

Abstract
Ovothiol is a histidine thiol derivative. The biosynthesis of ovothiol involves an extremely efficient trans-sulfuration strategy. The non-heme iron enzyme OvoA catalyzed oxidative coupling between cysteine and histidine is one of the key steps. Besides catalyzing the oxidative coupling between cysteine and histidine, OvoA also catalyzes the oxidation of cysteine to cysteine sulfinic acid (cysteine dioxygenase activity). Thus far, very little mechanistic information is available for OvoA-catalysis. In this report, we measured the kinetic isotope effect (KIE) in OvoA-catalysis using the isotopically sensitive branching method. In addition, by replacing an active site tyrosine (Tyr417) with 2-amino-3-(4-hydroxy-3-(methylthio) phenyl) propanoic acid (MtTyr) through the amber suppressor mediated unnatural amino acid incorporation method, the two OvoA activities (oxidative coupling between cysteine and histidine, and cysteine dioxygenase activity) can be modulated. These results suggest that the two OvoA activities branch out from a common intermediate and that the active site tyrosine residue plays some key roles in controlling the partitioning between these two pathways.
Graphical abstract
INTRODUCTION
Ergothioneine (5) and ovothiol (8 – 10) are histidine derivatives with a sulfur substitution at the ε-carbon or the δ-carbon of the histidine imidazole side-chain, respectively. Humans obtain ergothioneine from the diet through an ergothioneine-specific transporter,1 and ergothioneine has been suggested to play many beneficial roles in human health.2–3 Ovothiol is present in sea urchin eggs at millimolar concentrations and is assumed to protect the egg content during oxidative envelope maturation.4 Ergothioneine and ovothiol biosynthesis has been an intriguing question for decades.5–9 Their sulfur incorporation strategy differs from other reported natural product sulfur incorporation pathways.3,10–17 Three ergothioneine biosynthetic pathways were discovered in recent years (Scheme 1).18 Both the mycobacterial and the fungal pathways are aerobic biosynthetic pathways (Scheme 1A), in which the trans-sulfuration is achieved by non-heme iron enzyme-catalyzed oxidative C-S bond formation reactions (2 → 3 conversion catalyzed by EgtB19–20 or 2 → 4 conversion catalyzed by Egt121–23) and PLP-lyase catalyzed C-S cleavage (EgtE, Egt2-catalysis).24 In some anaerobic microbes, a rhodanase-like EanB enzyme incorporates sulfur into hercynine to produce ergothioneine using an activated “S” from cysteine desulfurase IscS (Scheme 1B).25
SCHEME 1.
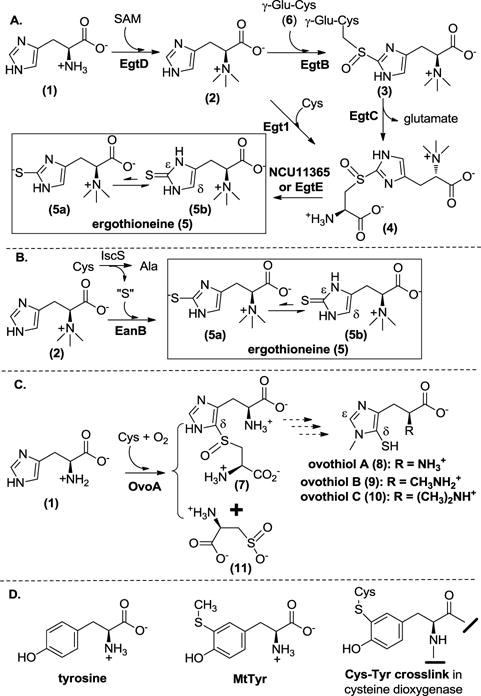
A. Two aerobic ergothioneine biosynthetic pathways in bacteria and fungi; B. The anaerobic ergothioneine biosynthetic pathway; C. The proposed ovothiol biosynthetic pathway. D. MtTyr used in this study and the Cys-Tyr crosslink in cysteine dioxygenase.
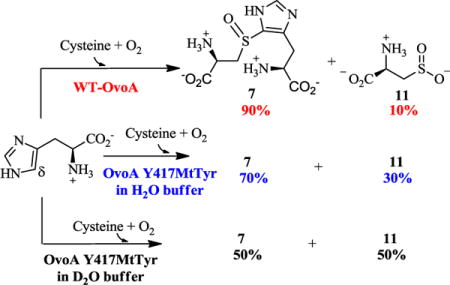
The enzyme responsible for the first step of ovothiol biosynthesis (OvoA) in Erwinia tasmaniensis has been identified (Scheme 1C).26 Similar to the EgtB and Egt1 enzymes in the aerobic ergothioneine biosynthetic pathways (Scheme 1A), OvoA is a mononuclear non-heme iron enzyme that catalyzes oxidative coupling between cysteine and histidine. The OvoA enzyme differs from Egt1 and EgtB-catalysis in both substrate selectivity and product C-S bond formation regioselectivity (Scheme 1A vs. 1C).27–28 More interestingly, when wild-type OvoA enzyme was characterized in vitro, besides the oxidative coupling of cysteine and histidine to produce the coupling product 7, cysteine sulfinic acid was also detected as a side-product (~ 10% of the product mixture, Scheme 1C).28 This is the first discovery of such activity in enzymes other than cysteine dioxygenase (CDO).29 The production of cysteine sulfinic acid is dependent on the presence of histidine. In the absence of histidine, cysteine is oxidized to cystine instead of cysteine sulfinic acid.28 It was thus proposed that the formation of the oxidative coupling product 7 and cysteine sulfinic acid 11 might be two pathways branched out from a common intermediate in OvoA-catalysis. A similar idea has also been proposed for EgtB-catalysis in ergothioneine biosynthesis based on the study on EgtB active site mutants.20, 30 Thus far, there has been no direct evidence supporting this mechanistic hypothesis. In this work, by taking the advantage that the wild-type OvoA produces two products simultaneously in a ratio of ~ 9:1,28 we use the isotopically sensitive branching method (Figure 1) to test this hypothesis.31–32
Figure 1.
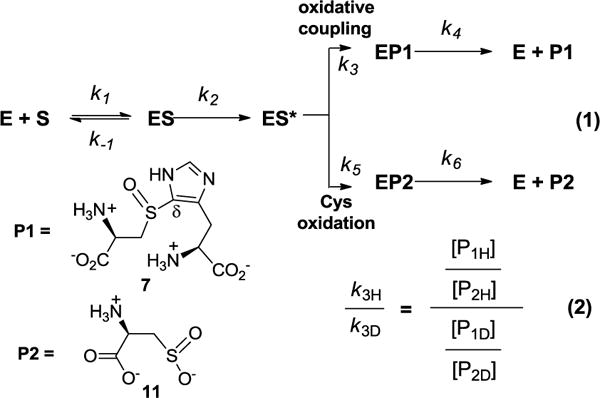
Use of the isotopically sensitive branching method in OvoA-mechanistic studies. P1 represents compound 7 and P2 represents cysteine sulfinic acid 11 in this study.
