

Abstract
Background & Aims
Hepatic ischemia-reperfusion injury (IRI), characterized by exogenous antigen-independent local inflammation and hepatocellular death, represents a risk factor for acute and chronic rejection in liver transplantation. We aimed to investigate the molecular communication involved in the mechanism of liver IRI.
Methods
We analyzed human liver transplants, primary murine macrophage cell cultures and IR-stressed livers in myeloid-specific heme oxygenase-1 (HO-1) gene mutant mice, for anti-inflammatory and cytoprotective functions of macrophage-specific HO-1/SIRT1 (sirtuin 1)/p53 (tumor suppressor protein) signaling.
Results
Decreased HO-1 expression in human post-reperfusion liver transplant biopsies correlated with a deterioration in hepatocellular function (serum ALT; p <0.05) and inferior patient survival (p <0.05). In the low HO-1 liver transplant biopsy group, SIRT1/Arf (alternative reading frame)/p53/MDM2 (murine double minute 2) expression levels decreased (p <0.05) while cleaved caspase 3 and frequency of TUNEL + cells simultaneously increased (p <0.05). Immunofluorescence showed macrophages were the principal source of HO-1 in human and mouse IR-stressed livers. In vitro macrophage cultures revealed that HO-1 induction positively regulated SIRT1 signaling, whereas SIRT1-induced Arf inhibited ubiquitinating activity of MDM2 against p53, which in turn attenuated macrophage activation. In a murine model of hepatic warm IRI, myeloid-specific HO-1 deletion lacked SIRT1/p53, exacerbated liver inflammation and IR-hepatocellular death, whereas adjunctive SIRT1 activation restored p53 signaling and rescued livers from IR-damage.
Conclusion
This bench-to-bedside study identifies a new class of macrophages activated via the HO-1–SIRT1–p53 signaling axis in the mechanism of hepatic sterile inflammation. This mechanism could be a target for novel therapeutic strategies in liver transplant recipients.
Lay summary
Post-transplant low macrophage HO-1 expression in human liver transplants correlates with reduced hepatocellular function and survival. HO-1 regulates macrophage activation via the SIRT1–p53 signaling network and regulates hepatocellular death in liver ischemia-reperfusion injury. Thus targeting this pathway in liver transplant recipients could be of therapeutic benefit.
Keywords: Heme oxygenase-1, Sirtuin 1, P53, Ischemia-reperfusion injury, Liver transplantation, Innate immunity, Myeloid-specific mutant mice
Graphical Abstract
Introduction
Ischemia-reperfusion injury (IRI) during liver resection, procurement and transplantation, characterized by sterile inflammation and hepatocellular death, represents a risk factor predisposing a patient to acute and chronic rejection.1 As one of the most challenging problems in transplantation, IRI contributes to the shortage of donor organs. The initiating events that account for tissue damage are not well understood. We previously found that Toll-like receptor 4 (TLR4) drives innate immune responses in IR-stressed murine livers.2 Recently, with the aim of developing therapeutic strategies to reduce IR-damage and improve clinical outcomes, we focused on a mechanistic roadmap of immunological events before and after reperfusion in liver transplant patients.3
Heme oxygenase-1 (HO-1; HMOX1), a rate-limiting enzyme catalyzing the conversion of heme into biliverdin, carbon monoxide, and iron, exerts anti-oxidative and anti-inflammatory functions. 4 We have reported on the cytoprotective effects of HO-1 overexpression using pharmacological modulators, Ad-based gene transfer and transfer of genetically-modified macrophages. 5–7 Although high “pre-transplant” HO-1 levels in human donor livers correlated with augmented graft injury,8 “postreperfusion” HO-1 features have not been analyzed in transplant patients. We detected infiltrating mononuclear cells as the primary source of HO-1 in IR-stressed mouse liver transplants, in parallel with low-to-undetectable HO-1 levels in hepatocytes,9 the predominant liver HO-1 producers under basal conditions (Fig. S1).
The p53 tumor suppressor protein (p53; TP53) regulates the expression of target genes in apoptosis, cell cycle, DNA repair and oncogenesis. Recent studies highlight an anti-inflammatory function of p53 in macrophage cultures, streptozotocin-induced diabetes and LPS-induced lung injury.10,11 Although exacerbated kidney damage was reported in IR-stressed p53 knockout (KO) mice,12 the opposite was found after chemical inhibitor or siRNA against p53 were applied in rat renal IRI.13,14 The role of p53 in liver IRI remains unclear. A number of proteins have been implicated in p53 regulation; for example, murine double minute 2 (MDM2) targets p53 for ubiquitin-related degradation, whereas p53 may be negatively self-regulated by MDM2 and MDM2- dependent ubiquitination. A tumor suppressor protein Arf (alternative reading frame; p19 in mouse; p14 in human) inactivates MDM2 E3 ligase activity to stabilize p53.15 Pharmacological induction of HO-1 increases p53 expression in human breast cancer cells,16 while Ad-mediated HO-1 upregulates p53 in vascular smooth muscle cells.17 The HO-1–p53 communication in macrophages has not been examined and the regulatory role of p53 in HO-1 anti-inflammatory phenotype remains unknown.
Sirtuin 1 (SIRT1), a NAD +-dependent type III histone/protein deacetylase, plays a key role in cellular senescence, inflammation and stress resistance. Anti-inflammatory effects of SIRT1 were observed in IR-stressed heart, liver, brain and kidney.18–21 Recent reports point towards the efficacy of SIRT1 deacetylation to suppress macrophage activation and inhibit NF-κB;22 other regulatory proteins, including AMPK, PGC1α and PPARα, have been implicated in anti-inflammatory function of SIRT1.23 The effect of SIRT1 on net cellular p53 activity in cellular stress may be cell-type specific.24 SIRT1 not only deacetylates and promotes p53 ubiquitin-related degradation,10,11 but may also upregulate Arf to inhibit p53 ubiquitin-related degradation.24 The role of SIRT1 in p53 signaling, leading to macrophage activation and inflammation, remains to be explored.
To gain insights into signaling regulation in hepatic IR-inflammation and transplant damage, we assessed how myeloid-specific HO-1 expression may affect macrophage activation. Our results from human liver transplants, murine models of hepatic IRI and cell culture systems identify HO-1–SIRT1–p53 axis as a novel regulator of macrophage activation under IR-stress.
