisons; Fig. 7B). No differences were detected for Arntl (H = 4.97, p = 0.29, Kruskal–Wallis), Atp2b3 (H = 7.08, p = 0.13, Kruskal–Wallis), Got2 (F(4,46) = 2.813, p = 0.03, ANOVA; p > 0.14, Tukey's multiple comparisons), Mrps31 (F(4,46) = 1.4, p = 0.25, ANOVA), or Ctsa (H = 8.79, p = 0.067, Kruskal–Wallis) compared with WT (Fig. 7A,C–F).
Figure 7.

PGC-1α-dependent transcripts are upregulated in the HDQ striatum at 6 months. Among the novel putative PGC-1α-dependent transcripts in the striatum, Wdr77 was significantly upregulated at 6 months in the HDQ 200/200, whereas Arntl, Atp2b3, Got2, Mrps31, and Ctsa were unaffected compared with WT (A–F). Among previously published PGC-1α-dependent genes, Idh3a, Phyh, Ak1, and Nceh1 were significantly upregulated in the HDQ 150/150 and/or 200/200 compared with WT. Cplx1, Nefh, St8sia1, and Stac2 are unchanged compared with WT (G–N). n = 1–3 animals/sample, n = 9–12 samples/genotype. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, one-way ANOVA followed by Tukey's multiple comparisons or Kruskal–Wallis followed by Dunn's test. Data are presented as mean + SEM.
We next tested whether previously identified PGC-1α-dependent transcripts were altered in the HDQ homozygous lines (Fig. 7G–N). Idh3a was significantly upregulated in the HDQ150/150 (H = 29.99, p < 0.0001, Kruskal–Wallis; p = 0.0004, Dunn's test) and HDQ200/200 (H = 29.99, p < 0.0001, Kruskal–Wallis; p = 0.0006, Dunn's test) striatum compared with WT (Fig. 7I).
Transcript levels for Phyh were significantly increased in mice HDQ150/150 CAG (H = 23.59, p < 0.0001, Kruskal–Wallis; p = 0.001, Dunn's test) compared with control (Fig. 7J). HDQ200/200 showed a significant increase in AK1 expression compared with control (H = 15.28, p = 0.004, Kruskal–Wallis; p = 0.04, Dunn's test; Fig. 7K). In addition, Nceh1 expression was significantly increased in the HDQ 150/150 (F(4,46) = 19.82, p < 0.0001, ANOVA; p < 0.0001, Tukey's multiple comparisons) and HDQ200/200 (F(4,46) = 19.82, p < 0.0001, ANOVA; p < 0.0001, Tukey's multiple comparisons) striata compared with WT (Fig. 7L). No differences among the groups were detected for the following transcripts: Cplx1 (H = 8.82, p = 0.066, Kruskal–Wallis), Nefh (H = 9.35, p = 0.053, Kruskal–Wallis), St8sia1 (F(4,46) = 3.45, p = 0.02, ANOVA; p > 0.29, Tukeys's multiple comparisons), or Stac2 (H = 11.41, p = 0.022, Kruskal–Wallis; p > 0.12, Dunn's multiple comparisons) at 6 months of age (Fig. 7G,H,M,N). These data indicate that, whereas PGC-1α transcript levels remained unchanged in these HDQ lines, several PGC-1α-dependent transcripts are upregulated with increased CAG repeat length.
Transcripts were then measured in the 6-month HDQ250/+ and HDQ310/+ striata. In the HDQ250/+ striatum, Cplx1 was significantly reduced (U = 23, p = 0.028, Mann–Whitney) and Idh3a was significantly increased (U = 17, p = 0.0073, Mann–Whitney) compared with control The following transcripts were unchanged in the HDQ250/+ 6-month striatum (Fig. 8A): Arntl (U = 46, p = 0.60, Mann–Whitney), Wdr77 (U = 50, p = 0.81, Mann–Whitney), Atp2b3 (t(19) = 0.25, p = 0.80, Mann–Whitney), Got2 (U = 53, p = 0.97, Mann–Whitney), Mrps31 (t(19) = 0.39, p = 0.70, Mann–Whitney), Ctsa (t(19) = 1.83, p = 0.083, unpaired t test), Nefh (U = 35, p = 0.17, Mann–Whitney), Phyh (t(19) = 1.28, p = 0.21, unpaired t test), Ak1 (t(19) = 1.79, p = 0.089, unpaired t test), Nceh1 (U = 28, p = 0.069, Mann–Whitney), St8sia1 (U = 27, p = 0.095, Mann–Whitney), and Stac2 (t(19) = 0.98, p = 0.34, unpaired t test; Fig. 8A).