In addition, in recent structural and biochemical characterizations of EgtB-catalysis, Tyr377 was proposed to be important for the oxidative C-S bond formation.20, 30 Thus far, there is no OvoA crystal structure information. We created a homology model of OvoA and this model suggests that Tyr417 in OvoA is the EgtB Tyr377 counterpart. By replacing Tyr417 in OvoA with 2-amino-3-(4-hydroxy-3-(methylthio) phenyl) propanoic acid (MtTyr, Scheme 1D) through the amber suppressor mediated unnatural amino acid incorporation method, we demonstrated that replacing Tyr417 with a tyrosine analog MtTyr can modulate the partitioning between the two OvoA-pathways: cysteine oxidation versus oxidative C-S bond formation.33 EgtB mechanistic models in literature imply that significant isotope effects might exist and an active site tyrosine residue might play a key role.20, 34 Due to these reasons, we have also repeated the KIE studies using OvoA MtTyr variant to test these hypotheses.
RESULTS
Use of the isotopically sensitive branching method in mechanistic studies
In cases where two competing pathways branch out from a common intermediate, such as some P-450 oxygenase/oxidase-catalyzed reactions, if the step immediately after the common intermediate in one pathway has kinetic isotope effect (KIE), KIE can be measured using the isotopically sensitive branching method.31–32 Previous studies in both OvoA and EgtB-catalysis led to the proposal that cysteine sulfinic acid 11 and oxidative coupling product 7 are produced by two pathways branched out from a common intermediate.20, 28, 30 As represented by a simple kinetic model in Figure 1, from a common intermediate (ES*) along the OvoA catalytic pathway, P1 is the oxidative coupling product 7 resulting from oxidative C-S bond formation branch, and P2 is cysteine sulfinic acid 11 produced from the sulfur oxidation branch. Because the relative amounts of P1 and P2 are directly proportional to the rate constants of the two steps at the branching point (k3 and k5 in Equaton 1, Figure 1), the isotope effect on k3 (k3H/k3D) can be measured by determining the ratio of the two products (P1 and P2) in two sets of reactions: a reaction with unlabeled substrates and a reaction with isotopically labeled substrates (Equation 2, Figure 1).
Measurement of the substrate deuterium KIE using the wild-type OvoA
To measure the KIE of the k3 step in the branch producing the oxidative coupling product 7, two approaches were used, one with isotopically labeled histidine (Figure 2A) and the other by conducting the reaction in D2O buffer (Figure 2B). We first carried out the isotopically sensitive branching experiment using [U-2H5]-histidine as the substrate. Two sets of parallel reactions were run under identical conditions (Figure 2). In the first reaction, unlabeled cysteine and histidine were used as substrates, while in the second reaction, [β-13C]-cysteine and [U-2H5]-histidine were used. After the reactions were complete, the two reaction mixtures were combined. Cysteine sulfinic acid 11 and the coupling product 7 were then characterized by mass spectrometry. We used mass spectrometry ion intensities to quantify the relative amounts of the products (7 vs. 7b and 11 vs. 11b). KIE was calculated according to Equation 2 in Figure 1. For KIE measurements, two batches of protein were used and for each batch of enzyme, two independent KIE measurements were performed. Therefore, any KIE value reported in this research is the average of at least four independent experiments.
Figure 2.
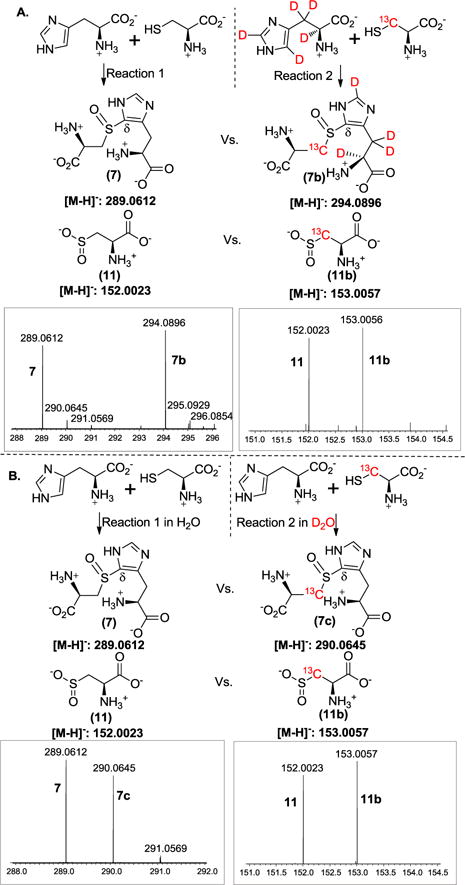
Measurement of the KIE using the isotopically sensitive branching method. A. Using [U-2H5]-histidine as the substrate. On the top are the conditions of the two reactions and at the bottom are the mass spectra of cysteine sulfinic acid 11 and the coupling product 7. B. Solvent KIE measurement by running the reactions in H2O buffer and D2O buffer, respectively. Any ion intensity presented here is the average height of twenty mass spectrometry scans.
Shown in Figure 2 are the spectra from one set of experiments. The m/z of 289.0612 (left panel of Figure 2A) corresponds to coupling product 7 ([M-H]− form) produced from the first reaction, while m/z of 294.0896 (left panel of Figure 2A) is the coupling product 7b ([M-H]− form) produced from the second reaction using [β-13C]-labeled cysteine and [U-2H5]-histidine as the substrates. The ratio of 7 and 7b ([P1H]/[P1D] = 0.88, Figure 2A) reflects the relative amounts of coupling products produced from these two reactions. Using a similar approach, the ratio of the cysteine sulfinic acids (11 and 11b) produced from these two reactions was accurately measured ([P2H]/[P2D] = 0.87, Figure 2A right panel). Based on the [P1H]/[P1D] and [P2H]/[P2D] (Figure 2), the KIE on k3 was calculated using Equation 2 (Figure 1). The average of four repeats was reported as the KIE here, to be kH/kD 1.01 ± 0.02. In addition to mass spectrometric studies, the same set of reactions were also characterized by 13C-NMR, which gave a consistent conclusion as that obtained from mass spectrometry analysis (Figure S1). There are at least two possible explanations for the close to unity KIE obtained in this study. This result implies the lack of a substrate primary deuterium KIE on the k3 step, suggesting that the C-H bond cleavage at the histidine δ-position is not the step at the branching point (k3 step in Figure 1). Alternatively, substrate isotope labeling affects both pathways (k3 and k5) at a comparable level, which will be discussed later.
