

Abstract
Populations of isogenic cells often respond coherently to signals, despite differences in protein abundance and cell state. Previously, we uncovered processes in the Saccharomyces cerevisiae pheromone response system (PRS) that reduced cell‐to‐cell variability in signal strength and cellular response. Here, we screened 1,141 non‐essential genes to identify 50 “variability genes”. Most had distinct, separable effects on strength and variability of the PRS, defining these quantities as genetically distinct “axes” of system behavior. Three genes affected cytoplasmic microtubule function: BIM1, GIM2, and GIM4. We used genetic and chemical perturbations to show that, without microtubules, PRS output is reduced but variability is unaffected, while, when microtubules are present but their function is perturbed, output is sometimes lowered, but its variability is always high. The increased variability caused by microtubule perturbations required the PRS MAP kinase Fus3 and a process at or upstream of Ste5, the membrane‐localized scaffold to which Fus3 must bind to be activated. Visualization of Ste5 localization dynamics demonstrated that perturbing microtubules destabilized Ste5 at the membrane signaling site. The fact that such microtubule perturbations cause aberrant fate and polarity decisions in mammals suggests that microtubule‐dependent signal stabilization might also operate throughout metazoans.
Keywords: cell‐to‐cell variability, genetic screen, MAP kinase, microtubules, noise
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Quantitative Biology & Dynamical Systems; Signal Transduction
Introduction
Cell signaling systems transmit information about the external environment, enabling cells to respond to extracellular signals. Accurate signal transmission and response of individual cells, and coherence in cell population response, are critical for the choreographed sequence of signal and response during embryonic development, and for regulated cell division and differentiation during tissue maintenance in the adult. Variability in cell responses is well recognized and widespread, from Escherichia coli infected with phages (Delbrück, 1945), to mammalian cells subjected to pro‐apoptotic signals (Spencer et al, 2009 and Appendix). However, the means by which cells transmit and respond to signals accurately, and so manifest coherent population responses, remain largely unknown.
We and others have studied cell‐to‐cell variability, using the cell fate decision system that controls mating in Saccharomyces cerevisiae, the pheromone response system (PRS) (Colman‐Lerner et al, 2005; Yu et al, 2006; Paliwal et al, 2007; Ricicova et al, 2013). The PRS has elements prototypic for many other signaling systems: It uses a GPCR, which, when bound by pheromone, couples via a G‐protein to a scaffold‐dependent MAPK cascade (Dohlman & Thorner, 2001; Fig 1). In this cascade, there are two partially redundant MAP kinases, Fus3 and Kss1, each able to activate downstream steps. After activation, receptors, G‐proteins, and the scaffold concentrate into a membrane patch (Suchkov et al, 2010; Ventura et al, 2014; Ismael et al, 2016) here called the signaling site. The cell converts extracellular ligand concentration into an occupancy measurement (Brent, 2009) by determining the ratio of ligand‐occupied to unoccupied receptors (Bush et al, 2016) and transmitting that information accurately, via negative feedback (Yu et al, 2008) and “push–pull” mechanisms (Andrews et al, 2016). Signaling causes outputs including induction of genes at appropriate levels (here called “system output”) that depend on a set of proteins that constitute the signaling arm of the PRS. Determination of the direction of a gradient of pheromone concentration, and subsequent growth toward a mating partner, depends on a partly overlapping set of proteins, the polarity determination arm of the system. Our previous work quantified system output by expression from PRS‐responsive and control reporter genes. It separated the cell‐to‐cell variability in output into two contributions. The first of these was from cell‐to‐cell variability in the pathway subsystem, P (includes all events upstream of the promoter of the reporter gene), quantified as η2(P) (η2 = variance/mean2), and here called “pathway variability”. The second contribution was from variability in events related to reporter gene expression, either due to (i) preexisting differences in the general capacity of cells to express genes into proteins, G, quantified as η2(G), and here called “variability in gene expression” or (ii) rapid‐acting changes in gene expression due to “intrinsic noise”, which we quantified as η2(γ). In this previous work, we made the assumption that cell‐to‐cell differences in (P) were composed of η2(L), (differences in L, the capacity component of the signal transmission subsystem at the start of the experiment) and η2(λ), (rapid‐acting changes in signal during the measurement) but we could not separate η2(L) and η2(λ) experimentally.
Figure 1. The signaling arm of the yeast pheromone response system (PRS).
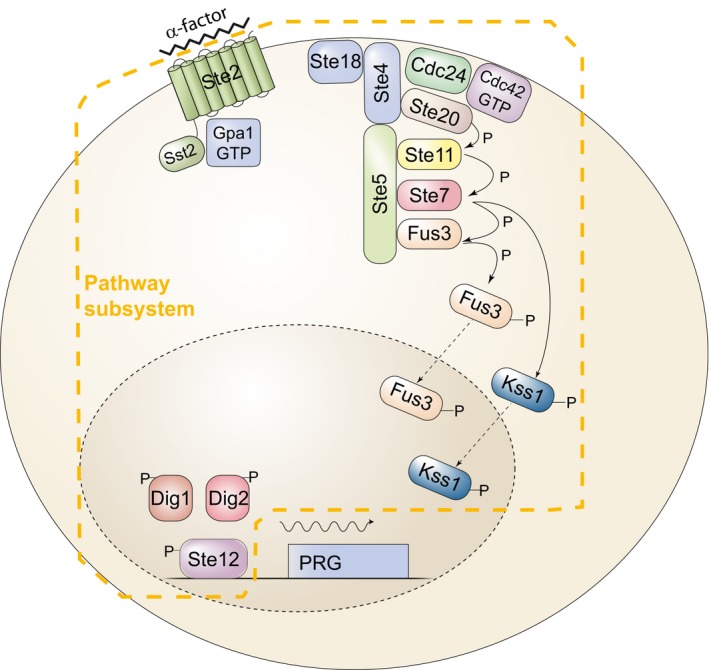
Binding of the ligand, α‐factor, to a seven‐helix transmembrane receptor, Ste2, in the MATa cell depicted, causes the dissociation of the α subunit of a trimeric G‐protein, Gpa1, from the βγ dimer, Ste4/Ste18. This event causes the recruitment to the plasma membrane of the scaffold protein Ste5, leading to the assembly and activation of the MAP kinase cascade (MAPKKK Ste11, MAPKK Ste7) and the detachment from the scaffold of the Erk1/2‐like MAPKs Fus3 and Kss1 (Dohlman & Thorner, 2001). In the cytoplasm, activated Fus3 and Kss1 phosphorylate targets including Ste5 (Bhattacharyya et al, 2006; Malleshaiah et al, 2010), and in the nucleus, they phosphorylate Dig1, Dig2, and Ste12 (Tedford et al, 1997). These events comprise the pathway subsystem, P; that is, the subsystem that transmits the signal to the promoters of inducible genes. Activation of Ste12 leads to the induction of approximately 100 pheromone‐responsive genes (PRGs) (Roberts et al, 2000) and their expression via the expression subsystem G (defined in the text).
Four lines of evidence show that cell‐to‐cell variation and pathway variability, η2(P), is under active control. First, pathway subsystem output P correlates negatively with gene expression capacity G, indicating a compensatory mechanism that reduces variability in system output (Colman‐Lerner et al, 2005). Second, mutations in either of the PRS MAPKs Kss1 and Fus3 affect η2(P), and do so differently (Colman‐Lerner et al, 2005). Third, maintenance of the matching dose‐response relationship between system output and system activity, which reduces the amplification of stochastic noise η2(λ) during signal transmission at an intermediate point requires the action of negative feedback from Fus3 (Yu et al, 2008). Fourth, we showed recently (Bush et al, 2016) that a push–pull mechanism suppresses cell‐to‐cell differences in signal‐dependent gene expression caused by changes in the abundance of the receptor. Here, we hypothesized that there might be additional mechanisms that regulate (or suppress) variability in transmitted signal.
Results
Large‐scale screen identifies genes whose products affect pathway variability
To identify genes that affected cell‐to‐cell variability, we first constructed a whole genome collection of yeast carrying the necessary reporters and mutations in each non‐essential gene. To do this, we extended established methods facilitating genetic crosses of arrayed collections (Tong et al 2004, see Appendix).
During our initial characterization of cell‐to‐cell variability phenotypes in our collection we found that, for many gene deletions, the patches of post‐sporulation segregants contained varying numbers of colonies of genetically variant haploids, likely arising from chromosomal mis‐segregation during meiosis (Hughes et al 2000), same sex diploid formation (Giaever & Nislow, 2014) and possibly also from mutations present in some cells in the starting collection. While such heterogeneity, if present, might have not had a large impact on phenotypes studied before in similar collections (Jonikas et al, 2009; Neklesa & Davis, 2009; Wolinski et al, 2009; Ayer et al, 2012), our measures of cell‐to‐cell variability were very sensitive to it. We thus generated our collection from clonal cultures derived from single colonies.
To screen the mutant collection, we optimized a flow cytometry adaptation of our microscopic methods to measure single‐cell responses. In this screen, we arrested cell cycle progression by inhibition of the Cdc28‐as2 mutant protein with the inhibitor 1NM‐PP1‐NM, incubated cells with pheromone in well plates, stopped the response by the addition of cycloheximide, and allowed time for the fluorophores to fully mature. We first tested this method on the reference strain. We extended our previous characterization of the dose response of pathway variability η2(P) to a broader range of different pheromone concentrations (0.1–30 nM). Consistent with previous microscopic measurements at just two doses (Colman‐Lerner et al, 2005), the fine‐grained dose response showed that η2(P) decreased monotonically with increasing pheromone (Fig EV1). We thus were reassured about using this approach to screen the arrayed mutant collection.
Figure EV1. Time‐dependent output and dose response of the reporter genes used in the screen.
-
A, BAverage fluorescence output of the pheromone‐inducible P PRM1 ‐mRFP reporter, O (A) and the constitutive P ACT1 ‐YFP reporter, G (B), in A.U., measured at four different doses over time.
-
CEstimating pathway variability (η2(P)). Panel shows a scatter plot, with one point per cell, of P PRM1 ‐mRFP vs. P ACT1 ‐YFP, in 500 cells stimulated with 20 nM pheromone for 3 h. The amount of pathway variability (η2(P) + η2(γ)) (a quantity very close to η2(P), see Appendix) is a measure of the average width of this distribution of plotted points about the identity line, drawn in pink.
-
DDose dependence of pathway variability and output. Plot shows pathway variability η2(P) + η2(γ) (blue), and output (gray) in SGA85 cells, as a function of pheromone dose after 180 min.
For the primary screen, we assayed 1,141 strains from the collection (996 randomly selected and 145 bearing a deletion in a non‐essential kinase or phosphatase (Appendix Table S2). We screened these for expression related variables (Table 1) that allowed us to compute pathway output (P) and/or variability in it (η2(P)), system output (O) and variability in it, and a proxy for gene expression capacity, G. Screened strains corresponded to more than 1/4 of the non‐essential yeast genes. Appendix Table S2 shows the numerical results.
Table 1.
Variables measured in isogenic cell populations
| Variable name | Short expression | Calculated as | |
|---|---|---|---|
| Median pheromone response system (PRS) output | O, <PPRM1‐mRFP> or <PPRM1‐mCherry> | Median (inducible RFP) | |
| Median constitutive or control output | G, <PACT1‐XFP> or <PBMH2‐XFP> | Median (constitutive XFP) | |
| Cell‐to‐cell variability in PRS output | η 2 (O), η2(PPRM1‐mRFP) or η2(mCherry) |
|
|
| Cell‐to‐cell variability in constitutive or control output (representing general gene expression capacity) | η 2 (G), η2(PACT1‐YFP) or η2(PBMH2‐YFP) |
|
|
| Cell‐to‐cell variability in signal transmission | η 2 (P), or η2(L + λ) | σ2(mRFP i/<mRFP> −YFP i/<YFP>) | |
| Signal strength | P | O/G |
σ2 is variance, μ is average, <B> means the average of B. Quantities in bold type are those used for selection and/or clustering analysis below.
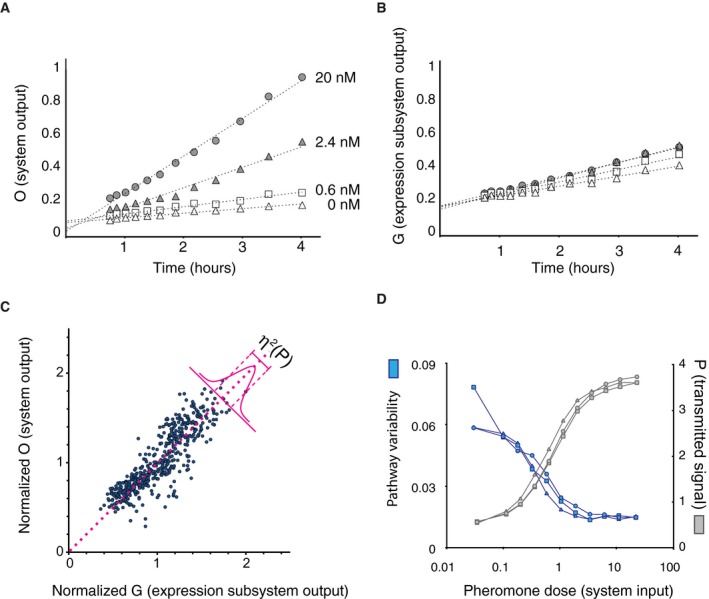
From these screened strains, we selected gene deletions for follow‐up “secondary screen” studies, based on their η2(P) and average output (O) phenotypes (Fig 2A–C). We chose selection thresholds that lay in the tails of the distributions of values measured for the 52 separate cultures of the reference strain included in the screen. From the low dose (0.6 nM pheromone) data, we selected mutants with high or low median pheromone system output (O) (Fig 2A) or high or low η2(P) (Fig 2B). From the high dose (20 nM pheromone) data, we only selected mutants that showed high η2(P) (Fig 2C). Figure 2D and E shows η2(P) vs. P, at 0.6 nM (D) and 20 nM (E) pheromone doses, for all measured strains.
