

Abstract
Purpose
Somatic mutations in IDH1/2 occur in ~20% of patients with myeloid neoplasms, including acute myeloid leukemia (AML). IDH1/2MUT enzymes produce D-2-hydroxyglutarate (D2HG), which associates with increases in DNA damage and improved responses to chemo/radiotherapy and PARP inhibitors in solid tumor cells. Whether this also holds true for IDH1/2MUT AML is not known.
Experimental Design
Well-characterized primary IDH1MUT, IDH2MUT and IDH1/2WT AML cells were analyzed for DNA damage and responses to daunorubicin, ionizing radiation and PARP inhibitors.
Results
IDH1/2MUT caused increased DNA damage and sensitization to daunorubicin, irradiation, and the PARP inhibitors olaparib and talazoparib in AML cells. IDH1/2MUT inhibitors protected against these treatments. Combined treatment with a PARP inhibitor and daunorubicin had an additive effect on the killing of IDH1/2MUT AML cells. We provide evidence that the therapy sensitivity of IDH1/2MUT cells was caused by D2HG-mediated downregulation of expression of the DNA damage response gene ATM and not by altered redox responses due to metabolic alterations in IDH1/2MUT cells.
Conclusions
IDH1/2MUT AML cells are sensitive to PARP inhibitors as monotherapy but especially when combined with a DNA-damaging agent such as daunorubicin, whereas concomitant administration of IDH1/2MUT inhibitors during cytotoxic therapy decrease the efficacy of both agents in IDH1/2MUT AML. These results advocate in favor of clinical trials of PARP inhibitors either or not in combination with daunorubicin in IDH1/2MUT AML.
Keywords: isocitrate dehydrogenase, acute myeloid leukemia, daunorubicin, olaparib, sensitivity
Introduction
Somatic mutations in genes encoding for isocitrate dehydrogenase 1 and 2 (IDH1/2MUT) occur in various types of cancer, such as glioma, cholangiocarcinoma and certain myeloid neoplasms, including AML, myelodysplastic syndromes and myeloproliferative neoplasms (1–5). Wild-type IDH1/2 (IDH1/2WT) converts isocitrate to α-ketoglutarate (αKG) with concomitant reduction of NADP+ to NADPH. IDH1/2MUT result in a neomorphic function, where IDH1/2MUT enzymes convert αKG and NADPH to 2-hydroxyglutarate (D2HG) and NADP+ (6). D2HG accumulation is oncogenic because it inhibits various αKG-dependent dioxygenases involved in epigenetic regulation, thus inducing cellular dedifferentiation and leukemogenesis (7,8). Appreciation of the causative role of IDH1/2MUT in AML formation and maintenance (9–11) led to the development of agents such as the IDH1MUT inhibitor ivosidenib (AG-120)(12) and the IDH2MUT inhibitor enasidenib (AG-221), which was recently FDA approved for the treatment of relapsed/refractory IDH2MUT AML (13,14). While ivosidenib or enasidenib monotherapy was effective in some patients with difficult-to-treat AML, the majority of treated patients either did not have deep responses, or did not have durable responses, indicating the need to combine these drugs with other anti-leukemic agent(s) (14–16).
Other effects of D2HG besides inhibition of αKG-dependent dioxygenases include the inhibition of the DNA repair enzyme alkB homolog (ALKBH) (17,18) and the DNA damage response proteins lysine-specific demethylase 4A/B (KDM4A/B) (19–21) and ataxia-telangiectasia mutated (ATM) (22). Decreased ATM function, due to mutational inactivation, transcriptional repression or posttranslational depletion, leads to decreased DNA double strand breaks (DSB) repair (23), increased DNA damage and sensitivity to DNA repair inhibitors, such as poly(ADP-ribose) polymerase (PARP) inhibitors, in prostate (24), breast (25), colorectal (26) and lung cancers (27) and lymphomas (28). Accordingly, compared to IDH1/2WT cells, IDH1/2MUT cells show increased levels of DNA damage and sensitization to olaparib and talazoparib, either as monotherapy or in combination with DNA-damaging agents (21,22,29,30). These results have been described using genetically engineered cancer cells or murine hematopoietic stem cells (HSC), but not using models relevant for human AML. We investigate the levels of DNA damage and sensitivity to PARP inhibitors and DNA damage-inducing chemotherapy in IDH1MUT, IDH2MUT and IDH1/2WT primary AML cells.