Measurement of the solvent deuterium KIE using wild-type OvoA
We also examined whether there is solvent KIE on the k3 step. Two sets of reactions were run in parallel (Figure 2B). In the first reaction, unlabeled cysteine and histidine were used as the substrates and the reaction was run in the H2O buffer, while in the second reaction, [β-13C]-cysteine and unlabeled histidine were used and the reaction was run in D2O buffer. After the two reactions were complete, the products were analyzed as described in Figure 2A. Based on the ratios of compounds 7 and 7c, and 11 and 11b (Figure 2B), the solvent deuterium KIE on k3 was calculated using Equation 2 (Figure 1), to be 1.29 ± 0.01. 13C-NMR analysis also gave results consistent to those obtained from mass spectrometry analysis (Figure S2). Recently, OvoA solvent KIE studies have been conducted using steady state kinetics and a small solvent KIE (1.2 ± 0.1) was reported.35
Characterization of the OvoA Y417F mutant
The detected solvent KIE, although small, is consistent with the idea that the formation of coupling product 7 and the oxidation of cysteine to cysteine sulfinic acid 11 are two branches of a common intermediate, with solvent exchangeable protons being part of the k3 step of the branch leading to the formation of the coupling product 7. Alternatively, the solvent may affect the two pathways to slightly different degrees, leading to the small KIE observed in this measurement. This observation immediately raises the question as to what could be the factors that govern the partitioning between the two pathways. Thus far, there is no reported OvoA crystal structure, even though the structure of Mycobacterium thermoresistibile EgtB in ergothioneine biosynthesis (Scheme 1A reaction) is available.20 The wild-type M. thermoresistibile EgtB does not have sulfur dioxygenase activity. However, upon mutation of an active tyrosine (Y377) to phenylalanine, the oxidation of γ-Glu-Cys to γ-Glu-Cysteine sulfinic acid becomes the dominant reaction,20, 30 while the EgtB Y377F mutant does maintain a very low level of oxidative C-S bond formation activity (~ 0.1% of the total activity). These results implied that an active site tyrosine residue might play a key role in modulating the partitioning between these two activities: oxidative coupling vs. cysteine dioxygenase activity.
We generated an OvoA structural model using the I-TASSER program (Figure S3).36 This analysis suggests that Y417 in OvoA might be the EgtB Y377 counterpart. To test this prediction, we generated the OvoA Y417F mutant (Figure S4) and characterized it by the same three assays reported in our earlier studies, oxygen consumption, 1H-NMR and 13C-NMR analysis (Figure S5–7). Similar to the case of EgtB studies, there was barely any coupling product formation in the OvoA Y417F reaction. This means that for this mutant, it would be challenging to obtain an accurate measurement of the KIE of the oxidative-coupling branch of Figure 1 by the isotopically sensitive branching method.
Replacing Y417 in OvoA with 2-amino-3-(4-hydroxy-3-(methylthio) phenyl) propanoic acid (MtTyr)
To resolve the above issue, we decided to replace Y417 with a tyrosine analog. In the cysteine dioxygenase structure, one of the important features is the presence of a Cys-Tyr crosslink (Scheme 1D).37–39 Such cross-link is not essential for CDO activity since the mutation of C93 to alanine or serine significantly reduces its activity, but does not abolish the CDO activity.40–45 Because wild-type OvoA has some level of cysteine dioxygenase activity, we decided to replace Y417 by MtTyr (Scheme 1D), a mimic of the Tyr-Cys cross-link in the cysteine dioxygenase active site. We first synthesized MtTyr enzymatically using an engineered variant of tyrosine phenol lyase (TPL).33 The identity of the resulting MtTyr was further confirmed by HPLC isolation and detailed characterization using 1H-NMR, 13C-NMR and high-resolution mass spectrometry (Figure S8–10).
Recently, we have developed a Methanococcus jannaschii tyrosyl amber suppressor tRNA (MjtRNATyrCUA)/tyrosyl-tRNA synthetase (MjTyrRS) pair to specifically recognize MtTyr.33 OvoA Y417MtTyr protein was overexpressed based on this system (Figure S11) and the identity was further verified by tandem mass spectrometry analysis (Figure S12). The iron content of OvoA Y417MtTyr variant was determined by the previously reported ferrozine assay used for the wild-type OvoA protein.46–47 The purified OvoA Y417MtTyr protein had 0.94 ± 0.04 equivalent of Fe, which is close to that of the wild-type OvoA, suggesting that the incorporation of MtTyr does not significantly disturb the metal-center.
The OvoA Y417MtTyr variant was then characterized by the three assays discussed earlier: oxygen consumption, 1H-NMR, and 13C-NMR analysis.28 Using a NeoFox oxygen electrode, we directly monitored the oxygen consumption rate. The kinetic parameters determined for OvoA Y417MtTyr variant were: kcat 1.14 ± 0.02 s−1, Km of cysteine 253 ± 25 μM and Km of histidine 506 ± 35 μM. Compared with wild-type OvoA,27 the kcat of OvoA Y417MtTyr variant is ~10-fold less (Figure S13). In the 1H-NMR assay, the peak at 7.75 ppm (Figure S14) correlates to the formation of compound 7. Interestingly, 13C-NMR results clearly indicated that OvoA Y417MtTyr variant behaves differently from wild-type OvoA (Figure 3A). In wild-type OvoA-catalysis, less than 10% of the products is cysteine sulfinic acid 11 (Figure S15). However, in OvoA Y417MtTyr catalysis, up to 30% of the products is cysteine sulfinic acid (Figure 3A and Figure S15). By changing Tyr417 to MtTyr, we can modulate the partitioning between the two branches of OvoA-catalysis: oxidative C-S bond formation and cysteine oxidation.
Figure 3.
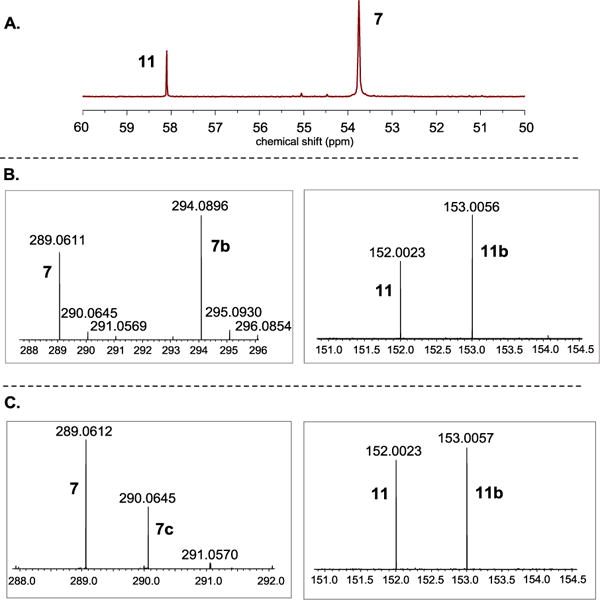
KIE measurement using OvoA Y417MtTyr variant as the enzyme. A. 13C-NMR spectra of the reaction mixture. B. Mass spectra of the coupling product 7 (left panel) and cysteine sulfinic acid 11 (right panel) by running the reactions with unlabeled substrates (histidine and cysteine) or labeled substrates ([U-2H5]-histidine and [β-13C]-cysteine), respectively. C. Mass spectra of the coupling product 7 (left panel) and cysteine sulfinic acid 11 (right panel) by running the reactions in H2O buffer or D2O buffer, respectively. Any ion intensity presented here is the average height of twenty mass spectrometry scans.