Figure 2. Selection of mutants for follow‐up studies.
-
APRS output, O (median mRFP signal), in 0.6 nM pheromone.
-
BEstimated pathway variability η2(P) in 0.6 nM pheromone
-
CEstimated η2(P) in 20 nM pheromone (see Appendix Table S1).
-
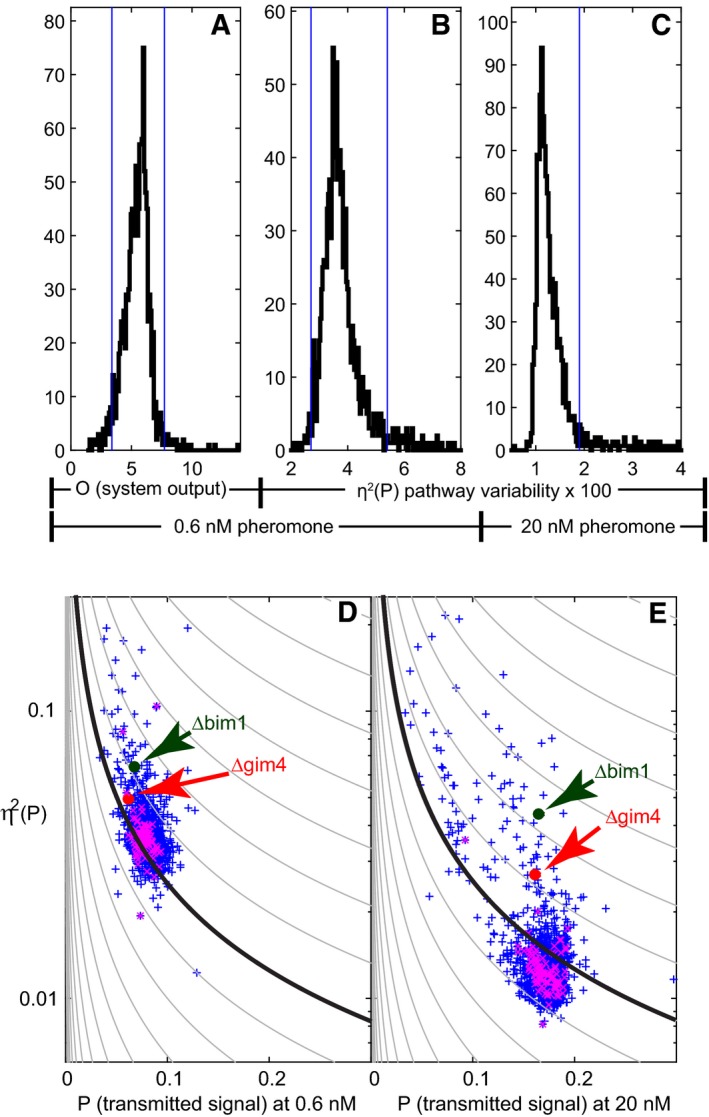
D, ESignaling variability vs. transmitted signal P (median mRFP/median YFP) for all 1,093 strains screened. Plots show an estimate of η2(P) vs. P for the same dataset displayed in Fig 4A–C. The contour lines show the expected dependence of variability on output for outputs proportional to a Poisson random variable (lower noise at higher outputs), with proportionality constants logarithmically spaced from 10−5 to 1. Purple Xs are independent replicates of the reference SGA 85 strain. Their spread gives an indication of the limits of this primary screen. The SGA 85 swarm lies below the 0.158 contour at 20 nM but above it at 0.6 nM, indicating that variability at the low dose is higher than expected from the same Poisson processes taking place at 20 nM. At 0.6 nM, Δbim1 and Δgim4 showed somewhat greater, and at 20 nM substantially greater, pathway variability than reference cells. See Appendix Table S2 for a list of all strains and their corresponding raw output and variability values.
For the secondary screen, we isolated three fresh independent haploid segregants and assayed them by flow cytometry as above (see Appendix for a complete description of primary and secondary screens). These screens identified 50 deletion strains (Table 2) that reproducibly showed changes in O or η2(P).
Table 2.
Genes found in the screen
| Gene name | Screen, criteria | Important for | Description |
|---|---|---|---|
| ARG82 | U,2 | AA metabolism | Inositol polyphosphate multikinase |
| ERV46 | U,1,4,5 | Cargo transport | ER vesicle protein, component of COPII complex; required for membrane fusion |
| HIS1 | U,1,4,5 | AA metabolism | ATP phosphoribosyltransferase |
| UGA1 | U,5 | AA metabolism | Gamma‐aminobutyrate (GABA) transaminase |
| SLA1 | U,2,3 | Actin binding | Cytoskeletal protein binding protein; required for assembly of the cortical actin cytoskeleton |
| SAP155 | K,2,3,5 | Cell cycle | Protein required for function of the Sit4 protein phosphatase |
| YER068C‐A | U,5 | Dubious open reading frame | Dubious open reading frame/overlaps with ARG5, ARG6 acetylglutamate kinase and N‐acetyl‐gamma‐glutamyl‐phosphate reductase |
| YIL032C | U,5 | Dubious open reading frame | Dubious open reading frame/next to BCY1 |
| ERG3 | U,1,5 | Ergosterol biosynthesis | C‐5 sterol desaturase |
| GAL83 | K,1 | Glucose repression | One of three possible beta‐subunits of the Snf1 kinase complex |
| GUP1 | U,2,3 | Glycerol metabolism, protein folding | Plasma membrane protein involved in remodeling GPI anchors |
| PPZ1 | K,5 | Ion homeostasis | Serine/threonine protein phosphatase Z, isoform of Ppz2p; involved in regulation of potassium transport, which affects osmotic stability, cell cycle progression, and halotolerance |
| FUS1 | U,5 | Mating | Membrane protein localized to the shmoo tip |
| AAT2 | U,5 | Metabolism | Cytosolic aspartate aminotransferase involved in nitrogen metabolism |
| PTC6 | K,2,3 | Metabolism | Mitochondrial type 2C protein phosphatase (PP2C) |
| GIM4 | U,5 | Microtubule chaperone/Protein folding | Subunit of the heterohexameric cochaperone prefoldin complex |
|
PAC10
GIM2 |
U,5 | Microtubule chaperone/Protein folding | Subunit of the heterohexameric cochaperone prefoldin complex |
| BIM1 | U,4,5 | Microtubule end binding | Microtubule plus‐end‐binding protein |
| MSH1 | U,5 | Mitochondrial homeostasis | Escherichia coli MutS homolog, binds DNA mismatches, required for mitochondrial function |
| BUB1 | K,4,5 | Mitosis | Protein kinase required for cell cycle checkpoint, delays entry into anaphase until kinetochores bound by opposing microtubules |
| ELM1 | K,2 | Morphogenesis | Serine/threonine protein kinase that regulates cellular morphogenesis |
| HSL1 | K,2 | Morphogenesis | Nim1‐related protein kinase; regulates the morphogenesis and septin checkpoints |
| NUP60 | U,5 | Nuclear transport | FG‐nucleoporin component of central core of the nuclear pore complex |
| SXM1 | U,4,5 | Nuclear transport | Nuclear transport factor (karyopherin) |
| CBR1 | U,5 | Respiration | Microsomal cytochrome β reductase |
| RTC3 | U,4,5 | RNA metabolism | Protein of unknown function involved in RNA metabolism |
| CKA1 | U,1,4,5 | Signaling | Alpha catalytic subunit of casein kinase 2 (CK2) |
| CKB1 | U,5 | Signaling | Beta regulatory subunit of casein kinase 2 (CK2) |
| CKB2 | K,1 | Signaling | Beta' regulatory subunit of casein kinase 2 (CK2) |
| FUS3 | K,3,5 | Signaling | Mitogen‐activated serine/threonine protein kinase (MAPK), part of PRS |
| HOG1 | K,3 | Signaling | Mitogen‐activated protein kinase involved in High Osmolarity (HOG) pathway |
| KSS1 | K,3 | Signaling | Mitogen‐activated protein kinase (MAPK); functions in PRS and signal transduction pathways that control filamentous growth and pheromone response |
| PBS2 | K,5 | Signaling | MAP kinase kinase of the HOG signaling pathway |
| SSK2 | K,3 | Signaling | MAP kinase kinase kinase of HOG1 signaling pathway |
| FAR1 | U,1 | Signaling/cell cycle/polarization | CDK inhibitor, nuclear anchor, recruited by Ste18‐Ste4 at polarity patch |
| SIP1 | U,4,5 | Signaling/glucose repression | Alternate beta‐subunit of the Snf1 protein kinase complex |
| KAR4 | U,5 | Signaling/mating | Transcription factor required for activation of some pheromone‐responsive genes |
| STE50 | U,1,4 | Signaling/mating | Adaptor protein, in PRS helps connect Ste20 MAPKKKK to Ste11 MAPKKK |
| SKY1 | K,3 | Splicing | SR protein kinase (SRPK); varied functions, regulates proteins involved in mRNA metabolism and cation homeostasis, helps some LexA fusion proteins bind operator |
| KIN3 | K,5 | Stress | Non‐essential serine/threonine protein kinase; possible role in DNA damage response |
| OCA1 | K,1 | Stress | Protein tyrosine phosphatase; required for cell cycle arrest in response to oxidative damage of DNA |
| CTK1 | K,4 | Transcription regulation | Catalytic (alpha) subunit of C‐terminal domain kinase I (CTDK‐I); phosphorylates RNA pol II |
| DEP1 | U,5 | Transcription regulation | Component of the Rpd3L histone deacetylase complex, variously needed for activation and repression, regulates DNA replication origin timing |
| SUM1 | U,2,4,5 | Transcription regulation | Transcriptional repressor that regulates middle‐sporulation genes; required for mitotic repression of middle‐sporulation‐specific genes; also acts as general replication initiation factor; involved in telomere maintenance, chromatin silencing |
| SWI5 | U,5 | Transcription regulation | Part of Mediator and Swi/Snf nucleosome remodeling complexes |
| UME6 | U,4,5 | Transcription regulation | Meiotic transcription regulator, DNA binding, recruits variously Sin3/Rpd3 repressor (HDAC) and Ime1 activator. |
| RPL12A | U,1,4 | Translation | Ribosomal 60S subunit protein L12A |
| RPL19B | U,1,4,5 | Translation | Ribosomal 60S subunit protein L19B |
| RPL34A | U,4,5 | Translation | Ribosomal 60S subunit protein L34A |
| ECM15 | U,1,4,5 | Unknown | Possibly tetrameric, non‐essential protein, unknown function |
| VPS64 | U,5 | Vacuole metabolism | Required for cytoplasmic proteins to enter vacuole |
Selection criteria codes (see Fig 2A–C): (1) O(0.6 nM) < 3.39; (2) O(0.6 nM) > 7.72; (3) η2(P(0.6 nM)) < 0.027; (4) η2(P(0.6 nM)) > 0.054; (5) η2(P(20 nM)) > 0.019. Genes from the strains in the unbiased (U) screen and the strains in the non‐essential kinase and phosphatase screen (K) screen that showed altered PRS system output (O), low or high pathway variability (η2(P)) at low pheromone (0.6 nM), or high variability (η2(P)) at high dose (20 nM). Table shows gene name, screen from which it was selected and selection criteria, overall functional class, and a brief description of its molecular role or activity.
We did an additional follow‐up “tertiary screen” on duplicate independent isolates of 44 of the haploid deletion strains (Appendix Table S6). For this screen, we used microscope‐based quantification of the fluorescent protein reporters. Although we did not seek to gain biological insight from observation of effects of these gene deletions on cell morphology, this microscope‐based quantification of fluorescence signal had two advantages. First, it allowed us to rule out the possibility that putative single‐cell values were actually derived from clumps of several cells. None of the mutant cultures we imaged was affected by these problems. Second, it allowed us to measure another variable, gene expression noise, η2(γ), by simultaneous quantitation of the two fluorescent protein reporters (CFP and mRFP) driven by P PRM1 (accurate CFP measurements were not possible in the flow cytometer). The tested mutants showed values of η2(γ) that were typical of the reference strain. The only significant differences were in O, η2(O), and η2(P).
Mutant genes define different axes of quantitative system behavior
To gain insight into the different phenotypes caused by these gene deletions, we grouped the mutant strains in the secondary screen using a hierarchical clustering approach based on the five variables we measured by flow cytometry, at low and high pheromone dose (Fig 3 and Appendix Table S2). Fourteen of the 19 cultures of the reference strain grouped together in one cluster (cluster I), one in cluster IIa, two in cluster IIIa, one in cluster IIIb, and one in cluster Vc. With a few exceptions (for example ∆ckb1, ∆his1, and ∆sky1) either all or all but one of the independent segregants bearing each gene deletion grouped in the same subcluster. Taken together with the results of the tertiary screen, these results show that differences in variability in strains with different gene deletions were due to the mutations. Since all 19 cultures of the SGA85 reference cells were isogenic, that five of these cultures grouped into different clusters highlight the fact that these high‐throughput flow cytometric assays sometimes perform inconsistently. Similarly, since our independent haploid segregants came from crosses with an otherwise isogenic MATα strain, we believe that the observed infrequent grouping of any single deletion's isolates into multiple clusters most likely reflects measurement anomalies rather than uncharacterized genetic differences between the MATa and MATα parents of the strains.