Methods
Patient population
Peripheral blood and bone marrow samples were obtained from AML patients treated in the Cleveland Clinic. Diagnosis was confirmed according to the 2008 WHO classification criteria. These samples were subjected to next-generation sequencing (NGS) and copy number variation (CNV) analysis targeting ~60 genes that are frequently mutated and/or lost in AML and genes involved in DNA damage response, including IDH1/2, TET2, ATM, BRCA1, BRCA2, XRCC2-5 and RAD50-52. Cancer and germline DNA was obtained from AML cells and paired CD3+ T cells or buccal swabs, respectively. Sequencing and bioinformatic analyses were conducted as previously described (4). Variant allelic frequencies (VAFs) were calculated as the fraction of mutated reads divided by the total number of reads for the gene. VAFs were adjusted to CNVs at the locus of each mutation. Informed consent was obtained from patients according to protocols approved by Cleveland Clinic Institutional Review Board and in accordance with the Declaration of Helsinki. Clinical details of the patients were obtained from their medical records.
Establishment of patient cohorts
From the AML patients genotyped by NGS, we selected those with the following somatic mutation configurations: IDH1MUT/IDH2WT/TET2+/+, IDH1WT/IDH2MUT/TET2+/+, IDH1WT/IDH2WT/TET2−/− or IDH1WT/IDH2WT/TET2+/+, hereafter referred to as IDH1MUT, IDH2MUT, TET2−/− and IDH1/2WT AML samples, respectively. Using copy number-adjusted VAFs, the clonal architecture of IDH1MUT, IDH2MUT and TET2−/− AML samples was reconstructed and cases wherein the classifying mutations were clonal/ancestral with a mutational load of >80% were selected (n = 5 primary human AML samples for each group). Only IDH1/2MUT that are known D2HG producers were included. TET2−/− patients included those with hemizygous or homozygous TET2 mutations (TET2MUT/− or TET2MUT/MUT).
In vitro culture
In all cell culture experiments, primary human AML cells from the aforementioned bone marrow samples were cultured in Iscove’s Modified Dulbecco’s Medium (Gibco, Life Technologies, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; HyClone, Thermo Fisher Scientific), 10 ng/ml interleukin-3, 50 ng/ml stem cell factor, 3 U/ml erythropoietin and 10 ng/ml granulocyte-macrophage colony-stimulating factor in 5% CO2 at 37° C and were simultaneously used in various experiments (Supplementary Figure S1). For colony formation assays (CFAs), cells pretreated in the presence or absence of AGI-5198 (the preclinical version of the IDH1MUT inhibitor ivosidenib (12)), AGI-6780 (the preclinical version of the IDH2MUT inhibitor enasidenib (31)), 10 mM D2HG or 5 μM N-acetyl cysteine (NAC) were seeded at a density of 1*104-1*106 cells/ml, in 3 ml Methocult methylcellulose medium (Stem Cell Technologies). The seeding density depended on the concentration of the cytotoxic agent. Cells were treated for 48 h with 10-50 nM daunorubicin, 200-1000 nM cytarabine, 10-50 μM 5-azacytidine, 1-10 μM decitabine (all for 48 h) or 2-6 Gy ionizing radiation (IR) using a 137Cs source. PARP inhibitors (0-25 μM olaparib or 0-25 nM talazoparib) were given during 48 h before the start of the CFA and for 7 days during the CFA. Thus, treatment with all cytotoxic agents lasted for at least 48 h, in which period >99% of investigated AML cells underwent at least one cell cycle (Supplementary Figure S2). Isogenic HCT116 IDH1WT/WT and IDH1WT/R132H knock-in cells, generated by AAV-targeting technology GENESIS (32), were kindly provided by Horizon Discovery Ltd and cell culture and CFAs were performed as described previously (30). Colonies (>50 cells) were counted at 7 days after treatment and results were analyzed to determine the clonogenic fraction. This is the number of colonies counted, divided by the number of cells plated and corrected for the plating efficiency, as described previously (30). Cell survival at 3 days after treatment was determined by MTT assays. MEL cells were cultured in RPMI1640 with L-glutamine, 10% FBS and 1% penicillin-streptomycin as described previously (33). AGI-5198 and AGI-6780 were purchased from MedChemExpress. D2HG, NAC, 5-azacytidine, cytarabine, daunorubicin, decitabine and MTT were purchased from Sigma-Aldrich. Olaparib (AZD-2281) and talazoparib (BMN-673) were purchased from SelleckChem.