In previous studies, it has been demonstrated that OvoA has flexible substrate selectivity. In addition, the product C-S bond formation regioselectivity can be modulated by the methylation state of the histidine amine group27 or the stereochemistry of the histidine stereocenter.35 When hercynine (2) or D-histidine replaced L-histidine (1) as the substrate, C-S bond formation regioselectivity changes from the histidine side-chain δ-position to ε-position. Since there is no OvoA crystal structure information to date, we cannot make concise conclusions on how the addition of methylthiol group from MtTyr affects the OvoA active site (e.g., hydrogen bonding network, active site waters, or even positions of active site residues). However, the disturbance is probably minimal since the reaction catalyzed by OvoA Y417 MtTyr variant still produces 7 and 11 as the products, and the C-S bond formation regioselectivity is unchanged.27, 35 Interestingly, the ratio between 7 and 11 changes from 9:1 with the wild-type OvoA to 7:3 with the OvoA Y417MtTyr variant. Because the amounts of these two products (7 & 11) in OvoA Y417MtTyr reaction are at comparable levels, we decided to further characterize it by the isotopically sensitive branching method.
Measurement of the substrate deuterium KIE using OvoA Y417MtTyr variant
Similar to the wild-type OvoA studies (Figure 2), unlabeled histidine and unlabeled cysteine were used as substrates for one reaction, and [U-2H5]-histidine and [β-13C]-cysteine were used as substrates for the other reaction. Shown in Figure 3 are the results from one set of experiments. The ratio of 7 and 7b ([P1H]/[P1D], Figure 3B) was 0.68, which reflects the amounts of cysteine sulfinic acid produced from these two reactions. The ratio of 11 and 11b ([P2H]/[P2D], Figure 3B) was 0.63, which reflects the amounts of the coupling products formed from these two reactions. From this ratio, KIE for k3 on the oxidative CS bond formation branch was calculated using Equation 2 (Figure 1). The average of four repeats were reported as the KIE here, to be 1.08 ± 0.01. The 13C-NMR assay results were also consistent with the mass spectrometry results (Figure S16).
Measurement of the solvent deuterium KIE using OvoA Y417MtTyr variant
We also conducted solvent deuterium KIE experiments using OvoA Y417MtTyr variant by following the protocol outlined in Figure 2B. The results are shown in Figure 3C. Based on the ratio of compounds 7 and 7c (1.95), and 11 and 11b (0.93), the solvent deuterium KIE on k3 was calculated using Equation 2 (Figure 1), to be 2.09 ± 0.02. Again, shown in Figure 3 is one of four sets of data used to calculate KIE. This solvent KIE provides another set of evidence supporting the hypothesis that cysteine sulfinic acid (11) and the oxidative coupling product (7) are produced by two pathways branched out from a common intermediate and that their ratio can be modulated. The 13C-NMR assays were also consistent with product ratio changes observed from mass spectrometry analysis (Figure S17). In the reaction catalyzed by the OvoA Y417MtTyr variant, the ratio of 11 to 7 increases from 3:7 in the reaction conducted in H2O buffer to approximately 1:1 when conducted in D2O buffer.
DISCUSSION
Mechanistic models
When Seebeck and coworkers first reported their discovery of the OvoA enzyme, a few mechanistic options were proposed, similar to those outlined in Scheme 2A.26 Later on, when it was discovered that OvoA exhibits cysteine dioxygenase activity,28 the question was raised immediately on which one is the first-half reaction: Scheme 2A vs. Scheme 2B. To form product 7, in Scheme 2A, oxidation of thiolate to sulfenic acid occurs first, while in Scheme 2B, the oxidative C-S bond formation is the first-half reaction. Thus far, there is no reported mechanistic or structural work on OvoA. However, EgtB structural information20 and mechanistic investigations using density function theory or quantum mechanics/molecular mechanics (QM/MM) method are both available.34, 48 OvoA and EgtB/Egt1 differ in both substrate selectivity and product C-S bond regioselectivity.19, 21, 26–28 However, the chemistries involved in these two biosynthetic pathways are similar (Scheme 1). For this reason, information from EgtB mechanistic studies serves as the guide when we discuss OvoA mechanistic models.
Scheme 2.
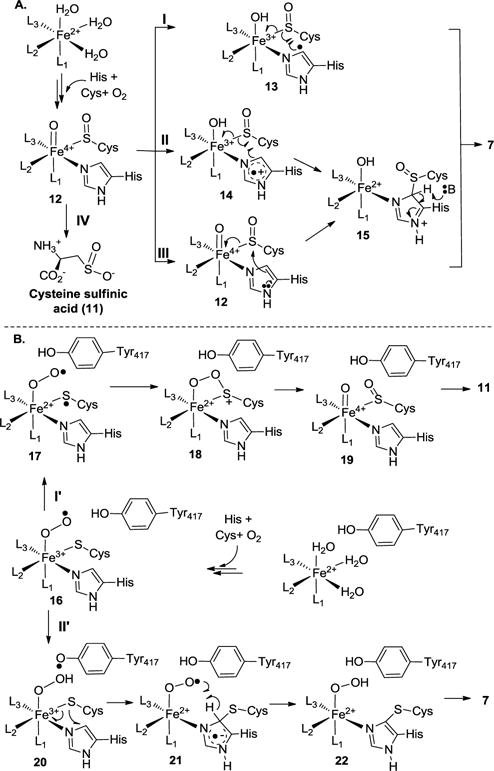
Proposed OvoA mechanistic models.
In Scheme 2A, upon substrate binding, two exchangeable ligands (H2O or OH−) are replaced by the substrates. At the same time, a vacant site is created for oxygen binding and activation. After oxygen is activated, the Fe(III)-superoxo intermediate reacts with the thiolate to form a cysteine sulfenic acid and an FeIV=O species (12, Scheme 2A). Several different ways might account for the subsequent oxidative C-S bond formation (two electron chemistry vs. one-electron chemistry, pathways I, II and III in Scheme 2A).26 These models were modified based on Seebeck’s initially mechanistic proposal and EgtB structural information.29, 49–50 In pathway I, the FeIV=O species abstracts a hydrogen atom from the histidine imidazole ring δ-carbon to produce an imidazole-based radical (13). Subsequent recombination between this radical and a thiol radical will lead to the formation of the oxidative coupling product 7. In pathway II, the FeIV=O species oxidizes the imidazole ring to produce an imidazole cation radical (14), followed by CS bond formation through a radical recombination. Recent computational work based on density function theory by Wei et al. suggests that Y377 participates in EgtB-catalysis by deprotonating the hercynine ε-position, and this step is the rate-limiting step in EgtB-catalysis.48 Therefore, in pathway II mechanistic model (Scheme 2A), Y417 may function as a Lewis acid/base (15 → 7 conversion, Scheme 2A). In pathway III, the reaction follows a two-electron chemistry. Nucleophilic attack by the imidazole ring on the sulfenic acid functional group leads to C-S bond formation (12 → 15). Subsequent deprotonation of the imidazole ring (15) by a base from the protein active site produces the final coupling product (7).