Figure 8.
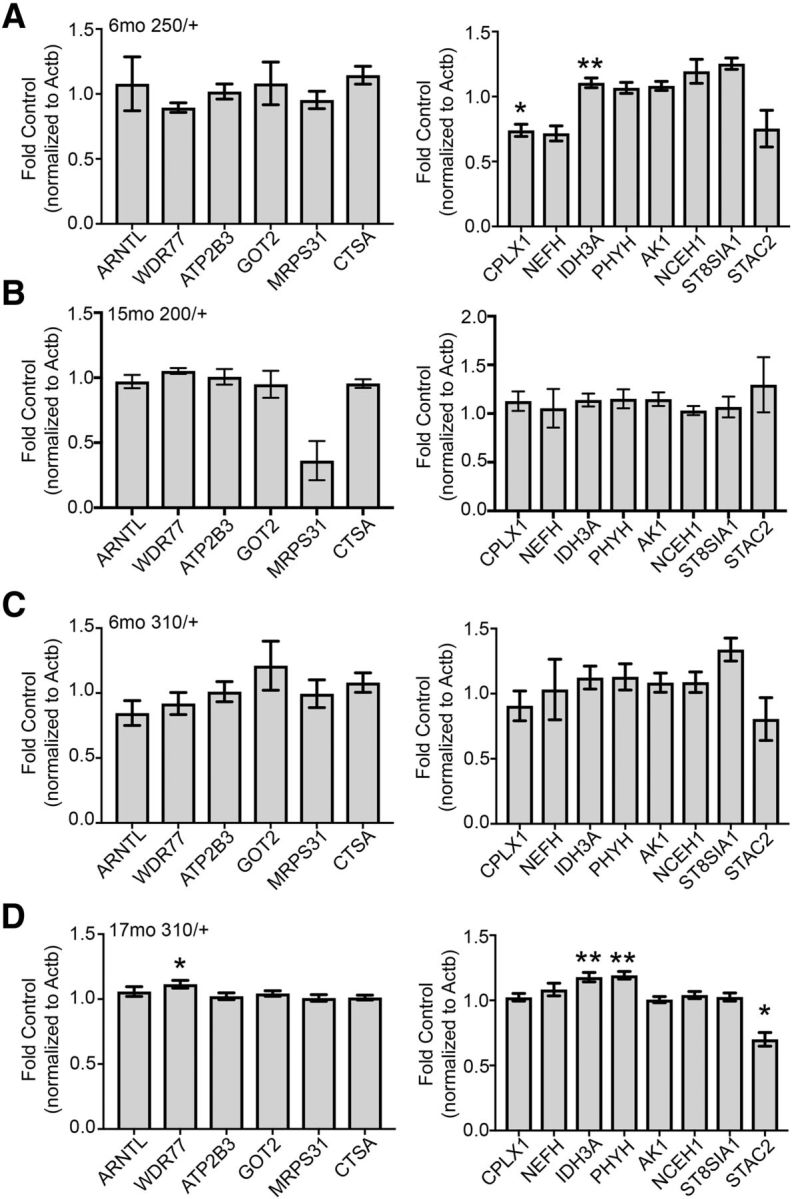
Age- and repeat-dependent upregulation of PGC-1α-dependent transcripts in the heterozygous HDQ striatum. A, Among the putative PGC-1α-dependent transcripts in the HDQ250/+ striatum, Cplx1 was significantly downregulated, whereas Idh3a was upregulated compared with control at 6 months. B, Expression of these transcripts was comparable to control in the HDQ200/+ striatum. C, There were no differences detected in PGC-1α-dependent transcripts in the 6-month HDQ 310/+ striatum. However, among these transcripts, Wdr77, Idh3a, and Phyh were significantly upregulated in this line at 17 months compared with control, whereas Stac2 was significantly reduced (D). n = 1–3 animals/sample, n = 9–12 samples/genotype in A and C; n = 7–9/genotype in B; n = 13–16/genotype in D. *p < 0.05; **p < 0.01, Student's t test or Mann–Whitney U test. Data are presented as mean ± SEM.