Methods
Clinical liver transplant study
Twenty-one adult primary orthotopic liver transplant (OLT) recipients were recruited under IRB protocol (13-000143; 5/10/2013 – 4/6/2015). Routine standard of care and immunosuppressive therapy was administered, as specified by UCLA liver transplant protocols. Protocol Tru-Cut needle biopsies (Bx) were obtained intra-operatively from the left lobe approximately 2 h after portal reperfusion (prior to abdomen closing). The demographic data and clinical parameters of recipients and donors are shown (Table S1).
Animals
Myeloid-specific HO-1 deficient (mHO-1 KO; C57BL/6) mice were generated by crossing floxed HO-1 KO mice25 with lysM (lysozyme M) Cre transgenic mice.26 Homozygous mice for floxed and Cre transgenic alleles (HO-1KOfl/fl, lysM Cre+/+) were used as mHO-1 KO mice, while HO-1fl/fl, lysM Cre−/− served as controls. Myeloid-specific HO-1 transgenic (mHO-1 Tg; C57BL/6) mice were generated by crossing floxed HO-1 Tg mice27 with lysM Cre transgenic mice.26 Homozygous mice for the floxed and Cre transgene alleles (HO-1fl/fl, lysM Cre+/+) were used as mHO-1 Tg mice.
Liver IRI model
We used an established mouse model of warm hepatic IRI.2 Animals were anesthetized, injected with heparin (100 U/kg), and an atraumatic clip was used to interrupt blood supply to the left/middle liver lobes. After 90 min of ischemia, the clamp was removed and mice were sacrificed after 6 h of reperfusion. Resveratrol ([Res], 25 mg/kg; Sigma-Aldrich, St. Louis, MO) or vehicle ([VHC], 15% ethanol) was administrated i.p. 1 h prior to ischemia. Res, a naturally occurring polyphenol, activates SIRT1 to regulate cell stress response, metabolism and survival by the deacetylation of target proteins.28
Cell isolations/in vitro cultures
Bone marrow-derived macrophages (BMDM) were cultured.29,30 The siRNA transfection was conducted using Lipofectamine Reagent (Invitrogen, San Diego, CA). In some experiments, cells were treated for 12 h with Res (100 μM) or VHC. The siRNA silencing was used to confirm Res effects were SIRT1-specific. BMDM were activated with LPS (100 ng/ml, 6 h).
Western blots
After protein concentration measurement, equal amount of protein was electrophoresed, blotted, incubated with antibodies and developed.29 To compare target protein expression in human OLT samples, densitometry quantification was conducted as follows. In a preliminary study, one of the Bx samples expressing all target proteins was assigned as a “reference” sample. Equal amounts of protein lysate from each sample were applied to each well/gel, and target band intensity was normalized to the reference sample expression in the same gel followed by normalization with β-actin.
Statistics
In mouse studies, differences between two or multiple groups were assessed by Student t test and one-way analysis of variance (ANOVA), followed by Tukey’s HSD (honest significant difference) test, respectively. For human data, continuous values were analyzed by Mann-Whitney U test and categorical variables by Fisher’s exact test. p values were considered statistically significant at <0.05. The cumulative survival rate was analyzed by Kaplan-Meier method, and differences between the groups were compared using a log-rank test.
For further details regarding the materials used, please refer to the supplementary data and CTAT table.
Results
HO-1 levels in human OLT post-reperfusion influence clinical outcomes
To focus on the role of HO-1 in human liver graft function, we measured HO-1 protein levels in post-transplant biopsies (Bx; 2 h post-reperfusion) from 21 primary OLT recipients. Based on Western blot-assisted HO-1 expression, liver Bx were divided into “high” vs. “low” HO-1 expression groups (n = 11 and n = 10, respectively, Fig. 1A). Serum alanine aminotransferase (sALT), aspartate aminotransferase (sAST) and total bilirubin (T-Bil) were analyzed (Fig. 1B–D). Patients with low HO-1 post-reperfusion had higher sALT at POD1-POD5 (p <0.05). Despite showing similar trends, sAST and T-Bil values did not reach statistical significance between the groups. To examine the relationship between HO-1 graft levels and clinical outcomes, we used a Kaplan-Meier method to analyze cumulative post-transplant survival with median follow-up of 712 days (range, 27–1,009 days). None of the patients underwent secondary liver transplant. As shown in Fig. 1E, cumulative survival in low HO- 1 expression was significantly worse (p <0.05) compared with high HO-1 expression group. The demographic data and clinical donor/recipient parameters are shown in Table S1. Statistical analysis found no correlation between HO-1 levels and any of the demographic or clinical parameters, including IRI pathophysiology factors, such as cold ischemia time or MELD score. Thus, low HO-1 levels in reperfused human liver grafts correlated with deteriorated hepatocellular function and inferior survival.
Fig. 1. Post-transplant hepatic HO-1 levels and OLT patient outcomes.

(A) Based on Western blot-assisted relative protein expression, twenty-one human liver transplant biopsies were identified as HO-1 “high” (n = 11) vs. “low” (n = 10). Representative HO-1 expression is shown in the bottom panel. (B–D) Serum ALT, AST and total bilirubin (T-Bil) levels at post-operative day 1–14 (POD 1–14). Solid line indicates high while dotted line low HO-1 OLT groups. Data are shown as mean ± SEM. #p <0.05 vs. high HO-1 (Mann-Whitney U test). (E) The cumulative probability of OLT survival (Kaplan-Meier method). Solid line indicates high and dotted line low HO-1 group. #p <0.05 vs. high HO-1 group (Log-rank test). (This figure appears in colour on the web.)
Low HO-1 levels in human OLT are accompanied by decreased SIRT1/p53 expression
To focus on HO-1–SIRT1–p53 crosstalk, we quantitated SIRT1 and p53 expression in “low” vs. “high” HO-1 human OLT. In the low HO-1 group, SIRT1 and p53 decreased (p <0.05, Fig. 2A, B), while cleaved caspase 3 (hepatocellular apoptosis indicator) simultaneously increased (p <0.05, Fig. 2C). Depressed p53 in low HO-1 Bx grafts was associated with lower MDM2 (downstream p53 effector) and p14Arf (p53 stabilizer) levels (p <0.05, Fig. 2D, E). Representative Western blot-assisted signaling profiles in human OLT are shown in Fig. 2F (high HO-1: case A/B; low HO-1: case P/Q).