Faponle et al. reported their EgtB mechanistic studies using a combination of quantum mechanics/molecular mechanics (QM/MM) approach.34 Results from this study led to a model very similar to the one in Scheme 2B, in which formation of a thioether is the first half of the reaction. In this model, after substrate binding, oxygen binds to the Fe2+ center leading to the first intermediate, an Fe3+-superoxo species (16). From Y417 and through the active site water network, a proton-coupled electron transfer process then reduces the superoxo species (16) to the Fe3+-hydroperoxo species (20) and at the same time, producing a Y417-based radical. A radical attack from the cysteine sulfur atom to the imidazole side-chain of histidine links the two substrates together through a thioether bond. Simultaneous to the C-S bond formation, the hydrogen atom of the hydroperoxo species relays back to Y417-based radical to regenerate tyrosine and produces an Fe2+-superoxo species (21). In the next step, Fe2+-superoxo (21) abstracts a hydrogen atom from the imidazole ring to regenerate the aromaticity and produces Fe2+-hydroperoxo intermediate (22). In this mechanistic model, Y417 is involved in redox-chemistries. In another recent report, Seebeck et al. suggested that Y377 in EgtB may function as Lewis acid/base.30
Currently available data from investigations on testing these mechanistic models
Pathway I in Scheme 2A is inconsistent with the current results because no primary KIE was observed when [U-2H5]-histidine was used as the substrate in this study. Similar conclusions were reached from previous steady-state kinetic analysis.35 In pathway II of Scheme 2A, oxidation of imidazole goes through a step-wise process. Recently, when fluoro-histidine was used in OvoA-catalysis, it was recognized as an OvoA substrate and its activity was comparable to that of histidine.35 Because the introduction of a fluoro-substitute affects the histidine reduction potential, the lack of obvious effects on OvoA activity suggests that OvoA does not follow this mechanistic model or this step is not the rate-limiting step if this mechanistic model is followed.35 Y417 might function as an acid to provide the proton in the 12 → 14 conversion step, while serves as a base in the 15 → 7 conversion. Recent calculation studies from Wei et al. based on density function theory led to a proposal close to the pathway II model (Scheme 2A). In the EgtB-model proposed by Wei et al., they predicted that the deprotonation step (e.g., 15 → 7) is the rate-limiting step and a substrate deuterium KIE as high as 5.7 was predicted.48 The deprotonation step is also part of pathway III in Scheme 2A. Such a large substrate deuterium KIE predicted from Wei et al.48 was not observed in our OvoA studies.
Wei et al. commented that it is possible that, mechanistically, OvoA and EgtB might behave differently.48 Obtaining OvoA structural information in the future will be important. Theoretical calculation and experimental characterizations could then be conducted on the same system for comparative studies. For the fluoro-histidine analog, there are no KIE studies yet, so it will be worthwhile to re-examine this substrate analog quantitatively by these KIE studies.
Our current results are partially consistent with the Scheme 2B model. In cysteine dioxygenase studies, it was suggested that the creation of a partial cysteine sulfur radical (17, Scheme 2B) is a key factor in directing the formation of the first S–O bond in the CDO catalytic cycle. It has also been suggested that this is the rate-limiting step along the cysteine oxidation pathway.29, 49–52 If the mechanistic model proposed by Faponle et al. on EgtB-catalysis is followed by OvoA-catalysis (Scheme 2B), the proton-coupled electron transfer from Y417 to Fe3+-superoxo species (16) serves as an efficient mechanism to divert the superoxo intermediate (16) from the cysteine oxidation pathway to the Fe3+-hydroperoxo intermediate (20) for the oxidative C-S bond formation. Therefore, the 16 → 20 conversion will be the critical step in controlling the partitioning between the cysteine dioxygenase and the oxidative C-S bond formation pathways. Indeed, in our OvoA studies reported here, the dominant activity of the OvoA Y417F mutant is the cysteine dioxygenase activity.
For EgtB Y377F mutant, a small solvent deuterium KIE was observed for the oxidative C-S bonding formation activity.30 Steady-state kinetic studies of wild-type OvoA indicated that there is a small solvent deuterium KIE (1.2 ± 0.1).35 This is very close to our solvent deuterium KIE (1.29 ± 0.01) measured using the isotopically sensitive branching method. Although there is a close match between the two KIEs, we should be cautious in making a direct link between these two measurements because our KIE studies measure the partitioning between the two pathways and solvent may affect both pathways. There were reports of solvent KIE studies for cysteine dioxygenase. However, the cysteine oxidation branch of OvoA-catalysis is different from the cysteine dioxygenase-catalysis characterized previously.29,49–52 In cysteine dioxygenase, cysteine serves as a bidendate ligand. In OvoA and EgtB, cysteine is monodendate and the histidine imidazole side-chain is the other ligand. In the absence of histidine, OvoA does not have any cysteine dioxygenase activity,28 which suggests that histidine ligation to the iron-center plays a crucial role in the cysteine oxidation branch of the OvoA-catalysis. Therefore, literature information related to cysteine dioxygenase-catalysis may not be directly applied to the cysteine oxidation branch of the OvoA-catalysis.
If the conclusions reached by Faponle et al. in their QM/MM EgtB study are also true for OvoA -catalysis, the 20 → 21 conversion step in Scheme 2B is rate-limiting.34 We did not observe substrate deuterium KIE (Figure 2). There might be at least three possible reasons. First, as Wei et al. suggested, OvoA may indeed be slightly different from EgtB-catalysis.48 Second, there may be a similar level of deuterium KIE for the proposed rate-limiting steps of the two branches (the 20 → 21 conversion and the 16 → 17 conversion). Third, it may also be possible that, in OvoA-catalysis, some key species (16, 17, and 20) are actually in equilibrium and what is observed in our kinetic isotopically sensitive branching studies reflects the change of their relative distribution upon isotope labeling.
Our studies using OvoA Y417MtTyr variant provided new insights into this novel conversion. Similar to the wild-type OvoA, OvoA Y417MtTyr variant takes histidine and cysteine as the substrates. In addition, cysteine dioxygenase activity and oxidative C-S coupling are still the only two observed activities. More importantly, the oxidative C-S bond formation regioselectivity of OvoA Y417MtTyr variant is also the same as that of the wild-type OvoA. Upon replacing Y417 in OvoA with MtTyr, the ratio between 7 and 11 changes from 9:1 in wild-type enzyme reaction to 7:3 in OvoA Y417MtTyr variant reaction. Unnatural tyrosine incorporation method clearly provided us with an efficient way to fine-tune the OvoA activities. In addition, for OvoA Y417MtTyr variant, the ratio between 7 and 11 further changes from 7:3 in H2O buffer to approximately 1:1 in D2O buffer. These changes were also reflected by the solvent deuterium KIE value, which is 2.09 ± 0.02 for the OvoA Y417MtTyr variant and 1.29 ± 0.01 for the wild-type OvoA. The other intriguing result is the observation of a small, yet reproducible isotope effect (1.08 ± 0.01) in OvoA Y417MtTyr variant when [U-2H5]-histidine was used as the substrate.
