

Abstract
Inositol 1,4,5-trisphosphate 3-kinase A (IP3K-A) regulates the level of the inositol polyphosphates, inositol trisphosphate (IP3) and inositol tetrakisphosphate to modulate cellular signaling and intracellular calcium homeostasis in the central nervous system. IP3K-A binds to F-actin in an activity-dependent manner and accumulates in dendritic spines, where it is involved in the regulation of synaptic plasticity. IP3K-A knockout mice exhibit deficits in some forms of hippocampus-dependent learning and synaptic plasticity, such as long-term potentiation in the dentate gyrus synapses of the hippocampus. In the present study, to further elucidate the role of IP3K-A in the brain, we developed a transgenic (Tg) mouse line in which IP3K-A is conditionally overexpressed approximately 3-fold in the excitatory neurons of forebrain regions, including the hippocampus. The Tg mice showed an increase in both presynaptic release probability of evoked responses, along with bigger synaptic vesicle pools, and miniature excitatory postsynaptic current amplitude, although the spine density or the expression levels of the postsynaptic density-related proteins NR2B, synaptotagmin 1, and PSD-95 were not affected. Hippocampal-dependent learning and memory tasks, including novel object recognition and radial arm maze tasks, were partially impaired in Tg mice. Furthermore, (R,S)-3,5-dihydroxyphenylglycine-induced metabotropic glutamate receptor long-term depression was inhibited in Tg mice and this inhibition was dependent on protein kinase C but not on the IP3 receptor. Long-term potentiation and depression dependent on N-methyl-d-aspartate receptor were marginally affected in Tg mice. In summary, this study shows that overexpressed IP3K-A plays a role in some forms of hippocampus-dependent learning and memory tasks as well as in synaptic transmission and plasticity by regulating both presynaptic and postsynaptic functions.
Introduction
Inositol 1,4,5-trisphosphate 3-kinase A (IP3K-A) has emerged as an important molecule for synaptic plasticity owing to its abilities to convert inositol trisphosphate (IP3) to inositol tetrakisphosphate (IP4) as well as to bind to F-actin and microtubules [1–6]. Although IP3K-A is thought to be an intracellular calcium (Ca2+) signaling regulator through the IP3 receptor (IP3R) pathway [1, 2], IP3K-A deletion in knockout (KO) mice does not change the net content of IP3 [7] but results in an increase in IP3 turnover in hippocampal synaptosomes [8]. The increased IP3 metabolism in the absence of IP3K-A appears to be due to improved inositol polyphosphate 5-phosphatase activity, which maintains the basal IP3-mediated Ca2+ release in the endoplasmic reticulum (ER) [8].
Previous studies showed that IP3K-A migrate toward F-actin-enriched dendritic spines via the N-terminal actin-binding domain [5] and that IP3K-A binds directly to activated Rac1 to modulate actin dynamics in dendritic spines [3]. Thus, overexpression of IP3K-A in cultured hippocampal neurons promotes dendritic spine formation through Rac1 interaction and actin remodeling. The N-terminus of IP3K-A can further interact with dendritic microtubules in an activity-dependent manner, and this interaction is inhibited by protein kinase A-dependent phosphorylation of Ser-119 within IP3K-A [4]. The IP3K-A kinase activity, however, is not essential for either the actin-binding [5] or spine-forming activities of IP3K-A [3]. Thus, IP3K-A exerts many physiological effects, including kinase activity-dependent Ca2+ homeostasis and kinase activity-independent neuronal cytoskeletal remodeling.
One way IP3K-A modulates neuronal function is by changing expression following neuronal activity. Hippocampal learning, such as water maze training, significantly enhances the expression of IP3K-A in the stratum radiatum of the CA1 and the molecular layer of the inferior blade of the dentate gyrus (DG) [9]. However, prolonged neuronal excitation by kainic acid treatment depletes the expression of IP3K-A in cerebral cortical neurons [10]. These data suggest that the expression of IP3K-A is regulated by neuronal activity in a cell type-dependent manner. Moreover, changes in IP3K-A expression levels affect some synaptic and cognitive functions. For example, IP3K-A KO mice show a loss of long-term potentiation (LTP) in granule cell synapses of the DG and a deficit in hippocampus-dependent learning tasks, such as novel object recognition [3], whereas CA1 pyramidal neuron synapses show an increase in LTP [7]. Recently, we demonstrated that IP3K-A KO mice also have a high level of innate fear and anxiety to aversive stimuli, suggesting a role for amygdala IP3K-A in associating environment with emotion [11].
In the present study, to further clarify the activity-dependent role of IP3K-A in brain function, we used a gain of function approach, generating a mouse line that conditionally overexpresses IP3K-A in the forebrain region of the mouse brain. Thus, we developed a tetracycline-inducible transgenic (Tg) mouse line that enables the transient overexpression of IP3K-A only in the excitatory neurons of the forebrain. Using these Tg mice, we demonstrated here that the inducible overexpression of IP3K-A in vivo impaired the performance of spatial memory tasks and blocked metabotropic glutamate receptor (mGluR)-dependent long-term depression (LTD) expression of CA1 pyramidal neuron synapses. The properties of basal synaptic transmission and synaptic plasticity were changed presynaptically as well as postsynaptically, revealing for the first time a presynaptic role of IP3K-A in vivo.
Materials and methods
Generation and maintenance of transgenic mouse
To generate the transgenic construct for TRE- IP3K-A Tg mouse, mouse IP3K-A cDNA was obtained by PCR amplification with plasmid templates using primers containing XbaI site at both end of primers. Forward primer: 5’-CAC CAT CTA GAA CCA TGA CCC TGC CCG GGC GCC C-3’; reverse primer: 5’-AAA AGT CTA GAG CCT CAT CTC TCA GCC AG-3’. PCR fragment was cloned into the XbaI site of placZ-GFP.gcg vector and the lacZ fragment was replaced with IP3K-A cDNA. The final 5.6Kb construct fragment was excised from the vector with AseI digestion, isolated from the vector backbone, purified by electro-elution, and microinjected into pronuclei of fertilized mouse eggs. Analysis of founder mice for transgene was performed by Southern blot and PCR using tail biopsies. To generate double transgenic mouse with CaMKIIα-rtTA2 and TRE-IP3K-A, TRE-IP3K-A Tg mice were crossed with CaMKIIα-rtTA2 mice (provided by Isabella M. Mansuy, [12]). Tail DNA was used for PCR genotyping with the following primers. Forward primer: 5’-TGG AAG GCG AGT CAT GGC AC-3’ and reverse primer: 5’-TCG TCA AGA GCG TCA GCA GGC AG-3’ for CaMKIIα-rtTA2 Tg mice. Forward primer: 5’-GCA GCT CGG AGC CTG AAC ACT ACT G-3’ and reverse primer: 5’-TCG TCA AGA GCG TCA GCA GGC AG-3’ for TRE- IP3K-A Tg mice. PCR mixtures was heated for 3 min at 94°C, followed by 35 cycles of 94°C for 30 sec, 56°C for 30 sec, and 72°C for 30 sec with a final extension of 72°C for 5 min. The PCR product of the TRE-IP3K-A Tg mouse was 733 bp from the transgene and 1446 bp from the mouse genome DNA that could be used for PCR internal control. To induce transgene expression, mice were dosed with 6 mg Doxycycline (Dox)/g of food. Dox-containing foods were prepared daily by mixing Dox (Sigma-Aldrich) with paste food (powdered food in sterile water, 50/50, w/v, in 2% sucrose). As WT mice, all three types of CaMKII-rtTA(−)/TRE-IP3K-A(−), -rtTA(+)/TRE-IP3K-A(−), and–rtTA(−)/TRE-IP3K-A(+) mice were used and CaMKII-rtTA(+)/TRE-IP3K-A(+) was used as Tg mice. They have all the same genetic background of c57BL/6 and all fed Dox-containing food. This study was approved by Institutional Animal Care and Use Committee of Korea Institute of Science & Technology (IACUC of KIST, 2017–004) and Korea University Institutional Animal Care and Use Committee (KUIACUC, 2016-0079-C1), and performed in accordance with the guidelines of Korea University.
Protein isolation and Western blot analysis
Mouse tissues were isolated and homogenized in ice-cold RIPA buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 1% β-mercaptoethanol) containing protease and phosphatase inhibitor cocktail (Roche Life Science). The solution was incubated on ice for 30 min, centrifuged at 15,000 × g for 10 min at 4°C and the supernatant was collected. Protein concentration was determined using the Bradford method (Bio-Rad). Bovine serum albumin was used as standard. Proteins were separated by electrophoresis on an 8–12% SDS-PAGE gel, transferred to a Nitrocellulose membrane (Whatman). Nonspecific binding was blocked by soaking in TBS-T buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.1% tween20) containing 5% skim milk for overnight at 4°C. Membranes were incubated with primary antibody for 3–6 h at room temperature (RT). After washing three times with TBS buffer, the membranes were incubated with donkey anti-mouse IgG antibody conjugated to HRP (Jackson ImmunoResearch) in TBS-T buffer containing 5% skim milk for 1 h at RT. The protein bands were visualized by using Western blot detection reagents (Thermo Scientific) and AGFA X-ray films. Quantification of band intensity was achieved by normalizing the protein level of each sample to the level of GAPDH (Santa Cruz Biotechnology).
Subcellular fractionation of hippocampal homogenates
Biochemical fractionation was performed according to the standard methods [13]). Hippocampal tissues were homogenized in ice-cold TEVP buffer (10 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 1 mM EGTA) containing 0.32 M sucrose and protease and phosphatase inhibitor cocktails (Roche Life Science). All processes were performed at 4°C or on-ice. The homogenate was centrifuged at 800 × g for 10 min to remove nuclei and large debris (P1). The supernatant (S1) was centrifuged at 9,200 × g for 15 min to obtain a crude synaptosomal fraction (P2) and a soluble cytosolic fraction (S2). P2 fraction was solubilized and centrifuged at 25,000 × g for 20 min to produce synaptosomal membrane (LP1) and synaptosomal cytosolic (LS1) fractions. LS1 fraction was centrifuged at 165,000 × g for 2 h to obtain synaptic vesicle-enriched fraction (LP2). An equal amount of fractionated protein was used for Western blot. SDS-PAGE was performed as described above.
Immunohistochemistry
Mice were anesthetized with urethane and perfused transcardially with TBS (pH 7.4) followed by 4% paraformaldehyde (PFA) in TBS. Brains were isolated, post-fixed in 4% PFA/TBS overnight at 4°C, and cryopreserved in 30% sucrose (w/v)/TBS solution at 4°C until brain subsided. Brains were embedded into optimal cutting temperature (OCT) compounds and frozen with dry-ice. The sections were made into a 40 μm thick cryostat and incubated at RT in TBS + 0.1% Triton X-100 containing 5% normal goat serum (VECTOR Laboratories, CA). The sections were then incubated with primary antibodies diluted in TBS + 0.1% Triton X-100 overnight at 4°C, washed with TBS 3 × 10min and then incubated with goat anti-mouse IgG secondary antibodies conjugated with Cy3 (Jackson ImmunoResearch, PA) for 45 min at RT. The sections were mounted on a slide glass with CRYSTAL (Biomeda, CA) and photographed with an LSM510 confocal microscope (Zeiss, Germany). The control sections were created by omitting the primary antibody without the fluorescent signal. Primary antibodies such as mouse anti-IP3K-A (1:1000, [3]) and rabbit anti-PSD95 (1:500, Zymed) were used.
Golgi staining
2-month-old mice fed Dox-containing food for 2 weeks were used for Golgi staining. Littermate mice were sacrificed by deep anesthesia and transcardial perfusion with 4% PFA. The brains were post-fixed in 4% paraformaldehyde for 24 h and then stained with a rapid Golgi protocol. Briefly, the blocks containing DG were rinsed with 0.1 N sodium cacodylate buffer and stored in 2.25% potassium dichromate and 0.4% osmium tetroxide solution for 4 days in the dark at 20°C. The blocks were then transferred to a 0.75% silver nitrate solution and maintained in a dark room at 20°C for 3 days. After the process for electron microscopy (dehydration through a graded series of ethanol and then propylene oxide, followed by embedding in Epon/Araldite), coronal sections (80 μm thick) were cut from the embedded samples using a sliding microtome. The sections were re-embedded on glass slides and allowed to cure. Fully impregnated dentate granule cells and CA1 pyramidal cells were randomly selected in each animal and observed under a light microscope (AxioSkop; Zeiss).