Fig. 2. HO-1 levels in human OLT correlate with SIRT1, p53, cleaved caspase 3, MDM2, and p14Arf levels.

Twenty-one human liver transplant biopsies were divided, based on the relative HO-1 expression, into “high” (n = 11) and “low” (n = 10) groups (Fig. 1A). (A–E) Relative expression of SIRT1 (A; mean: 2.78 vs. 1.09), p53 (B, mean: 1.11 vs. 0.63), cleaved caspase 3 (C; mean: 0.66 vs. 1.13), MDM2 (D; mean: 0.80 vs. 0.46) and p14Arf (E; mean: 0.69 vs. 0.36). Data are shown in dot plots; bars indicate mean ± SEM. #p <0.05 high (n = 11) vs. low (n = 10) HO-1 (Mann-Whitney U test). (F) Representative Western blot-assisted OLT expression of HO-1, SIRT1, p53, cleaved caspase 3, MDM2 and p14Arf (A/B: high HO-1; P/Q: low HO-1).
HO-1 is expressed primarily by IR-stressed liver macrophages
To identify the dominant HO-1 producing cell in liver IRI, we performed immunofluorescent staining of human post-transplant Bx. As shown in Fig. 3A, HO-1 (red) expression co-localized with CD68 (blue), indicating that macrophages are the principal producers of HO-1 in IR-stressed human OLT. Consistent with Fig. 1B–D and Fig. 2C, low HO-1 group displayed increased frequency of TUNEL + cells (green) compared to the high HO-1 group. The CD68/HO-1 co-expression in the liver was confirmed in two mouse models of cold IRI/OLT (18 h cold storage) and warm IRI (90 min ischemia) (Fig. 3B).
Fig. 3. Frequency of TUNEL + cells in human OLT is negatively correlated with macrophage-expressing HO-1.

(A) Immunofluorescence staining of CD68 (blue, macrophage), HO-1 (red), TUNEL (green) and merged image in high vs. low HO-1 human OLT. Representative of three in each group is shown. (B) Immunofluorescence staining of CD68 (red), HO-1 (green), DAPI (blue) and merged image in IR-stressed mouse livers. Upper panels: 6 h reperfusion/OLT (18 h cold storage). Lower panels: 6 h reperfusion/90 min warm ischemia. Representative of three in each group is shown. (This figure appears in colour on the web.)
HO-1 upregulates SIRT1/p53 while inhibiting macrophage activation in vitro
Having identified that macrophages are the principle producer of HO-1 in IR-stressed liver, we focused on dissecting the regulatory function of HO-1 upon SIRT1/p53 signaling in LPS-stimulated BMDM from WT or myeloid-specific HO-1 transgenic (mHO-1 Tg) mice. As shown in Fig. 4A, B, HO-1 overexpression in BMDM increased SIRT1, p19, p53 and p53-downstream effectors (MDM2, PUMA, Noxa, p21) while decreasing macrophage activation markers (p-Stat1, iNOS) compared with HO-1 proficient counterparts. Consistent with human OLT (Fig. 2), this indicates that HO-1 suppresses mouse BMDM activation while upregulating SIRT1/p53 and p19 expression. Although anti-inflammatory HO-1, SIRT1 and p53 molecules control macrophage activation,10,22,31 it is unknown whether, and how, SIRT1 and p53 may function in a HO-1-enriched environment, with SIRT1 influencing p53 activity in a cell-specific manner.24 Indeed, myeloid-specific HO-1 overexpression ameliorated liver IRI, suppressed inflammation and enhanced SIRT1/p19/p53/MDM2 expression (Fig. S2 and unpublished).
Fig. 4. HO-1 upregulates SIRT1–p53 signaling in BMDM.

BMDM from WT or mHO-1 Tg mice were stimulated with LPS (100 ng/ml, 6 h). (A) Western blot-assisted detection of HO-1, SIRT1, p19, p53, MDM2, PUMA and p-Stat1 (Tyr701). β-actin expression served as an internal control for normalization. The values under the bands represent relative ratios of normalized intensity compared to WT/cells + LPS. Representative of three experiments is shown. (B) Quantitative RT-PCR-assisted detection of mRNA coding for SIRT1, Noxa, p21 and iNOS. Data normalized to B2M gene (n = 4/group) are presented as mean ± SD. *p <0.05 vs. WT/cells + LPS (Student t test).
SIRT1 depresses macrophage activation and upregulates p53 expression in vitro
To further dissect SIRT1 function in macrophage regulation and p53 signaling, we explored LPS-stimulated BMDM cultures supplemented with Res (SIRT1 inducer) or siRNA-SIRT1. Western blot analysis (Fig. 5A) showed that the addition of Res suppressed macrophage activation markers (p-Stat1, p-IkBα, iNOS), a pattern that reversed after SIRT1 silencing. Notably, SIRT1 activation in vitro increased expression of p53 and its downstream effector proteins (MDM2/PUMA) even though SIRT1 decreased the acetylation ratio (Ac-/total-p53 = 0.03 (Res) vs. 4.12 [siRNA-SIRT1]). In addition, SIRT1 activation increased p19, which inactivates MDM2 E3 ligase and sustains p53 activity. We confirmed these findings after SIRT1 knockdown, which had opposite effects to those seen in Res-treated BMDM (Fig. 5B). As shown in Fig. 5C, SIRT1 decreased macrophage activation (iNOS, IL-1β, MCP1, CCL5, CXCL10, GzmB) while upregulating p53-downstream effectors (Noxa, p21; p <0.05). Thus, despite p53 deacetylation, SIRT1 induction was able to enhance p53 signaling and suppress macrophage activation.
Fig. 5. SIRT1 depresses macrophage activation and upregulates p53 signaling in vitro.

LPS-stimulated BMDM (100 ng/ml, 6 h) with/without Res (SIRT1 inducer, 100 μM, 12 h) or siRNA SIRT1 were screened for activation markers. (A and B) Western blot-assisted detection of SIRT1, p-Stat1 (Tyr701), p-IkBα (Ser32), IkBα, iNOS, p19, p53, Ac-p53 (Lys379), MDM2, and PUMA. Band intensities were quantified with ImageJ and normalized by dividing target band intensity by that of housekeeping β-actin. The values under the bands represent relative ratios of normalized intensity, compared to cells + LPS. Representative of three experiments is shown. (C) Quantitative RT-PCR-assisted detection of mRNA coding for SIRT1, Noxa, p21, iNOS, IL-1β, MCP1, CCL5, CXCL10 and GzmB. Data normalized to B2M gene expression (n = 4/group) are presented as the mean ± SD. *p <0.05 vs. cells + LPS (one-way ANOVA).