Mutating Tyr417 to MtTyr changes both the pKa and reduction potential.33, 53–55 In a small molecular model system that mimics the Tyr-Cys crosslink, the pKa of 2-(methylthio)-p-cresol was lowered by ~ 0.8 units relative to p-cresol, and the reduction potential of 2-(methylthio)-p-cresol was lower than that of p-cresol by ~ 370 mV.56 In OvoA MtTyr variant, both the pKa and reduction potential of MtTyr are less than that of tyrosine. If the Scheme 2B model is followed, it predicts an outcome favoring more oxidative-coupling product formation. Surprisingly, the oxidative coupling product decreases from 90% with wild-type OvoA to roughly 70% with OvoA Y417MtTyr variant. Thus, if OvoA-catalysis follows the mechanistic model showed in Scheme 2B, a likely explanation to account for the above results is that MtTyr substitution also affects the cysteine dioxygenase activity. This is possible because in cysteine dioxygenase, although the active tyrosine is not directly involved in redox chemistry, the formation of the Tyr-Cys does improve cysteine dioxygenase activity by nearly 10-fold.29 In OvoA Y417MtTyr variant reaction, the amount of oxidative coupling product further decreases from ~70% in H2O buffer to ~50% in D2O buffer. If MtTyr does affect both pathways, the observed solvent KIE suggests that MtTyr modulates these two pathways to different degrees at the critical steps of the two branches (k3 vs. k5, Figure 1).
Our studies clearly indicate that MtTyr incorporation can be used to modulate OvoA-catalysis. However, because both pKa and reduction of MtTyr are different from that of tyrosine,33, 53–55 it is challenging to differentiate between the contributions of these two factors from just the current sets of studies. By using another tyrosine analog, 3-methoxytyrosine, we can differentiate the contributions of pKa or reduction potential in cytochrome c oxidase studies.57 3-methoxytyrosine has almost the same pKa as tyrosine. However, its reduction potential is different than that of tyrosine by almost 200 mV.57 In the future, further characterization using 3-methoxytyrosine may provide additional mechanistic information related to OvoA- and EgtB-catalysis. All of the mechanistic models discussed in Scheme 2 were proposed based on related studies in EgtB-catalysis. As commented by Wei et al. recently, it is possible that, mechanistically, OvoA and EgtB might behave differently.48 Therefore, in the near future, given by the recently reported EgtB structural20 and theoretical calculation studies,34 similar types of KIE studies about EgtB-catalysis will be worthwhile.
In conclusion, our results presented here clearly indicate that we can modulate the two activities in OvoA-catalysis by replacing the active site tyrosine using a tyrosine analog. Our current results partially align with the Scheme 2B mechanistic model, while more characterizations in the future will be needed in order to accurately interpret the mechanistic implications of these KIE results.
EXPERIMENTAL SECTION
General
The codon optimized OvoA gene (DNA sequences are shown in Supporting Information) was inserted into the KpnI/XhoI multiple cloning site of pASK-IBA5plus to give recombinant plasmid pPL-OvoA wild-type. OvoA wild-type protein was overexpressed and prepared as reported previously.28 1H-NMR and 13C-NMR spectra were recorded on a Varian 500 MHz VNMRS. High-performance liquid chromatography (HPLC) was performed using an Agilent 1200 Series separations module. The enzyme reaction samples were analyzed by LC-MS on a LTQ-FT-ICR mass spectrometer (Thermo Scientific). The OvoA protein sequencing was conducted on a QExactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific). Oxygen consumption kinetic assay was carried out by a NeoFox oxygen electrode. Chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA). L-Cysteine (3-13C, 99% purity) and [U-2H5]-histidine (ring-2,4-D2; α, β, β-D3, 98% purity) were purchased from Cambridge Isotope Laboratories, Inc. Q5®Site-Directed Mutagenesis Kit was purchased from New England BioLabs. Strep-Tactin resins were purchased from IBA life science.
Measurement of the substrate deuterium KIE using wild-type OvoA
For the KIE measurement, three OvoA enzymatic reactions were conducted:
Reaction 1: OvoA reaction using unlabeled histidine and unlabeled cysteine. The reaction mixture contained 20 mM air-saturated KPi buffer in H2O at pH 8.0, 10 mM histidine, 10 mM cysteine, 3 mM DTT, 1 mM sodium ascorbate, and 8 μM iron-reconstituted OvoA. The reaction was run aerobically at 25°C for 2 h. After the reaction was complete, the protein was removed by ultrafiltration.
Reaction 2: OvoA reaction using unlabeled histidine and [β-13C]-cysteine. The reaction mixture contained 20 mM air-saturated KPi buffer in H2O at pH 8.0, 10 mM histidine, 10 mM [β-13C]-cysteine, 3 mM DTT, 1 mM sodium ascorbate, and 8 μM iron-reconstituted OvoA. The reaction was run aerobically at 25°C for 2 h. After the reaction was complete, the protein was removed by ultrafiltration.
Reaction 3: OvoA reaction using [U-2H5]-histidine and [β-13C]-cysteine. The reaction mixture contained 20 mM air saturated KPi H2O buffer at pH 8.0, 10 mM [U-2H5]-histidine, 10 mM [β-13C]-cysteine, 3 mM DTT, 1 mM sodium ascorbate, and 8 μM iron-reconstituted OvoA. The reaction was run aerobically at 25°C for 2 h. After the reaction was complete, the protein was removed by ultrafiltration.
The reaction mixtures of reaction 1 and reaction 3 were combined and then analyzed by LC-MS (negative ion mode). A HYPERCARB column (Thermo scientific) was employed and the gradient was: 0-3 min, 95% B; 3-12 min, 95% B - 30% B; 12-14 min, 30% B; 14-17 min, 30% B - 95% B; 17-20 min, 95% B at a flow rate of 0.2 mL/min. Solvent A: H2O, adjusted to pH 9.2 by ammonia; Solvent B: Acetonitrile, adjusted to pH 9.2 by ammonia. High resolution mass spectra of compounds 7 and 11 were recorded. The ion intensity of each compound was an average of twenty mass spectrometry scans. The ratios of 7:7b, 7:7c and 11: 11b were calculated based on the ion intensity of corresponding compounds obtained from the same LC-MS run. Then it was used to calculate the isotope effect with the equations showed in Figure 1. This set of experiments were repeated four times.
Reaction 2 and reaction 3 were also analyzed by 13C-NMR spectroscopy by mixing 400 μL reaction mixture with 200 μL D2O to prepare the NMR samples.
Measurement of the solvent deuterium KIE using wild-type OvoA
For the OvoA solvent deuterium KIE, we have also conducted three OvoA enzymatic reactions:
Reaction 4: OvoA reaction in H2O using unlabeled histidine and unlabeled cysteine. The reaction mixture contained 20 mM air-saturated KPi H2O buffer at pH 8.0, 10 mM histidine, 10 mM cysteine, 3 mM DTT, 1 mM sodium ascorbate, and 8 μM iron-reconstituted OvoA. The reaction was run aerobically at 25°C for 2 h. After the reaction was complete, the protein was removed by ultrafiltration.