Enzyme activity measurements
Quantitative enzyme cytochemistry (metabolic mapping) of AML cells was performed and analyzed as described previously (30,34,35). The specific NADP+-dependent IDH1/2 activity (EC# 1.1.1.42), NAD+-dependent IDH3 activity (EC# 1.1.1.43) and NADP+-dependent activity of glucose-6-phosphate dehydrogenase (G6PD; EC# 1.1.1.49) were determined against 10 mM isocitrate or glucose-6-phosphate (Serva) and 3 mM NAD+ or 0.8 mM NADP+ (Boehringer) in the presence of nitrotetrazolium blue chloride (Sigma-Aldrich). Incubation was performed at 37° C for 60 min. Control reactions were performed in the absence of substrate but in the presence of cofactors to assess non-specific enzyme activity. To detect the impact of cytoplasmic IDH1MUT and mitochondrial IDH2MUT on NADP+-dependent IDH1/2 activity, 1-methoxy-5-methylphenazinium methylsulfate (methoxy-PMS) and 5-methylphenazinium methylsulfate (PMS, both Sigma) were used, respectively, because the former does not pass mitochondrial membranes while the latter does (35). Photomicrographs were made on a Leica microscope at 40× magnification using Qwin software.
Mass spectrometry analysis
Enantiomer-specific mass spectrometry analysis of D2HG levels of AML cell lysates was performed as described before (36).
Cellular NADP+, NADPH, GSH, GSSG and ROS measurements
AML cells were analyzed using a colorimetric NADP+:NADPH ratio assay (Abcam), a fluorometric GSH:GSSG ratio assay (Abcam) and a fluorometric CellROX Deep Red ROS assay (Life Technologies), in 96-well plates using a POLARStar Galaxy microplate reader (BMG Labtech).
Quantitative real-time (qRT-PCR)
qRT-PCR was performed as previously described (37). Each sample was assayed in triplicate and normalized to ABL expression (38). Primers are listed in Supplementary Table S1.
Analysis of ATM protein levels and siRNA against ATM
ATM protein expression was measured by immunoblotting using primary antibodies against ATM (Genetex) and β-Actin (Cell Signaling Technology). Immunoblots were analyzed using a Li-Cor Odyssey system (Li-Cor Biotechnology). Two sets of “Silencer Select” siRNAs against ATM mRNA (s530444 and s5304445) and one negative control siRNA (#4390843) were obtained from Life Technologies and transfected into AML cells using standard protocols. siRNA efficacy was confirmed by immunoblotting against ATM. ATM-siRNA s530444 was selected to be used in CFAs.
Analysis of TCGA data
IDH1, IDH2 and TET2 mutational data and ATM, IDH1, IDH2 and G6PD mRNA expression data (RNASeq v2 RSEM or RPKM) for AML, low-grade glioma, and glioblastoma cases were extracted from The Cancer Genome Atlas (TCGA) via cBioPortal (39,40) and correlated with each other as described previously (41).