EM analysis
Mice were anesthetized deeply with sodium-pentobarbital and transcardially perfused with normal saline before 2% PFA, and 2.5% glutaraldehyde in 0.1M phosphate buffer (pH 7.4). The pre-fixed brain was removed and stored in the same fixative overnight at 4°C. Hippocampus were dissected from pre-fixed brain and then washed with the same buffer several times. Tissues were post-fixed with 2% osmium tetroxide for 2 h, dehydrated through an ascending ethanol series and embedded with Epon mixture. Polymerized blocks were fine trimmed into the hippocampal region of the blocks and sections were cut to 70 nm thickness using ultramicrotome. Electron micrographs containing synapses and synaptic vesicles randomly selected in the region of interest at hippocampus were taken under transmission electron microscope at the accelerating voltage of 75 kV (Hitachi H-7650).
Hippocampal slice preparation and electrophysiological recordings
From 10 days after the offspring was born, paste food containing Dox was given to the mother, and the transfer of Dox to the offspring was confirmed by Western blot method (S1 Fig). This administration continued until the day of the electrophysiological experiment. Hippocampal slices (300 zμm thick, male mice 3–5 weeks old) were prepared from both sides of male mouse brain. In brief, hippocampal containing region was dissected out using cold oxygenated (95% O2/5% CO2) artificial CSF (aCSF): 124 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4H2O, 11 mM Glucose, 0.5 mM CaCl2H2O, 5 mM MgCl2, pH 7.3. The slices were kept on the surface of the cell culture insert in an incubation chamber provided with humidified oxygen for continuous storage, and at least one hour after dissection, one or two slices were transferred to the recording chamber. Recording aCSF has 2.5 mM Ca2+ and 1.3 mM Mg2+, and the other components are the same as those of the dissecting aCSF. For field excitatory postsynaptic potential (fEPSP) measurement, single bipolar metal electrode (FHC, Bowdoinham, ME) was placed on one side of the CA1 dendritic region and the glass recording electrode filled with aCSF was positioned on the other side of 300–500 μm. Stimulating currents were delivered from the constant current isostimulator (SC-100, WECO) every 30 sec. To measure input-output synaptic responses, seven different stimulus strength of currents (10 to 190 pA) were applied twice and the slope of fEPSP were calculated. During this step, about 40–50% of the maximum stimulus current intensity, which showed the population spike response of fEPSP, was chosen as the sub-threshold stimulus for both basal and LFS or theta-burst stimulus protocol. LFS for LTD was given 900 pulses at 1Hz and single theta-burst protocol was composed of 10 bursts at 4 Hz where each burst consists of 5 pulses at 100 Hz. In case of four theta-burst stimulations for strong LTP, each burst was repeated at 5 min interval. fEPSP was collected with Dagan amplifier (EX-1), filtered at 2 kHz, and stored on a PC hard disk.
The miniature excitatory postsynaptic current (mEPSC) was obtained by whole-cell recording. For whole-cell recordings, healthy pyramidal neurons were selected from the CA1 pyramidal cell body layer by a 40-fold objective infrared differential interference contrast (IR-DIC) microscope (Olympus BX51W1). For α-amino-3-hydroxy-5-methylisoxazolepropionate receptor (AMPAR)-mediated mEPSC measurement, 1 μM tetrodotoxin (TTX), 100 μM DL-2-amino-5-phosphonovaleric acid (APV), 100 μM picrotoxin, and 10 μM bicuculline were added in aCSF. For mEPSC, intracellular solution has the following components: 130 mM Cs-methanesulfonate, 20 mM CsCl, 10 mM HEPES, 2 mM MgATP, 0.3 mM Na3GTP, 0.2 mM EGTA, pH 7.3 (about 300 mOsm). The cell was held at -68mV and recording was performed at 30°C. Traces of 500 ms were collected at 3 sec intervals, filtered at 2 kHz, and amplified with Axopatch-1D patch clamp (Axon Instruments, USA). Cells with a series resistance greater than 15 MΩ were discarded. Most of the chemicals were purchased from Sigma and DHPG, TTX, Rö-31-8220 mesylate, calphostin C, 2-APB, KN62, KN93, and KN92 were from Tocris.
All data acquisition and analysis, except for miniature current, were performed with custom software written in Axobasic 3.1 (Axon Instruments). Representing values are expressed as Mean ± SEM. Miniature currents were analyzed using Mini Analaysis Program (v. 5.6.7, Synaptosoft Inc).
Behavioral tests
Behavioral experiments were initiated after 2 weeks of Dox-containing food (6 mg/g of food) in mice aged 2 to 4 months, and Dox administration continued until the end of the experiments.
Open field test
A square black box (40 wide × 40 deep × 27 cm high) was placed in the center of a well-lit room and surrounded by black curtains. Background noise (60 dB) was provided by the white noise generator. Animal activity was measured by an automatic video tracking system (SmarTrack®; Smartech, Madison, WI). Animals were transferred to the experimental room 30 min before the start of test. The mouse was placed in the open field box and its locomotor activity (total distance) was measured with an automatic tracking system for 10 min. Spatial characteristics (percentage of time spent in the center or marginal area of open field box) were also measured.
Object recognition test
Two objects not larger than animals and immovable by animals were placed 10 cm from the edge of square black box (40 wide × 40 deep × 27 cm high). After fully handled, the mice were exposed to test arena for 10 min one day before the object recognition sample test was performed. The object recognition test consists of two types of trials: sample trial (first day, lasting 10 min) and retention trial (second and fourth day, lasting 5 min). In the sample trial, two identical objects (A and A) were placed in the box and the mouse was placed in the center of the test arena. The total time spent exploring both objects was measured. In the retention trial after 24 h from the sample trial, one of the two identical objects in the sample trial was changed to new object (A and B) and the time spent exploring objects was measured. In the retention trial after 72 h from the sample trial, the changed object in the 24 h retention trial was changed to another object (A and C). The preference, which is the ratio of exploring time to new and old objects, was used as a measure.
Radial arm maze test
To test spatial working memory, the radial maze test was conducted as described previously, with slight modifications [14]. The floor of the maze was made of white Plexiglas, and the wall (17 cm high) was consisted of transparent Plexiglas. Each arm (6 × 28 cm) radiated from a circular central starting platform (20 cm in diameter). A small well (1 cm deep and 1cm in diameter) containing 0.2 ml of 10% sucrose solution was placed at the distal end of each arm as rewards. The maze was elevated 60 cm above the floor and placed in a room with a number of extramaze cues, including the experimenter. Three days before pretraining, animals were habituated to a restricted water supply and fed with 10% sucrose solution for one hour every day. Pretraining was composed of three days. On the first pretraining day, each mouse was exposed to the central starting platform and allowed to explore and to habituate to the maze. On the second pretraining day, the mouse was placed in the central platform and allowed to consume rewards scattered over the arms for 5 min. On the third pretraining day, the mouse was placed at the distal end of each arm and allowed to obtain the reward from each well. A trial was finished after the subject approached to the bait and returned to the central platform, irrespective of its consuming the solution. This trial was conducted in all eight arms for each mouse. After three days of pretraining, 14 days of maze acquisition begun. During the acquisition session, all eight arms were baited with sucrose solution. The mouse was placed on the central platform and allowed to obtain all eight rewards within 5 min. A session was terminated immediately after all eight rewards had been consumed or if 5 min had elapsed, whichever comes first. An arm choice was defined as traveling more than 10 cm from the central platform. The session was conducted once a day and data were presented as blocks of two training sessions. Spatial working memory errors were measured by the number of visits to the same arm more than once, where the reward has already been obtained = number of revisits). For each training session, the error rate was calculated from the number of revisits divided by the total number of visits to any of the arms.
Statistical analysis
Statistical analyses were conducted using SPSS v. 12 for Windows (IBM corp., Amonk, NY, USA). For data comparing two groups and electrophysiology data, unpaired two-tailed Student’s t-test was performed, otherwise stated. Experiments with repeatedly measured data such as behavior tests and PPF ratio were analyzed by two-way repeated measures ANOVA (SPSS) with appropriate post hoc tests. α = 0.05 was used to show significance (asterisk) at P < 0.05. Values are expressed as Mean ± SEM.
Results
Generation of IP3K-A overexpressing Tg mice
The Tg mice were generated using the Tet-On system and reverse tetracycline-controlled transactivator (rtTA) protein was expressed under the control of Ca2+/calmodulin-dependent protein kinase II alpha (CaMKIIα) promoter activation [12]. The CaMKIIα promoter restricted rtTA protein expression to the forebrain, and the semisynthetic tetracycline, Dox, bound to rtTA and induced transcriptional activation of the IP3K-A gene containing the tetracycline responsive element (Fig 1A). The IP3K-A expression in the hippocampus was analyzed at Dox concentrations of 0, 2, 6 mg per gram of food. The IP3K-A expression was consistently higher in Tg mice fed Dox at 6 mg/g of food than in mice fed Dox at 2 mg/g of food (S2 Fig). The expression of IP3K-A in the hippocampus was increased approximately 3-fold, peaking 14 days after the mice were provided with chow containing Dox. The net induction of the IP3K-A protein was higher in the hippocampal CA3 region than in the CA1 region (Fig 1B and 1C). When mice were fed Dox-free chow, IP3K-A expression decreased slowly, returning to expression levels similar to those in wild-type (WT) mice at 6–7 weeks (S3 Fig). Enhanced expression of IP3K-A was detected in various regions of the Tg mouse brain, including the olfactory bulb, cortex, hippocampus, and striatum, but not in the cerebellum (Fig 1D). These results indicated that an IP3K-A Tg mouse line with reversible induction of IP3K-A in the forebrain region was successfully generated.
Fig 1. Generation of transgenic mice conditionally overexpressing IP3K-A.

(A), Schematic diagram of the Tet-On regulatory system. Reverse tetracycline-controlled transactivator (rtTA) is expressed under the control of the tissue-specific promoter CaMKIIα. The rtTA protein binds and activates transcription of the tetracycline response element (TRE) only in the presence of Dox. (B), IP3K-A transgene expression in a Dox-dependent manner. IP3K-A expression was determined using Western blot of hippocampal lysates. (C and D), Immunohistochemical detection of IP3K-A protein. Coronal hippocampal sections (C) and sagittal whole brain sections (D) of WT and Tg mice fed Dox for 2 weeks immunofluorescence stained for IP3K-A (red) and Hoechst 33342 (blue). The induction pattern of the IP3K-A protein in the hippocampus was analyzed by subtracting the WT signal from the Tg signal. Scale bar, 200 μm (C) and 2 mm (D). Ctx, cortex; Hc, hippocampus; Ob, olfactory bulb; Cpu, caudate putamen; Th, thalamus; Cb, cerebellum.
Hippocampal-dependent learning and memory deficits in Tg mice
To gain insight into the relationship between IP3K-A overexpression and cognitive function, we investigated behaviors in Tg mice. First, an open-field test was conducted to assess general motor performance and anxiety levels in the Tg mice. There were no significant differences between WT and Tg mice in the total distance traveled (Fig 2A, t(19) = −0.444, p = 0.662) or percentage of time spent in the central and marginal areas of the open-field test apparatus (Fig 2B, center: t(19) = 1.278, margin: t(19) = −1.281, p = 0.216). These results suggested that transient IP3K-A overexpression does not have a significant effect on general motor activity or basal anxiety-like behavior in mice. To observe the effects of IP3K-A overexpression on learning and memory, we subjected Tg mice to a novel object recognition task, which is hippocampus-dependent. There was significant difference in novel objects recognition preference between groups, but the group did not affect trend of change in novel objects recognition preference (Fig 2C, condition, F(2,50) = 78.630, p < 0.05; group, F(1,25) = 7.148, p < 0.05; condition × group, F(2,50) = 2.797, p = 0.105, repeated measures ANOVA). Post hoc test revealed that Tg mice spent significantly less time exploring novel objects than WT mice did during the 24 h retention test; however, few effects were observed during the 72 h retention task (Fig 2C, t(1,25) = 6.773, p < 0.05, unpaired two-tailed Student’s t-test). We also used a radial arm maze test, another hippocampal-dependent learning task, to assess spatial learning and memory abilities of mice. Both WT and Tg mice showed a gradual decrease in the error rate, that is, re-entry into an arm already visited during the daily trial. Repeated measures ANOVA revealed that there is no difference in error rate between groups and the group did not affect the error rate trend (Fig 2D, session, F(6,150) = 9.255, p < 0.05; group, F(1,25) = 2.781, p = 0.108; session × group, F(6,150) = 0.999, p = 0.428). However, the Tg mice tended to show higher error rates than WT mice did in early sessions. Therefore, we also measured the total number of errors as a percentage of total visits to each arm and found that Tg mice showed significantly higher error rates than WT mice did during the early phase of learning session (S1–S3) (Fig 2E, t(25) = −2.458, p < 0.05, unpaired two-tailed Student’s t-test). Analysis of the number of visits showed similar trend as error rate results. The repeated ANOVA test showed that there was no difference in the total number of visits between groups and the group did not affect the total number of visits trend (session, F(6,150) = 7.862, p < 0.01; group, F(1,25) = 3.228, p = 0.084; session × group, F(6,150) = 1.259, p = 0.280). However, the Tg mice tended to show higher number of visits than WT mice did in early sessions. Therefore, we also analyzed differences between early sessions (S1–S3) and late sessions (S4–S7), just as error rate. Consistent with error rate results, Tg mice showed higher visits than WT mice did in the early sessions (t(25) = −2.620, p < 0.05, unpaired two-tailed Student’s t-test). These results suggest that that Tg mice exhibited learning retardation, but not complete impairment in hippocampal-dependent learning.