Reaction 5: OvoA reaction in H2O using unlabeled histidine and [β-13C]-cysteine. The reaction mixture contained 20 mM air-saturated KPi H2O buffer at pH 8.0, 10 mM histidine, 10 mM [β-13C]-cysteine, 3 mM DTT, 1 mM sodium ascorbate, and 8 μM iron-reconstituted OvoA. The reaction was run aerobically at 25°C for 2 h. After the reaction was complete, the protein was removed by ultrafiltration.
Reaction 6: OvoA reaction in D2O using unlabeled histidine and [β-13C]-cysteine. The reaction mixture contained 20 mM air-saturated KPi D2O buffer at pH 8.0, 10 mM histidine, 10 mM of [β-13C]-cysteine, 3 mM DTT, 1 mM sodium ascorbate, and 8 μM iron-reconstituted OvoA. The reagents were dissolved in KPi D2O buffer and the enzyme was also prepared in D2O buffer. The reaction was run aerobically at 25°C for 2 h. After the reaction was complete, the protein was removed by ultrafiltration.
Reaction 4 and Reaction 6 mixture were combined and the ratios (7:7c and 11: 11b) were measured directly using LC-MS as described in the previous section to calculate the solvent KIE using the equations showed in Figure 1. This set of experiments were repeated four times as well. Reaction 5 and reaction 6 were also analyzed by 13C-NMR spectroscopy by mixing 400 μL reaction mixture with 200 μL D2O to prepare the NMR samples.
Production of OvoA Y417MtTyr variant using the amber-suppressor method
Site-directed mutagenesis based on the pPL-OvoA wild-type construct was carried out to create pPL-OvoA Y417MtTyr by primer pair PL1316/PL1317 (primer sequences are shown in the Supporting Information). For the pPL-OvoA Y417MtTyr construct, the Y417 codon was mutated to TAG. pBK-MtTyrRS was constructed by one of us recently.33 To overexpress OvoA Y417MtTyr, plasmid pPL-OvoA Y417MtTyr was co-transformed with pBK-MtTyrRS into E. coli BL21. Single colony was inoculated into 100 mL LB media supplemented with 100 μg/mL ampicillin and 12.5 μg/mL chloramphenicol and incubated overnight at 37°C. 10 mL of seed cells were transferred into 1 L of LB media supplemented with 0.1 mM ferrous ammonium sulfate, 100 μg/mL ampicillin and 12.5 μg/mL chloramphenicol. When the OD600 reached 1.2, MtTyr was added into the culture medium at a final concentration of 1 mM. At the same time, 0.5 μg/mL anhydrotetracycline (AHT) and 0.02% L-arabinose were added to induce pPL-OvoA Y417MtTyr and pBK-MtTyrRS, respectively. Cells were incubated at 25°C for an additional 14 h and harvested by centrifugation. The OvoA Y417MtTyr variant protein was then purified anaerobically using the protocols for wild-type OvoA.28
For enzymatic characterization and KIE measurement, OvoA Y417MtTyr variant reactions were conducted by following the same procedures used for wild-type OvoA enzyme.
Supplementary Material
Acknowledgments
This work is supported in part by grants from the National Institutes of Health (R01 GM093903) and the National Science Foundation (CHE-1309148) to P.L., and grant from National Natural Science Foundation of China (31670030, 31628004) to C.Z. and P.L. L.C. and C.Z. are supported by fellowship from China Scholarship Council. We thank Prof. Sean Elliott for constructive comments.
Footnotes
Supporting Information. Protein production, purification and characterization details, product isolation, characterization (NMR, MS results), EgtB/OvoA sequence alignment and OvoA structural model created by I-TASSER are included as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Grundemann D, Harlfinger S, Golz S, Geerts A, Lazar A, Berkels R, Jung N, Rubbert A, Schomig E. Proc Natl Acad Sci U S A. 2005;102:5256–5261. doi: 10.1073/pnas.0408624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paul BD, Snyder SH. Cell Death Differ. 2010;17:1134–1140. doi: 10.1038/cdd.2009.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mueller EG. Nat Chem Biol. 2006;2:185–194. doi: 10.1038/nchembio779. [DOI] [PubMed] [Google Scholar]
- 4.Turner E, Hager LJ, Shapiro BM. Science. 1988;242:939–941. doi: 10.1126/science.3187533. [DOI] [PubMed] [Google Scholar]
- 5.Genghof DS. J Bacteriol. 1970;103:475–478. doi: 10.1128/jb.103.2.475-478.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genghof DS, Van Damme O. J Bacteriol. 1968;95:340–344. doi: 10.1128/jb.95.2.340-344.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reinhold VN, Ishikawa Y, Melville DB. J Bacteriol. 1970;101:881–884. doi: 10.1128/jb.101.3.881-884.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steenkamp DJ, Weldrick D, Spies HS. Eur J Biochem. 1996;242:557–566. doi: 10.1111/j.1432-1033.1996.0557r.x. [DOI] [PubMed] [Google Scholar]
- 9.Vogt RN, Spies HS, Steenkamp DJ. Eur J Biochem. 2001;268:5229–5241. doi: 10.1046/j.0014-2956.2001.02444.x. [DOI] [PubMed] [Google Scholar]
- 10.Fontecave M, Ollagnier-de-Choudens S, Mulliez E. Chem Rev. 2003;103:2149–2166. doi: 10.1021/cr020427j. [DOI] [PubMed] [Google Scholar]
- 11.Hand CE, Honek JF. J Nat Prod. 2005;68:293–308. doi: 10.1021/np049685x. [DOI] [PubMed] [Google Scholar]
- 12.Fahey RC. Annu Rev Microbiol. 2001;55:333–356. doi: 10.1146/annurev.micro.55.1.333. [DOI] [PubMed] [Google Scholar]
- 13.Lin CI, McCarty RM, Liu HW. Chem Soc Rev. 2013;42:4377–4407. doi: 10.1039/c2cs35438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Chen S, Xu T, Taghizadeh K, Wishnok JS, Zhou X, You D, Deng Z, Dedon PC. Nat Chem Biol. 2007;3:709–710. doi: 10.1038/nchembio.2007.39. [DOI] [PubMed] [Google Scholar]
- 15.Knerr PJ, van der Donk WA. Annu Rev Biochem. 2012;81:479–505. doi: 10.1146/annurev-biochem-060110-113521. [DOI] [PubMed] [Google Scholar]
- 16.Dunbar KL, Scharf DH, Litomska A, Hertweck C. Chem Rev. 2017;117:5521–5577. doi: 10.1021/acs.chemrev.6b00697. [DOI] [PubMed] [Google Scholar]