The p19/p53 signaling axis is critical in anti-inflammatory SIRT1 function in vitro
We investigated whether p19 function is necessary and sufficient for enhancement of p53 signaling by SIRT1 in BMDM cultures. As shown in Fig. S3A, p19 knockdown (siRNA) in Res-treated cells decreased p53, MDM2, and PUMA, compared with cells + LPS + Res, or cells + LPS + Res + siRNA (scrambled) settings. RT-PCR revealed that p19 knockdown in Res-treated cells impaired the ability of SIRT1 to increase p53 signaling (Noxa, p21) and suppress macrophage activation (iNOS, CXCL10) (p <0.05, Fig. S3B). The efficacy of Res to decrease ubiquitinylated-p53 was diminished when p19 was silenced (Fig. 6A), indicating that SIRT1 inhibits p53 ubiquitination in a p19-dependent manner. Thus, p19 is an essential downstream mediator of SIRT1 to enhance p53 by protecting it from ubiquitination, and attenuating inflammation in vitro.
Fig. 6. SIRT1 inhibits macrophage activation through p19/p53 signaling.

(A–C) BMDM stimulated with LPS (100 ng/ml, 6 h) were treated with Res (100 μM, 12 h), siRNA (p53) or siRNA (p19). (A) 25 μg of protein lysate from BMDM were divided by immunoprecipitation into ubiquitinated (UB-) and non-ubiquitinated (nonUB-) proteins and analyzed by Western blotting. Representative images (upper panel) and densitometry quantification (lower panel) are shown. *p <0.05 vs. cells + LPS, #p <0.05 vs. cells + LPS + Res, n = 3/group (one-way ANOVA). (B) Western blot-assisted detection of SIRT1, p53, PUMA, p-IkBα (Ser32) and IkBα in LPS-stimulated BMDM. Band intensities were normalized by dividing target band intensity by that of β-actin. The values under the bands represent relative ratios of normalized intensity compared to that of cells + LPS. Representative of three experiments is shown. (C) Quantitative RT-PCR-assisted detection of mRNA coding for SIRT1, Noxa, p21, iNOS, IL-1β, MCP1 and CCL5. Data were normalized to B2M gene expression (*p <0.05 vs. cells + LPS, #p <0.05 vs. cells + LPS + Res, n = 4/group, one-way ANOVA). (D and E) Livers in WT mice underwent 90 min of warm ischemia followed by 6 h reperfusion. (D) Western blot-assisted detection of SIRT1, p19, p53, MDM2 and PUMA. The values under the bands represent relative ratios of normalized intensity compared to that of IR. Representative of three experiments is shown. (E) Quantitative RT-PCR-assisted detection of mRNA coding for Noxa. Data normalized to HPRT gene expression (*p <0.05 vs. IR, #p <0.05 vs. IR + VHC, n = 4/group, one-way ANOVA) are presented as mean ± SD.
The next question was whether p53 is necessary for suppressing macrophage activation by SIRT1. The p53 knockdown (siRNA) enhanced p-IkBα levels in LPS-stimulated BMDM. Furthermore, the p53 knockdown impaired the efficacy of SIRT1 to decrease p-IkBα in BMDM + LPS + Res cultures. The impact of p19 knockdown in BMDM + LPS ± Res cultures showed a similar trend to that of p53 knockdown (Fig. 6B). RT-PCR confirmed that p19 and p53 knockdown suppressed downstream p53 signaling (Noxa, p21) while enhancing macrophage activation (iNOS, IL-1β, MCP1, CCL5) with or without SIRT1 enhancement (p <0.05, Fig. 6C). Thus, the p19/p53 axis plays a key role in SIRT1-mediated suppression of macrophage activation.
To confirm our in vitro findings, we then examined SIRT1/p19/p53 expression at 6 h of reperfusion in mouse livers subjected to 90 min of ischemia. Unlike in untreated IR or VHC treated controls, pre-treatment with Res (25 mg/kg i.p. for 1 h) ameliorated hepatic IRI (Fig. S4A, B), lowered mRNA levels coding for IL-1β, MCP1 (Fig. S4C), accompanied by an upregulation of p19/p53 and downstream MDM2, PUMA and Noxa effector molecules (Fig. 6D, E).
SIRT1 activation rescues anti-inflammatory phenotype and p53 signaling in HO-1-deficient macrophages
Up to this point, we have shown that: i) human OLT characterized by low HO-1 expression exhibited depressed SIRT1/p14Arf/p53/MDM2 levels (Fig. 2) and inferior clinical outcomes (Fig. 1B–E); ii) HO-1 downregulated macrophage activation while increasing SIRT1/p19/p53 (Fig. 4); and iii) SIRT1 inhibited macrophage activation in p19/p53-dependent manner (Fig. 6A–C). In line with these findings, we hypothesized that SIRT1–p53 signaling may underlie anti-inflammatory HO-1 macrophage function. First, we explored whether HO-1 deficient macrophages are indeed lacking SIRT1/p53, and then whether SIRT1 activation may rescue such a deficit and reinstate an anti-inflammatory phenotype. As shown in Fig. 7A–C, genetic disruption of HO-1 in BMDM decreased SIRT1, p19, p53, MDM2, Noxa and p21 levels, while enhancing p-Stat1, iNOS, TNFα, MCP1, IL-1β, GzmB, and IL-12p40. Moreover, Res supplement in HO-1-deficient macrophages restored SIRT1/p53 and attenuated excessive macrophage activation, indicating that anti-inflammatory HO-1 function is indeed mediated by SIRT1–p53 signaling.
Fig. 7. Macrophage HO-1 deficiency depresses SIRT1–p53 signaling and augments inflammation, while SIRT1 activation restores p53/anti-inflammatory phenotype in BMDM.