Fig 2. Spatial learning and memory deficits in IP3K-A Tg mice.

In the open-field test, WT and IP3K-A Tg mice showed no difference in total distance traveled (A) and time spent exploring the margins or the center of the field (B) (WT, n = 11; Tg, n = 10). (C), However, IP3K-A Tg mice showed a mild impairment in object recognition memory. Mice were exposed to two identical or different objects (A-A, A-B, and A-C) during the sample, 24 h retention, and 72 h retention trials, respectively. The percentage of the total time that was spent investigating the novel object during each trial period was quantified as the preference percentage (WT, n = 11; Tg, n = 10). (D and E), Compared with WT mice, Tg mice showed increased error rates in the radial arm maze test. Tg mice tended to show a higher error rate in early sessions (S1–S3) (D) and showed a significant higher error rate in early phase (S1–S3), but not late phase (S4–S7) (E) (WT, n = 15; Tg, n = 12).
Changes in basal properties of CA3-to-CA1 synaptic transmission in Tg mice
To examine the basal properties of synaptic transmission in CA1 synapses, we first examined evoked synaptic responses. To measure the synaptic input-output (I-O) relationship, we analyzed the slope of the field excitatory postsynaptic potential (fEPSP) and the fiber volley amplitude generated by activating CA3 Shaffer collateral axonal fibers within the stratum radiatum area of the CA1 in hippocampal slices (Fig 3A, left plot). Each slice was activated at seven incremental input stimulus intensities and the linear regression slope values of all seven points of the fEPSP slope versus the fiber volley amplitude were plotted (Fig 3A, right plot). The mean of the linear regression slope values in Tg mice was significantly higher than that in WT mice, indicating that the CA1 synapse of Tg mouse had greater evoked synaptic transmission efficacy. This result may be due to enhanced postsynaptic functions or presynaptic properties, such as synaptic α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor (AMPAR) function, excitability or release probability. Next, we investigated the activity-dependent presynaptic release probability by measuring the paired-pulse facilitation (PPF) ratio of fEPSP slopes at the CA1 synapse using inter-pulse intervals of 25 to 250 msec. PPF is a type of short-term presynaptic plasticity that indirectly reflects the presynaptic release probability (Fig 3B). We observed significant difference in PPF ratio between groups and between intervals, but the group did not affect trend of change in PPF ratio (Fig 3B, interval, F(4,704) = 99.743, p < 0.01; group, F(1,176) = 12.149, p < 0.01; interval × group, F(4,704) = 1.382, p = 0.248, repeated measures ANOVA). Post hoc test revealed that the Tg mice showed significantly reduced PPF at all intervals, suggesting a higher activity-dependent release probability at the CA3 presynaptic terminals of Tg mice (Fig 3B, 30 msec: t(151.011) = 2.490, p < 0.05, 50 msec: t(144.589) = 3.236, p < 0.01, 100 msec: t(157.519) = 3.452, p < 0.05, 150 msec: t(161.517) = 3.441, p < 0.01, 250 msec: t(162.976) = 2.998, p < 0.01, unpaired two-tailed Student’s t-test). To our knowledge, this is the first report showing that IP3K-A expression affects activity-dependent release of presynaptic terminals. To further examine activity-independent presynaptic- and postsynaptic functions in CA1 synapses, we measured mEPSCs in the presence of tetrodotoxin, a sodium channel blocker (Fig 3C). Overexpression of IP3K-A led to a significant increase in the amplitude of the mEPSC (Fig 3D) but not in the frequency, rise, or decay kinetics (Fig 3E–3G) of mEPSCs, suggesting higher density or conductance of postsynaptic glutamate receptors or an increase in the quantal content of presynaptic vesicles at CA1 synapses in Tg mice. Overall, these results suggest that both presynaptic and postsynaptic properties of CA1 pyramidal neurons are altered by IP3K-A overexpression.
Fig 3. Basal physiological properties of hippocampal CA1 synapses in IP3K-A Tg young mice.

(A), Input-output relationship of basal evoked synaptic transmission. Left, the field EPSP slopes were measured in response to six-step incremental stimulus current intensity per hippocampal slice. Scale bars: 0.5 mV, 5msec. Right, linear regression (LR) was performed on the slope values from each slice in the left I-O plot. Mean ± SEM. (WT, 1.92 ± 0.05, n = 86 slices, 19 mice; Tg, 2.10 ± 0.06, n = 97, 18; *p < 0.05). (B), Paired-pulse ratio of fEPSP responses. The ratio of the 2nd fEPSP slope (R2) to the 1st fEPSP slope (R1) was measured at the CA3-CA1 synapse of the hippocampal slice. WT, n = 85 slices, 19 mice; Tg, n = 94, 18; *p < 0.05 at the five intervals measured. Scale bars: 0.5 mV, 30 msec. (C-G), mEPSC properties. (C), Scale bars for all sample traces: 50 μA, 50 msec. (D), Measurements of amplitude. Mean ± SEM. (WT, 13.5 ± 0.2 pA; Tg, 15.1 ± 0.4 pA; **p < 0.001). (E), Frequency (WT, 1.87 ± 0.21 Hz; Tg, 1.91 ± 0.36 Hz; p = 0.92). (F), Rise time (WT, 1.66 ± 0.02 msec; Tg, 1.65 ± 0.03 msec; p = 0.74). (G), Decay time constant (WT, 2.36 ± 0.03 msec; Tg, 2.39 ± 0.04 msec; p = 0.57). The mEPSCs were measured from WT (n = 24 cells, 4 mice) and Tg mice (n = 22, 3).
Normal dendritic spine morphology in Tg mice
Given that LTP expression induces translocation of IP3K-A into F-actin enriched dendritic spines [4] and that IP3K-A-deficient mice show impaired LTP at hippocampal DG granule cell synapses [3], we investigated whether the overexpression of IP3K-A changed dendritic spine morphology. The dendritic spines were visualized by Golgi staining of hippocampal DG granule cells and CA1 pyramidal cells. A spine was defined as a dendritic protrusion 0.5 to 3 μm in length, with or without a head (Fig 4A and 4B). No significant difference was detected in spine density between Tg (DG granule cells, 12.13 ± 0.38 spines/10 μm; CA1 pyramidal cells, 12.4 ± 0.43 spines/10 μm) and WT mice (DG granule cells, 11.88 ± 0.68 spines/10 μm; CA1 pyramidal cells, 13.15 ± 1.22/10 μm). In addition, the expression levels of the synaptic proteins NR2B, synaptotagmin 1, and PSD-95 were not different between WT and Tg mice (Fig 4C and 4D). To determine whether any compensatory expression occurred in Tg mice, we also examined expression levels of other postsynaptic and presynaptic proteins but found none with any significant change (S5 Fig). These results indicated that overexpression of IP3K-A does not specifically affect the density of dendritic spines or levels of the examined synaptic proteins, suggesting that the basal IP3K-A expression is sufficient for homeostasis of spine morphology generation and maintenance.
Fig 4. Normal density of dendritic spines and expression of synaptic molecules in IP3K-A Tg mice.
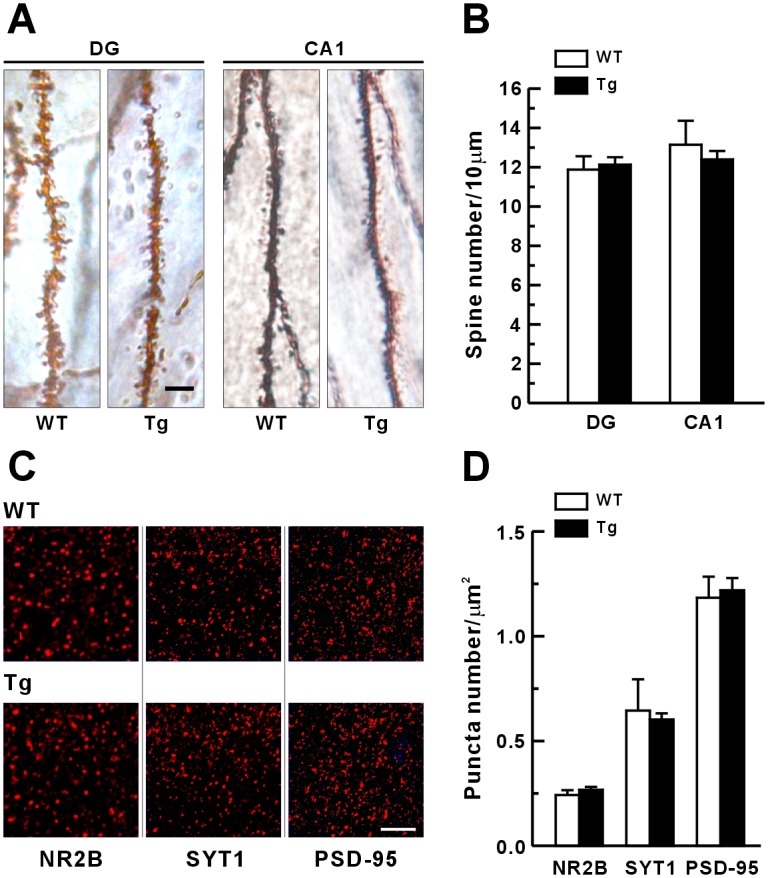
(A), Representative images of Golgi-stained dendritic segments of CA1 and DG neurons in WT and Tg mice. Scale bar, 5 μm. (B), Quantification of dendritic spine density. Mean ± SEM. (WT, n = 60 neurons, 5 mice; Tg, n = 45, 4). (C), Representative images of immunohistochemistry of NR2B (C1), synaptotagmin 1 (SYT1; C2), and PSD-95 (C3) at the apical dendritic region of hippocampal CA1 pyramidal cells. Scale bar, 5 μm. (D), Quantification of immunofluorescent puncta of NR2B, SYT1 and PSD-95.
Increase in presynaptic vesicle pool size in Tg mice
Although previous studies and our data show the existence of IP3K-A in the postsynaptic fraction [15, 16] and in the presynaptic vesicle fraction of the mouse hippocampus (Fig 5A), we further investigated whether presynaptic changes occurred in IP3K-A Tg mice using electron microscopy (EM). We used EM ultrastructural analysis to determine whether IP3K-A overexpression affected the total pool of synaptic vesicles, which could change the release probability of neurotransmitters. The results showed that the number of synaptic vesicles in CA1 synapses of Tg mice was increased compared with that of WT mice (Fig 5B and 5C). This finding suggested that IP3K-A overexpression may be involved in maintaining larger synaptic vesicle pools in terminals, which would lead to a higher presynaptic release probability.
Fig 5. Subcellular localization of IP3K-A and increased number of synaptic vesicles in IP3K-A Tg mice.
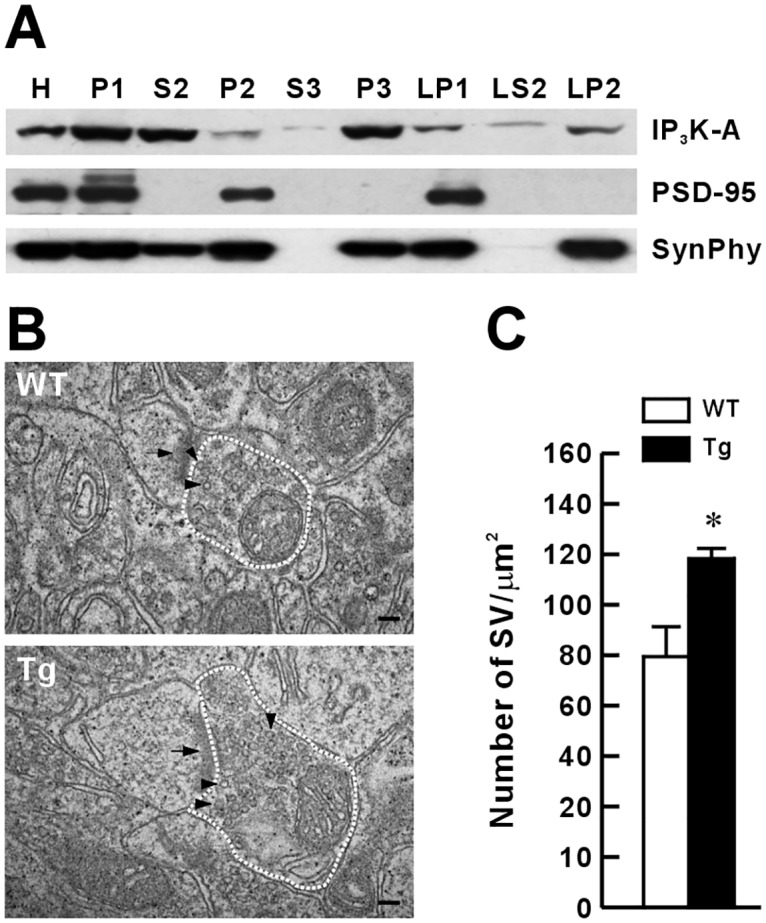
(A), Distribution pattern of IP3K-A in the adult mouse brain subcellular fractions. PSD-95 and synaptophysin (SynPhy) were used as controls. H, homogenates; P1, nuclei and large debris; P2, crude synaptosomes; S2, supernatant after P2 precipitation; S3, cytosol; P3, light membranes; LP1, synaptosomal membranes; LS2, synaptosomal cytosol; LP2, synaptic vesicle-enriched fraction. (B), Representative EM images of hippocampal CA1 synapses in WT and Tg mice. Scale bar: 100 mm. (C), Quantification of the number of synaptic vesicles per area (WT, n = 60 synapses, 3 mice; Tg, n = 60, 3). Presynaptic terminals with well-defined postsynaptic density (arrows) were chosen for analysis. Dashed lines indicated the presynaptic area for the analysis of synaptic vesicle (arrowheads) density per area.