- 17.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruszczycky MW, Liu HW. Nature. 2017;551:37–38. doi: 10.1038/551037a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seebeck FP. J Am Chem Soc. 2010;132:6632–6633. doi: 10.1021/ja101721e. [DOI] [PubMed] [Google Scholar]
- 20.Goncharenko KV, Vit A, Blankenfeldt W, Seebeck FP. Angew Chem Int Ed. 2015;54:2821–2824. doi: 10.1002/anie.201410045. [DOI] [PubMed] [Google Scholar]
- 21.Hu W, Song H, Her AS, Bak DW, Naowarojna N, Elliott SJ, Qin L, Chen XP, Liu PH. Org Lett. 2014;16:5382–5385. doi: 10.1021/ol502596z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bello MH, Barrera-Perez V, Morin D, Epstein L. Fungal Genet Biol. 2012;49:160–172. doi: 10.1016/j.fgb.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Pluskal T, Ueno M, Yanagida M. PLoS One. 2014;9:e97774. doi: 10.1371/journal.pone.0097774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song H, Hu W, Naowarojna N, Her AS, Wang S, Desai R, Qin L, Chen XP, Liu PH. Sci Rep. 2015;5:11870. doi: 10.1038/srep11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burn R, Misson L, Meury M, Seebeck FP. Angew Chem Int Ed. 2017;56:12508–12511. doi: 10.1002/anie.201705932. [DOI] [PubMed] [Google Scholar]
- 26.Braunshausen A, Seebeck FP. J Am Chem Soc. 2011;133:1757–1759. doi: 10.1021/ja109378e. [DOI] [PubMed] [Google Scholar]
- 27.Song H, Leninger M, Lee N, Liu PH. Org Lett. 2013;15:4854–4857. doi: 10.1021/ol402275t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song H, Her AS, Raso F, Zhen ZB, Huo YD, Liu PH. Org Lett. 2014;16:2122–2125. doi: 10.1021/ol5005438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joseph CA, Maroney MJ. Chem Commun. 2007:3338–3349. doi: 10.1039/b702158e. [DOI] [PubMed] [Google Scholar]
- 30.Goncharenko KV, Seebeck FP. Chem Commun. 2016;52:1945–1948. doi: 10.1039/c5cc07772a. [DOI] [PubMed] [Google Scholar]
- 31.Jones JP, Korzekwa KR, Rettie AE, Trager WF. J Am Chem Soc. 1986;108:7074–7078. [Google Scholar]
- 32.Korzekwa KR, Trager WF, Gillette JR. Biochem. 1989;28:9012–9018. doi: 10.1021/bi00449a009. [DOI] [PubMed] [Google Scholar]
- 33.Zhou Q, Hu MR, Zhang W, Jiang L, Perrett S, Zhou JZ, Wang JY. Angew Chem Int Ed. 2013;52:1203–1207. doi: 10.1002/anie.201207229. [DOI] [PubMed] [Google Scholar]
- 34.Faponle AS, Seebeck FP, de Visser SP. J Am Chem Soc. 2017;139:9259–9270. doi: 10.1021/jacs.7b04251. [DOI] [PubMed] [Google Scholar]
- 35.Mashabela GT, Seebeck FP. Chem Commun (Camb) 2013;49:7714–7716. doi: 10.1039/c3cc42594k. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simmons CR, Liu Q, Huang QQ, Hao Q, Begley TP, Karplus PA, Stipanuk MH. J Biol Chem. 2006;281:18723–18733. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]
- 38.McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN. Proc Natl Acad Sci U S A. 2006;103:3084–3089. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye S, Wu X, Wei L, Tang DM, Sun P, Bartlam M, Rao ZH. J Biol Chem. 2007;282:3391–3402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]
- 40.Davies CG, Fellner M, Tchesnokov EP, Wilbanks SM, Jameson GNL. Biochem. 2014;53:7961–7968. doi: 10.1021/bi501277a. [DOI] [PubMed] [Google Scholar]
- 41.Siakkou E, Rutledge MT, Wilbanks SM, Jameson GNL. Biochim Biophys Acta, Proteins Proteomics. 2011;1814:2003–2009. doi: 10.1016/j.bbapap.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 42.Li W, Blaesi EJ, Pecore MD, Crowell JK, Pierce BS. Biochem. 2013;52:9104–9119. doi: 10.1021/bi4010232. [DOI] [PubMed] [Google Scholar]
- 43.Dominy JE, Hwang J, Guo S, Hirschberger LL, Zhang S, Stipanuk MH. J Biol Chem. 2008;283:12188–12201. doi: 10.1074/jbc.M800044200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arjune S, Schwarz G, Belaidi AA. Amino Acids. 2015;47:55–63. doi: 10.1007/s00726-014-1843-7. [DOI] [PubMed] [Google Scholar]
- 45.Njeri CW, Ellis HR. Arch Biochem Biophys. 2014;558:61–69. doi: 10.1016/j.abb.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 46.Stookey LL. Anal Chem. 1970;42:779–781. [Google Scholar]
- 47.Gibbs CR. Anal Chem. 1976;48:1197–1201. [Google Scholar]
- 48.Wei WJ, Siegbahn PE, Liao RZ. Inorg Chem. 2017;56:3589–3599. doi: 10.1021/acs.inorgchem.6b03177. [DOI] [PubMed] [Google Scholar]
- 49.Kumar D, Thiel W, de Visser SP. J Am Chem Soc. 2011;133:3869–3882. doi: 10.1021/ja107514f. [DOI] [PubMed] [Google Scholar]
- 50.Aluri S, de Visser SP. J Am Chem Soc. 2007;129:14846–14847. doi: 10.1021/ja0758178. [DOI] [PubMed] [Google Scholar]
- 51.Tchesnokov EP, Faponle AS, Davies CG, Quesne MG, Turner R, Fellner M, Souness RJ, Wilbanks SM, de Visser SP, Jameson GN. Chem Commun (Camb) 2016;52:8814–8817. doi: 10.1039/c6cc03904a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blaesi EJ, Fox BG, Brunold TC. Biochem. 2014;53:5759–5770. doi: 10.1021/bi500767x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee YK, Whittaker MM, Whittaker JW. Biochem. 2008;47:6637–6649. doi: 10.1021/bi800305d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amorati R, Catarzi F, Menichetti S, Pedulli GF, Viglianisi C. J Am Chem Soc. 2008;130:237–244. doi: 10.1021/ja075554h. [DOI] [PubMed] [Google Scholar]
- 55.Whittaker MM, Chuang YY, Whittaker JW. J Am Chem Soc. 1993;115:10029–10035. [Google Scholar]
- 56.Itoh S, Takayama S, Arakawa R, Furuta A, Komatsu M, Ishida A, Takamuku S, Fukuzumi S. Inorg Chem. 1997;36:1407–1416. doi: 10.1021/ic961144a. [DOI] [PubMed] [Google Scholar]
- 57.Yu Y, Zhou Q, Wang L, Liu XH, Zhang W, Hu MR, Dong JS, Li JS, Lv XX, Ouyang HL, Li H, Gao F, Gong WM, Lu Y, Wang JY. Chem Sci. 2015;6:3881–3885. doi: 10.1039/c5sc01126d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.