ɣH2AX immunofluorescence staining and measurements
DNA DSBs were determined using immunofluorescence staining of ɣH2AX (Millipore). The number of ɣH2AX+ foci per cell was quantified from deconvoluted stacks of photomicrographs using custom-made software, as described previously (4).
Statistical analysis
Data were processed and analyzed using R and visualized using GraphPad Prism. Two-sided tests were used with significance defined as α < 0.05.
Results
Clinical characterization of primary AML cells
The clinical, cytogenetic and molecular characteristics of the selected IDH1MUT, IDH2MUT, TET2−/− and IDH1/2WT AML patient samples (n = 5 for each group) are shown in Supplementary Tables S2-4. The clinical characteristics of the selected IDH1MUT and IDH2MUT AML patients were representative for those described in a previous cohort study of IDH1/2MUT AML patients (4).
IDH1/2MUT decrease ATM expression and increase DNA DSBs
Motivated by earlier reports that genetically engineered and primary IDH1/2MUT cancer cells have decreased levels of ATM expression (22) and increased levels of DNA damage (21,22,29,30), we investigated these phenomena in primary human AML cells. We observed decreased ATM mRNA and protein expression in IDH1/2MUT AML cells as compared to IDH1/2WT AML cells. Administration of an IDH1/2MUT inhibitor restored ATM expression in IDH1/2MUT AML cells. ATM mRNA expression in TET2−/− cells was not significantly lower than in TET2+/+ AML cells (Figure 1A). We determined D2HG concentrations in cell lysates of each IDH1/2MUT sample. D2HG concentrations were higher in IDH1MUT AML cells than in IDH2MUT AML cells, as has been described previously (42), and were potently suppressed by AGI-5198 and AGI-6780, respectively (Figure 1B). Using TCGA data, we confirmed that ATM mRNA expression is severely decreased in IDH1MUT AML and not significantly decreased in IDH2MUT and TET2−/− AML (Figure 1C). We observed more ɣH2AX+ foci (which recognize DNA DSBs) in IDH1/2MUT than in IDH1/2WT AML cells under steady-state conditions. Furthermore, the number of ɣH2AX+ foci was higher in IDH1/2MUT than in IDH1/2WT AML cells after IR or daunorubicin treatment. To confirm the causal relationship between IDH1/2MUT and increased levels of DNA damage, we pretreated IDH1/2MUT cells with an IDH1/2MUT inhibitor prior to treatment with IR or daunorubicin, which reversed the number of ɣH2AX+ foci in IDH1/2MUT to levels observed in IDH1/2WT AML cells in a time-dependent fashion (Figure 1D).
Figure 1. IDH1/2MUT increase DNA DSBs and sensitize AML cells to PARP inhibitors.
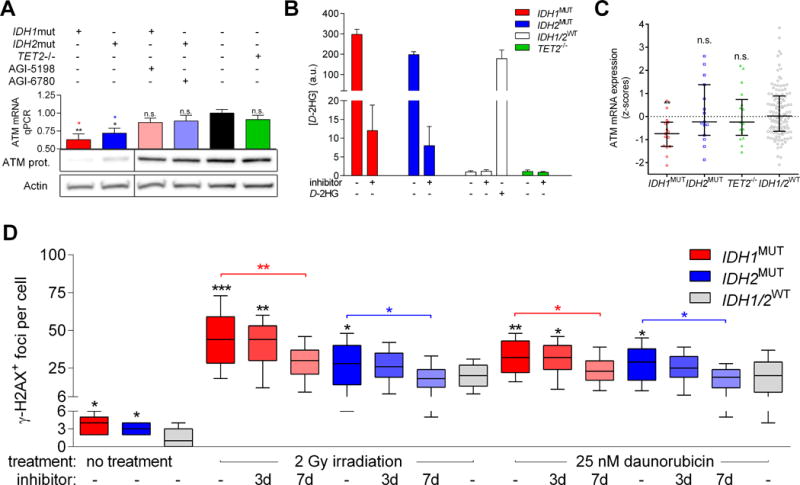