Enhancement of the very early phase of NMDAR-dependent LTP in Tg mice
To explore the role of IP3K-A overexpression in synaptic plasticity, we induced NMDAR-dependent LTD using single-pulse low-frequency stimulation (LFS-LTD) at CA1 synapses in young WT and Tg mice (Fig 6A) while measuring the PPF ratio (40 msec interval). A similar level of LFS-LTD was induced in both WT and Tg mice without significant changes in PPF during LTD expression, suggesting that NMDAR-dependent LTD is expressed mainly postsynaptically in both types of mice. For NMDAR-dependent LTP induction, single-theta-burst (Fig 6B) and four-theta-burst (Fig 6C) stimulation protocols were used to induce protein synthesis-independent and protein synthesis-dependent processes, respectively. Compared with WT mice, Tg mice exhibited enhanced LTP only during the very early phase induced by either protocol. The PPF ratio measurements following single theta-burst stimulation indicated that Tg and WT mice showed a similar amount of enhanced presynaptic release probability during the very early phase of LTP (Fig 6B, lower panel), suggesting an additional transiently enhanced postsynaptic function in Tg mice. In four-theta-burst LTP, a more marked contribution of postsynaptic function might explain the early increase of LTP in Tg mice because the PPF change in Tg mice was much less than that in WT mice (Fig 6C, lower panel). These results suggested that overexpressed IP3K-A in Tg mice is involved in a higher postsynaptic contribution to the early phase expression of NMDAR-dependent LTP compared with that of LTP in WT mice while having no significant role in NMDAR-dependent LTD.
Fig 6. Normal fEPSP-LTD and transient increment of early LTP in young IP3K-A Tg mice.
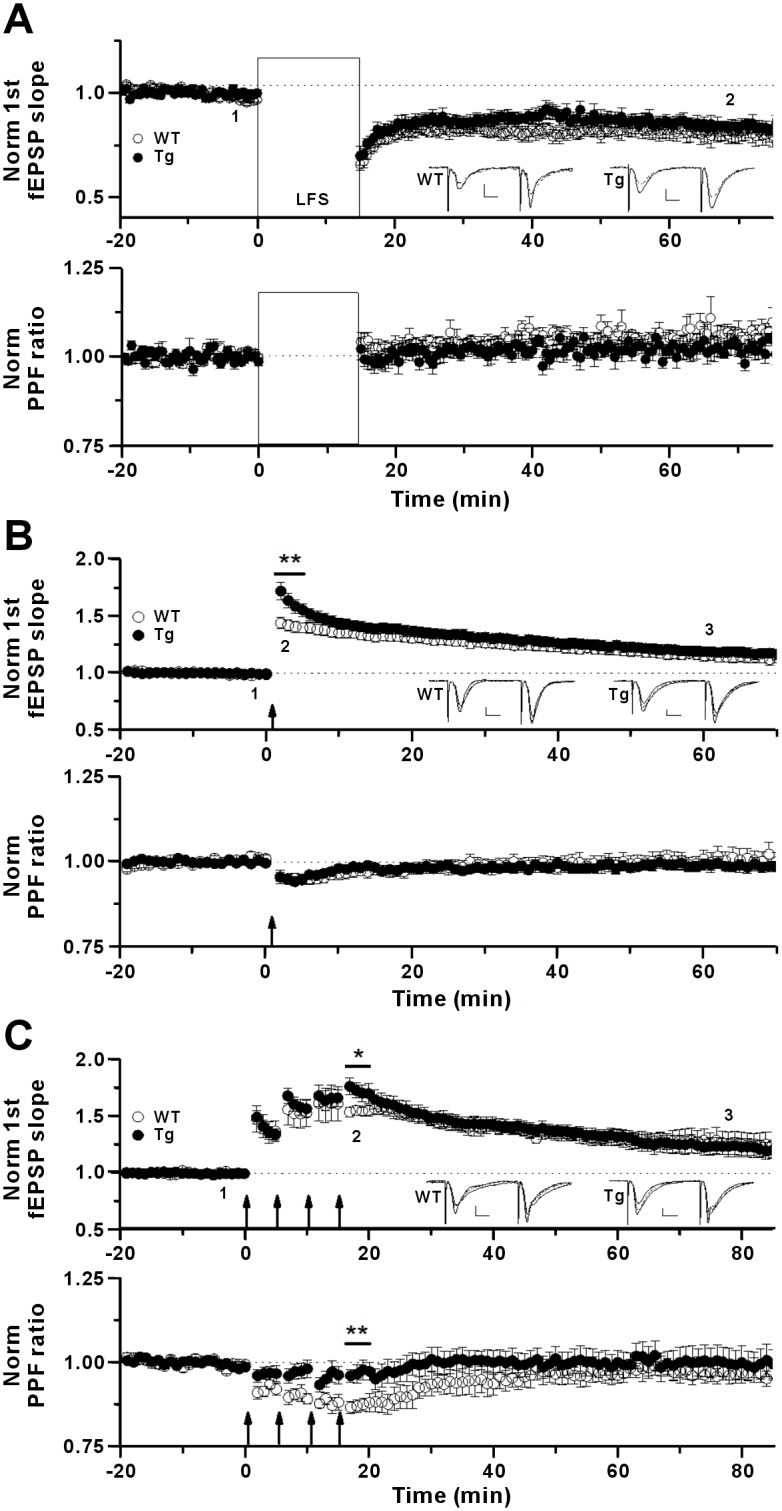
(A), LFS-induced LTD. Upper plot, LTD was induced by LFS protocol (1 Hz, 900 pulses) at the CA1 dendritic region of the hippocampal slice. The slope of fEPSP was 81.1 ± 1.5% (WT, n = 10 slices, 4 mice) and 84.0 ± 1.0% (Tg, n = 10, 4) at 60–65 min on the time axis (p = 0.11). Inlet traces are sample traces measured at baseline (1, thin line) and at 65 min (2, dashed line), respectively. Scale bars: 0.5 mV, 8 msec. Lower plot shows the normalized PPF ratio of the fEPSP slopes. PPF was measured at 60 to 65 min after induction of LTD (WT, 1.06 ± 0.80E-2; Tg, 1.02 ± 0.79E-2). (B), Single theta-burst induced LTP. Upper plot, LTP was induced with a single theta-burst protocol. Significant difference was observed immediately for 2–6 min after the theta-burst protocol (2, bar): WT, 141.5 ± 2.1%, n = 15; Tg, 160.6 ± 2.8%, n = 20; **p < 0.001. At 58–62 min (3), WT, 115.9 ± 2.0%; Tg, 118.9 ± 1.9% of baseline; p = 0.28. Inlet traces are sample traces indicating baseline (1, thin line), early peak (2, thick line) and late phase (3, dashed line). Scale bars for all sample traces: 0.5 mV, 8 msec. Lower plot shows the normalized PPF ratio of the fEPSP slopes. At 2–6 min, WT, 0.95 ± 0.62E-2; Tg, 0.95 ± 0.60E-2. At 58–62min, WT, 1.00 ± 0.98E-2; Tg, 0.99 ± 0.50E-2. (C), Four theta-burst induced LTP. LTP was induced with four theta-burst protocol. Significant difference was observed immediately after the theta-burst protocol (2, bar): WT, 155.0 ± 2.3%, n = 6; Tg, 169.1 ± 4.2%, n = 8; p = 0.027. At 78–82 min (3), WT, 125.3 ± 4.5%; Tg, 123.5 ± 3.1% of baseline; p = 0.74. Inlet traces are sample traces indicating baseline (1, thin line), early peak (2, thick line) and late phase (3, dashed line). Scale bars: 0.5 mV, 8 msec. Lower plot shows the normalized PPF ratio of the fEPSP slopes. At 17–20 min (2), WT, 0.87 ± 0.012; Tg, 0.94 ± 0.014; **p = 0.002. At 78–82 min (3), WT, 0.97 ± 0.011; Tg, 0.99 ± 0.021.
Inhibition of mGluR-dependent DHPG-LTD and alteration in the postsynaptic contribution to LTD expression in Tg mice
As shown in Fig 6A, LFS-LTD was normal in Tg mice; moreover, paired-pulse LFS-LTD was not affected in these mice (S6 Fig). These results suggested that NMDAR-dependent postsynaptic plasticity is relatively spared in Tg mice. Because LFS activates both NMDARs and mGluRs [17, 18], we examined mGluR-dependent LTD by applying (R,S)-3,5-dihydroxyphenylglycine (DHPG), a group I mGluR agonist [19] to hippocampal slices of young mice. The results showed that DHPG-LTD was induced in WT mice with a small decrease in the PPF ratio, suggesting a strong postsynaptic contribution to LTD expression despite some increase in the presynaptic release probability. DHPG-LTD was inhibited in Tg mice (Fig 7A, top plot). The results also indicated that, compared with WT mice, Tg mice showed a significant reduction (by approximately 5%) in the PPF of basal synaptic transmission (Fig 7A, middle plot, and Fig 7E), suggesting an increased basal presynaptic release probability, consistent with the result shown in Fig 3B. This indicated that overexpressed IP3K-A is involved in the enhanced presynaptic release process at the basal level. An analysis of LTD (Fig 7A, top plot) together with normalized PPF (Fig 7A, bottom plot) in Tg mice revealed a change in the postsynaptic IP3K-A contribution to DHPG-LTD expression. The normalized PPF ratio after DHPG washout was strongly reduced in Tg mice compared with that in WT mice, indicating enhanced presynaptic release, which might inhibit DHPG-LTD expression (Fig 7F). However, the early phase of DHPG-LTD (10–30 min after DHPG washout) was normally expressed despite the reduced PPF ratios, suggesting a strong postsynaptic contribution to the early phase of DHPG-LTD in Tg mice. During the late phase (> 40 min after DHPG washout), DHPG-LTD was blocked in Tg mice, while the PPF ratio reduction was similar to that of the early phase, indicating a decay in the postsynaptic contribution (Fig 7A, bottom plot, and Fig 7F).
Fig 7. Impairment of DHPG-LTD in young IP3K-A Tg mice.