Figure 3. Cluster analysis of 50 genes identified as affecting variability and or pheromone response output.
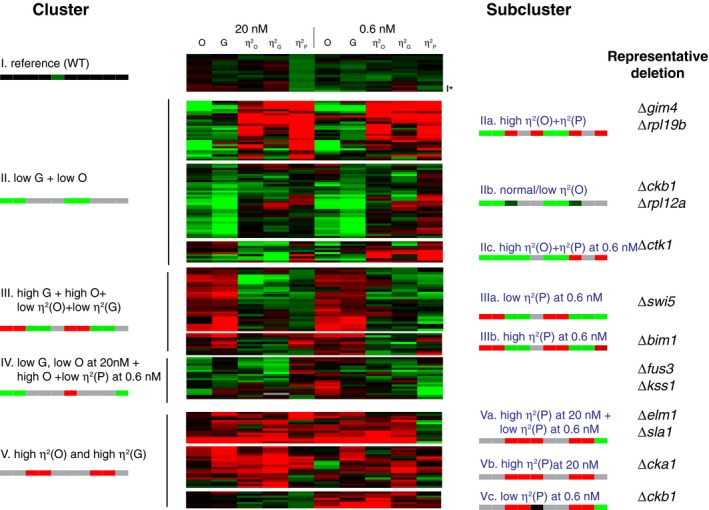
Hierarchical clustering of values derived from flow cytometry measurements from 198 cell populations (19 replicates for reference strain SGA85, four independent segregants each for 17 deletions from the kinases or phosphatase set and three independent segregants each for 37 deletions from the unbiased set). We used the Pearson correlation metric to assess distance between strains and the average linkage method to form clusters. Before clustering, we first log‐transformed the data and then median centered each row (each strain). Each strain had the following 10 measurements (five after induction with 20 nM pheromone and five after induction with 0.6 nM pheromone): O (pheromone system output), G (gene expression output), and η2(O), η2(G) and η2(P), the three cell‐to‐cell variability measurements. The panel shows these values as a “heat map”, from red (higher than the median) to black (equal to the median) to green (lower than the median). The signature pattern for each cluster or subcluster is represented with a color bar with 10 blocks, one for each measurement (gray indicates that that the measurement may take any value). Rightmost column shows representative deletion strains for each subcluster. The asterisk next to the last row of the reference cluster indicates the data are from Δfus1, which did not differ from reference in this re‐assay. Appendix Table S3 shows the raw data and Appendix Table S4 lists the clustered, log‐transformed and median‐centered dataset. Appendix Table S6 shows microscope data that complement these flow cytometer data for 44 of these 50 mutant strains selected for clustering.Source data are available online for this figure.
We noted that the pathway and gene expression output variables (O and G) were often affected by different genes than the “cell‐to‐cell variability” variables (η2(O), η2(G), η2(P)). For example, cluster II was comprised of all the entries with low pathway output (O) and low gene expression output (G). Within this cluster, there were three subclusters: strains with high or unchanged η2(O) and η2(P) (IIa), strains with low or unchanged η2(O) (IIb), and strains with high or unchanged η2(O) and η2(P) at the 0.6 nM dose (IIc). Another example was cluster III, which contained strains with predominantly high pathway output O and gene expression output G, but low or unchanged η2(O) and η2(G). Within cluster III, subclusters IIIa and IIIb were defined, respectively, by low η2(P) and high η2(P), both at the low pheromone dose. Such genetic independence strongly suggests the existence of distinct, independent mechanisms affecting the two types of quantitative phenotypes (mean output and variability) and disfavor an interpretation in which variability is inextricably linked to output strength. From this screen, mean and variability emerged as independent axes of system behavior, subject to independent regulation, in the sense that they were often independently affected by genetic changes (see Discussion).
This analysis also revealed that processes that affected variability appeared to be different at low and high doses of pheromone. This was evidenced in the subclusters within cluster V. Cluster V grouped strains with high cell‐to‐cell variability in both system output and gene expression output. Subcluster Va contained strains with high η2(P) at high pheromone and low η2(P) at low pheromone. In contrast, strains in subcluster Vb showed high, unchanged, or low η2(P) at high or low pheromone. Strains in subcluster Vc had moderately high or unchanged η2(P) at low pheromone and low η2(P) at high pheromone.
Another result of the clustering analysis was that mutations in related genes showed similar patterns of change in their set of quantitative measurements. This was expected and yet reassuring. For example, deletions of duplicated paralogs of ribosomal protein genes were grouped in subclusters IIa and IIb (distinguished by their different variability phenotypes) and those for the two PRS MAPKs, FUS3 and KSS1, grouped together in cluster IV. By analogy with dataset clustering studies based on gene expression data and other phenotypes (including our own, Colman‐Lerner et al 2001; Wolfe et al 2005), we expect that the gene deletions that shared cluster or subcluster membership might function in the same processes.
Three gene deletions with higher signaling variability affect microtubule function
Of the 50 “variability genes” so identified, six (FUS1, FUS3, KSS1, FAR1, KAR4, and STE50) were known components of the pheromone response system or induced by it, three of which (FUS3, KSS1, and STE50) we had previously shown to affect pathway variability (Colman‐Lerner et al, 2005; Pincus et al, 2013). The other 44 included genes involved in cell cycle regulation (11), gene expression—transcription, RNA processing, and nuclear pore transport (12 genes, including NUP60)—metabolism—amino acid synthesis and mitochondrial function (10)—morphogenesis—actin, tubulin, and their regulation (7)—other (2) and unassigned function (3).
Three genes that affected η2(P) were known to affect microtubule function. These were GIM4 and PAC10/GIM2, whose products form part of a six‐protein prefoldin complex needed for tubulin supply, and BIM1, whose product mediates attachment of cytoplasmic microtubule plus ends to the signaling site. We selected two of these genes, BIM1 (two out of three in cluster IIIa) and GIM4 (GIM4 two out of three in cluster IIa) as candidate genes to explore a possible relationship between microtubule function and signal variability. Although deletions of both BIM1 and GIM4 caused elevated η2(P) in the primary screen at both low and high doses, Δbim1 did not show elevated η2(P) at low doses in the secondary screen, but showed elevation at both doses in the tertiary screen. We again took these differences in measured η2(P) values as likely indicating the limitations of such measurements via the relatively high‐throughput culture in multiwell plate/flow cytometry assays rather than arising from otherwise cryptic genetic variability among isolates.
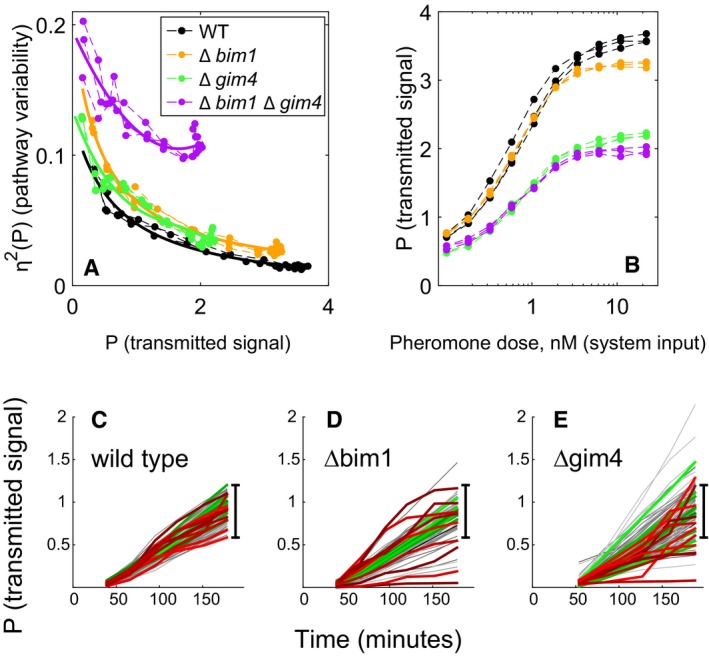
However, to address the above possibility, and to get around any possible effect of uncharacterized genetic heterogeneity among independent haploid deletion strains resulting from independent meioses, we remade these strains without meiosis, in a clean genetic background, an independently constructed BY4741 derivative, equivalent to the reference strain (Appendix). From this strain (GPY4000), we constructed ∆bim1 and ∆gim4 single deletion strains, and a ∆bim1 ∆gim4 double mutant. We characterized the behavior of these newly generated mutant strains in repeated fine‐grained dose‐response flow cytometry assays. Figure 4 shows the results. Pathway variability as a function of transmitted signal, η2(P) vs. P, was increased similarly in both deletion strains, across all pheromone doses (Fig 4A). In contrast, transmitted signal P as a function of pheromone dose was relatively unaffected in ∆bim1 but reduced by ~30% in ∆gim4 (Fig 4B). Reductions in P at a given dose merely indicate that signal transmission is less efficient in the mutant strain; it might still occur by the same process. In ∆bim1 ∆gim4 cells, the increase in pathway variability η2(P) was more than twice as large as the measured effect of the two individual deletions. This synergistic genetic interaction suggested that the two gene products acted through distinct mechanisms to affect η2(P) (see for example Fisher 1918; Boone et al 2007).
Figure 4. Increased cell‐to‐cell variability and distinct time‐dependent trajectories in Δbim1 and Δgim4 mutants.
-
A, BDeletions of BIM1 and GIM4 in clean genetic background increase signaling variability at all outputs. Data were collected from dose‐response flow cytometry measurements of reference (GPY4000), Δbim1 (GPY4001), Δgim4 (GPY4031), and ∆bim1 ∆gim4 (GPY4036) (bearing the P PRM1 ‐mCherry and P BMH2 ‐YFP reporters) stimulated for 3 h with the indicated pheromone doses. (A) η2(P) as a function of P. Solid curves are best fits of a rational polynomial model to the measurements from each strain. These models tend toward infinity as transmitted signal tends toward zero, where we expect very large relative variability. (B) P as a function of pheromone dose. Data correspond to measurements at different doses measured on the same day (three replicates).
-
C–EAccumulated signal P vs. time in reference and microtubule perturbed cells. Transmitted signal P vs. time in individual cells of the reference (GPY123, “wild type”), Δbim1 (GPY4144) and Δgim4 (GPY4150) strains (N = 103, 102, 118, respectively). We induced the PRS by addition of 20 nM pheromone to the medium and imaged cells every 30 min as previously done (Colman‐Lerner et al, 2005 and Gordon et al, 2007). In all populations, about 5% of the cells did not respond to pheromone induction. Traces correspond to pathway output (inducible P PRM1 ‐mCherry signal/constitutive P ACT1 ‐CFP signal) from individual cells followed over time. For each strain, we colored the 10 most stable trajectories green, and the 10 least stable (crooked) trajectories red. The black bar shows the full range of WT responses, for comparison.
Source data are available online for this figure.
To better characterize the signaling phenotype of the ∆bim1 and ∆gim4 mutants, we used time‐lapse microscopy to measure total system output (O) and transmitted signal (P) in single cells tracked over time. Figure 4C–E shows plots of P in single cells over time (“trajectories”) after stimulation with 20 nM pheromone. The reference strain (“wild‐type”) cell trajectories were clustered relatively tightly (Fig 4C); in contrast, the trajectories in the Δbim1 (Fig 4D) and Δgim4 (Fig 4E) populations were loosely spread. To the naked eye, the Δbim1 and Δgim4 cells differed from each other in terms of how common trajectories with extremely high or low Ps were. In Δgim4, the trajectories presented a broader distribution of trajectories around the mean. In contrast, in Δbim1 populations, most cells had trajectories matching the reference strain, but a number of “outlier” cells showed P very far from the mean.
To quantify these differences, we developed a distributional measure called the median progressive spread (MPS) based on the progressive spread distribution (PSD, see Appendix); For a symmetric distribution, the MPS is essentially the interquartile range). The MPS for the reference strain population was 0.22 (95% CI 0.19–0.23); for Δbim1 cells, it was 0.24 (95% CI 0.17–0.31, not distinguishable from reference), while for Δgim4 cells, it was 0.36 (95% CI 0.32–0.37). The MPS thus shows that the distribution of trajectories in Δgim4 cells is in fact significantly broader overall than the reference, while, for Δbim1 cells, the breadth of the core distribution is indistinguishable from the reference.
We then analyzed the stability of the trajectories over time. In all populations, a few cells showed unstable increases in P visually evidenced as erratic or “crooked” trajectories [red traces in Fig 4C–E)]. To assess changes in the occurrence of crooked trajectories we defined a crookedness index (IC, see Appendix). We set an IC threshold of 0.3, above which we consider trajectories to be crooked. By this measure, 11% of the trajectories in the ∆bim1 set and 16% in the ∆gim4 set were crooked, in contrast to 6% in the reference strain population.
Both reference and ∆bim1 cells showed crooked trajectories above and below the mean value of P. In contrast, in the ∆gim4 cells, 13 out of 17 (76.5%) cells showing crooked trajectories had values of P below the mean. Since Δgim4 cells also showed a reduction in system output (O), this result suggested that cells that have their signal lowered by loss of prefoldin are prone to exhibiting unstable trajectories. Thus, for Δgim4 cells, a substantial fraction of the increased η2(P) might be a secondary consequence of a primary defect in signal transmission strength, while for ∆bim1 cells, IC and η2(P) are independent of changes in signal transmission strength.