(A) IDH1MUT, IDH2MUT, IDH1/2WT, and TET2−/− primary AML cells (n=5 for each group) were incubated in the presence or absence of 1 μM AGI-5198 (IDH1MUT inhibitor) or AGI-6780 (IDH2MUT inhibitor) for 7 days, harvested, and analyzed for ATM protein expression by immunoblotting. β-Actin served as loading control. Lanes were reordered horizontally for clarity. ATM mRNA expression by qRT-PCR was also measured in these cells. (B) D-2HG levels as determined by enantiomer-specific mass spectrometry in cell lysates of 106 cells. Values were normalized to the D-2HG concentration of untreated IDH1/2WT cells and are shown as arbitrary units. (C) ATM mRNA expression data was taken from The Cancer Genome Atlas (TCGA) database and plotted on the basis of the IDH1, IDH2 and TET2 mutational status. Statistical comparisons were made using the one-way ANOVA test, comparing each group with the IDH1/2WT group, with Dunnett correction for multiple comparisons. (D) IDH1/2MUT and IDH1/2WT primary human AML cells (n=5 for each group) were incubated in the presence or absence of 1 μM IDH1/2MUT inhibitor for 3 days or 7 days and pretreated with either 2 Gy IR or 25 nM daunorubicin. Cells were immunocytochemically stained for ɣH2AX/DSBs and DAPI/DNA content. Numbers of ɣH2AX+ foci per cell are shown (20 cells per patient sample). P values were obtained using one-way ANOVA on the difference between patient samples, using Tukey’s correction for multiple comparisons.
IDH1/2MUT sensitize AML cells to PARP inhibitors
The relationship between increased DNA damage, decreased ATM function and sensitivity to PARP inhibitors (24–28,43) prompted us to compare the responses of IDH1/2MUT and IDH1/2WT AML cells to the PARP inhibitors olaparib and talazoparib. After treatment with olaparib or talazoparib, the surviving fraction of IDH1/2MUT AML cells was lower than that of IDH1/2WT AML cells in CFAs (Figure 2A-B). To investigate whether a causal relationship existed between IDH1/2MUT and this sensitization to PARP inhibitors, we pretreated IDH1MUT AML cells with AGI-5198 and IDH2MUT AML cells with AGI-6780 before cytotoxic treatment (Figure 2C). Pharmacological inhibition of IDH1/2MUT for at least 7 days protected IDH1/2MUT AML cells against PARP inhibitors (Figure 2D-G). In addition, IDH1/2MUT inhibitors did not affect the sensitivity of IDH1/2WT AML cells to PARP inhibitors (Figure 2H-I). We also observed reversible sensitivity to PARP inhibitors using another model of isogenic IDH1WT/R132H HCT116 cells, as compared to IDH1WT/WT HCT116 cells (Supplementary Figure S3).
Figure 2. IDH1/2MUT sensitize AML cells to the PARP inhibitors olaparib and talazoparib.