(A), Induction and expression of DHPG-LTD. Top plot, DHPG-LTD was significantly inhibited in IP3K-A Tg mice compared with WT (measured at 61–70 min (grey bar); WT, 86.5 ± 0.1% of baseline, n = 20 slices; Tg, 97.7 ± 0.4%, n = 18 slices; ***p < 0.001). Middle plot, the mean PPF ratio of the baseline of Tg mice was significantly lower than that of WT (WT, 1.69 ± 0.34E-2; Tg, 1.61 ± 0.38E-2; ***p < 0.001). Bottom plot, The normalized PPF ratio after DHPG washout was significantly reduced in Tg mice compared with that of WT (measured at 61–70 min (grey bar); WT, 0.93 ± 0.20E-2; Tg, 0.81 ± 0.42E-2; ***p < 0.001). (B), DHPG-LTD in Rö-31-8220 pre-incubated slices. 1 μM Rö-31-8220 was applied throughout the entire recording period. Top plot, DHPG (50 μM) application for 10 min induced LTD in CA1 synapses in WT and Tg mice (measured at 61–70 min; WT, 83.9 ± 0.3% of baseline, n = 14; Tg, 81.4 ± 0.3%, n = 15). Middle plot, the mean PPF ratio of the baseline of Tg mice was significantly higher than that of WT (WT, 1.58 ± 0.25E-2; Tg, 1.70 ± 0.18E-2; ***p < 0.001). Bottom plot, normalized PPF ratio after DHPG washout was significantly reduced in both WT and Tg mice (measured at 61–70 min; WT, 0.75 ± 0.01; ***p < 0.001; Tg, 0.80 ± 0.01; ***p < 0.001). (C), DHPG-LTD in calphostine C pre-incubated slices. 1 μM calphostin C was applied throughout the entire recording period. Top plot, DHPG (50 μM) application induced LTD in CA1 synapses in WT and Tg mice (measured at 61–70 min; WT, 77.3 ± 0.2% of baseline, n = 6, p < 0.001; Tg, 80.0 ± 0.7%, n = 6, p < 0.001). Middle plot, themean PPF ratio of the baseline of Tg mice was similar to that of WT (WT, 1.63 ± 0.30E-2; Tg, 1.66 ± 0.02E-2; p < 0.001). Bottom plot, After DHPG washout, the normalized PPF ratio was almost unchanged compared to the baseline of WT and Tg mice (measured at 61–70 min; WT, 1.00 ± 0.01; Tg, 0.97 ± 0.01; p = 0.004). (D), DHPG-LTD in 2-APB pre-incubated slices. 16 μM 2-APB was applied throughout the entire recording period. Top plot, DHPG (50 μM) application did not induce LTD in CA1 synapses in WT and Tg mice (measured at 61–70 min; WT, 101.0 ± 0.5% of baseline, n = 18; Tg, 98.8 ± 0.3%, n = 20). Middle plot, the mean PPF ratio of the baseline of Tg mice was similar to that of WT (WT, 1.57 ± 0.40E-2; Tg, 1.55 ± 0.16E-2; p < 0.001). Bottom plot, the normalized PPF ratio after DHPG washout was reduced compared with that of baseline in WT and Tg mice (measured at 61–70 min; WT, 0.74 ± 0.78E-2; p < 0.001; Tg, 0.83 ± 0.39E-2, p < 0.001). Inlet traces are sample traces at scale of 0.5 mV/8 msec. All inlet traces are sample traces. Scale bars: 0.8 mV, 8 msec. (E), Summary plot of basal PPF ratio data before and after drug treatments shown in (A–D). (F), Summary plot of normalized PPF ratio data at 61–70 min period after DHPG-LTD induction shown in (A–D).
In summary, these results suggested that DHPG-LTD is expressed both presynaptically and postsynaptically in WT mice, but the postsynaptic activity overcomes the presynaptic activity, resulting in LTD. In Tg mice, the early phase of DHPG-LTD is expressed by a strong postsynaptic component, which overcomes the presynaptic effect, and the late phase of DHPG-LTD is inhibited, possibly due to the decay of the postsynaptic component while the enhanced presynaptic contribution is maintained. Therefore, overexpressed IP3K-A appears to contribute both presynaptically and postsynaptically to DHPG-LTD, in which a change of postsynaptic IP3K-A contribution determines the level of DHPG expression, with an inhibitory role of overexpressed presynaptic IP3K-A.
IP3K-A overexpression reveals differential roles of PKC in presynaptic release probability and a postsynaptic role of PKC in DHPG-LTD
Activation of the mGluR signaling pathway produces diacylglycerol (DAG) and IP3 from phosphatidylinositol 4,5-bisphosphate (PIP2) via phospholipase C (PLC) activation [20, 21]. Both DAG and IP3-mediated Ca2+ efflux from the ER activate PKC, which is involved in the presynaptic release machinery [22, 23]. Because it is unclear how these signaling molecules work with one another, we looked at the roles of the PKC and IP3R pathways in synaptic transmission and in DHPG-LTD of CA1 synapses. To examine the role of the PKC pathway, the PKC inhibitor Rö-31-8220 (1 μM) was applied to hippocampal slices prior to DHPG administration. The results showed that pretreatment with Rö-31-8220 significantly reduced the baseline PPF ratio by approximately 7% (Fig 7A, middle plot, and Fig 7E, WT without Rö-31-8220, 1.69 ± 0.34E-2 vs Fig 7B, middle plot, WT with Rö-31-8220, 1.58 ± 0.25E-2; p < 0.001), indicating that inhibiting basal PKC activity increased presynaptic release in WT mice. The effect of Rö-31-8220 on PPF in WT mice was similar to that produced by overexpressing IP3K-A in Tg mice in the absence of Rö-31-8220 (i.e., approximately 5% reduction in the PPF ratio, Fig 7E). However, DHPG-LTD expression was not affected by Rö-31-8220 in WT mice, although the reduction in PPF was much greater (approximately 25%; Fig 7B, bottom plot, and Fig 7F). This finding suggested that PKC inhibition in WT mice facilitates the postsynaptic contribution to DHPG-LTD to overcome the effect of enhanced presynaptic release. Our results also showed that Rö-31-8220 in Tg mice increased the baseline PPF ratio by approximately 4% (Fig 7E and 7A, middle plot, Tg without Rö-31-8220, 1.61 ± 0.38E-2 vs Fig 7B, middle plot, Tg with Rö-31-8220, 1.70 ± 0.18E-2; p < 0.001), indicating that inhibiting PKC in Tg mice reduced basal presynaptic release, such that basal PPF in Tg mice was now similar to that in WT mice. This finding indicated that the increased basal presynaptic release caused by overexpressed IP3K-A is PKC-dependent. We also found that the blockade of DHPG-LTD in Tg mice was removed by Rö-31-8220, resulting in normal LTD expression while maintaining PPF ratios reduced by approximately 20% (Fig 7B and 7F). This result suggested that the PKC inhibitor may remove the postsynaptic contribution decay in Tg mice.
To confirm the effects of PKC inhibition, calphostin C (1 μM), which blocks the DAG-binding C1 domain of PKC, was also examined. We found that calphostin C pre-application reduced the baseline PPF ratio by approximately 4% in WT mice (Fig 7E and 7A, middle plot, WT without calphostin C, 1.69 ± 0.34E-2 vs Fig 7C, middle plot, WT with calphostin C, 1.63 ± 0.30E-2; p < 0.001), which was consistent with the effect of Rö-31-8220. We also found that DHPG-LTD was expressed normally in the presence of calphostin C in WT mice with no change in PPF during LTD at 60–70 min (Fig 7C and 7F, 1.00 ± 0.63E-2 of baseline). This result indicated that DHPG-LTD is not affected by calphostin C and that LTD is expressed postsynatptically. Our results further showed that calphostin C increased the baseline PPF ratio in Tg mice by approximately 3% (Fig 7E and 7A, middle plot, Tg without calphostin C, 1.61 ± 0.38E-2 vs Fig 7C, middle plot, Tg with calphostin C, 1.66 ± 0.02E-2; p < 0.001), which is similar to the change od induced by Rö-31-8220. Calphostin C also blocked the inhibitory effect of IP3K-A overexpression on DHPG-LTD expression with little change in PPF (Fig 7C and 7F). The effects of calphostin C and Rö-31-8220 on DHPG-LTD were similar except for their presynaptic effects in both WT and Tg mice.
In summary, both PKC inhibitors, Rö-31-8220 and calphostin C, revealed differential roles of PKC in basal presynaptic release. Basal PKC activity inhibited release probability in WT mice but facilitated it in Tg mice. The mechanism for this altered PKC role in the release process is unknown. The effect of the inhibitors on PPF during DHPG-LTD differed, with Rö-31-8220 enhancing PPF while calphostin C did not. By contrast, both PKC inhibitors blocked the inhibitory effect of IP3K-A overexpression on DHPG- LTD in Tg mice, in which PKC might play a role in the decay of the postsynaptic contribution to DHPG-LTD expression. However, PKC inhibition did not affect DHPG-LTD in WT mice, consistent with previous study results [24–30]. Taken together, these results suggested that overexpression of IP3K-A changes the roles of PKC in both presynaptic and postsynaptic processes during basal synaptic transmission and in DHPG-LTD.
Inhibitory role of IP3R signaling in presynaptic release and DHPG-LTD
To further investigate the change in the IP3 signaling pathway induced by PLC in DHPG-LTD, we applied 2-aminoethoxydiphenyl borate (2-APB, 16 μM), an IP3-sensitive Ca2+ release inhibitor working as an IP3R antagonist, before the addition of DHPG (Fig 7D). The results indicated that 2-APB treatment reduced the baseline PPF in WT mice by approximately 7% (Fig 7E and 7A, middle plot, WT without 2-APB, 1.69 ± 0.34E-2 vs Fig 7C, middle plot, WT with 2-APB, 1.57 ± 0.40E-2; p < 0.001), similar to the reduction induced by the PKC inhibitors. This suggested that the IP3R, like PKC, negatively regulates basal presynaptic release in WT mice. Our results also showed that 2-APB slowly blocked DHPG-LTD in WT mice while causing a relatively greater reduction in PPF (Fig 7D, bottom plot, and Fig 7F). Therefore, the IP3R antagonist-induced blockade of DHPG-LTD may be caused by enhanced presynaptic release, although a postsynaptic contribution cannot be excluded especially during the early phase of LTD. We also found that 2-APB reduced the basal PPF ratio in Tg mice by approximately 4% (Fig 7E and 7A, middle plot, Tg without 2-APB, 1.61 ± 0.38E-2 vs Fig 7C, middle plot, Tg with 2-APB, 1.55 ± 0.16E-2; p < 0.001), which was an effect opposite to that of the PKC inhibitors. This finding suggested that presynaptic IP3Rs continue to negatively modulate presynaptic release in Tg mice. Application of 2-APB also blocked DHPG-LTD expression and reduced PPF in Tg mice (Fig 7D and 7F). These results revealed a role for presynaptic IP3Rs in DHPG-LTD in both WT and Tg mice, although they did not exclude a postsynaptic function.
All three drugs (Rö-31-8220, calphostin C, and 2-APB) blocked changes in the fEPSP I-O plot in Tg mice (S7 Fig). The relevance of CaMKII in the DHPG-LTD process was tested using pre-application of specific CaMKII inhibitors, KN62 or KN93; however, no significant effect on the expression of DHPG-LTD was observed (S8 Fig). This finding suggested that the protocol for DHPG-LTD used here did not appear to activate the CaMKII signaling pathway.
Discussion
Previous studies using IP3K-A KO mice showed that IP3K-A targets to F-actin-enriched dendritic spines and is involved in LTP and spatial memory [3]. The F-actin bundling activity of IP3K-A is much higher in immature than in mature neurons [3, 8]. In the present study, to further investigate the morphological and physiological role of IP3K-A in neurons, we generated Tg mice overexpressing IP3K-A in the brain by using a Tet-On inducible system (Fig 1). Unlike the results following IP3K-A overexpression in cultured hippocampal neurons, transient overexpression of IP3K-A in adult mice did not alter the formation of dendritic spines in hippocampal CA1 and DG neurons (Fig 2A and 2B). However, Tg mice showed impairments in spatial learning and memory (Fig 2C–2E) and in the late phase of DHPG-LTD in hippocampal CA1 synapses (Fig 7A), with a mild enhancement of the very early phase of theta-burst LTP (Fig 6B and 6C). These Tg mice further demonstrated that overexpression of IP3K-A resulted in an increase in the presynaptic vesicle pool size and release probability and in the inhibition of mGluR-dependent LTD. This is the first report to suggest that, along with its known postsynaptic roles in the central nervous system, IP3K-A modulates synaptic transmission and plasticity presynaptically.
Transient retardation of hippocampal-dependent memory
Our results from novel object recognition and radial arm maze tests suggested that overexpression of IP3K-A attenuated hippocampal-dependent working memory retention transiently rather than permanently. In the novel object recognition test, there was a significant impairment in working memory retention during the 24 h, but not the 72 h, test in Tg mice. There was a higher error rate during the early rather than the late phase (Fig 2E; S1–S3 Figs) of the spatial working memory test in Tg mice. In both tasks, the early retardation shown in Tg mice was attenuated as the mice performed more learning trials. These results were not due to overall motor deficit or alterations in emotional states because there were no differences between WT and Tg mice in the open field test. The abnormal regulation of synaptic plasticity shown in the physiological experiment, particularly the impairment of early NMDAR-dependent LTP (Fig 6B and 6C), could provide a clue to the mechanisms underpinning the transient behavioral deficit we observed. A causal relationship between enhanced hippocampal LTP and memory deficit has been provided by the disruption of genes encoding the synaptic scaffold PSD-95 [31] or ionotropic GluR2 subunit [32] or by a recently developed mouse model of Alzheimer’s disease [33]. However, impaired DHPG-LTD did not appear to interfere with normal retention performance during the late phase of learning, consistent with previous studies showing a relatively minor influence of a LTD deficit on hippocampal-dependent learning, such as that examined using the water maze [34–36]. Therefore, the transient working memory deficit found in Tg mice could be caused in part by altered LTP and LTD within related neuronal circuitry, including in the hippocampus.