Impairment of the microtubule bridge between the nucleus and the signaling site causes increased signaling variability, while its elimination diminishes pathway output
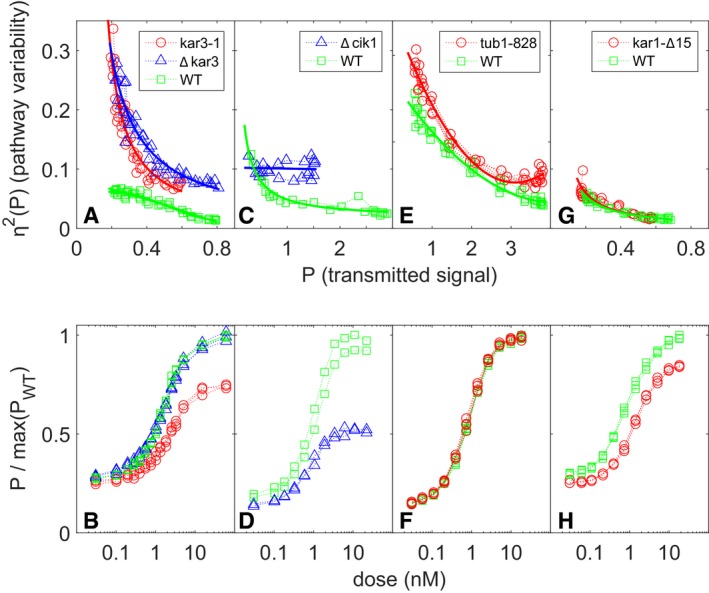
When the PRS is activated, receptors, G‐proteins, the Ste5 scaffold, and other membrane proteins localize to a membrane signaling site. Polarized growth from this site causes the cells to form a mating projection, and the cells to adopt an overall morphology known as a “shmoo”. Cytoplasmic microtubules form a bridge connecting the Spindle Pole Body (SPB) on the nuclear membrane to the site at the shmoo tip (Maddox et al, 1999, 2000, 2003). To better understand the effect of the ∆bim1 and ∆gim4 mutations, we used genetic and chemical means to perturb specific aspects of microtubule function (Fig 5). We first tested the effect of mutations known to affect microtubule‐generated pulling forces. At the plasma membrane, the microtubule bridge connecting the signaling site to the nucleus alternately grows and shrinks. This is due to the fact that microtubule plus ends at the plasma membrane alternate between binding membrane‐attached Bim1 and the Kar3‐Cik1 complex. Bim1 binds plus ends of polymerizing microtubules, thus lengthening the microtubule bridge and generating a “push” force on the nucleus. The Δbim1 mutation disrupts pushing, and, as shown in Fig 4, increases η2(P) without greatly diminishing average P. Kar3/Cik1 binds plus ends and actively depolymerizes them, thus generating a “pull” force on the nucleus. Figure 5 shows the effects of deletions in KAR3 or CIK1 (Fig 5A–D). For comparable values of pathway output (P), both perturbations markedly increased variability in transmitted signal, η2(P) (Fig 5A and C). ∆kar3 did not affect the average transmitted signal (P) (Fig 5B). By contrast, ∆cik1 caused a strong reduction in P (Fig 5D).
Figure 5. Cell‐to‐cell variability in signal transmission in cells with mutations affecting microtubule end function.
-
A–Hη2(P) vs. P and dose response in: (A, B) kar3‐1 (SGA108, the “rigor mutant”), ∆kar3 (SGA2015), and WT cells (SGA103) (three replicates each); (C, D) ∆cik1 (GPY4123) and WT cells (GPY4000) (two replicates); (E, F) tub1‐828 expressing and WT cells (GPY1858) (four replicates); and (G, H) kar3‐∆15 (SGA109) and WT cells (SGA103) (three replicates). In (A, C, E, G), the x‐axis values are the transmitted signal P. In (B, D, F, H), the y‐axis values are P divided by the maximum value of P observed for the WT strain in each.
Source data are available online for this figure.
We next disrupted microtubule polymerization and de‐polymerization by expression of a dominant negative variant of α‐tubulin, encoded by the TUB1‐828 allele. Tub1‐828 generates “frozen” plus ends, unable to depolymerize or polymerize (Anders & Botstein, 2001). To do so, we constructed a strain carrying an estradiol‐inducible TUB1‐828 construct driven by the GAL1 promoter (see Materials and Methods and Louvion et al, 1993). As shown in Fig 5E and F, the very low baseline Tub1‐828 expression in the absence of estradiol increased η2(P) at all pheromone doses tested. Notably, this low level, Tub1‐828‐expression caused no changes in transmitted signal (P) (Fig 5F). These results showed that altering microtubule function can increase variability in transmitted signal without affecting mean signal strength.
In Δbim1 and Δkar3 cells, the microtubule bridge was frequently detached from the plasma membrane (Maddox et al, 1999, 2000). In light of this behavior, we considered the hypothesis that the existence of the bridge alone would be sufficient to maintain normal levels of η2(P), and that generation of pulling and pushing forces might be irrelevant. To test this idea, we constructed a kar3‐1 variant of the reference strain. Kar3‐1 localizes to the signaling site and binds microtubule plus ends, but can neither actively depolymerize nor release them (because these microtubules can neither lengthen nor contract, Kar3‐1 is referred to as a “rigor” mutant). The rigor mutation increased η2(P) by at least a factor of three, as much as the full ∆kar3 lesion; it also caused a ~30% reduction in transmitted signal (Fig 5A and B). These results show that, to reduce η2(P), microtubules not only have to be attached to the plasma membrane, but must also be able to alternatively push and pull on the nucleus and exert force.
Next, we tested the effects of preventing the attachment of microtubules to the SPB. To do that, we replaced KAR1 with the kar1‐Δ15 allele (Appendix Table S1). KAR1 encodes a component of the SPB. The kar1‐Δ15 allele expresses a C‐terminally truncated variant of Kar1. During the pheromone response, the Kar1 C terminus is the main site of minus‐end microtubule anchoring to the MTOC (Pereira et al, 1999); its absence leads to detachment of cytoplasmic microtubules from the nucleus and from the signaling site (Erlemann et al, 2012). The kar1‐∆15 mutation was without effect on η2(P) (Fig 5G), although it caused a reduction in pathway output (P) (Fig 5H).
Like kar1‐∆15, some of the microtubule perturbations above (∆gim4, ∆cik1, kar3‐1) also caused decreases in P. We thus hypothesized that a microtubule bridge is required for pathway output to reach its maximal levels. In this view, even a malfunctioning bridge allows P to reach high values (as seen with the ∆kar3, ∆bim1, and Tub1‐828 perturbations), albeit with a concomitant increase in η2(P). Conversely, the absence of the bridge constrains P without impacting η2(P) (as in kar1‐∆15).
To test the hypothesis above, we used a chemical treatment that causes the disappearance of visible microtubule structures in pheromone‐treated cells. We used a combination of the microtubule‐polymerization inhibitors benomyl and nocodazole, each at close‐to‐saturation concentrations (see Materials and Methods and Palframan et al, 2006). Chemical disruption of microtubules caused a decrease in P at doses of pheromone higher than the EC50 (Fig EV2A) and did not affect η2(P) (Fig EV2B). These results mirrored the kar1‐∆15 results. Since both perturbations caused the disappearance of the microtubule bridge, these results support the view that the absence of the bridge diminishes the strength of the transmitted signal P vs. dose, but causes no changes in signal variability η2(P) vs. P, while presence of a bridge that cannot exert push and pull forces results in normal P but increased variability in it.
Figure EV2. Chemical inhibitors of microtubule function attenuate signal but do not affect pathway variability.
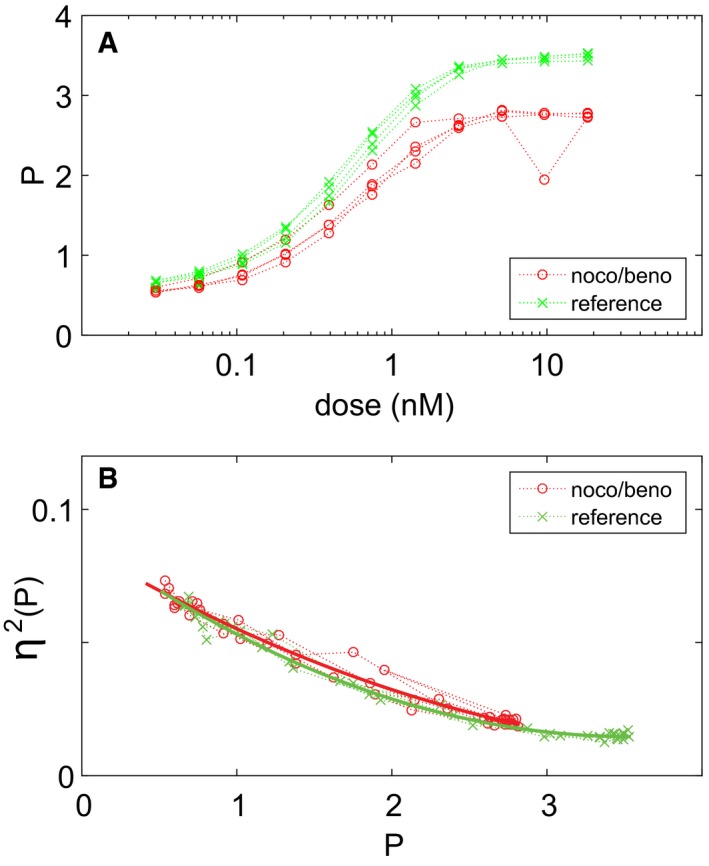
- Plot shows P vs. dose.
- Plot shows η2(P) vs. P.
Nucleus‐to‐signaling‐site distance does not correlate with pathway output
Υeast cells actively position both the cell nucleus and the pheromone‐inducible genes within it in relation to the signaling site. During the pheromone response, the nucleus remains seemingly “anchored” in a cytoplasmic volume at the base of the mating projection (often referred to as the “base of the shmoo tip”). Casolari et al (2005) showed that 49 pheromone‐inducible nuclear genes became associated with the nuclear pore complex after pheromone stimulation and that one of them, FIG2, translocated to the region of the nuclear periphery closest to the shmoo tip. More recently, Randise‐Hinchliff et al (2016) showed that PRM1 and other pheromone‐inducible genes translocate to the same spot, as long as they are bound by the activated pheromone‐responsive transcription factor Ste12. The relationship between this localization and signal transmission is unclear. Maeder et al (2007) showed that, in cells exposed to saturating isotropic pheromone, the concentration of active Fus3 (phosphorylated or “P‐Fus3”, see Fig 1), was maximal at the signaling site and decreased toward the base of the shmoo tip. We had observed that at doses of pheromone near the EC50 the nuclei are localized closer to the signaling site than they are at saturating doses (Appendix Fig S6).
Given the above and that a variability gene found in our screen, NUP60, encodes a nuclear pore protein required for pheromone‐responsive gene recruitment to the nuclear periphery, we wondered whether the position of the Ste12 bound genes within the nucleus and within the gradient might affect or even regulate signal transmission. Since nuclear positioning depends on the integrity and proper function of the microtubule bridge, we hypothesized that microtubule perturbations that increased signal variability might do so by altering the position of the nucleus within the P‐Fus3 gradient in the cell, which in turn might affect the average strength and/or the variability of the transmitted signal received by the pheromone‐responsive genes.
We therefore tested whether in mutant cells in which the nucleus was unattached to the signaling site, pathway output (P) would be weaker when the nucleus wandered further from the signaling site, and stronger when closer. To do so, we measured nuclear positioning and its relation to pathway output (P) in ∆bim1, ∆gim4, kar3‐1, and reference cells. Our measurements showed clear and distinct effects of the three perturbations on nuclear positioning. However, we did not see a correlation between the position of the nucleus and the strength of transmitted signal (see Appendix Fig S6). These results argued against the idea that the signal received at pheromone‐induced genes depended on the distance between the nucleus and the signaling site (see Discussion).
Genetic bypass of Ste5 recruitment suppresses the effect of microtubule perturbations on pathway variability
We next sought to identify the steps in the signaling pathway at which microtubule perturbation increased signal variability. To do so, we performed “bypass” experiments made possible by work by Pryciak and collaborators that demonstrated graded ectopic activation of the PRS at different downstream steps by expression of different artificial activators (Takahashi & Pryciak, 2008). In this system, expression of PRS activator proteins was driven by P GAL1, whose activity was controlled in turn by the above‐described estrogen‐responsive Gal4 derivative (Gal4‐ER‐VP16, Louvion et al, 1993). Figure 6 shows results of experiments using two artificial activators: native Ste4, whose expression mimics dissociated (i.e., active) Gβγ dimers, and Ste5‐CTM, a fusion of the Ste5 scaffold with a transmembrane domain, whose expression mimics membrane‐recruited (i.e., activated) Ste5 (Pryciak & Huntress, 1998). To prevent interference from basal activation of the native PRS, this last strain was Δste5. In the Ste4 activator strains, ∆bim1 and ∆gim4 caused increases in η2(P) (Fig 6A), the same effects they had when we stimulated the PRS with pheromone (see Fig 4). In Ste5‐CTM cells, the ∆bim1 perturbation did not increase η2(P), while the increase caused by ∆gim4 was smaller than in the Ste4 strain. These results suggested that the microtubule‐dependent process(es) affected by the ∆bim1 perturbation, and possibly by the ∆gim4 perturbation, increased pathway variability downstream of Ste4, but at or upstream of recruitment of Ste5 to the membrane by Ste4. Given that Ste5 activation by membrane recruitment is an early signaling‐site event that is not immediately relatable to known microtubule roles, this was a surprising finding.