To determine whether 200+ CAG repeats had an influence on transcript levels at an advanced age, these genes were tested in the striatum of mice heterozygous for 200CAG repeats in the htt gene; no significant differences were detected in the 15-month HDQ200/+ striatum (Fig. 8B): Arntl (t(14) = 0.51, p = 0.62, unpaired t test), Wdr77 (U = 17, p = 0.14, Mann–Whitney), Atp2b3 (t(14) = 0.10, p = 0.92, unpaired t test), Got2 (t(14) = 0.46, p = 0.65, unpaired t test), Mrps31 (U = 21, p = 0.46, Mann–Whitney), Ctsa (t(14) = 0.72, p = 0.48, unpaired t test), Cplx1 (t(14) = 1.22, p = 0.24, unpaired t test), Nefh (t(14) = 0.26, p = 0.80, unpaired t test), Idh3a (t(14) = 1.97, p = 0.069, unpaired t test), Phyh (t(14) = 1.47, p = 0.16, unpaired t test), Ak1 (U = 16, p = 0.11, Mann–Whitney), Nceh1 (t(14) = 0.55, p = 0.59, unpaired t test), St8sia1 (t(14) = 0.59, p = 0.56, unpaired t test), and Stac2 (U = 26, p = 0.61, Mann–Whitney).
Regarding the HDQ310/+ striatum, all transcripts were unaffected at 6 months (Fig. 8C): Arntl (t(21) = 1.43, p = 0.17, unpaired t test), Wdr77 (t(21)=0.58, p = 0.57, unpaired t test), Atp2b3 (t(21) = 0.12, p = 0.91, unpaired t test), Got2 (U = 58, p = 0.65, Mann–Whitney), Mrps31 (t(21) = 0.04, p = 0.97, unpaired t test), Ctsa (t(21) = 0.94, p = 0.36, unpaired t test), Cplx1 (t(21) = 0.66, p = 0.51, unpaired t test), Nefh (t(21) = 0.12, p = 0.90, unpaired t test), Idh3a (U = 46, p = 0.24, Mann–Whitney), Phyh (U = 48, p = 0.29, Mann–Whitney), Ak1 (U = 53, p = 0.45, Mann–Whitney), Nceh1 (U = 54, p = 0.49, Mann–Whitney), St8sia1 (t(20) = 1.96, p = 0.065, unpaired t test), and Stac2 (t(21) = 0.77, p = 0.45, unpaired t test).
To determine whether these transcripts were affected with age, these transcripts were measured in the HDQ310/+ striatum at 17 months of age (Fig. 8D). Compared with control, Stac2 was significantly downregulated (t(27) = 2.61, p = 0.015, unpaired t test), whereas the following three genes were significantly upregulated (Fig. 8D): Wdr77 (t(27) = 2.72, p = 0.01, unpaired t test), Idh3a (t(27) = 3.15, p = 0.0039, unpaired t test), and Phyh (t(27) = 3.40, p = 0.0021). The remaining transcripts were unchanged (Fig. 8D): Arntl (t(27) = 0.87, p = 0.39, unpaired t test), Atp2b3 (t(27) = 0.42, p = 0.67, unpaired t test); Got2 (t(27) = 1.26, p = 0.22, unpaired t test), Mrps31 (t(27) = 0.20, p = 0.84, unpaired t test), Ctsa (t(27) = 0.39, p = 0.70, unpaired t test), Cplx1 (U = 73, p = 0.18, Mann–Whitney), Nefh (t(27) = 1.017, p = 0.32, unpaired t test), Ak1 (U = 103, p = 0.98, Mann–Whitney), Nceh1 (t(27) = 0.78, p = 0.44, unpaired t test), and St8sia1 (t(27) = 0.43, p = 0.67, unpaired t test).
Discussion
Studies indicate reductions in PGC-1α expression and PGC-1α-dependent transcripts in HD patients and rodent models (Cui et al., 2006; Weydt et al., 2006; Török et al., 2015). mHtt can repress the expression of PGC-1α directly at its promoter by interacting with the upstream regulators CREB-TAF4 (Cui et al., 2006); activation of the CREB-TAF4-PGC-1α pathway is able to rescue mitochondrial function, cell viability, and morphological and motor performance of in vitro and in vivo HD models (Cui et al., 2006; Wareski et al., 2009; Tsunemi et al., 2012). In addition, reductions in CREB-PGC-1α-dependent transcription contribute to NMDAR-mediated excitotoxicity in cortical and striatal cultures when mHtt is present (Okamoto et al., 2009). Striatal MSNs are a highly vulnerable in HD and, although a significant reduction in PGC-1α transcript is detected in isolated MSNs from an HD mouse model (Cui et al., 2006), there has been no investigation into the causal relationship between PGC-1α loss in this cell type and the behavioral phenotype in disease.