Localization of IP3K-A in presynaptic terminals and enhancement of basal presynaptic release and vesicle pool size following IP3K-A overexpression
Schaffer collateral-CA1 synapses in Tg mice showed a significant decrease in PPF reflecting an increased presynaptic release probability (Fig 3B). The biochemical data in the present study revealed that IP3K-A was detected in synaptic fractions, including crude synaptosomes, synaptic membranes, and synaptic vesicle fractions, of the mouse hippocampus, providing strong evidence for the presynaptic presence of IP3K-A. Therefore, we looked at the size of the presynaptic vesicle pools to determine whether a change there underpinned change in the presynaptic release probability. The results showed that the number of synaptic vesicles in the hippocampal CA3 terminals of Tg mice was significantly increased compared with that of the control mice, as assessed by EM analysis (Fig 5B and 5C). This is consistent with a previous immuno-EM ultrastructural study showing that IP3K-A is localized at axon terminals and associated with synaptic vesicles [15]. However, the mechanism whereby overexpressed IP3K-A modulates the vesicle pool size remains unknown.
Previous studies also showed that IP3K-A immunoreactivity is associated with the spine apparatuses and plasmalemma of pyramidal neurons in the cortex and hippocampus and is located in the dendritic spines of Purkinje and basket cells in the cerebellum [15], suggesting a postsynaptic function of IP3K-A. Our results showed that IP3K-A overexpression did not affect the spine density of mature neurons in adult mice, consistent with a previous study that found that overexpression or deletion of IP3K-A has few effects on synaptic formation, spine density, and synaptic contacts of mature neurons [8]. However, in immature neurons, overexpression of IP3K-A increases the number of dendritic protrusions [8]. Although the postsynaptic function of IP3K-A is still unclear, IP3K-A may play a role in the trafficking of AMPAR-containing postsynaptic vesicles.
PKC role is changed by IP3K-A overexpression during both basal presynaptic release and DHPG-LTD
Stimulation of mGluR activates PLC, leading to the production of DAG and IP3 from PIP2. The PKC enzymes are activated by intracellular signals such as increased DAG [22] or Ca2+ [23]. Regarding the role of PKC activity, our results showed that the basal presynaptic release probability is reduced by PKC activity in WT mice but is enhanced in Tg mice. Furthermore, DHPG-LTD is expressed PKC-independently in WT mice, consistent with previous studies [24–30]. PKC activation after PLC-induced DAG and IP3 signaling appears to not be essential for the formation of stable DHPG-LTD in WT mice. However, DHPG-LTD is PKC-dependent in Tg mice, in which basal PKC activity may be increased. Overexpression of IP3K-A changed the role of PKC activity at both presynaptic sites, such that activated PKC increased presynaptic intracellular Ca2+ levels via an unknown mechanism and was involved in the decay of the postsynaptic contribution to DHPG-LTD.
DAG signaling by diacylglycerol kinase iota (DGKι) has been recently implicated in mGluR-dependent LTD, suggesting that DGKι-induced DAG elimination promotes the reduction of transmitter release via inhibiting PKC activity [37]. However, there are studies showing that PLC and PKC can work separately in diverse signaling conditions. In spinal sensory synapses, DHPG-LTD was prevented by preincubation with thapsigargin, cyclopiazonic acid, or 2-APB, but not by ryanodine, a Ca2+-induced Ca2+ release inhibitor, and PKC inhibitors [28], which is consistent with our results. In basolateral amygdala synapses, DHPG-LTD was induced via a PLC-dependent but PKC-independent mechanism, whereas the priming action of mGluR5Rs on basolateral amygdala LTP was both PLC- and PKC-dependent. It has also been shown that presynaptic muscarinic M1 receptor activation is necessary for PKC activation during DHPG-LTD [38]. These results, including ours, suggest that PKC roles change dynamically depending on the stimulating input pathways during synaptic transmission, and plasticity, such as during DHPG-LTD.
IP3R signaling role in presynaptic release process is unaltered in Tg mice
Neurotransmitter release from presynaptic terminals is altered by intracellular Ca2+ levels, which are regulated by the influx of extracellular Ca2+ ions and the release and uptake of intracellular Ca2+ stores [39, 40]. The Ca2+ release from the ER is mediated by ryanodine receptors and IP3Rs, which are gated by intracellular Ca2+ and IP3, respectively [41]. Thus, IP3K-A might be associated with Ca2+ efflux from the ER. However, our results with 2-APB, IP3R antagonist, showed that IP3R signaling was negatively involved in presynaptic release in both WT and Tg mice and that IP3K-A overexpression did not significantly change the role of IP3R in this process.
When the mGluR signaling pathway is activated by DHPG, the amount of IP3 is expected to increase transiently due to the subsequent IP3K-A activity. Our results showed that the presynaptic release probability was mildly increased during DHPG-LTD in WT mice (Fig 7A). When IP3R was inhibited, the increased presynaptic release probability was much greater during DHPG-LTD than it was during basal synaptic transmission (Fig 7D, middle plot, and Fig 7E). In addition, either IP3K-A overexpression or IP3R inhibition increased the presynaptic release probability during DHPG-LTD. It is expected that IP3K-A overexpression would deplete IP3, resulting in IP3R inhibition. However, it is unknown how inhibition of IP3R leads to the observed increase in presynaptic release probability. One possibility is that the increased IP4 level, following IP3K-A-mediated conversion of IP3, may augment Ca2+ influx from the extracellular fluid through activation of voltage-gated Ca2+ channels [42], providing better conditions for presynaptic release when IP3R is inhibited by an antagonist or IP3 depletion. Another possibility is that overexpression of IP3K-A may lead to changes in the activity of proteins that modulate intracellular Ca2+ levels or in the metabolism of inositol polyphosphates, both of which would increase release probability. In IP3K-A KO mice, synaptosomes display a shortened duration of both IP3 signals and IP3-dependent Ca2+ transients due to the compensatory up-regulation of IP3 phosphatase and the ER Ca2+ ATPase pump 2b proteins, which reduce intracellular Ca2+ levels [8]. If these compensatory regulations work in the opposite direction in IP3K-A-overexpressing neurons, a transient increase in intracellular Ca2+ levels may increase the release probability of Tg neurons.
The roles of postsynaptic IP3Rs in synaptic plasticity are quite complex and vary depending on the types of neurons. First, IP3Rs play a role in the Ca2+ increase in dendrites and by somatic and nuclear Ca2+ signals [43]. Second, both the GluA2 exit from the ER and the synaptic insertion, and the synaptic depression by the concomitant synaptic GluA1 endocytosis, depend on IP3R- and ryanodine receptor-controlled Ca2+ release in medium spiny neuron cultures [44, 45] and in the suppression of AMPAR currents by orexin-A in retinal ganglion cells [46]. Third, postsynaptic IP3R is also involved in AMPAR insertion during spike timing-dependent plasticity [47] and in both muscarinic and cholinergic enhancement of AMPAR-mediated transmission in CA1 pyramidal neurons [48, 49]. The mechanisms underlying the roles of postsynaptic IR3Rs remain to be determined in the DHPG-LTD model, especially in Tg mice.
Differential roles of overexpressed IP3K-A during DHPG-LTD and NMDAR-dependent LTP
Increases in presynaptic release probability and postsynaptic Ca2+ release from stores have been suggested to explain early LTP expression [50]. Our results examining LTP in WT mice could be explained by an enhanced Ca2+-dependent presynaptic neurotransmission as shown by changes in PPF (Fig 6B and 6C). However, our results in Tg mice suggested both postsynaptic and presynaptic contributions to LTP. In single-theta-burst LTP experiments, early LTP in Tg mice was greater than that in WT mice, while presynaptic PPF changes were similar, indicating an additional postsynaptic role in Tg mice (Fig 6B). Results with four-theta-burst LTP further indicated that early LTP was primarily expressed postsynaptically in Tg mice because the PPF reduction was much less in Tg mice than in WT mice (Fig 6C). Therefore, these results with Tg mice revealed an additional postsynaptic role of overexpressed IP3K-A for the enhancement of early LTP. Because postsynaptic IP4, but not IP3, can increase LTP [42], it is possible that a mechanism involving IP4 might enhance early LTP in Tg mice.
An analysis of LTD with the PPF change also provided a clue to the source of LTD expression. In WT mice, induction of LTD appeared immediately after DHPG washout, and PPF was reduced during most of the DHPG-LTD (Fig 7A, by approximately 7% at 60–70 min, p < 0.0001). The PPF reduction should have caused an increase in the fEPSP, but LTD was still induced. This suggested that both presynaptic and postsynaptic components are involved in the expression of DHPG-LTD and that inhibitory postsynaptic processes can overcome the potential increase in fEPSP induced by presynaptic components, resulting in a net reduction in the postsynaptic AMPAR current for DHPG-LTD expression. Compared with WT mice, Tg mice displayed a reduction in DHPG-LTD (Fig 7A, upper panel) and PPF (Fig 7A, lower panel, and Fig 7F) in CA1 synapses of young mice, while NMDAR-dependent LTD was normal (Fig 6A). This finding in Tg mice indicated that LTD expression consists of different phases, with both presynaptic and postsynaptic contributions. Relative to that in WT mice, during the early phase of LTD in Tg mice, a larger, lasting PPF reduction (reduced approximately 19% at 70 min) appeared immediately, which might inhibit LTD expression via increased presynaptic release. However, early LTD still occurred, suggesting strong postsynaptic components for early LTD expression. Blockade of LTD developed slowly while PPF was reduced, suggesting a decay of the early postsynaptic contribution. These results indicated that overexpressed IP3K-A can increase synaptic function either presynaptically or postsynaptically with different temporal kinetics. In addition, the results of our PKC inhibitor experiments suggested a role of PKC in the decay of the early postsynaptic contribution to DHPG-LTD in Tg mice.
Interaction of IP3K-A, IP3R, and PKC with one another
Reduced presynaptic release probability [51, 52] and postsynaptic AMPAR endocytosis have been suggested [44, 45, 51, 53] as mechanisms for DHPG-LTD expression. Our results showing that a molecule can play different roles depending on the phase of LTD add complexity to the understanding of the DHPG-LTD expression mechanism. Our results indicated that both IP3K-A-dependent presyntaptic and postsynaptic components are involved in the early phase of DHPG-LTD in WT mice, but the postsynaptic contribution is reduced in the late phase. The PKC inhibitors did not block DHPG-LTD in WT mice but blocked the effect of overexpressed IP3K-A in DHPG-LTD expression, which suggested that PKC is activated in the overexpressed IP3K-A signaling pathway. The effects of the IP3R antagonist differed from those of the PKC inhibitors. The IP3R antagonist blocked DHPG-LTD in both WT and Tg mice without blocking the reduction of PPF during DHPG-LTD. These results suggested that the PKC activity-dependent pathway and the IP3R-dependent pathway are differentially regulated by overexpressed IP3K-A during DHPG-LTD expression. Such a timely modulation has been shown in an Aplysia model system, where induction of long-term facilitation comprises the early short-term state, in which spontaneous release increases, and the intermediate-term stage, in which increases of postsynaptic IP3 and intracellular Ca2+ result in the recruitment of AMPAR clusters [54].
A summary of the DHPG-LTD model based on the results of the present study follows. In WT mice, the initial strength of the postsynaptic component is maintained constant, continuously overcoming the presynaptic component. In Tg mice, the strength of both the presynaptic and postsynaptic components is increased relative to that in WT mice. However, the much stronger postsynaptic component decays over a time, a process that is PKC-dependent. Thus, overexpressed IP3K-A appears to enhance both presynaptic and postsynaptic components as well as the late decay of the postsynaptic component in DHPG-LTD. Therefore, our results in Tg mice indicated that DHPG-LTD is expressed by a dynamic regulation of both presynaptic and postsynaptic function. Without these Tg mice, these dynamic regulatory roles of IP3K-A on synaptic function might not have been discovered.
In conclusion, overexpression of IP3K-A in the forebrain of the mouse increased the presynaptic vesicle pool of CA3 neuron terminals, presynaptic release probability, and basal postsynaptic AMPAR currents without significant effects on the spine density or morphology of CA1 neurons. The PKC-independent mGluR-LTD in WT mice was converted to PKC-dependent in mice overexpressing IP3K-A. The very early period of NMDAR-dependent LTP was selectively enhanced in Tg mice. The synaptic transmission of hippocampal CA1 neurons in WT mice appeared to be modulated by interactions among IP3K-A, PKC, and IP3R activities, which changed depending on the input signaling path. In IP3K-A overexpression, DHPG-LTD is impaired by mechanisms that are PKC- and IP3R-dependent. The present results using a gain of function approach together with those of previous KO studies show that abnormal expression of IP3K-A reveals unique and complex roles of IP3K-A in NMDAR- and mGluR-dependent synaptic plasticity models and some hippocampal-dependent learning tasks.