Figure 6. Microtubule perturbations affect pathway variability η2(P) and transmitted signal P at or upstream of the Ste5 recruitment step.
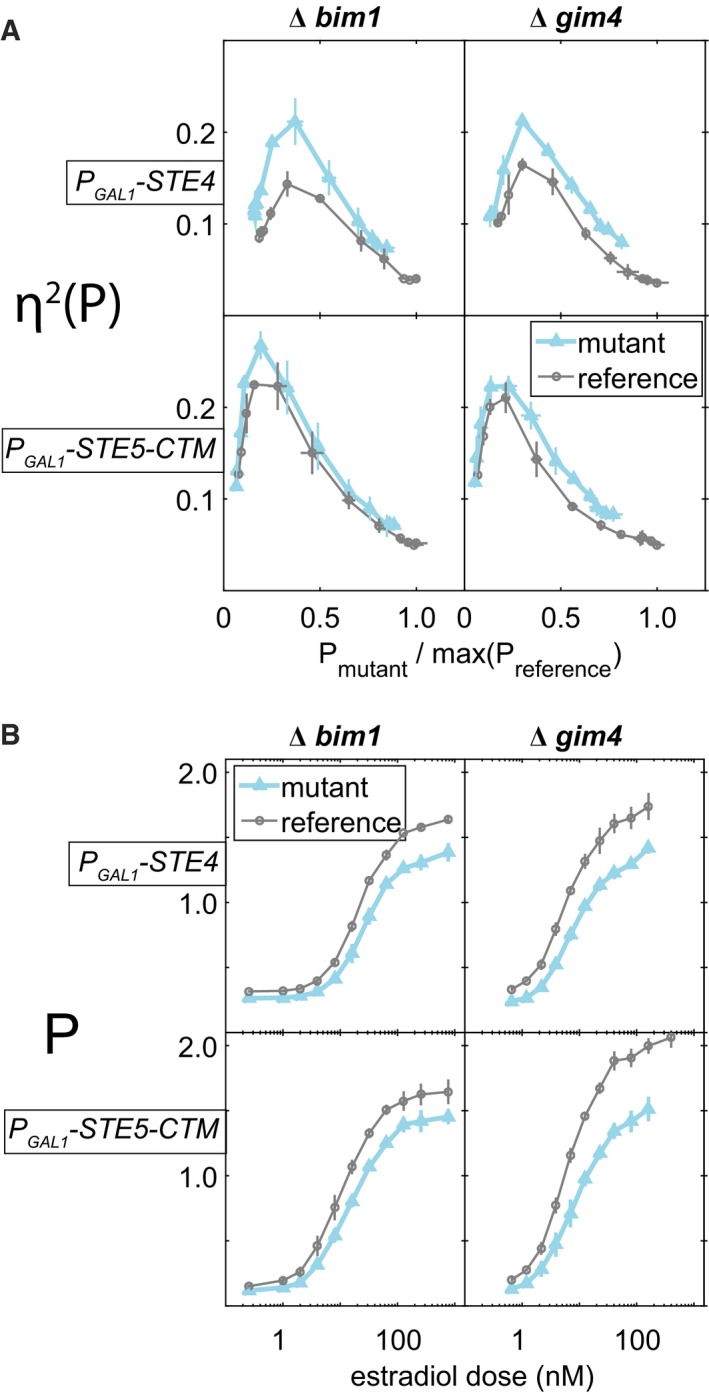
- Y‐axis shows pathway variability η2(P), x‐axis shows signal strength P, normalized by the maximum P observed for each reference strain. This normalization allows comparison between strains with different activation points. There are three replicate cultures of each mutant and reference strain.
- X‐axis values are estradiol dose, y‐axis values are P (same measurements as in x‐axis of panel A, but here un‐normalized). Reductions in these values are thus also reflected in reduced ranges of P, relative to reference, for plots in (A). Figure 4B shows corresponding reduced response of ∆bim1 and ∆gim4 mutants to normal pheromone induction.
By contrast, the effects of ∆bim1 and ∆gim4 on transmitted signal strength, P, were not suppressed by Ste5‐CTM (Fig 6B). ∆bim1 caused a decrease in P in the artificial activator system that was greater than in the pheromone‐activated PRS (see Fig 4), while ∆gim4 caused a decrease of similar magnitude. These reductions in P were also present in the Ste5‐CTM activator strains. These results suggested that ∆bim1 and ∆gim4 affected signal strength (P) by mechanisms distinct from those that mediated their effects in signal variability (η2(P)).
Induced signaling variability by microtubule perturbations requires Fus3
These bypass experiments were consistent with the idea that the increased pathway variability caused by the ∆bim1 and ∆gim4 perturbations are due to effects on membrane recruitment by Ste4 of the Ste5 MAP kinase cascade scaffold. To be activated, the Fus3 MAPK must be bound to the Ste5 scaffold, while the other MAPK, Kss1, does not require such association (Fig 1 and legend). We therefore suspected that the effect of microtubule perturbations might propagate preferentially via Fus3. Thus, we measured η2(P) in ∆fus3 or ∆kss1 cells. Notably, in ∆bim1 and ∆gim4 strains, the ∆fus3 deletion suppressed, at all doses, the increased η2(P) (Fig 7A, top panels), while the ∆kss1 deletion exacerbated the defect of ∆bim1 (Fig 7A, bottom panels). The fact that deletion of FUS3 eliminated the increase in pathway variability caused by the ∆gim4 and ∆bim1 mutations showed that the increased variability was not a secondary consequence of a generalized increase in variability in cells with disrupted microtubule function, but rather reflected an effect of these mutations on the operation of the PRS. These results demonstrate that microtubule perturbations increase pathway variability η2(P) by specifically impacting signaling by the MAP kinase Fus3.
Figure 7. Fus3 is required for deletions of GIM4 and BIM1 to increase cell‐to‐cell variability.
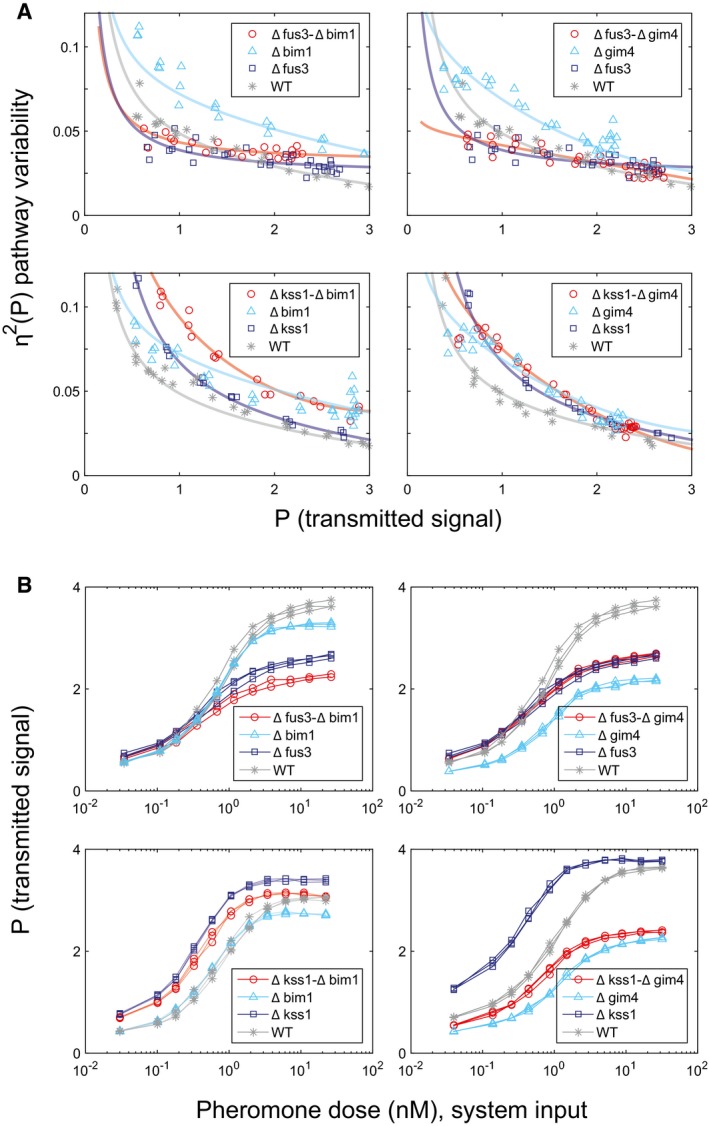
- Deletions of FUS3 and KSS1 have distinct effects on the increased cell‐to‐cell variability caused by deletions of GIM4 and BIM1. We induced the PRS in the indicated strains by addition of 20 nM pheromone to the medium and measured reporter activity after 3 h. Y‐axis in each panel shows η2(P) as a function of P. Deletion strains derive from the reference WT GPY4000. Δbim1 and Δgim4 strains show increased η2(P) relative to WT (P‐values are both less than 10−5), while Δfus3 ∆bim1 and Δfus3 ∆gim4 strains show η2(P) not distinguishable from WT (P‐values are 0.341 and 0.095, respectively). However, ∆bim1 Δkss1 and ∆gim4 Δkss1 cells show higher η2(P) relative to WT (P‐values are both less than 10−5). The double mutant Δbim1 Δkss1 had significantly higher η2(P) than either Δkss1 or Δbim1 alone (P‐values are both 10−5 or less), while the double mutant Δgim4 Δkss1 had essentially the same η2(P) as either Δkss1 or Δgim4 alone (P‐values are 0.341 and 0.106, respectively). This shows that Fus3 protein is required for increased η2(P). (All strains have three replicates except for one of the double deletions, Δfus3 ∆bim1, which has two replicates.) To compare the piecewise‐linear curves implied by the data from each pair of strains, we use an Area Between the Curves (ABC) metric, with each area estimated by trapezoidal integration. We then obtain the P‐values by resampling over the replicates under the null hypothesis that the two strains are in fact the same; the P‐value is the number of times that the resampled ABC was at least as far from the median as the realized ABC.
- P vs. dose for the datasets of panel (A).
Source data are available online for this figure.
In its effects on P, diminution caused by the deletion Δfus3 was additive to the diminution caused by Δbim1. By contrast, the deletion Δkss1 counteracted the diminution caused by Δbim1, so that P for the double mutant Δkss1‐Δbim1 was above the reference at all but the highest doses (Fig 7B, left panels). The reduction of signal strength due to ∆gim4 (Fig 7B, top right) was smaller in cells that were also ∆fus3, whose reduction in P was the same as in ∆fus3 GIM4 + cells. By contrast, in Δgim4 cells, the additional deletion Δkss1 does not enhance signal strength, but in fact reduces it (Fig 7B, bottom right). These results further support the idea that the mechanism(s) that affect pathway variability is (are) distinct from those affecting signal transmission strength.
Perturbation of microtubule function increases variability in establishment and maintenance of the initial signaling site
The above experiments suggested that the ∆bim1 and ∆gim4 perturbations operated at the Ste5 membrane recruitment/retention step to cause variability in signal transmitted by Fus3. We thus sought to directly observe the dynamics of Ste5 membrane localization in the natural PRS and in the perturbed strains. We constructed strains that expressed Ste5 fused at its C terminus to three tandem copies of YFP (Ste5‐YFP‐ YFP‐YFP, here called Ste5‐YFP). In previous work, we had used this construct to show that, in cells in the G1 phase of the cell cycle, cytosolic Ste5‐YFP translocated to the plasma membrane isotropically within 2–3 min of exposure to isotropic pheromone, followed, after 2–30 min, by clustering of the Ste5‐YFP signal into the signaling site (Fig 8A; Ventura et al, 2014). Here, we constructed reference cells that contained this construct, and otherwise isogenic derivatives carrying the ∆bim1, ∆gim4, and tub1‐828 expression perturbations. We exposed these cells to a saturating concentration of isotropic pheromone and monitored them by fluorescence confocal microscopy for up to 3 h.
Figure 8. Microtubule perturbations cause Ste5 patches to form less reliably, delay patch formation, and cause patches to persist for less time.
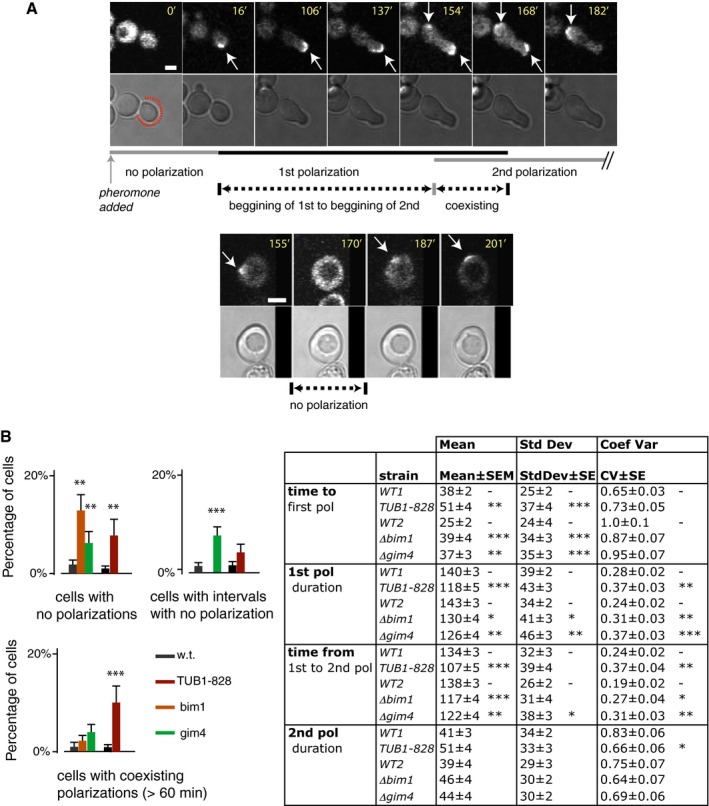
- Single‐cell measurements of Ste5 patch dynamics. Arrows indicate first and second Ste5 patches. Images show examples of a cell with two co‐existing Ste5 patches (top) and a cell with an interval without detectable Ste5 patches between the first and second patch (bottom). The lines below mark the dynamic features we quantified: time to 1st patch (polarization), duration of 1st and 2nd polarization, interval in which 1st and 2nd polarization overlap, or gap between them, and time from 1st to 2nd polarization. Scale bar: 2 μm.