A loss of PGC-1α throughout the body and neuron-specific deletion of PGC-1α in mice leads to robust motor deficits including hyperactivity, impaired coordination, and gait abnormalities by 1 month of age (Lin et al., 2004; Lucas et al., 2012, 2014a). Prominent vacuolizations are also present throughout the PGC-1α-null cortex and striatum of these mice at 1 month of age (Lin et al., 2004; Lucas et al., 2012). We show here that deletion of PGC-1α from MSNs leads to hypoactivity (instead of hyperactivity) and lack of vacuolizations at an advanced (instead of developmental) age, indicating that dysfunction in other cell types may contribute to these abnormalities in the PGC-1α-null mice (Lucas et al., 2010, 2014b). In fact, MSNs show relatively low levels of PGC-1α expression with respect to these cell types (Cowell et al., 2007; Fig. 2), suggesting that cell types more highly enriched in PGC-1α rely heavily on its presence throughout development versus adulthood, conferring more robust behavioral dysfunction. Studies in which PGC-1α is deleted in CaMKIIα-positive or PV-positive cell types suggest this as well, with the former exhibiting vacuolizations (Ma et al., 2010) and the latter exhibiting nest-building, learning, and neuronal firing deficits at 3 months of age (Ma et al., 2010; Dougherty et al., 2014; Lucas et al., 2014a; Bartley et al., 2015).
Previous reports of decreased PGC-1α expression in HD have formed the basis for the idea that the PGC-1α-null mouse could serve as an appropriate model to understand the pathogenesis of HD. Contrary to our predictions, we did not observe any reductions in PGC-1α expression in the striatum of the HDQ mice. It is important to note that we did not measure PGC-1α protein content or investigate the possibility of posttranslational modifications of PGC-1α. However, with respect to the striatum-specific roles of PGC-1α, our previous studies demonstrated many differences between the transcriptional profiles of the PGC-1α-null mouse and the R6/2 model of HD (Lucas et al., 2012). Therefore, the data presented here do not support the idea that striatal PGC-1α deficiency alone can generate an HD-like behavioral phenotype. However, it would be interesting to determine whether concurrent PGC-1α ablation in the cortex, another region affected in HD, could cause the emergence of more severe motor symptoms.
We considered the possibility that the huntingtin-mediated effects on PGC-1α expression and/or function could be dependent on age. Most HD knock-in mouse models, including the 140CAG line, exhibit late-onset motor phenotypes (Menalled, 2005; Rising et al., 2011). In theory, reductions in PGC-1α in MSNs of late-onset HD models could contribute to hypoactivity because this phenotype is seen in RGS9LCre:PGC-1αfl/fl mice. Although we did not detect any changes in striatal PGC-1α expression in the HDQ allelic series, including aged HDQ mice, it is possible that decreases in PGC-1α occur in select neuronal subsets. Because MSNs of the indirect pathway are more vulnerable relative to those of the direct pathway in HD (Galvan et al., 2012), it is attractive to speculate that MSNs of the indirect pathway are more vulnerable to a loss of PGC-1α. Cre-recombinase expression under the RGS9L promoter affects MSNs of both the direct and indirect pathways. Further investigation into the reliance of direct and indirect pathway MSNs on PGC-1α and how inhibition of postsynaptic targets is influenced by these neurons is needed.
Although markers of MSNs remain unaffected by a loss of PGC-1α, several previously identified PGC-1α-dependent cortical transcripts (Lucas et al., 2014a) were reduced in the conditional knock-out striatum, including Cplx1 and Nefh. Several studies have indicated that Cplx1 is important in synchronous neurotransmitter release (Martin et al., 2011; Lin et al., 2013; Lucas et al., 2014a; Chang et al., 2015); from these data, we hypothesize that synchronous release of GABA from MSNs to the globus pallidus and/or substantia nigra pars reticulata is impaired. Further, due to a reduction in the axonal stabilizing protein Nefh and previously published data indicating reduced neuritic branching of PGC-1α-null striatal cultures (Lin et al., 2004), it is possible that axonal projections to these downstream regions are compromised and synapse number may be reduced. Further studies to investigate MSN morphology and synapse number in these downstream brain regions are warranted.