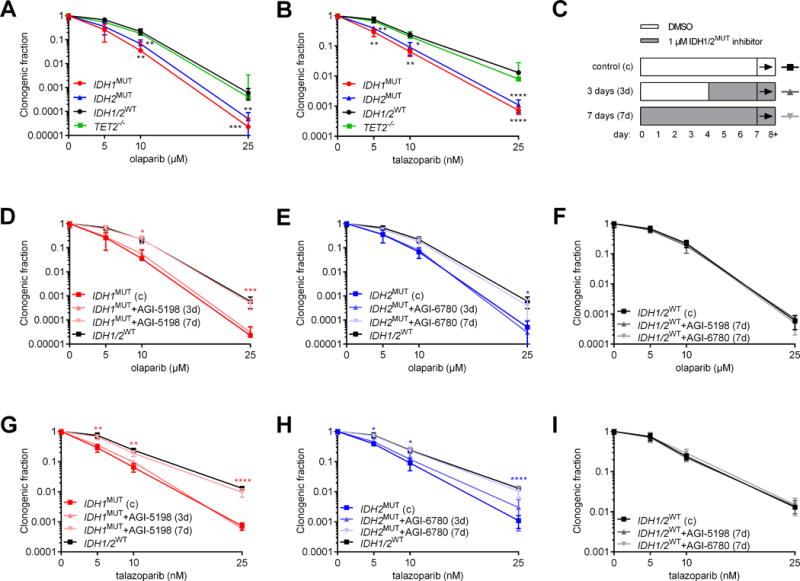
(A-B) Colony-forming assays with IDH1MUT, IDH2MUT, IDH1/2WT or TET2−/− primary AML cells (n=5 for each group) after 48 h pretreatment with, and during 7 days after plating with (A) 0-25 μM olaparib or (B) 0-25 nM talazoparib. (C) Pretreatment schedules for the IDH1MUT inhibitor (AGI-5198) or the IDH2MUT inhibitor (AGI-6780) for 3 or 7 days shown in panels D-I. Cells were exposed to daunorubicin or IR on day 7 and subsequently pretreated with daunorubicin for 48 h or irradiated and plated for colony-forming assays. Throughout, squares are for control conditions, upward triangles for 3 days inhibition and downward triangles for 7 days inhibition. (C-H) Same as in (A-B), but after pretreatment or not with an IDH1/2MUT inhibitor for the indicated period. Data are mean±SD from 3 independent experiments. The clonogenic fraction is the number of colonies counted, divided by the number of cells plated and corrected for the plating efficiency. Y-axes are on a logarithmic scale. Data obtained in control conditions are from the same experiments. Black significance indicators compare the indicated group with IDH1/2WT AML cells. Colored significance indicators compare the indicated group with its IDH1/2MUT inhibitor-untreated counterpart. P values are indicated as * <.05; ** <.01; ***<0.005; ****<0.001.
IDH1/2MUT sensitize AML cells to irradiation and daunorubicin
Given that IDH1/2MUT decrease the DNA damage response and cause sensitivity to PARP inhibitors, we hypothesized that IDH1/2MUT also sensitize AML cells to other DNA damage-inducing agents. Relative to IDH1/2WT and TET2−/− AML cells, we observed a significantly reduced surviving fraction of IDH1/2MUT AML cells after treatment with daunorubicin or IR in CFAs (Figure 3A-B). In addition, pharmacological inhibition of IDH1/2MUT for at least 7 days protected IDH1/2MUT AML cells, but not IDH1/2WT AML cells, against subsequent treatment with daunorubicin or IR (Figure 3C-H). We confirmed these results in isogenic IDH1WT/WT and IDH1WT/R132H HCT116 colorectal cancer cells (Supplementary Figure S4). Pretreatment with the ROS scavenger NAC during 3 days did not affect the survival of IDH1/2MUT AML cells after treatment with daunorubicin or IR (Supplementary Figure S5). We did not observe survival differences between IDH1/2MUT and IDH1/2WT AML cells after treatment with cytarabine, 5-azacytidine or decitabine, which are antimetabolites and hypomethylating agents but do not induce DNA damage. We also did not observe survival differences between IDH1/2MUT and IDH1/2WT AML cells after treatment with daunorubicin or IR in short-term (3-day) cell viability assays that do not capture the long-term effects of treatment-induced DNA damage as adequately as CFAs (44)(Supplementary Figure S6).
Figure 3. IDH1/2MUT sensitize AML cells to irradiation and daunorubicin.