BMDM were harvested from HO-1 proficient (WT) or myeloid-specific HO-1 deficient (mHO-1 KO) mice and activated with LPS (100 ng/ml, 6 h). In some experiments, cells were pretreated with Res (100 μM) or VHC for 12 h. (A) Western blot-assisted detection of HO-1, SIRT1, p19, p53, Ac-p53 (Lys379), MDM2, and p-Stat1 (Tyr701). β-actin expression as internal control was used for normalization. The values under the bands represent the relative ratio of normalized intensity compared to WT/cells + LPS. Representative of three experiments is shown. (B) Quantitative RT-PCR-assisted detection of SIRT1, Noxa, p21, iNOS, TNFα, MCP1, IL-1β, GzmB and IL-12p40. Data normalized to B2M gene expression (n = 4/group). (C) ELISA-assisted detection of MCP1 in BMDM culture medium (pg/μl, n = 4/group). Data are presented as mean ± SD. *p <0.05 vs. WT/cells + LPS, #p <0.05 vs. HO-1 deficient/cells + LPS + Res (one-way ANOVA).
Myeloid-specific HO-1 deficiency exacerbates liver IRI while SIRT1 rescues IR-livers by restoring p19/p53 signaling
To determine the impact of macrophage HO-1–SIRT1–p53 axis in vivo, groups of myeloid-specific HO-1-deficient mice were subjected to hepatic IR-stress (90 min). By 6 h of reperfusion, ischemic livers in mHO-1 KO animals displayed severe sinusoidal congestion, vacuolization and hepatocellular necrosis, compared with HO-1 proficient livers (Fig. 8A). These correlated with Suzuki’s histological grading of liver IRI and serum enzyme levels (p <0.05, Fig. 8B). Myeloid HO-1 deficiency increased the frequency of hepatic TUNEL + cells, liver-infiltrating macrophages (CD11b) and neutrophils (Ly6G) as well as oxidative stress marker (4HN), compared with controls (Fig. S5A–D). Western blots (Fig. 8C), ELISA (Fig. 8D) and RT-PCR (Fig. 8E) revealed that HO- 1 deficiency correlated with decreased SIRT1/p19/p53 and downstream mediators (MDM2/Noxa) while enhancing MCP1/TNFα levels. This data indicates that myeloid HO-1 plays a dominant role in managing liver IR-hepatocellular death, inflammation and oxidative stress. Consistent with in vitro data (Fig. 7A, B), exacerbated liver damage in mHO-1 KO mice was attenuated after treatment with Res (but not VHC) (p <0.05, Fig. 8A, B). Moreover, IR-stressed livers in mHO-1 KO mice conditioned with Res showed decreased frequency of TUNEL + cells/infiltrating leukocytes along with diminished 4HN (Fig. S5A–D) and MCP1/TNFα (p <0.05, Fig. 8D–E) expression; and restored otherwise blunted SIRT1–p53 signaling (Fig. 8C). These results support the contention that SIRT1 activation is sufficient to reinstate the anti-inflammatory phenotype in an IR-stressed mHO-1-deficient environment.
Fig. 8. Myeloid-specific HO-1 deficiency depresses SIRT1–p53 and augments IRI, while SIRT1 activation restores p53 and attenuates liver IRI/inflammation in mHO-1 KO mice.

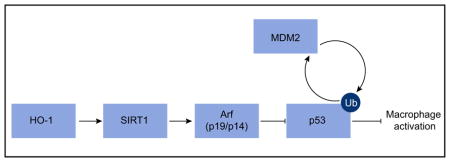
Groups of control and mHO-1 KO mice were subjected to warm liver IRI. Some mice were treated with Res (25 mg/kg i.p. at −1 h) or VHC. (A) Representative H&E staining (original magnification, 100×; n = 4–6/group). (B) Suzuki’s histological grading of liver IRI and sALT/AST [IU/L] (n = 4–6/group). (C) Western blot-assisted detection of SIRT1, p19, p53, Ac-p53 (Lys379) and MDM2. β-actin expression serves as an internal control. The values under the bands represent the relative ratio of normalized intensity compared to IR/Control. Representative of three experiments is shown. (D) ELISA-assisted detection of serum MCP1 (pg/ml, n = 3–4/group) (E) Quantitative RT-PCR-assisted detection of mRNA coding for SIRT1, Noxa, TNFα and MCP1. Data normalized to HPRT gene expression (n = 4/group) are presented as mean ± SD. *p <0.05 vs. IR/Control, #p <0.05 vs. IR/mHO-1 KO + Res (one-way ANOVA). (F) Schematic illustration of macrophage regulation by HO-1–SIRT1–p53 axis. HO-1 upregulates SIRT1 (#1), whereas SIRT1-induced Arf (p19 in mouse, p14 in human) (#2) inhibits MDM2 E3 ligase activity to stabilize p53 (#3). Consequently, p53 tumor suppressor protein upregulated by HO1/SIRT1/p19 axis (#1–3) attenuates macrophage activation (#4). DEG: ubiquitin-degradation of p53 by MDM2. (This figure appears in colour on the web.)
Discussion
In this study, encompassing human OLT, a mouse model of liver IRI, and murine BMDM culture, we have identified a novel HO-1– SIRT1–p53 signaling axis in macrophage activation during IR-stress. Although IRI represents a major clinical problem, remarkably few studies have been conducted in human transplant recipients to understand its mechanistic underpinnings. To the best of our knowledge, this is the first report linking post-transplant HO-1 levels with the severity of hepatocellular damage in transplant patients. Notably, low post-reperfusion HO-1 levels, accompanied by decreased SIRT1/p53 expression, correlated with augmented hepatocellular death (caspase 3/TUNEL), deteriorated OLT function (sALT) and decreased cumulative patients’ survival. Failure to detect statistically significant differences in sAST and T-Bil levels may have been due to the limited number of subjects.
HO-1 may serve as a cytoprotective molecule and stress-inducible heat shock protein (hsp32).32 Although our patients underwent optimized transplant surgery and peri-operative care with comparable cold ischemia times (Table S1, p = 0.32), the exacerbated hepatocellular damage in low HO-1 Bx may have resulted from individual inability to upregulate HO-1 response in IR-stressed liver. Indeed, post/pre-transplant HO-1 mean expression ratio in the low HO-1 group was depressed compared with the high HO-1 group (0.90 and 1.74, respectively), implying that low HO-1 Bx livers failed to trigger HO-1 increase under IR-stress. Although aging and obesity are poor-prognosis factors, there was no correlation between post-transplant HO-1 levels and donor/recipient demographic parameters, including age and BMI in our study. Short guanine-thymine nucleotide repeats polymorphism located in the promotor region on chromosome 22q13.1 of human HO-1 was shown to enhance its expression in response to oxidative stress.33 However, controversy exists as to whether short repeats polymorphism in human livers associates with superior graft function.8,34 More studies on putative factors influencing human HO-1 responses are warranted.