- Bar graph plots show qualitative defects observed: cells with no polarization, cells with gaps between first and second polarizations and cells with overlapping (coexisting) polarizations. Error bars represent the standard error as calculated by bootstrapping (104 resamples), and asterisks indicate significant difference from WT as calculated by Fisher's exact test for count data. Table shows quantitative defects. Data correspond to the mean ± SEM, the standard deviation ± SE and the coefficient of variation (standard deviation divided by the mean) ± SE. Standard errors were calculated by bootstrapping (104 resamples), and significant differences from WT were calculated by permutation tests (104 permutations). Values for the probability P of the observed data under the null hypothesis that each mutant strain is no different from WT (WT1 vs. TUB1‐828, and WT2 vs. Δbim1 and Δgim4) are shown by asterisks: *P < 0.05; **P < 0.01; ***P < 0.001. Experiments were done in three biological replicates (N = 3). As no important differences were observed among replicates, cells were pooled for the analysis. At least 80 cells of each strain were quantified (N > 80).
In cells of the reference strain, for the first 45–120 min, a Ste5‐YFP patch was visible as a crescent at the tip of the growing shmoo. In most cells, the shmoo tip ceased growing after 45–120 min. In such cells, a second site of polarized growth formed later in a different location. At that location, before polarized growth was apparent, a premonitory Ste5‐YFP patch appeared. This second site became progressively brighter while the first site faded (Fig 8A). Cells bearing all three microtubule perturbations showed several common changes. First, a greater number of cells failed to form a signaling site (Fig 8B). This result is consistent with the fact that about 5% of Δbim1 and Δgim4 cells did not induce the P PRM1 ‐mCherry PRS reporter. Second, in perturbed cells that did form a signaling site, the formation time was greater on average, with more variability between cells. Third, the first site in perturbed strains lasted a shorter time on average and varied more between cells. Fourth, the time from initiation of the first site until appearance of the second was on average shorter and more variable than in WT. By contrast, the perturbed strains showed no changes in the average and variability of duration of the second patch.
These observations showed that cells with microtubule perturbations had difficulties establishing and maintaining their first signaling sites, but not their second sites. This indicates that the three microtubule perturbations we used did not cause a generalized cellular effect that impacted signaling‐site formation but rather that they specifically affected the formation of the first signaling site. These results suggest that formation and stability of the second site do not require the microtubule bridge. Overall, these results indicate that microtubule perturbations affect recruitment and retention of Ste5 and are consistent with the idea that their effects on signal are due to their effects on Ste5 function at the signaling site.
Discussion
We present here the results of a hunt for mutants that affect variability in cell signaling responsive to the activation of a surface receptor by its cognate ligand (Fig 1). For this study, we created a whole genome collection of S. cerevisiae strains, each of which bore the reporter genes required to quantify signaling variables, as well as a deletion in a non‐essential gene. We used this collection to screen more than 1,100 non‐essential yeast genes, comprising more than 1/4 of the non‐essential genome, including all non‐essential protein kinases and phosphatases. We isolated and studied carefully 50 mutants. To our knowledge, this was the first large‐scale genetic screen for genes affecting variability in receptor‐responsive cell signaling phenotypes. Cluster analysis of the reporter gene phenotypes revealed groups of non‐essential genes that affected receptor‐responsive signaling in distinct ways, suggesting common mechanisms of action for genes within each group.
Some deletions specifically affected pathway variability (cell‐to‐cell variation in transmitted signal) and others signaling strength (mean transmitted signal), showing that these two aspects of the signal depended on different sets of genes (Fig 3). In this sense, our study defined two quantitative traits, signal strength and pathway variability, as independent axes of system behavior, affected independently by genes, and that also might be affected independently by chemical and environmental perturbations. The fact that mutations exist that can specifically affect the amount of pathway variability suggests that adaptive evolution, for example for increased signaling accuracy, may have shaped the value of this quantitative trait, in the yeast pheromone response and in other signaling systems.
Our screen extends previous work identifying genes that affect variability, as well as mean value of quantitative phenotypes. In S. cerevisiae, work by Raser & O'Shea (2004) showed that deletion of genes whose products participate in the Swi/Snf, Ino80, and SAGA chromatin remodeling complexes increased cell‐to‐cell variability in expression of a P PHO5 reporter gene. Chromatin remodeling is required to induce PHO5 and this result suggests that proper chromatin remodeling is required to suppress variability. Our previous work (Colman‐Lerner et al, 2005) showed that mutations in the two PRS MAP kinases differentially affected cell‐to‐cell variability in signal transmitted to pheromone‐responsive P PRM1 reporter genes. Our work here showed that the Fus3 kinase was required for the signal variability caused by microtubule perturbations, and suggested that proper assembly of microtubule plus ends at the signaling site is needed to suppress variability. Work by El‐Samad, Madhani, and coworkers (McCullagh et al, 2010) showed that deletion of DIG1, but not its paralog DIG2, increased both mean expression and cell‐to‐cell variability in uninduced pheromone‐responsive P FUS1 and P AGA1 reporters, suggesting that Dig2 may have lost the ability to decrease variability after the whole genome duplication early in the species history. Work by Yvert and coworkers (Ansel et al, 2008) in different strains mapped three loci that increased cell‐to‐cell variability in expression of a P MET17 reporter; surprisingly, one of these loci, crossed into S288C, reduced variability, suggesting that this locus might regulate variability more directly, rather than functioning in a process that affects it. Numerous studies in plants and animals, including our work in Caenorhabditis elegans, have revealed genes and alleles that affect variability in gene expression (Fraser & Schadt, 2010; Jimenez‐Gomez et al, 2011; Mendenhall et al, 2017) and variability in other quantitative traits from plant height, flowering, and leaf number, to the number of somatic cells present in fresh cow milk (Hall et al, 2007; Ansel et al, 2008; Ordas et al, 2008; Fraser & Schadt, 2010; Jimenez‐Gomez et al, 2011; Makumburage & Stapleton, 2011; Landers & Stapleton, 2014). These results make the point that there is much to be learned about mechanisms that affect variability in quantitative traits, and about circumstances in which allelic differences that increase or decrease this variability have general or intelligible effects on organismic fitness.
This work uncovered an unexpected contribution of cytosolic microtubules to pathway variability and signaling strength. In normally signaling cells, a microtubule bridge connects the signaling site on the cell membrane with the SPB in the nuclear envelope. Cells with perturbations in microtubule function showed increased variability in transmitted signal and/or reduced mean signaling strength. In particular, cells with deletions in BIM1 and in GIM4 showed greater cell–cell variability in transmitted signal in populations of single cells at a single time point. Monitoring the “trajectories” of accumulation of pathway output in these mutant cells over time revealed a broader distribution of transmitted signal and, in some cells, “jumps” in accumulated pathway output, evidenced by crooked trajectories, defining erratic operation of the signaling system.
The effect of microtubule perturbations on pathway variability was specific to the operation of the native pheromone response system. For example, pathway variability was not affected by some microtubule perturbations when the signal was triggered by artificially membrane‐anchored Ste5 (Fig 6). Furthermore, direct microscopic observation of Ste5 at the signaling site showed that, in microtubule perturbed cells, the initial Ste5 patches at the signaling site were slower to form and more likely to disappear (Fig 8). Future experiments using faster maturing and shorter lived fluorescent protein derivatives might enable temporal correlation of transmitted signal increase and decrease with formation and loss of Ste5 patches. In the absence of such data, our results showed that perturbations that increased pathway variability impaired Ste5 accumulation at the signaling site and that their effects on variability were suppressed by artificial recruitment of Ste5 to the membrane. Moreover, variability in signaling caused by the perturbations required the MAP Kinase Fus3: in cells that lacked Fus3, the Δbim1 and Δgim4 perturbations did not increase pathway variability (Fig 7). These two facts suggest that microtubule perturbations cause variable signaling by Fus3, when activated by the MAPKK Ste7 and the MAPKKK Ste11 in complex with membrane‐recruited Ste5.
We propose that small irregularities in Ste5 recruitment or Fus3 signaling caused by microtubule perturbations might become amplified into larger differences in Ste5/Fus3 dependent signaling by positive feedback. Figure 9 shows some of the stimulatory reactions by which irregularities in Fus3 signaling can be amplified, including the stimulated membrane recruitment of Cdc42, which has previously been shown to stimulate its own activity. (Kozubowski et al, 2008; Johnson et al, 2011). In this view, disruption of plus‐end function of microtubules near the site might cause differences in delivery of signaling components; such differences would then become amplified. In controlled systems, including servo systems (Hanzen, 1934) and electronic amplifiers (Armstrong, 1914; Franklin, 1914; Meissner, 1919), positive feedbacks can improve performance, but without damping or negative feedback, such systems are prone to instability (Bennett, 1979). In the PRS, impact of positive feedbacks on system performance is further affected by the fact that system components are in limited supply [for example there are only ~2,000 molecules of the Gβ, Ste4, per cell (Thomson et al, 2011)], so that the autostimulatory molecular events that generate a robust second signaling site can only occur after a first site falls apart.
Figure 9. Model for origin of pathway variability.
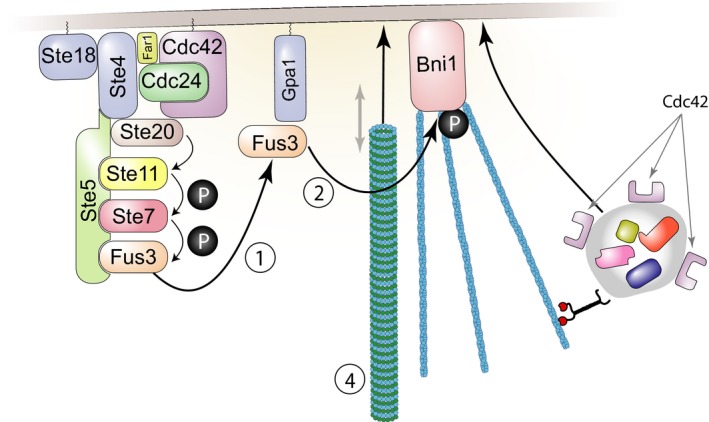
Our work suggests that disruption of microtubule plus‐end function near the signaling site by perturbations such as the Δbim1 mutation causes variability in Ste5 recruitment or Fus3 signaling. Figure shows stimulatory reactions at the signaling site by which irregularities in Ste5 recruitment or Fus3 signaling could be amplified. (1) Fus3 is phosphorylated and activated due to the operation of the PRS. Some phosphorylated Fus3 binds to the Gpa1 subunit of the dissociated G‐protein. (2) Gpa1‐bound Fus3 activates a protein, Bni1, which nucleates formation of actin cables. (3) Additional proteins involved in signaling, cell polarization, and cell fusion, including Cdc42 and Fus2 (Paterson et al, 2008, not shown) are then trafficked to the membrane as cargo carried along the actin cables. Activated Cdc42 stimulates its own activation (Kozubowski et al, 2008; Johnson et al, 2011). (4) Microtubule plus ends are captured by Kar3/Cik1 associated with Gpa1 (not shown) and Kar9/Bim1‐complexed plus ends can be walked down the actin cables to the signaling site by motor proteins including Myo2 (not shown). Microtubules, actin cables, and cargo proteins are larger than the approximate scale used here would suggest. Appendix and Appendix Fig S7 describe additional stimulatory reactions and positive feedbacks that might contribute to irregular signaling at the site.
The mechanisms by which microtubules increase transmitted signal strength and reduce signal variability may be distinct. Some perturbations that impaired (but did not eliminate) the microtubule bridge increased pathway variability but did not affect the strength of the transmitted signal (∆bim1, ∆kar3, TUB1‐828‐expression), while two perturbations that eliminated the microtubule bridge (kar1‐∆15 and microtubule depolymerizing drugs) did not affect pathway variability but reduced transmitted signal strength. This finding is consistent with the initial results of the genetic screen, in which transmitted signal strength and pathway variability can be affected differently by different mutations (Fig 3). These effects on pathway variability are not apparent at low doses. This fact suggests that cells may have a microtubule‐independent way to transmit signal that is weaker than the normal mode, but that functions with low pathway variability as well. Such an alternative mode of signaling might be helpful if attempts to signal by the microtubule‐dependent mode had failed. The Ste5 dynamics results (Fig 8) are consistent with this idea: cells form first a Ste5 patch that is impaired by microtubule perturbations and then a second patch that is impervious to them. This hypothesis could be tested: it predicts that the Ste5 patches should behave normally under perturbations that eliminate the microtubule bridge (kar1‐∆15 and microtubule depolymerizing drugs). In any case, a weaker transmitted signal in the absence of microtubules might enable cells to delay cell fusion until the nucleus is properly tethered to the membrane fusion site, a required step for zygotic nuclear fusion.