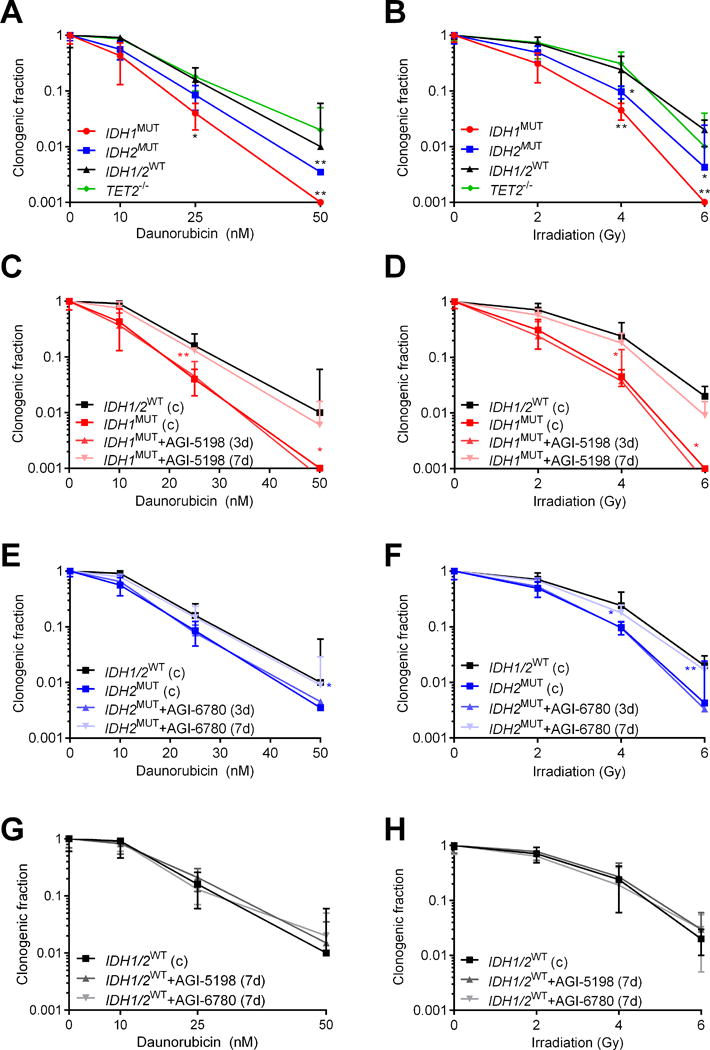
Colony-forming assays with IDH1MUT, IDH2MUT, IDH1/2WT or TET2−/− primary AML cells (n=5 for each group) after treatment with (A) 0-50 nM daunorubicin for 48 h or (B) 0-6 Gy ionizing radiation (IR). (C-H) Same as in (A-B), but after pretreatment or not with an IDH1/2MUT inhibitor for the indicated period according to the pretreatment schedule shown in Figure 2C. Data are mean±SD from 3 independent experiments. The clonogenic fraction is the number of colonies counted, divided by the number of cells plated and corrected for the plating efficiency. Y-axes are on a logarithmic scale. Data obtained in control conditions are from the same experiments. Black significance indicators compare the indicated group with IDH1/2WT AML cells. Colored significance indicators compare the indicated group with its IDH1/2MUT inhibitor-untreated counterpart. P values are indicated as * <.05; ** <.01.
PARP inhibitors further sensitize IDH1/2MUT AML cells to cytotoxic therapy
We hypothesized that combined treatment with a PARP inhibitor and a DNA-damaging agent has additive effects on IDH1/2MUT AML cells. Combined treatment with olaparib or talazoparib and daunorubicin was more lethal to both IDH1/2WT and IDH1/2MUT AML cells than daunorubicin treatment alone, but the effect was significantly larger in IDH1/2MUT AML cells (Figure 4).
Figure 4. PARP inhibitors and daunorubicin have additive effects in IDH1/2MUT AML cells.