Although post-reperfusion HO-1 levels correlated with hepatocellular function and clinical outcomes, it is premature to conclude that graft HO-1 influenced patient survival by suppressing IRI per se. First, the direct link between IRI severity and patient/graft survival remains somewhat controversial.35,36 However, it is noteworthy that pharmacological HO-1 induction attenuated cholestasis in rats,37 one of the major IRI complications. Second, although survival analysis demonstrated significant differences between “high” vs. “low” HO-1 groups, we recognize that our retrospective study in a limited patient cohort (n = 21) needs to be confirmed prospectively with a larger number of OLT recipients.
Our novel finding on HO-1 phenotype predisposing human OLT to IR-insult provides a solid springboard for a more in-depth mechanistic assessment of HO-1 signaling in well-controlled murine settings. Consistent with our previous studies,9 we first confirmed liver macrophages (infiltrating and tissue-resident) as major HO-1 producers, both in human OLT and IR-stressed mouse livers. Here, we focused on HO-1 signaling in mouse BMDM because: i) circulating monocytes are instrumental in the mechanism of liver IRI;1,38 ii) adoptive transfer of BMDM was sufficient to recreate hepatocellular damage in otherwise IRI-resistant CD11b-DTR mice;38 iii) number of liver-resident Kupffer cells drastically decreased under IR-stress, with a simultaneous massive influx of circulating macrophages;39 and iv) circulating monocytes can generate self-renewing Kupffer cells.40 Consistent with the recipient myeloid HO-1 deletion worsening hepatocellular damage in a murine OLT model (not shown), our findings support the significance of HO-1 expressing circulating macrophages in the mechanism associated with liver IRI.
SIRT1, also referred to as a “longevity gene”, has been shown to extend lifespan and protect cells from an array of stresses.41 To induce SIRT1, we used Res, a small molecule SIRT1 activator with antioxidant, cardioprotective and anticancer properties.42 Being aware of SIRT1-independent off-target Res effects,43 we used siRNA silencing to confirm that our findings in BMDM cultures were SIRT1-dependent. In a recent study, HepG2 cells became more susceptible to hypoxia/reoxygeneration-induced apoptosis after SIRT1 knockdown,44 indicating an anti-apoptotic role of SIRT1 in hepatocytes. Thus, cytoprotection after SIRT1 induction in mHO-1 KO livers (Fig. 8A, B) may not only result from recreating anti-inflammatory macrophage function, but also from hepatocyte SIRT1 activation.
Having demonstrated that i) myeloid-specific HO-1 deletion augmented hepatocellular death (Fig. 8A, B); and ii) SIRT1 mediated a cytoprotective function of macrophage HO-1 (Fig. 7A–C), the idea that the myeloid HO-1/SIRT1 axis controls sterile inflammation and tissue damage in IR-stressed mouse livers is strengthened. However, Cui et al. reported that hepatocyte-specific SIRT1 KO mice displayed enhanced hepatocyte NF-κB activity and resistance to apoptosis in endotoxemic liver injury model.45 As hepatocyte SIRT1 function against apoptosis may be disease and microenvironment dependent, future studies in hepatocyte-specific SIRT1 deficient strains are warranted. Although worseliver IRI in mHO-1 KO mice can be firmly attributed to HO-1 deficiency in circulating macrophages, we are aware that mHO-1 KO mice underwent Cre-mediated deletion of HO-1 under the control of the LysM promoter, which is expressed in all myeloid cells (26). Therefore, both macrophages and neutrophils may have contributed to deteriorated hepatocellular function. As HO-1 regulates neutrophil activation,46 the role of neutrophil HO-1 in liver IRI awaits further studies.
Although acetylation affects the function of the p53 molecule, multiple factors may control p53 signaling. Chae et al. reported that endogenous stress of reactive oxygen species in embryonic stem cells induced by 2-ME removal, enhanced p53 function in a SIRT1-dependent manner.47 By focusing on SIRT1–p53 molecular communication at the macrophage level, our study, to the best of our knowledge, is the first to document that SIRT1 upregulates p53. Thus, screening downstream molecules, including MDM2 to determine p53 function, is warranted. Unlike in macrophages, SIRT1 activation downregulated p53 in primary hepatocyte cultures (data not shown). Therefore, it is essential to determine the role of SIRT1 on p53 in individual cell types, tissues and in vivo models. Our finding on anti-inflammatory p53 signaling in the liver is important in a broad context of anti-tumor p53 activities. Indeed, adenoviral p53 gene therapy (ADVEXIN®) is currently under clinical evaluation in patients with malignancies. As p53 activation may cause cell cycle inhibition, apoptosis or cardiac dysfunction,48 future p53-specific in vivo manipulations in IR-stressed livers are warranted.
With a clinical hint from human OLT samples, we have identified a novel SIRT1-Arf-p53-MDM2 network in macrophage activation and liver IRI. Collectively, our human andmurine findings support the following model of mechanistic integration between HO-1, SIRT1 and p53 signaling (Fig. 8F). HO-1 positively regulates SIRT1 (#1) while SIRT1-induced Arf (p19 in mouse, p14 in human) (#2) inhibits MDM2 E3 ligase activity to stabilize p53 (#3). Consequently, p53 tumor suppressor protein upregulated by HO1-SIRT1-p19 axis (#1–3) attenuates macrophage activation (#4).
This bench-to-bedside study identifies a new class of HO-1–SIRT1–p53 signaling axis in the mechanism of macrophage activation and sterile inflammation in liver transplantation. The immune phenotypes and pathways described here are likely to be applicable to other types of liver inflammation, including non-alcoholic steatohepatitis, an emerging global epidemic projected to become the leading indication for liver transplantation.
Supplementary Material
Highlights.
HO-1 is expressed primarily by ischemia-stressed liver macrophages.
HO-1 regulates macrophage activation via SIRT1–p53 signaling.
HO-1 regulates hepatocellular death in liver ischemia-reperfusion injury.
Low macrophage HO-1 levels correlate with reduced human liver transplant function.