How might microtubules strengthen signal transmission? Just as they facilitate transport to other membrane sites (Maekawa et al, 2003; Cavalli et al, 2005; Foe & von Dassow, 2008), microtubules might facilitate transport of signaling molecules to the signaling site. Here, we considered a non‐exclusive alternative idea, that the microtubule bridge might position the nucleus (Maddox et al, 2003) at an optimal location with respect to the signaling site and a gradient of activated MAP kinase emanating from the site (Maeder et al, 2007). We found a lack of correlation between distance to the signaling site and pathway output in cells with destabilized or frozen nuclear positioning (Appendix Fig S6). However, it is possible that some of the perturbations we used might have interfered with a MAP kinase gradient. For example, the frequent forays of the unattached nucleus in ∆bim1 (Maddox et al, 2003) might stir the cytosol and disrupt a gradient. It thus remains possible that in unperturbed cells nuclear positioning strengthens signal by positioning the nucleus higher up a signal gradient.
Our current means to perturb the system and monitor its operation are not sufficient to elucidate how normal operation of cytoplasmic microtubules helps the cell transmit signal of constant strength. Too much remains unknown. At the signaling site, there are too many different proteins operating, too many positive cross‐regulatory interactions, too many simultaneously occurring mechanical processes like cargo delivery and membrane fusion that are now insufficiently understood. It is as if we had tried to understand the smooth function of an electric motor by monitoring frequency and timing of sounds it made after disrupting the operation of particular bearings, bushings, and shafts. In this light, analysis of microtubule effects on signal transmission is a classic “inverse problem”, for which inferences from doable experiments are limited and insufficient to fully describe the system under investigation (Brenner, 2010). Within these limits, however, our genetics‐powered quantitative physiological experimentation enabled us to identify the proteins involved, and this in turn helped us constrain models for their function. Moreover, as in the motor analogy above, different kinds of noises may well identify aspects or axes of system function dependent on different proteins and molecular events, and so perhaps contribute to future insight.
It is possible that cytoplasmic microtubule function may affect pathway variability in metazoans, for which coherent population fate and polarity decisions are needed for forming and maintaining correct tissue architecture. For example, vertebrate orthologs of yeast BIM1, MAPRE1‐3/EB1‐3 (Su & Qi, 2001) interact with APC (Adenoma Polyposis Coli), which is required for radial glial cells polarization, and for them to support birth and migration of cortical neurons (Yokota et al, 2009). APC is a tumor suppressor, frequently inactivated in colorectal and other epithelial cancers (Kinzler & Vogelstein, 1996; Vogelstein et al, 2013). At the minus end, lesions in proteins that connect microtubules to the microtubule organizing center (in particular those affecting the Nesprin‐1 and Nesprin‐2 isoforms encoded by SYNE1 and SYNE2 (reviewed by Gundersen & Worman, 2013) contribute to formation of solid tumors in which tumor development requires incorrect polarity decisions [e.g., squamous cell carcinomas of the head and neck (Stransky et al, 2011)]. Exome sequencing reveals considerable coding sequence polymorphism in genes encoding the BIM1 ortholog MAPRE/EB1, and in APC, and other microtubule end‐interacting proteins in the human population (Fu et al, 2013), raising the possibility that different allelic forms of microtubule end proteins might have different quantitative effects on variability in cell decisions in response to signals, and so affect cancer incidence. In this light, the existence of alleles affecting pathway variability may help motivate development of genetic and pharmacological interventions aimed at reducing it.
Materials and Methods
We performed DNA manipulations including PCR and subcloning as described (Ausubel et al, 1987–2017). We cultured and manipulated yeast as described (Ausubel et al, 1987–2017; Guthrie & Fink, 1991). Unless otherwise noted, we grew cells in synthetic dextrose complete (SDC) media consisting of Brent Supplemental Media (MP Biomedicals, Solon, OH), yeast nitrogen base without amino acids and ammonium sulfate (BD, Franklin Lakes, NJ), and dextrose (Sigma‐Aldrich, St Louis, MO).
Analysis of cell‐to‐cell variability
We performed the analysis as in Colman‐Lerner et al (2005). Briefly, we considered the system output for any given cell Oi, determined by the abundance of a fluorescent protein inducible by the pheromone response system, to be the product of (i) the average pathway subsystem output per unit time, Pi (which varies with input pheromone dose αF), (ii) the expression subsystem output Ei, and (iii) the duration of stimulation ΔT (Colman‐Lerner et al, 2005), as follows:
We considered Pi and Ei to be the sum of the capacity of the subsystem in each cell (Li and Gi) plus stochastic fluctuations in the operation of each subsystem during the course of an experiment (λi and γi). Thus,
We defined the cell‐to‐cell variability in system output as the normalized variance of Oi, η2(O), decomposable into the sum of individual sources and a correlation term (Colman‐Lerner et al, 2005), as follows,
In the WT, in the deletion strains, and mutants used in the manuscript, we measured output and cell‐to‐cell variability of each reporter; η2(γ), gene expression noise [“intrinsic noise” (Elowitz et al, 2002)]; as well as η2(P) (η2(L) + η2(λ)), cell‐to‐cell variability of the pheromone response system. We measured η2(γ) as the variance in the difference of the normalized abundance of the two fluorescent proteins driven by identical copies of P PRM1. We estimated cumulative signal transmitted P in individual cells as the normalized signal from the pheromone‐inducible P PRM1 reporter (O) divided by the signal from the constitutive control promoter, (P ACT1 or P BMH2, depending on the strain) (O/G). We estimated η2(P) as the variance in the difference between the normalized abundances of two fluorescent proteins, one driven by the P PRM1 and the other by the constitutive, pheromone‐independent promoter (P ACT1 or P BMH2, depending on the strain) (σ2(mRFP i/<mRFP> −YFP i/<YFP>)). This variance is actually equal to η2(P) + η2(γ) (see Appendix), but η2(γ) was low enough in the WT and the mutants in which we measured it to assume that it may be neglected (Colman‐Lerner et al, 2005).
Construction of Heterozygous Diploid Deletion Variability Collection and its use to generate sets of haploid deletion strains for screening
We constructed a MATα strain, SGA88, which carried two pheromone‐inducible reporter genes, one constitutive reporter gene, a bar1‐ mutation which blocked a protease that removed pheromone from the extracellular medium, and a cdc28‐as2 mutation which allowed us to block the inhibition of the pheromone response by the cell cycle machinery by adding to the cells a chemical inhibitor of the mutant protein kinase. In SGA88, all of these genetic elements and the MATa marker were linked to individually selectable recessive (nutritional auxotrophy) or dominant (antibiotic resistance) markers. We mated SGA88 to a fresh instance of the original (“1.0”) haploid deletion collection (Chu and Davis, 2008, a gift of Amy Chu) to create the Pesce Heterozygous Deletion Diploid Variability collection (PHDDV collection), comprised of more than 4,100 diploid strains. In these diploid strains, three dominant resistance markers: hygB R, G418 R, nat R, and two recessive markers, his3 and leu2, allowed selection of genetic elements, while two dominant sensitivity markers: canavanines (due to the CAN1 allele) and thialysineS (due to the LYP1 allele) allowed selection against unsporulated diploids. We then sporulated different members of the HDDV collection on appropriate selective media to generate haploids that bore the deletion and the other genetic markers needed for the screen. We picked these as individual small colonies on selective plates and assayed individual cultures grown from these colonies.
To screen for mutants that affected cell‐to‐cell variability in pathway output, we grew cells in log phase (< 3–106 cells/ml) for at least 14 h. This step is in contrast to the standard practice of diluting carbon‐exhausted cultures 4–6 h prior to measuring them. By relying on exponential phase cultures we minimized undesired variability in PRS output arising from strain‐to‐strain and day‐to‐day differences in time to enter the exponential growth phase. We exposed our cultures for 3 h to two different pheromone concentrations (0.6 nM or 20 nM) and 10 μM cdc28‐as2 inhibitor 1‐NM‐PP1. We then added 50 μg/ml cycloheximide to inhibit protein synthesis and allowed for existing translated fluorescent protein molecules to mature (Colman‐Lerner et al, 2005; Gordon et al, 2007). To aid the mutant screen and follow‐up experiments, we measured the maturation times of mRFP (strain collection) and mCherry (follow‐up experiments) after blocking protein synthesis with cyclohexamide as in Gordon et al (Gordon et al, 2007). Measured 1/2 time to maturation was 120 min (mRFP) and 45 min (mCherry).
We measured fluorescence signal from the P PRM1 ‐mRFP and P ACT1 ‐YFP reporters by cytometry (BD LSRII with HTS auto‐sampling attachment) and calculated or estimated parameters of interest, such as system output Oi and cell‐to‐cell variability in signal transmission, η2(P), as described above. We then verified (by cytometry) altered behaviors in three additional clonal isolates from the same mating, as described above. We confirmed by PCR in a random strain from the set of four for the presence of the expected deletion and the absence of the wild‐type coding sequence. We checked this strain by image cytometric fluorescence microscopy at the two different doses to confirm lack of aggregation and to measure P PRM1 ‐CFP signal. Measurement of CFP signal allowed us to determine if the mutants affected η2(γ). As described, η2(γ) was a small contributor to cell‐to‐cell differences in gene expression and no mutant affected it.
Data availability
Datasets sufficient to reproduce all plots in this paper are provided as Source Data files or Appendix tables. The flow cytometry data for individual strains in the screen are available at the Dryad Digital Repository, https://doi.org/10.5061/dryad.67bc0 and at http://authors.fhcrc.org/1202/ with DOI: 10.6076/j77d2s8q.
Author contributions
CGP designed and constructed the strain collection, performed the screen, analyzed the results, and characterized the selected mutants. DR assisted in this work. CGP and SZ designed and performed the microtubule perturbation experiments. MVR and AB designed, performed and analyzed the Ste5‐YFP patch experiments. WJP analyzed the numerical data, developed statistics for describing distributional shape, system stability, and comparing datasets in Figs 6 and 7. RCY contributed to experimental design, data analysis, and earlier versions of the manuscript. RB, CGP, and AC‐L directed and guided the work and its interpretation. CGP, WJP, AC‐L, and RB wrote the paper and guarantee the integrity of its results.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Appendix Table S1
Appendix Table S2
Appendix Table S3
Appendix Table S4
Appendix Table S6
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are grateful to Amy Tong, Charlie Boone, Guri Giaever, and Corey Nislow for strains, plasmids, advice, and previously unpublished information that aided construction of the whole genome deletion reporter collection, Steve Andrews, Alexander Mendenhall, and Alan Bush; to SGD staff, including Mike Cherry and Stacia Engel, for valuable discussions throughout; and to Christopher Neils (UW) and Joerg Stelling (ETHZ) and Anne Stapleton (NCSU) for valuable discussions on control theory and canalizing genes. Work was supported by R01 GM097479 to RB and PICT2013‐2210 from the Argentine Agency of Research and Technology to ACL. Earlier work received support from grants R01 GM086615 to RB and RCY and from P50 HG002370 to RB.