As a transcriptional coactivator, PGC-1α does not bind directly to DNA, but rather requires the presence of other factors for regulating gene expression, raising the possibility that gene expression changes in the HDQ mice occur independently of changes in PGC-1α. Although not shown, we identified via WebGestalt an enrichment of specificity protein 1 (SP1)-binding sites in the promoter regions of genes downregulated in the PGC-1α-null striatum. Studies have demonstrated that mHtt is able to suppress SP1-dependent transcription by preventing it from binding to DNA in postmortem tissue of presymptomatic and symptomatic HD patients and in in vitro and in vivo HD models (Dunah et al., 2002; Li et al., 2002; Chen-Plotkin et al., 2006).
PGC-1α is notably a master regulator of metabolic and mitochondrial function, so the link between polymorphisms in the PGC-1α-interacting transcription factor nuclear respiratory factor 1 (NRF-1) and HD is also of interest. Several metabolic transcripts (Got2, Mrps31, Ak1, Idh3a, and Phyh) are reduced in the conditional knock-out striatum. Metabolic function may be reduced in MSNs of the conditional knock-out line due to a downregulation in these transcripts collectively. We find that a number of PGC-1α-dependent genes are upregulated in homozygous (6 month) and heterozygous (17 month) knock-in mice with increasing repeat length, some of which are metabolic (Ak1, Idh3a, and Phyh). Interestingly, the overexpression of PGC-1α can prevent the aggregation of mHtt in mouse models (Tsunemi et al., 2012) and we and others have shown that PGC-1α overexpression can induce a large number of genes involved in mitochondrial function and antioxidant defense (Cowell et al., 2009; Wareski et al., 2009; Ciron et al., 2012; Mudò et al., 2012; Lucas et al., 2014a), calcium buffering (Lucas et al., 2014a), and synaptic plasticity (Cheng et al., 2012; Lucas et al., 2014a). It is possible that providing high levels of exogenous PGC-1α or increased expression of its interacting factors is capable of driving different subsets of genes than endogenous programs. It is also possible that upregulation of PGC-1α-interacting transcription factors to elevate transcript levels in the HDQ lines here may be pathogenic. In fact, in light of the neuronal toxicity observed in some Parkinson's disease (PD) (Ciron et al., 2012; Clark et al., 2012) and Alzheimer's disease (Dumont et al., 2014) models, caution is warranted with long-term overexpression of PGC-1α and its interacting transcription factors for general use. Future experiments are required to better elucidate the cell-specific transcriptional response to disease to design strategies for neuroprotection.
Because PGC-1α polymorphisms influence the age of onset of HD, so too do they influence the onset of PD (Clark et al., 2011), a neurodegenerative disease marked by dyskinesia. PGC-1α dysfunction is linked to several PD candidate genes, including Parkin/Park2, Pink1, and DJ-1 (Dauer and Przedborski, 2003; Zhong and Xu, 2008; Shin et al., 2011; Choi et al., 2013). PARIS, a substrate for Parkin, represses PGC-1α expression in the striatum of PD mouse models and human patients (Shin et al., 2011). PARIS acts similarly on NRF-1 (Shin et al., 2011) to affect transcription. It would be interesting to test these same PGC-1α-dependent transcripts in the striatum or substantia nigra of these PD models. In addition, polymorphisms in NRF-1 itself are able to influence age of onset in HD (Taherzadeh-Fard et al., 2011). Our previous studies have shown that NRF-1 is a potential mediator of PGC-1α-dependent regulation of both Nefh and Cplx1 in the cortex (McMeekin et al., 2016) and enrichment of NRF-1 binding sites has been reported in the promoter region of Nefh by CHIP-seq (Satoh et al., 2013). Further investigation into the overlap in SP1 and NRF-1 with PGC-1α-dependent transcription in MSNs and in age-related movement disorders will shed light on gene programs affected in these disease states and potential ways to improve motor function.