Acknowledgments
Financial support
This work was supported by NIH grants PO1 AI120944, RO1 DK062357, DK107533, DK102110 (to JWKW); NIH RO1 ES016959, R56 ES016959-06 (to JAA); and The Dumont-UCLA Research Foundation.
We are grateful to Drs. Enrique Rozengurt and Steve Smale (UCLA) for helpful comments. We thank Ko Takanashi (UCLA-TPCL) for immunohistochemical assistance.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jhep.2017.08.010.
Conflict of interest
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Please refer to the accompanying ICMJE disclosure forms for further details.
Authors’ contributions
KN, JAA and JWKW – study concept and design; KN, SK and TF – acquisition of experimental data; KN and SK – surgical procedures; BK – assistance in molecular assays; MZ and JAA – generation/characterization of mHO-1 KO/Tg mice; RS, ND, EFR and AZ – clinical liver transplant samples; EFR, JAA and RWB – discussants, manuscript review; KN and JWKW – drafted manuscript; RWB, JAA and JWKW – obtained funding; all authors have read and edited the manuscript.
References
Author names in bold designate shared co-first authorship
- 1.Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation-from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10:79–89. doi: 10.1038/nrgastro.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 3.Sosa RA, Zarrinpar A, Rossetti M, Lassman CR, Naini BV, Datta N, et al. Early cytokine signatures of ischemia/reperfusion injury in human orthotopic liver transplantation. JCI Insight. 2016;1:e89679. doi: 10.1172/jci.insight.89679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 5.Amersi F, Buelow R, Kato H, Ke B, Coito AJ, Shen XD, et al. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest. 1999;104:1631–1639. doi: 10.1172/JCI7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ke B, Shen XD, Gao F, Ji H, Qiao B, Zhai Y, et al. Adoptive transfer of ex vivo HO-1 modified bone marrow-derived macrophages prevents liver ischemia and reperfusion injury. Mol Ther. 2010;18:1019–1025. doi: 10.1038/mt.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang J, Shen XD, Yue S, Zhu J, Gao F, Zhai Y, et al. Adoptive transfer of heme oxygenase-1 (HO-1)-modified macrophages rescues the nuclear factor erythroid 2-related factor (Nrf2) antiinflammatory phenotype in liver ischemia/reperfusion injury. Mol Med. 2014;20:448–455. doi: 10.2119/molmed.2014.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geuken E, Buis CI, Visser DS, Blokzijl H, Moshage H, Nemes B, et al. Expression of heme oxygenase-1 in human livers before transplantation correlates with graft injury and function after transplantation. Am J Transplant. 2005;5:1875–1885. doi: 10.1111/j.1600-6143.2005.00960.x. [DOI] [PubMed] [Google Scholar]
- 9.Kato H, Amersi F, Buelow R, Melinek J, Coito AJ, Ke B, et al. Heme oxygenase- 1 overexpression protects rat livers from ischemia/reperfusion injury with extended cold preservation. Am J Transplant. 2001;1:121–128. [PubMed] [Google Scholar]
- 10.Zheng SJ, Lamhamedi-Cherradi SE, Wang P, Xu L, Chen YH. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes. 2005;54:1423–1428. doi: 10.2337/diabetes.54.5.1423. [DOI] [PubMed] [Google Scholar]
- 11.Liu G, Park YJ, Tsuruta Y, Lorne E, Abraham E. P53 Attenuates lipopolysac-charide- induced NF-kappaB activation and acute lung injury. J Immunol. 2009;182:5063–5071. doi: 10.4049/jimmunol.0803526. [DOI] [PubMed] [Google Scholar]
- 12.Sutton TA, Hato T, Mai E, Yoshimoto M, Kuehl S, Anderson M, et al. P53 is renoprotective after ischemic kidney injury by reducing inflammation. JASN. 2013;24:113–124. doi: 10.1681/ASN.2012050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molitoris BA, Dagher PC, Sandoval RM, Campos SB, Ashush H, Fridman E, et al. SiRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. JASN. 2009;20:1754–1764. doi: 10.1681/ASN.2008111204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelly KJ, Plotkin Z, Vulgamott SL, Dagher PC. P53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: protective role of a p53 inhibitor. JASN. 2003;14:128–138. doi: 10.1097/01.asn.0000040596.23073.01. [DOI] [PubMed] [Google Scholar]
- 15.Alarcon-Vargas D, Ronai Z. P53-Mdm2–the affair that never ends. Carcino-genesis. 2002;23:541–547. doi: 10.1093/carcin/23.4.541. [DOI] [PubMed] [Google Scholar]
- 16.Kim DH, Song NY, Kim EH, Na HK, Joe Y, Chung HT, et al. 15-deoxy-Delta 12,14-prostaglandin J(2) induces p53 expression through Nrf2-mediated upregulation of heme oxygenase-1 in human breast cancer cells. Free Radic Res. 2014;48:1018–1027. doi: 10.3109/10715762.2014.897343. [DOI] [PubMed] [Google Scholar]
- 17.Liu XM, Chapman GB, Wang H, Durante W. Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation. 2002;105:79–84. doi: 10.1161/hc0102.101369. [DOI] [PubMed] [Google Scholar]
- 18.Thirunavukkarasu M, Penumathsa SV, Koneru S, Juhasz B, Zhan L, Otani H, et al. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: Role of nitric oxide, thioredoxin, and heme oxygenase. Free Radic Biol Med. 2007;43:720–729. doi: 10.1016/j.freeradbiomed.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rickenbacher A, Jang JH, Limani P, Ungethum U, Lehmann K, Oberkofler CE, et al. Fasting protects liver from ischemic injury through Sirt1-mediated downregulation of circulating HMGB1 in mice. J Hepatol. 2014;61:301–308. doi: 10.1016/j.jhep.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 20.Ren J, Fan C, Chen N, Huang J, Yang Q. Resveratrol pretreatment attenuates cerebral ischemic injury by upregulating expression of transcription factor Nrf2 and HO-1 in rats. Neurochem Res. 2011;36:2352–2362. doi: 10.1007/s11064-011-0561-8. [DOI] [PubMed] [Google Scholar]