Supporting information
IP3K-A overexpression was induced in the hippocampus of young mice by Dox administration through breast-feeding. Following electrophysiological analysis in the 4-week-old WT and Tg mice brain, CA1 and CA3 tissues were analyzed using Western blot.
(TIF)
Tg mice fed Dox at the indicated dose for 2 weeks. IP3K-A protein expression of brain sagittal section was detected with anti-IP3K-A and Cy3-conjugated secondary antibody.
(TIF)
Brain lysates were immunoblotted with IP3K-A and β-actin antibodies. The mice were fed Dox-containing food for 0, 3, 6, 9, 12, or 15 days, and after 15 days of feeding Dox-containing food, fed Dox-free food for 7, 14, 21, 28, 35, 42 or 49 days, respectively. Hippocampal lysates were prepared at each time point. Time-dependent expression of the IP3K-A protein was analyzed by immunoblotting with IP3K-A and β-actin antibodies (A) and quantified (B).
(TIF)
Tg mice showed increased total visits in the radial arm maze test. Tg mice tended to show increased total visits in early sessions (S1-S3) (A) and showed a higher number of visits in early phase (S1-S3), but not late phase (S4-S7) (B) (WT, n = 15; Tg, n = 12).
(TIF)
Hippocampal lysates from adult WT or Tg mice were analyzed using Western blot with several antibodies of synaptic molecules (left). Densitometric quantification normalized with GAPDH did not show any significant difference between WT and Tg mice except for IP3K-A (right, n = 4).
(TIF)
(A1–A3), 1 μM Rö-31-8220 pre-incubation. (A1), fEPSP slope vs. presynaptic fiber volley amplitude plot. (A2), linear regression slope values of each slice from the fEPSP I-O plot (A1). WT, 2.34 ± 0.26, n = 12 slices; Tg, 2.25 ± 0.11, n = 15; p = 0.72. (A3), the PPF ratios at five intervals showed no significant difference between WT and Tg mice. (B1–B3), 1 μM Calphostin C pre-incubation. (B1), fEPSP slope vs. presynaptic fiver volley amplitude plot. (B2), linear regression slope values of each slice from the fEPSP I-O plot (left). WT, 2.05 ± 0.37, n = 6; Tg, 1.97 ± 0.16, n = 6; p = 0.84. (B3), the PPF ratios at five intervals showed no significant difference between WT and Tg mice. (C1–C3), 16 μM 2-APB pre-incubation. (C1), fEPSP slope vs. presynaptic fiver volley amplitude plot. (C2), linear regression slope values of each slice from the fEPSP I-O plot (C1). WT, 2.18 ± 0.17, n = 15; Tg, 2.45 ± 0.19, n = 20; p = 0.32. (C3), the PPF ratios at five intervals showed no significant difference between WT and Tg mice.
(TIF)
Upper plot, PP-LFS produced LTD of young CA1 synapses of both WT and Tg mice. LTD was induced by PP-LFS protocol (1Hz, 900 paired-pulses at 40 msec interval) at the CA1 dendritic region of the hippocampal slice. The slope of fEPSP was 86.9 ± 1.4% (WT, n = 8 slices) and 83.3 ± 0.6% (Tg, n = 10) at 60–65 min (grey bar, p = 0.11). Lower plot shows the normalized PPF ratio of the fEPSP slopes and the average values at 60–65 min were statistically compared (WT, 1.04 ± 0.57E-2; Tg, 1.09 ± 0.55E-2, p < 0.0001).
(TIF)
(A), Basal I-O plot in KN62 (or KN93) pre-incubated slices. Left, fEPSP slope vs. presynaptic fiver volley amplitude plot. Control (0.5% DMSO or 2 μM KN92), n = 23 slices; KN62 or KN93, n = 28. Right, linear regression slope values of each slice from the fEPSP I-O plot (left). WT, 2.18 ± 0.16; Tg, 2.25 ± 0.12, n = 15; p = 0.71. (B), Paired pule ratio in KN62 (or KN93) pre-incubated slices. PPF ratios at the five intervals indicated no significant difference between wild type and OX mutant mice. (C), DHPG-induced LTD in KN62 (or KN93) pre-incubated slices. 2HM KN62 (or KN93) or control 0.5% DMSO (or 2μM KN92) was applied during the whole recording period. 10 minute application of DHPG (50μM) caused LTD of CA1 synapses in wild type mice (measured at 51–60 min, control: 81.4 ± 0.4% of baseline, n = 23, p < 0.001, KN62/KN93: 79.4 ± 0.3%, n = 28, p < 0.001).
(TIF)
Acknowledgments
This work was supported by National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning (NRF-2017M3C7A1048566, NRF-2017M3C7A1079692 to H. Kim; NRF-2016M3C7A1905119 to C-H. Kim) and KIST Institutional Program (2E28412 to C-H. Kim)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning (NRF-2017M3C7A1048566, NRF-2012M3A9B6055378, NRF-2017M3C7A1079692 to H. Kim; NRF-2016M3C7A1905119 to C-H. Kim) and KIST Institutional Program (2E28412 to C-H. Kim).
References
- 1.Erneux C, De Smedt F, Moreau C, Rider M, Communi D. Production of recombinant human brain type I inositol-1,4,5-trisphosphate 5-phosphatase in Escherichia coli. Lack of phosphorylation by protein kinase C. Eur J Biochem. 1995;234(2):598–602. Epub 1995/12/01. . [DOI] [PubMed] [Google Scholar]
- 2.Erneux C, Lemos M, Verjans B, Vanderhaeghen P, Delvaux A, Dumont JE. Soluble and particulate Ins(1,4,5)P3/Ins(1,3,4,5)P4 5-phosphatase in bovine brain. Eur J Biochem. 1989;181(2):317–22. Epub 1989/05/01. . [DOI] [PubMed] [Google Scholar]
- 3.Kim IH, Park SK, Hong ST, Jo YS, Kim EJ, Park EH, et al. Inositol 1,4,5-trisphosphate 3-kinase a functions as a scaffold for synaptic Rac signaling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29(44):14039–49. Epub 2009/11/06. doi: 10.1523/JNEUROSCI.2483-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee D, Lee HW, Hong S, Choi BI, Kim HW, Han SB, et al. Inositol 1,4,5-trisphosphate 3-kinase A is a novel microtubule-associated protein: PKA-dependent phosphoregulation of microtubule binding affinity. The Journal of biological chemistry. 2012;287(19):15981–95. Epub 2012/03/06. doi: 10.1074/jbc.M112.344101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schell MJ, Erneux C, Irvine RF. Inositol 1,4,5-trisphosphate 3-kinase A associates with F-actin and dendritic spines via its N terminus. The Journal of biological chemistry. 2001;276(40):37537–46. Epub 2001/07/27. doi: 10.1074/jbc.M104101200 . [DOI] [PubMed] [Google Scholar]
- 6.Ashour DJ, Pelka B, Jaaks P, Wundenberg T, Blechner C, Zobiak B, et al. The catalytic domain of inositol-1,4,5-trisphosphate 3-kinase-a contributes to ITPKA-induced modulation of F-actin. Cytoskeleton (Hoboken). 2015;72(2):93–100. Epub 2015/01/27. doi: 10.1002/cm.21208 . [DOI] [PubMed] [Google Scholar]
- 7.Jun K, Choi G, Yang SG, Choi KY, Kim H, Chan GC, et al. Enhanced hippocampal CA1 LTP but normal spatial learning in inositol 1,4,5-trisphosphate 3-kinase(A)-deficient mice. Learn Mem. 1998;5(4–5):317–30. Epub 1999/08/24. [PMC free article] [PubMed] [Google Scholar]
- 8.Windhorst S, Minge D, Bahring R, Huser S, Schob C, Blechner C, et al. Inositol-1,4,5-trisphosphate 3-kinase A regulates dendritic morphology and shapes synaptic Ca2+ transients. Cell Signal. 2012;24(3):750–7. Epub 2011/11/29. doi: 10.1016/j.cellsig.2011.11.010 . [DOI] [PubMed] [Google Scholar]
- 9.Kim IH, Park SK, Sun W, Kang Y, Kim HT, Kim H. Spatial learning enhances the expression of inositol 1,4,5-trisphosphate 3-kinase A in the hippocampal formation of rat. Brain Res Mol Brain Res. 2004;124(1):12–9. Epub 2004/04/20. doi: 10.1016/j.molbrainres.2003.12.016 . [DOI] [PubMed] [Google Scholar]
- 10.Sun W, Kang Y, Kim IH, Kim EH, Rhyu IJ, Kim HJ, et al. Inhibition of rat brain inositol 1,4,5-trisphosphate 3-kinase A expression by kainic acid. Neuroscience letters. 2006;392(3):181–6. Epub 2005/10/18. doi: 10.1016/j.neulet.2005.09.048 . [DOI] [PubMed] [Google Scholar]
- 11.Chung S, Kim IH, Lee D, Park K, Kim JY, Lee YK, et al. The role of inositol 1,4,5-trisphosphate 3-kinase A in regulating emotional behavior and amygdala function. Sci Rep. 2016;6:23757 Epub 2016/04/08. doi: 10.1038/srep23757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michalon A, Koshibu K, Baumgartel K, Spirig DH, Mansuy IM. Inducible and neuron-specific gene expression in the adult mouse brain with the rtTA2S-M2 system. Genesis. 2005;43(4):205–12. Epub 2005/12/13. doi: 10.1002/gene.20175 . [DOI] [PubMed] [Google Scholar]
- 13.Hallett PJ, Collins TL, Standaert DG, Dunah AW. Biochemical fractionation of brain tissue for studies of receptor distribution and trafficking. Curr Protoc Neurosci. 2008;Chapter 1:Unit 1 16 Epub 2008/04/23. doi: 10.1002/0471142301.ns0116s42 . [DOI] [PubMed] [Google Scholar]
- 14.Miyakawa T, Yared E, Pak JH, Huang FL, Huang KP, Crawley JN. Neurogranin null mutant mice display performance deficits on spatial learning tasks with anxiety related components. Hippocampus. 2001;11(6):763–75. Epub 2002/01/29. doi: 10.1002/hipo.1092 . [DOI] [PubMed] [Google Scholar]
- 15.Yamada M, Kakita A, Mizuguchi M, Rhee SG, Kim SU, Ikuta F. Specific expression of inositol 1,4,5-trisphosphate 3-kinase in dendritic spines. Brain research. 1993;606(2):335–40. Epub 1993/03/26. . [DOI] [PubMed] [Google Scholar]
- 16.Yamada M, Kakita A, Mizuguchi M, Rhee SG, Kim SU, Ikuta F. Ultrastructural localization of inositol 1,4,5-trisphosphate 3-kinase in rat cerebellar cortex. Brain research. 1992;578(1–2):41–8. Epub 1992/04/24. . [DOI] [PubMed] [Google Scholar]
- 17.Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18(6):969–82. Epub 1997/06/01. . [DOI] [PubMed] [Google Scholar]
- 18.Nicoll RA, Oliet SH, Malenka RC. NMDA receptor-dependent and metabotropic glutamate receptor-dependent forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neurobiology of learning and memory. 1998;70(1–2):62–72. Epub 1998/10/01. doi: 10.1006/nlme.1998.3838 . [DOI] [PubMed] [Google Scholar]
- 19.Fitzjohn SM, Kingston AE, Lodge D, Collingridge GL. DHPG-induced LTD in area CA1 of juvenile rat hippocampus; characterisation and sensitivity to novel mGlu receptor antagonists. Neuropharmacology. 1999;38(10):1577–83. Epub 1999/10/26. . [DOI] [PubMed] [Google Scholar]
- 20.Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. The Journal of biological chemistry. 1992;267(19):13361–8. Epub 1992/07/05. . [PubMed] [Google Scholar]
- 21.Aramori I, Nakanishi S. Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron. 1992;8(4):757–65. Epub 1992/04/01. . [DOI] [PubMed] [Google Scholar]
- 22.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258(5082):607–14. Epub 1992/10/23. . [DOI] [PubMed] [Google Scholar]
- 23.Luo JH, Weinstein IB. Calcium-dependent activation of protein kinase C. The role of the C2 domain in divalent cation selectivity. The Journal of biological chemistry. 1993;268(31):23580–4. Epub 1993/11/05. . [PubMed] [Google Scholar]
- 24.Mockett BG, Guevremont D, Wutte M, Hulme SR, Williams JM, Abraham WC. Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31(20):7380–91. Epub 2011/05/20. doi: 10.1523/JNEUROSCI.6656-10.2011 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnabel R, Kilpatrick IC, Collingridge GL. Protein phosphatase inhibitors facilitate DHPG-induced LTD in the CA1 region of the hippocampus. British journal of pharmacology. 2001;132(5):1095–101. Epub 2001/02/28. doi: 10.1038/sj.bjp.0703905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhardwaj SK, Ryan RT, Wong TP, Srivastava LK. Loss of dysbindin-1, a risk gene for schizophrenia, leads to impaired group 1 metabotropic glutamate receptor function in mice. Front Behav Neurosci. 2015;9:72 Epub 2015/04/11. doi: 10.3389/fnbeh.2015.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camodeca N, Breakwell NA, Rowan MJ, Anwyl R. Induction of LTD by activation of group I mGluR in the dentate gyrus in vitro. Neuropharmacology. 1999;38(10):1597–606. Epub 1999/10/26. . [DOI] [PubMed] [Google Scholar]