Mol Syst Biol. (2018) 14: e7390
References
- Anders KR, Botstein D (2001) Dominant‐lethal alpha‐tubulin mutants defective in microtubule depolymerization in yeast. Mol Biol Cell 12: 3973–3986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SS, Peria WJ, Yu RC, Colman‐Lerner A, Brent R (2016) Push‐pull and feedback mechanisms can align signaling system outputs with inputs. Cell Syst 3: 444–455 e442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansel J, Bottin H, Rodriguez‐Beltran C, Damon C, Nagarajan M, Fehrmann S, Francois J, Yvert G (2008) Cell‐to‐cell stochastic variation in gene expression is a complex genetic trait. PLoS Genet 4: e1000049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong EH (1914) Wireless receiving system. Alexandria, VA: USPTO; [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1987. –2017) Current protocols in molecular biology. New York, NY: John Wiley & Sons, Inc; [Google Scholar]
- Ayer A, Fellermeier S, Fife C, Li SS, Smits G, Meyer AJ, Dawes IW, Perrone GG (2012) A genome‐wide screen in yeast identifies specific oxidative stress genes required for the maintenance of sub‐cellular redox homeostasis. PLoS One 7: e44278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett S (1979) A history of control engineering. Stevenage, UK: IEEE, Peter Peregrinus; [Google Scholar]
- Bhattacharyya RP, Remenyi A, Good MC, Bashor CJ, Falick AM, Lim WA (2006) The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science 311: 822–826 [DOI] [PubMed] [Google Scholar]
- Brenner S (2010) Sequences and consequences. Philos Trans R Soc Lond B Biol Sci 365: 207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent R (2009) Cell signaling: what is the signal and what information does it carry? FEBS Lett 583: 4019–4024 [DOI] [PubMed] [Google Scholar]
- Boone C, Bussey H, Andrews BJ (2007) Exploring genetic interactions and networks with yeast. Nat Rev Genet 8: 437–449 [DOI] [PubMed] [Google Scholar]
- Bush A, Vasen G, Constantinou A, Dunayevich P, Patop IL, Blaustein M, Colman‐Lerner A (2016) Yeast GPCR signaling reflects the fraction of occupied receptors, not the number. Mol Syst Biol 12: 898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casolari JM, Brown CR, Drubin DA, Rando OJ, Silver PA (2005) Developmentally induced changes in transcriptional program alter spatial organization across chromosomes. Genes Dev 19: 1188–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli V, Kujala P, Klumperman J, Goldstein LS (2005) Sunday driver links axonal transport to damage signaling. J Cell Biol 168: 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman‐Lerner A, Chin TE, Brent R (2001) Yeast Cbk1 and Mob2 activate daughter‐specific genetic programs to induce asymmetric cell fates. Cell 107: 739–750 [DOI] [PubMed] [Google Scholar]
- Colman‐Lerner A, Gordon A, Serra E, Chin T, Resnekov O, Endy D, Pesce CG, Brent R (2005) Regulated cell‐to‐cell variation in a cell‐fate decision system. Nature 437: 699–706 [DOI] [PubMed] [Google Scholar]
- Chu AM, Davis RW (2008) High‐throughput creation of a whole‐genome collection of yeast knockout strains. Methods Mol Biol 416: 205–220 [DOI] [PubMed] [Google Scholar]
- Delbrück M (1945) The burst size distribution in the growth of bacterial viruses (bacteriophages). J Bacteriol 50: 131–135 [DOI] [PubMed] [Google Scholar]
- Dohlman HG, Thorner JW (2001) Regulation of G protein‐initiated signal transduction in yeast: paradigms and principles. Annu Rev Biochem 70: 703–754 [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS (2002) Stochastic gene expression in a single cell. Science 297: 1183–1186 [DOI] [PubMed] [Google Scholar]
- Erlemann S, Neuner A, Gombos L, Gibeaux R, Antony C, Schiebel E (2012) An extended gamma‐tubulin ring functions as a stable platform in microtubule nucleation. J Cell Biol 197: 59–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA (1918) The correlation between relatives on the supposition of Mendelian inheritance. Trans. R. Soc. Edin. 52: 399–433 [Google Scholar]
- Foe VE, von Dassow G (2008) Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. J Cell Biol 183: 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin CS, MSWT Company (1914) Receivers for use in wireless telegraphy and telephony. London, UK: I.P. Office; [Google Scholar]
- Fraser HB, Schadt EE (2010) The quantitative genetics of phenotypic robustness. PLoS One 5: e8635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, Nickerson DA, Bamshad MJ, NHLBI Exome Sequencing Project , Akey JM (2013) Analysis of 6,515 exomes reveals the recent origin of most human protein‐coding variants. Nature 493: 216–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Nislow C (2014) The yeast deletion collection: a decade of functional genomics. Genetics 197: 451–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Colman‐Lerner A, Chin TE, Benjamin KR, Yu RC, Brent R (2007) Single‐cell quantification of molecules and rates using open‐source microscope‐based cytometry. Nat Methods 4: 175–181 [DOI] [PubMed] [Google Scholar]
- Gundersen GG, Worman HJ (2013) Nuclear positioning. Cell 152: 1376–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR (1991) Methods in enzymology, guide to yeast genetics and molecular biology. San Diego, CA: Academic Press; [PubMed] [Google Scholar]
- Hall MC, Dworkin I, Ungerer MC, Purugganan M (2007) Genetics of microenvironmental canalization in Arabidopsis thaliana . Proc Natl Acad Sci USA 104: 13717–13722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzen HL (1934) Theory of servo‐mechanisms. J Franklin Inst 218: 279–331 [Google Scholar]
- Hughes TR, Roberts CJ, Dai H, Jones AR, Meyer MR, Slade D, Burchard J, Dow S, Ward TR, Kidd MJ, Friend SH, Marton MJ (2000) Widespread aneuploidy revealed by DNA microarray expression profiling. Nat Genet 25: 333–337 [DOI] [PubMed] [Google Scholar]
- Ismael A, Tian W, Waszczak N, Wang X, Cao Y, Suchkov D, Bar E, Metodiev MV, Liang J, Arkowitz RA, Stone DE (2016) Gbeta promotes pheromone receptor polarization and yeast chemotropism by inhibiting receptor phosphorylation. Sci Signal 9: ra38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez‐Gomez JM, Corwin JA, Joseph B, Maloof JN, Kliebenstein DJ (2011) Genomic analysis of QTLs and genes altering natural variation in stochastic noise. PLoS Genet 7: e1002295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JM, Jin M, Lew DJ (2011) Symmetry breaking and the establishment of cell polarity in budding yeast. Curr Opin Genet Dev 21: 740–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, Schuldiner M (2009) Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323: 1693–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87: 159–170 [DOI] [PubMed] [Google Scholar]
- Kozubowski L, Saito K, Johnson JM, Howell AS, Zyla TR, Lew DJ (2008) Symmetry‐breaking polarization driven by a Cdc42p GEF‐PAK complex. Curr Biol 18: 1719–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landers DA, Stapleton AE (2014) Genetic interactions matter more in less‐optimal environments: a Focused Review of “Phenotype uniformity in combined‐stress environments has a different genetic architecture than in single‐stress treatments” (Makumburage and Stapleton, 2011). Front Plant Sci 5: 384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvion JF, Havaux‐Copf B, Picard D (1993) Fusion of GAL4‐VP16 to a steroid‐binding domain provides a tool for gratuitous induction of galactose‐responsive genes in yeast. Gene 131: 129–134 [DOI] [PubMed] [Google Scholar]
- Maddox P, Chin E, Mallavarapu A, Yeh E, Salmon ED, Bloom K (1999) Microtubule dynamics from mating through the first zygotic division in the budding yeast Saccharomyces cerevisiae . J Cell Biol 144: 977–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox PS, Bloom KS, Salmon ED (2000) The polarity and dynamics of microtubule assembly in the budding yeast Saccharomyces cerevisiae . Nat Cell Biol 2: 36–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox PS, Stemple JK, Satterwhite L, Salmon ED, Bloom K (2003) The minus end‐directed motor Kar3 is required for coupling dynamic microtubule plus ends to the cortical shmoo tip in budding yeast. Curr Biol 13: 1423–1428 [DOI] [PubMed] [Google Scholar]
- Maeder CI, Hink MA, Kinkhabwala A, Mayr R, Bastiaens PI, Knop M (2007) Spatial regulation of Fus3 MAP kinase activity through a reaction‐diffusion mechanism in yeast pheromone signalling. Nat Cell Biol 9: 1319–1326 [DOI] [PubMed] [Google Scholar]
- Maekawa H, Usui T, Knop M, Schiebel E (2003) Yeast Cdk1 translocates to the plus end of cytoplasmic microtubules to regulate bud cortex interactions. EMBO J 22: 438–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makumburage GB, Stapleton AE (2011) Phenotype uniformity in combined‐stress environments has a different genetic architecture than in single‐stress treatments. Front Plant Sci 2: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleshaiah MK, Shahrezaei V, Swain PS, Michnick SW (2010) The scaffold protein Ste5 directly controls a switch‐like mating decision in yeast. Nature 465: 101–105 [DOI] [PubMed] [Google Scholar]
- McCullagh E, Seshan A, El‐Samad H, Madhani HD (2010) Coordinate control of gene expression noise and interchromosomal interactions in a MAP kinase pathway. Nat Cell Biol 12: 954–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner D (1919) Einrichtung zur Erzeugung elektrischer Schwingungen. Berlin, Germany: Gesellschaft für Drahtlose Telegraphie mbH, D.P.‐u. Markenamt; [Google Scholar]
- Mendenhall A, Crane MM, Tedesco PM, Johnson TE, Brent R (2017) Caenorhabditis elegans genes affecting interindividual variation in life‐span biomarker gene expression. J Gerontol A Biol Sci Med Sci 72: 1305–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa TK, Davis RW (2009) A genome‐wide screen for regulators of TORC1 in response to amino acid starvation reveals a conserved Npr2/3 complex. PLoS Genet 5: e1000515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordas B, Malvar RA, Hill WG (2008) Genetic variation and quantitative trait loci associated with developmental stability and the environmental correlation between traits in maize. Genet Res (Camb) 90: 385–395 [DOI] [PubMed] [Google Scholar]
- Palframan WJ, Meehl JB, Jaspersen SL, Winey M, Murray AW (2006) Anaphase inactivation of the spindle checkpoint. Science 313: 680–684 [DOI] [PubMed] [Google Scholar]
- Paliwal S, Iglesias PA, Campbell K, Hilioti Z, Groisman A, Levchenko A (2007) MAPK‐mediated bimodal gene expression and adaptive gradient sensing in yeast. Nature 446: 46–51 [DOI] [PubMed] [Google Scholar]
- Paterson JM, Ydenberg CA, Rose MD (2008) Dynamic localization of yeast Fus2p to an expanding ring at the cell fusion junction during mating. J Cell Biol 181: 697–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira G, Grueneberg U, Knop M, Schiebel E (1999) Interaction of the yeast gamma‐tubulin complex‐binding protein Spc72p with Kar1p is essential for microtubule function during karyogamy. EMBO J 18: 4180–4195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus D, Ryan CJ, Smith RD, Brent R, Resnekov O (2013) Assigning quantitative function to post‐translational modifications reveals multiple sites of phosphorylation that tune yeast pheromone signaling output. PLoS One 8: e56544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryciak PM, Huntress FA (1998) Membrane recruitment of the kinase cascade scaffold protein Ste5 by the Gbetagamma complex underlies activation of the yeast pheromone response pathway. Genes Dev 12: 2684–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randise‐Hinchliff C, Coukos R, Sood V, Sumner MC, Zdraljevic S, Meldi Sholl L, Garvey Brickner D, Ahmed S, Watchmaker L, Brickner JH (2016) Strategies to regulate transcription factor‐mediated gene positioning and interchromosomal clustering at the nuclear periphery. J Cell Biol 212: 633–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK (2004) Control of stochasticity in eukaryotic gene expression. Science 304: 1811–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricicova M, Hamidi M, Quiring A, Niemistö A, Emberly E, Hansen CL (2013) Dissecting genealogy and cell cycle as sources of cell‐to‐cell variability in MAPK signaling using high‐throughput lineage tracking. Proc Natl Acad Sci USA 110: 11403–11408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CJ, Nelson B, Marton MJ, Stoughton R, Meyer MR, Bennett HA, He YD, Dai H, Walker WL, Hughes TR, Tyers M, Boone C, Friend SH (2000) Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science 287: 873–880 [DOI] [PubMed] [Google Scholar]
- Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK (2009) Non‐genetic origins of cell‐to‐cell variability in TRAIL‐induced apoptosis. Nature 459: 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, Auclair D, Berger MF, Saksena G, Guiducci C et al (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333: 1157–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LK, Qi Y (2001) Characterization of human MAPRE genes and their proteins. Genomics 71: 142–149 [DOI] [PubMed] [Google Scholar]
- Suchkov D, DeFlorio R, Draper E, Ismael A, Sukumar M, Arkowitz R, Stone D (2010) Polarization of the yeast pheromone receptor requires its internalization but not actin‐dependent secretion. Mol Biol Cell 21: 1737–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Pryciak PM (2008) Membrane localization of scaffold proteins promotes graded signaling in the yeast MAP kinase cascade. Curr Biol 18: 1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedford K, Kim S, Sa D, Stevens K, Tyers M (1997) Regulation of the mating pheromone and invasive growth responses in yeast by two MAP kinase substrates. Curr Biol 7: 228–238 [DOI] [PubMed] [Google Scholar]
- Thomson TM, Benjamin KR, Bush A, Love T, Pincus D, Resnekov O, Yu RC, Gordon A, Colman‐Lerner A, Endy D, Brent R (2011) Scaffold number in yeast signaling system sets tradeoff between system output and dynamic range. Proc Natl Acad Sci USA 108: 20265–20270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L et al (2004) Global mapping of the yeast genetic interaction network. Science 303: 808–813 [DOI] [PubMed] [Google Scholar]
- Ventura AC, Bush A, Vasen G, Goldín MA, Burkinshaw B, Bhattacharjee N, Folch A, Brent R, Chernomoretz A, Colman‐Lerner A (2014) Utilization of extracellular information before ligand‐receptor binding reaches equilibrium expands and shifts the input dynamic range. Proc Natl Acad Sci USA 111: E3860–E3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339: 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe CJ, Kohane IS, Butte AJ (2005) Systematic survey reveals general applicability of “guilt‐by‐association” within gene coexpression networks. BMC Bioinformatics 6: 227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolinski H, Petrovic U, Mattiazzi M, Petschnigg J, Heise B, Natter K, Kohlwein SD (2009) Imaging‐based live cell yeast screen identifies novel factors involved in peroxisome assembly. J Proteome Res 8: 20–27 [DOI] [PubMed] [Google Scholar]
- Yokota Y, Kim WY, Chen Y, Wang X, Stanco A, Komuro Y, Snider W, Anton ES (2009) The adenomatous polyposis coli protein is an essential regulator of radial glial polarity and construction of the cerebral cortex. Neuron 61: 42–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Xiao J, Ren X, Lao K, Xie XS (2006) Probing gene expression in live cells, one protein molecule at a time. Science 311: 1600–1603 [DOI] [PubMed] [Google Scholar]
- Yu RC, Pesce CG, Colman‐Lerner A, Lok L, Pincus D, Serra E, Holl M, Benjamin K, Gordon A, Brent R (2008) Negative feedback that improves information transmission in yeast signalling. Nature 456: 755–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Appendix Table S1
Appendix Table S2
Appendix Table S3
Appendix Table S4
Appendix Table S6
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
Datasets sufficient to reproduce all plots in this paper are provided as Source Data files or Appendix tables. The flow cytometry data for individual strains in the screen are available at the Dryad Digital Repository, https://doi.org/10.5061/dryad.67bc0 and at http://authors.fhcrc.org/1202/ with DOI: 10.6076/j77d2s8q.