- 21.Sener G, Tugtepe H, Yuksel M, Cetinel S, Gedik N, Yegen BC. Resveratrol improves ischemia/reperfusion-induced oxidative renal injury in rats. Arch Med Res. 2006;37:822–829. doi: 10.1016/j.arcmed.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 22.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25:1939–1948. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 24.Chua KF, Mostoslavsky R, Lombard DB, Pang WW, Saito S, Franco S, et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76. doi: 10.1016/j.cmet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 25.Mamiya T, Katsuoka F, Hirayama A, Nakajima O, Kobayashi A, Maher JM, et al. Hepatocyte-specific deletion of heme oxygenase-1 disrupts redox homeostasis in basal and oxidative environments. Tohoku J Exp Med. 2008;216:331–339. doi: 10.1620/tjem.216.331. [DOI] [PubMed] [Google Scholar]
- 26.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 27.Hull TD, Bolisetty S, DeAlmeida AC, Litovsky SH, Prabhu SD, Agarwal A, et al. Heme oxygenase-1 expression protects the heart from acute injury caused by inducible Cre recombinase. Lab Invest. 2013;93:868–879. doi: 10.1038/labinvest.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung S, Yao H, Caito S, Hwang JW, Arunachalam G, Rahman I. Regulation of SIRT1 in cellular functions: role of polyphenols. Arch Biochem Biophys. 2010;501:79–90. doi: 10.1016/j.abb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamo N, Ke B, Busuttil RW, Kupiec-Weglinski JW. PTEN-mediated Akt/beta-catenin/Foxo1 signaling regulates innate immune responses in mouse liver ischemia/reperfusion injury. Hepatology. 2013;57:289–298. doi: 10.1002/hep.25958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, et al. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol. 2008;294:G498–505. doi: 10.1152/ajpgi.00482.2007. [DOI] [PubMed] [Google Scholar]
- 31.Ke B, Shen XD, Ji H, Kamo N, Gao F, Freitas MC, et al. HO-1-STAT3 axis in mouse liver ischemia/reperfusion injury: regulation of TLR4 innate responses through PI3K/PTEN signaling. J Hepatol. 2012;56:359–366. doi: 10.1016/j.jhep.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi AM, Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol. 1996;15:9–19. doi: 10.1165/ajrcmb.15.1.8679227. [DOI] [PubMed] [Google Scholar]
- 33.Hirai H, Kubo H, Yamaya M, Nakayama K, Numasaki M, Kobayashi S, et al. Microsatellite polymorphism in heme oxygenase-1 gene promoter is associated with susceptibility to oxidant-induced apoptosis in lymphoblastoid cell lines. Blood. 2003;102:1619–1621. doi: 10.1182/blood-2002-12-3733. [DOI] [PubMed] [Google Scholar]
- 34.Zhang ZY, Guan J, Li H, Zhou ZQ, Zhou GW. Heme oxygenase-1 promoter polymorphism protects liver allograft. Indian J Surg. 2016;78:14–19. doi: 10.1007/s12262-015-1309-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berberat PO, Friess H, Schmied B, Kremer M, Gragert S, Flechtenmacher C, et al. Differentially expressed genes in postperfusion biopsies predict early graft dysfunction after liver transplantation. Transplantation. 2006;82:699–704. doi: 10.1097/01.tp.0000233377.14174.93. [DOI] [PubMed] [Google Scholar]
- 36.Ali JM, Davies SE, Brais RJ, Randle LV, Klinck JR, Allison ME, et al. Analysis of ischemia/reperfusion injury in time-zero biopsies predicts liver allograft outcomes. Liver Transpl. 2015;21:487–499. doi: 10.1002/lt.24072. [DOI] [PubMed] [Google Scholar]
- 37.Donner MG, Topp SA, Cebula P, Krienen A, Gehrmann T, Sommerfeld A, et al. HbG200-mediated preinduction of heme oxygenase-1 improves bile flow and ameliorates pericentral downregulation of Bsep and Mrp2 following experimental liver ischemia and reperfusion. Biol Chem. 2013;394:97–112. doi: 10.1515/hsz-2012-0153. [DOI] [PubMed] [Google Scholar]
- 38.Ji H, Liu Y, Zhang Y, Shen XD, Gao F, Busuttil RW, et al. T-cell immunoglobulin and mucin domain 4 (TIM-4) signaling in innate immune-mediated liver ischemia-reperfusion injury. Hepatology. 2014;60:2052–2064. doi: 10.1002/hep.27334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yue S, Zhou H, Wang X, Busuttil RW, Kupiec-Weglinski JW, Zhai Y. Prolonged ischemia triggers necrotic depletion of tissue-resident macrophages to facilitate inflammatory immune activation in liver ischemia reperfusion injury. J Immunol. 2017;198:3588–3595. doi: 10.4049/jimmunol.1601428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. 2016;7:10321. doi: 10.1038/ncomms10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tome-Carneiro J, Larrosa M, Gonzalez-Sarrias A, Tomas-Barberan FA, Garcia- Conesa MT, Espin JC. Resveratrol and clinical trials: the crossroad from in vitro studies to human evidence. Curr Pharm Des. 2013;19:6064–6093. doi: 10.2174/13816128113199990407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan H, Jihong Y, Feng Z, Xiaomei X, Xiaohan Z, Guangzhi W, et al. Sirtuin 1- mediated inhibition of p66shc expression alleviates liver ischemia/reperfusion injury. Crit Care Med. 2014;42:e373–e381. doi: 10.1097/CCM.0000000000000246. [DOI] [PubMed] [Google Scholar]
- 45.Cui X, Chen Q, Dong Z, Xu L, Lu T, Li D, et al. Inactivation of Sirt1 in mouse livers protects against endotoxemic liver injury by acetylating and activating NF-kappaB. Cell Death Dis. 2016;7:e2403. doi: 10.1038/cddis.2016.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X, Schwacha MG, Chaudry IH, Choudhry MA. Heme oxygenase-1 protects against neutrophil-mediated intestinal damage by down-regulation of neutrophil p47phox and p67phox activity and O2- production in a two-hit model of alcohol intoxication and burn injury. J Immunol. 2008;180:6933–6940. doi: 10.4049/jimmunol.180.10.6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chae HD, Broxmeyer HE. SIRT1 deficiency downregulates PTEN/JNK/FOXO1 pathway to block reactive oxygen species-induced apoptosis in mouse embryonic stem cells. Stem Cells Dev. 2011;20:1277–1285. doi: 10.1089/scd.2010.0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, et al. P53- induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.