- 28.Heinke B, Sandkuhler J. Signal transduction pathways of group I metabotropic glutamate receptor-induced long-term depression at sensory spinal synapses. Pain. 2005;118(1–2):145–54. Epub 2005/09/28. doi: 10.1016/j.pain.2005.08.004 . [DOI] [PubMed] [Google Scholar]
- 29.Chen A, Hu WW, Jiang XL, Potegal M, Li H. Molecular mechanisms of group I metabotropic glutamate receptor mediated LTP and LTD in basolateral amygdala in vitro. Psychopharmacology. 2017;234(4):681–94. Epub 2016/12/29. doi: 10.1007/s00213-016-4503-7 . [DOI] [PubMed] [Google Scholar]
- 30.Rush AM, Wu J, Rowan MJ, Anwyl R. Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J Neurosci. 2002;22(14):6121–8. Epub 2002/07/18. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, et al. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396(6710):433–9. Epub 1998/12/16. doi: 10.1038/24790 . [DOI] [PubMed] [Google Scholar]
- 32.Gerlai R, Henderson JT, Roder JC, Jia Z. Multiple behavioral anomalies in GluR2 mutant mice exhibiting enhanced LTP. Behavioural brain research. 1998;95(1):37–45. Epub 1998/10/01. . [DOI] [PubMed] [Google Scholar]
- 33.Jolas T, Zhang XS, Zhang Q, Wong G, Del Vecchio R, Gold L, et al. Long-term potentiation is increased in the CA1 area of the hippocampus of APP(swe/ind) CRND8 mice. Neurobiology of disease. 2002;11(3):394–409. Epub 2003/02/15. . [DOI] [PubMed] [Google Scholar]
- 34.Kim JI, Lee HR, Sim SE, Baek J, Yu NK, Choi JH, et al. PI3Kgamma is required for NMDA receptor-dependent long-term depression and behavioral flexibility. Nature neuroscience. 2011;14(11):1447–54. Epub 2011/10/25. doi: 10.1038/nn.2937 . [DOI] [PubMed] [Google Scholar]
- 35.Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S, et al. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron. 2008;58(1):104–17. Epub 2008/04/11. doi: 10.1016/j.neuron.2008.01.039 . [DOI] [PubMed] [Google Scholar]
- 36.Yokoi M, Kobayashi K, Manabe T, Takahashi T, Sakaguchi I, Katsuura G, et al. Impairment of hippocampal mossy fiber LTD in mice lacking mGluR2. Science. 1996;273(5275):645–7. Epub 1996/08/02. . [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Seo J, Nair R, Han S, Jang S, Kim K, et al. DGKiota regulates presynaptic release during mGluR-dependent LTD. EMBO J. 2011;30(1):165–80. Epub 2010/12/02 doi: 10.1038/emboj.2010.286 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamsler A, McHugh TJ, Gerber D, Huang SY, Tonegawa S. Presynaptic m1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(4):1618–23. Epub 2010/01/19. doi: 10.1073/pnas.0912540107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simkus CR, Stricker C. The contribution of intracellular calcium stores to mEPSCs recorded in layer II neurones of rat barrel cortex. The Journal of physiology. 2002;545(Pt 2):521–35. Epub 2002/11/29. doi: 10.1113/jphysiol.2002.022103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lauri SE, Bortolotto ZA, Nistico R, Bleakman D, Ornstein PL, Lodge D, et al. A role for Ca2+ stores in kainate receptor-dependent synaptic facilitation and LTP at mossy fiber synapses in the hippocampus. Neuron. 2003;39(2):327–41. Epub 2003/07/23. . [DOI] [PubMed] [Google Scholar]
- 41.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21(1):13–26. Epub 1998/08/11. . [DOI] [PubMed] [Google Scholar]
- 42.Szinyei C, Behnisch T, Reiser G, Reymann KG. Inositol 1,3,4,5-tetrakisphosphate enhances long-term potentiation by regulating Ca2+ entry in rat hippocampus. The Journal of physiology. 1999;516 Pt 3):855–68. Epub 1999/04/14. doi: 10.1111/j.1469-7793.1999.0855u.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bengtson CP, Bading H. Nuclear calcium signaling. Advances in experimental medicine and biology. 2012;970:377–405. Epub 2012/02/22. doi: 10.1007/978-3-7091-0932-8_17 . [DOI] [PubMed] [Google Scholar]
- 44.Lu W, Khatri L, Ziff EB. Trafficking of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA) receptor subunit GluA2 from the endoplasmic reticulum is stimulated by a complex containing Ca2+/calmodulin-activated kinase II (CaMKII) and PICK1 protein and by release of Ca2+ from internal stores. The Journal of biological chemistry. 2014;289(27):19218–30. Epub 2014/05/17. doi: 10.1074/jbc.M113.511246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pick JE, Khatri L, Sathler MF, Ziff EB. mGluR long-term depression regulates GluA2 association with COPII vesicles and exit from the endoplasmic reticulum. The EMBO journal. 2017;36(2):232–44. Epub 2016/11/20. doi: 10.15252/embj.201694526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng C, Deng QQ, Liu LL, Wang MY, Zhang G, Sheng WL, et al. Orexin-A differentially modulates AMPA-preferring responses of ganglion cells and amacrine cells in rat retina. Neuropharmacology. 2015;93:80–93. Epub 2015/02/07. doi: 10.1016/j.neuropharm.2015.01.016 . [DOI] [PubMed] [Google Scholar]
- 47.Mihalas S. Calcium messenger heterogeneity: a possible signal for spike timing-dependent plasticity. Frontiers in computational neuroscience. 2011;4:158 Epub 2011/01/25. doi: 10.3389/fncom.2010.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez de Sevilla D, Nunez A, Borde M, Malinow R, Buno W. Cholinergic-mediated IP3-receptor activation induces long-lasting synaptic enhancement in CA1 pyramidal neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28(6):1469–78. Epub 2008/02/08. doi: 10.1523/JNEUROSCI.2723-07.2008 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernandez de Sevilla D, Buno W. The muscarinic long-term enhancement of NMDA and AMPA receptor-mediated transmission at Schaffer collateral synapses develop through different intracellular mechanisms. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30(33):11032–42. Epub 2010/08/20. doi: 10.1523/JNEUROSCI.1848-10.2010 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emptage NJ, Reid CA, Fine A, Bliss TV. Optical quantal analysis reveals a presynaptic component of LTP at hippocampal Schaffer-associational synapses. Neuron. 2003;38(5):797–804. Epub 2003/06/12. . [DOI] [PubMed] [Google Scholar]
- 51.Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25(11):2992–3001. Epub 2005/03/18. doi: 10.1523/JNEUROSCI.3652-04.2005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rammes G, Palmer M, Eder M, Dodt HU, Zieglgansberger W, Collingridge GL. Activation of mGlu receptors induces LTD without affecting postsynaptic sensitivity of CA1 neurons in rat hippocampal slices. The Journal of physiology. 2003;546(Pt 2):455–60. Epub 2003/01/16. doi: 10.1113/jphysiol.2002.033514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanderson TM, Collingridge GL, Fitzjohn SM. Differential trafficking of AMPA receptors following activation of NMDA receptors and mGluRs. Mol Brain. 2011;4:30 Epub 2011/07/29. doi: 10.1186/1756-6606-4-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin I, Udo H, Rayman JB, Puthanveettil S, Kandel ER, Hawkins RD. Spontaneous transmitter release recruits postsynaptic mechanisms of long-term and intermediate-term facilitation in Aplysia. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(23):9137–42. Epub 2012/05/24. doi: 10.1073/pnas.1206846109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
IP3K-A overexpression was induced in the hippocampus of young mice by Dox administration through breast-feeding. Following electrophysiological analysis in the 4-week-old WT and Tg mice brain, CA1 and CA3 tissues were analyzed using Western blot.
(TIF)
Tg mice fed Dox at the indicated dose for 2 weeks. IP3K-A protein expression of brain sagittal section was detected with anti-IP3K-A and Cy3-conjugated secondary antibody.
(TIF)
Brain lysates were immunoblotted with IP3K-A and β-actin antibodies. The mice were fed Dox-containing food for 0, 3, 6, 9, 12, or 15 days, and after 15 days of feeding Dox-containing food, fed Dox-free food for 7, 14, 21, 28, 35, 42 or 49 days, respectively. Hippocampal lysates were prepared at each time point. Time-dependent expression of the IP3K-A protein was analyzed by immunoblotting with IP3K-A and β-actin antibodies (A) and quantified (B).
(TIF)
Tg mice showed increased total visits in the radial arm maze test. Tg mice tended to show increased total visits in early sessions (S1-S3) (A) and showed a higher number of visits in early phase (S1-S3), but not late phase (S4-S7) (B) (WT, n = 15; Tg, n = 12).
(TIF)
Hippocampal lysates from adult WT or Tg mice were analyzed using Western blot with several antibodies of synaptic molecules (left). Densitometric quantification normalized with GAPDH did not show any significant difference between WT and Tg mice except for IP3K-A (right, n = 4).
(TIF)
(A1–A3), 1 μM Rö-31-8220 pre-incubation. (A1), fEPSP slope vs. presynaptic fiber volley amplitude plot. (A2), linear regression slope values of each slice from the fEPSP I-O plot (A1). WT, 2.34 ± 0.26, n = 12 slices; Tg, 2.25 ± 0.11, n = 15; p = 0.72. (A3), the PPF ratios at five intervals showed no significant difference between WT and Tg mice. (B1–B3), 1 μM Calphostin C pre-incubation. (B1), fEPSP slope vs. presynaptic fiver volley amplitude plot. (B2), linear regression slope values of each slice from the fEPSP I-O plot (left). WT, 2.05 ± 0.37, n = 6; Tg, 1.97 ± 0.16, n = 6; p = 0.84. (B3), the PPF ratios at five intervals showed no significant difference between WT and Tg mice. (C1–C3), 16 μM 2-APB pre-incubation. (C1), fEPSP slope vs. presynaptic fiver volley amplitude plot. (C2), linear regression slope values of each slice from the fEPSP I-O plot (C1). WT, 2.18 ± 0.17, n = 15; Tg, 2.45 ± 0.19, n = 20; p = 0.32. (C3), the PPF ratios at five intervals showed no significant difference between WT and Tg mice.
(TIF)
Upper plot, PP-LFS produced LTD of young CA1 synapses of both WT and Tg mice. LTD was induced by PP-LFS protocol (1Hz, 900 paired-pulses at 40 msec interval) at the CA1 dendritic region of the hippocampal slice. The slope of fEPSP was 86.9 ± 1.4% (WT, n = 8 slices) and 83.3 ± 0.6% (Tg, n = 10) at 60–65 min (grey bar, p = 0.11). Lower plot shows the normalized PPF ratio of the fEPSP slopes and the average values at 60–65 min were statistically compared (WT, 1.04 ± 0.57E-2; Tg, 1.09 ± 0.55E-2, p < 0.0001).
(TIF)
(A), Basal I-O plot in KN62 (or KN93) pre-incubated slices. Left, fEPSP slope vs. presynaptic fiver volley amplitude plot. Control (0.5% DMSO or 2 μM KN92), n = 23 slices; KN62 or KN93, n = 28. Right, linear regression slope values of each slice from the fEPSP I-O plot (left). WT, 2.18 ± 0.16; Tg, 2.25 ± 0.12, n = 15; p = 0.71. (B), Paired pule ratio in KN62 (or KN93) pre-incubated slices. PPF ratios at the five intervals indicated no significant difference between wild type and OX mutant mice. (C), DHPG-induced LTD in KN62 (or KN93) pre-incubated slices. 2HM KN62 (or KN93) or control 0.5% DMSO (or 2μM KN92) was applied during the whole recording period. 10 minute application of DHPG (50μM) caused LTD of CA1 synapses in wild type mice (measured at 51–60 min, control: 81.4 ± 0.4% of baseline, n = 23, p < 0.001, KN62/KN93: 79.4 ± 0.3%, n = 28, p < 0.001).
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.