Footnotes
This work was supported by the National Institute of Mental Health (Grant 5K01MH077955-05) and the National Institute of Neurological Disorders and Stroke (Grants 5R01NS070009-05 and R01 NS101958-02) at the National Institutes of Health and by the Tourette Syndrome Association. Behavioral studies were performed with equipment and training provided by the Evelyn F. McKnight University of Alabama–Birmingham (UAB) Neurobiology Behavior Core and the UAB Neuroscience Behavioral Assessment Core (NIH Grant P30NS047466). We thank the Center for Neurodegeneration and Experimental Therapeutics for access to the confocal microscope.
The authors declare no competing financial interests.
References
- Bartley AF, Lucas EK, Brady LJ, Li Q, Hablitz JJ, Cowell RM, Dobrunz LE (2015) Interneuron transcriptional dysregulation causes frequency-dependent alterations in the balance of inhibition and excitation in hippocampus. J Neurosci 35:15276–15290. 10.1523/JNEUROSCI.1834-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Reim K, Pedersen M, Neher E, Brose N, Taschenberger H (2015) Complexin stabilizes newly primed synaptic vesicles and prevents their premature fusion at the mouse calyx of held synapse. J Neurosci 35:8272–8290. 10.1523/JNEUROSCI.4841-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Calingasan NY, Yang L, Hennessey T, Johri A, Beal MF (2010) Impairment of PGC-1alpha expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington's disease following chronic energy deprivation. Hum Mol Genet 19:3190–3205. 10.1093/hmg/ddq229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, Mattson MP (2012) Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat Commun 3:1250. 10.1038/ncomms2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Sadri-Vakili G, Yohrling GJ, Braveman MW, Benn CL, Glajch KE, DiRocco DP, Farrell LA, Krainc D, Gines S, MacDonald ME, Cha JH (2006) Decreased association of the transcription factor Sp1 with genes downregulated in Huntington's disease. Neurobiol Dis 22:233–241. 10.1016/j.nbd.2005.11.001 [DOI] [PubMed] [Google Scholar]
- Chinsomboon J, Ruas J, Gupta RK, Thom R, Shoag J, Rowe GC, Sawada N, Raghuram S, Arany Z (2009) The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc Natl Acad Sci U S A 106:21401–21406. 10.1073/pnas.0909131106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Batchu VV, Schubert M, Castellani RJ, Russell JW (2013) A novel PGC-1α isoform in brain localizes to mitochondria and associates with PINK1 and VDAC. Biochem Biophys Res Commun 435:671–677. 10.1016/j.bbrc.2013.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciron C, Lengacher S, Dusonchet J, Aebischer P, Schneider BL (2012) Sustained expression of PGC-1α in the rat nigrostriatal system selectively impairs dopaminergic function. Hum Mol Genet 21:1861–1876. 10.1093/hmg/ddr618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Reddy S, Zheng K, Betensky RA, Simon DK (2011) Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson's disease. BMC Med Genet 12:69. 10.1186/1471-2350-12-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Silvaggi JM, Kiselak T, Zheng K, Clore EL, Dai Y, Bass CE, Simon DK (2012) Pgc-1α overexpression downregulates Pitx3 and increases susceptibility to MPTP toxicity associated with decreased bdnf. PLoS One 7:e48925. 10.1371/journal.pone.0048925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell RM, Blake KR, Russell JW (2007) Localization of the transcriptional coactivator PGC-1alpha to GABAergic neurons during maturation of the rat brain. J Comp Neurol 502:1–18. 10.1002/cne.21211 [DOI] [PubMed] [Google Scholar]
- Cowell RM, Talati P, Blake KR, Meador-Woodruff JH, Russell JW (2009) Identification of novel targets for PGC-1alpha and histone deacetylase inhibitors in neuroblastoma cells. Biochem Biophys Res Commun 379:578–582. 10.1016/j.bbrc.2008.12.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D (2006) Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127:59–69. 10.1016/j.cell.2006.09.015 [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y (2006) Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci U S A 103:15254–15259. 10.1073/pnas.0601758103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39:889–909. 10.1016/S0896-6273(03)00568-3 [DOI] [PubMed] [Google Scholar]
- Dougherty SE, Bartley AF, Lucas EK, Hablitz JJ, Dobrunz LE, Cowell RM (2014) Mice lacking the transcriptional coactivator PGC-1α exhibit alterations in inhibitory synaptic transmission in the motor cortex. Neuroscience 271:137–148. 10.1016/j.neuroscience.2014.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont M, Stack C, Elipenahli C, Jainuddin S, Launay N, Gerges M, Starkova N, Starkov AA, Calingasan NY, Tampellini D, Pujol A, Beal MF (2014) PGC-1α overexpression exacerbates β-amyloid and tau deposition in a transgenic mouse model of Alzheimer's disease. FASEB J 28:1745–1755. 10.1096/fj.13-236331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D (2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296:2238–2243. 10.1126/science.1072613 [DOI] [PubMed] [Google Scholar]
- Ehrlich ME. (2012) Huntington's disease and the striatal medium spiny neuron: cell-autonomous and non-cell-autonomous mechanisms of disease. Neurotherapeutics 9:270–284. 10.1007/s13311-012-0112-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan L, André VM, Wang EA, Cepeda C, Levine MS (2012) Functional differences between direct and indirect striatal output pathways in Huntington's disease. J Huntingtons Dis 1:17–25. 10.3233/JHD-2012-120009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng MY, Tallaksen-Greene SJ, Detloff PJ, Albin RL (2007) Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington's disease. J Neurosci 27:8989–8998. 10.1523/JNEUROSCI.1830-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Zhang J, Tallaksen-Greene S, Crowley MR, Crossman DK, Morton AJ, Van Groen T, Kadish I, Albin RL, Lesort M, Detloff PJ (2016) Allelic series of Huntington's disease knock-in mice reveals expression discorrelates. Hum Mol Genet 25:1619–1636. 10.1093/hmg/ddw040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP (2005) PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3:e101. 10.1371/journal.pbio.0030101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li X-J (2002) Interaction of huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 22:1277–1287. 10.1128/MCB.22.5.1277-1287.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, et al. (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119:121–135. 10.1016/j.cell.2004.09.013 [DOI] [PubMed] [Google Scholar]
- Lin MY, Rohan JG, Cai H, Reim K, Ko CP, Chow RH (2013) Complexin facilitates exocytosis and synchronizes vesicle release in two secretory model systems. J Physiol 591:2463–2473. 10.1113/jphysiol.2012.244517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Markwardt SJ, Gupta S, Meador-Woodruff JH, Lin JD, Overstreet-Wadiche L, Cowell RM (2010) Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1alpha. J Neurosci 30:7227–7235. 10.1523/JNEUROSCI.0698-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Dougherty SE, McMeekin LJ, Trinh AT, Reid CS, Cowell RM (2012) Developmental alterations in motor coordination and medium spiny neuron markers in mice lacking pgc-1α. PLoS One 7:e42878. 10.1371/journal.pone.0042878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Dougherty SE, McMeekin LJ, Reid CS, Dobrunz LE, West AB, Hablitz JJ, Cowell RM (2014a) PGC-1α provides a transcriptional framework for synchronous neurotransmitter release from parvalbumin-positive interneurons. J Neurosci 34:14375–14387. 10.1523/JNEUROSCI.1222-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Reid CS, McMeekin LJ, Dougherty SE, Floyd CL, Cowell RM (2014b) Cerebellar transcriptional alterations with purkinje cell dysfunction and loss in mice lacking PGC-1α. Front Cell Neurosci 8:441. 10.3389/fncel.2014.00441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Li S, Lucas EK, Cowell RM, Lin JD (2010) Neuronal inactivation of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) protects mice from diet-induced obesity and leads to degenerative lesions. J Biol Chem 285:39087–39095. 10.1074/jbc.M110.151688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS (2011) Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol 21:97–105. 10.1016/j.cub.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMeekin LJ, Lucas EK, Meador-Woodruff JH, McCullumsmith RE, Hendrickson RC, Gamble KL, Cowell RM (2016) Cortical PGC-1α-dependent transcripts are reduced in postmortem tissue from patients with schizophrenia. Schizophr Bull 42:1009–1017. 10.1093/schbul/sbv184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB. (2005) Knock-in mouse models of Huntington's disease. NeuroRx 2:465–470. 10.1602/neurorx.2.3.465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudò G, Mäkelä J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Mälkiä A, Bonomo A, Kairisalo M, Aguirre JA, Korhonen L, Belluardo N, Lindholm D (2012) Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Sci 69:1153–1165. 10.1007/s00018-011-0850-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR, Lipton SA (2009) Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 15:1407–1413. 10.1038/nm.2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman Z, Gold SJ, Potenza MN, Cowan CW, Ni YG, He W, Wensel TG, Nestler EJ (1999) Cloning and characterization of RGS9–2: a striatal-enriched alternatively spliced product of the RGS9 gene. J Neurosci 19:2016–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos EM, et al. (2012) Population stratification may bias analysis of PGC-1α as a modifier of age at huntington disease motor onset. Hum Genet 131:1833–1840. 10.1007/s00439-012-1205-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rising AC, Xu J, Carlson A, Napoli VV, Denovan-Wright EM, Mandel RJ (2011) Longitudinal behavioral, cross-sectional transcriptional and histopathological characterization of a knock-in mouse model of Huntington's disease with 140 CAG repeats. Exp Neurol 228:173–182. 10.1016/j.expneurol.2010.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe GC, El-Khoury R, Patten IS, Rustin P, Arany Z (2012) PGC-1α is dispensable for exercise-induced mitochondrial biogenesis in skeletal muscle. PLoS One 7:e41817. 10.1371/journal.pone.0041817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J, Kawana N, Yamamoto Y (2013) Pathway analysis of ChIP-seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul Syst Bio 7:139–152. 10.4137/GRSB.S13204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM (2011) PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell 144:689–702. 10.1016/j.cell.2011.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalardy L, Zadori D, Plangar I, Vecsei L, Weydt P, Ludolph AC, Klivenyi P, Kovacs GG (2013) Neuropathology of partial PGC-1α deficiency recapitulates features of mitochondrial encephalopathies but not of neurodegenerative diseases. Neurodegener Dis 12:177–188. 10.1159/000346267 [DOI] [PubMed] [Google Scholar]
- Taherzadeh-Fard E, Saft C, Andrich J, Wieczorek S, Arning L (2009) PGC-1alpha as modifier of onset age in huntington disease. Mol Neurodegener 4:10. 10.1186/1750-1326-4-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taherzadeh-Fard E, Saft C, Akkad DA, Wieczorek S, Haghikia A, Chan A, Epplen JT, Arning L (2011) PGC-1alpha downstream transcription factors NRF-1 and TFAM are genetic modifiers of huntington disease. Mol Neurodegener 6:32. 10.1186/1750-1326-6-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Matias S, Dugue GP, Mainen ZF, Costa RM (2014) Balanced activity in basal ganglia projection pathways is critical for contraversive movements. Nat Commun 5:4315. 10.1038/ncomms5315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom R, Rowe GC, Jang C, Safdar A, Arany Z (2014) Hypoxic induction of vascular endothelial growth factor (VEGF) and angiogenesis in muscle by truncated peroxisome proliferator-activated receptor γ coactivator (PGC)-1α. J Biol Chem 289:8810–8817. 10.1074/jbc.M114.554394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Török R, Kónya JA, Zádori D, Veres G, Szalárdy L, Vécsei L, Klivényi P (2015) mRNA expression levels of PGC-1α in a transgenic and a toxin model of Huntington's disease. Cell Mol Neurobiol 35:293–301. 10.1007/s10571-014-0124-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR (2012) PGC-1α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med 4:142ra97. 10.1126/scitranslmed.3003799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareski P, Vaarmann A, Choubey V, Safiulina D, Liiv J, Kuum M, Kaasik A (2009) PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J Biol Chem 284:21379–21385. 10.1074/jbc.M109.018911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR (2006) Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab 4:349–362. 10.1016/j.cmet.2006.10.004 [DOI] [PubMed] [Google Scholar]
- Weydt P, Soyal SM, Gellera C, Didonato S, Weidinger C, Oberkofler H, Landwehrmeyer GB, Patsch W (2009) The gene coding for PGC-1alpha modifies age at onset in Huntington's disease. Mol Neurodegener 4:3. 10.1186/1750-1326-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Soyal SM, Landwehrmeyer GB, Patsch W; European Huntington Disease Network (2014) A single nucleotide polymorphism in the coding region of PGC-1α is a male-specific modifier of huntington disease age-at-onset in a large european cohort. BMC Neurol 14:1. 10.1186/1471-2377-14-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Xu J (2008) Synergistic activation of the human MnSOD promoter by DJ-1 and PGC-1alpha: regulation by SUMOylation and oxidation. Hum Mol Genet 17:3357–3367. 10.1093/hmg/ddn230 [DOI] [PubMed] [Google Scholar]