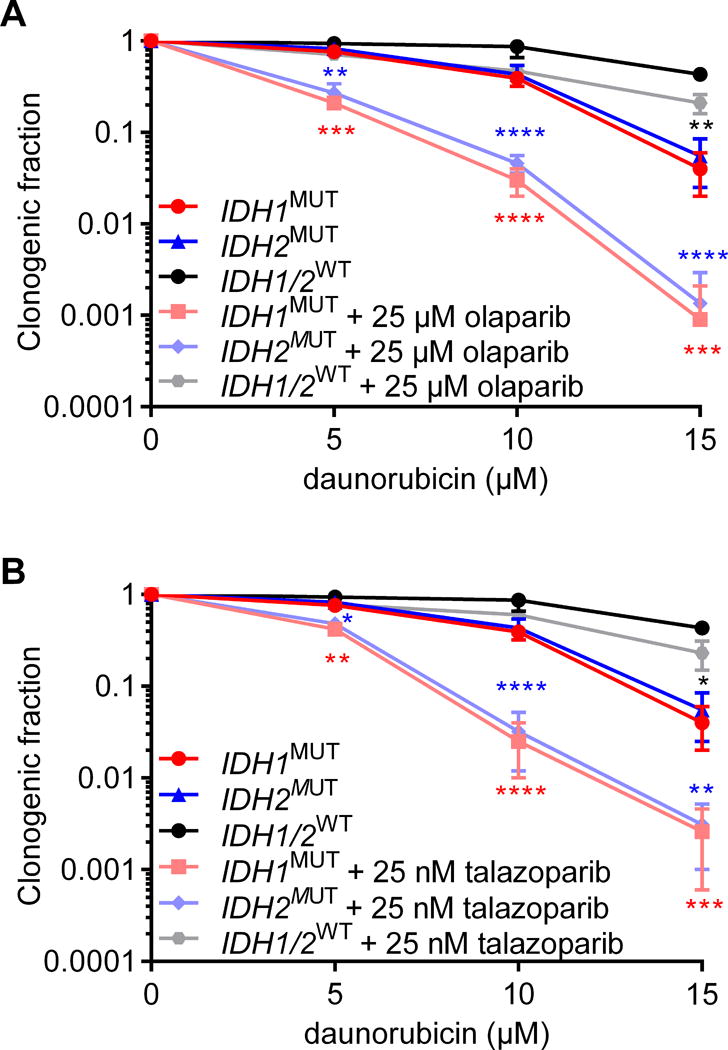
Colony-forming assays with IDH1MUT, IDH2MUT and IDH1/2WT primary AML cells (n=5 for each group) after 48 h pretreatment before plating and during 7 days after plating with 0-25 μM olaparib or 0-25 nM talazoparib and with 0-15 μM daunorubicin. Data are mean±SD from 3 independent experiments. The clonogenic fraction is the number of colonies counted, divided by the number of cells plated and corrected for the plating efficiency. Y-axes are on a logarithmic scale. Black significance indicators compare the indicated group with IDH1/2WT AML cells. Colored significance indicators compare the indicated group with its IDH1/2MUT inhibitor-treated counterpart. P values are indicated as * <.05; ** <.01; *** <.005; **** <.001.
Increased sensitivity to DNA-damaging agents in IDH1/2MUT AML cells is associated with decreased ATM expression
To investigate causality between IDH1/2MUT, ATM suppression and therapy sensitivity, we knocked down ATM in AML cells using siRNA (Supplementary Figure S7). ATM knockdown did not affect the sensitivity of IDH1/2MUT AML cells to daunorubicin or IR (Figure 5A-D) but sensitized IDH1/2WT AML cells to these treatments (Figure 5E-F). After 7 days of pretreatment with D2HG, untransfected IDH1/2WT AML cells were sensitized to daunorubicin or IR, but IDH1/2WT AML cells were not further sensitized when ATM was knocked down (Figure 5G-H). IDH1/2MUT inhibitors protected untransfected IDH1/2MUT AML cells against daunorubicin or IR (Figure 3C-F), but did not protect IDH1/2MUT AML cells when ATM was knocked down (Figure 5I-J). Another siRNA with a lower knockdown efficiency of siRNA sensitized IDH1/2WT AML cells less for daunorubicin or IR (Supplementary Figure S8).
Figure 5. ATM knockdown sensitizes IDH1/2WT AML cells to cytotoxic treatment, but not in the presence of D-2HG or IDH1/2MUT.
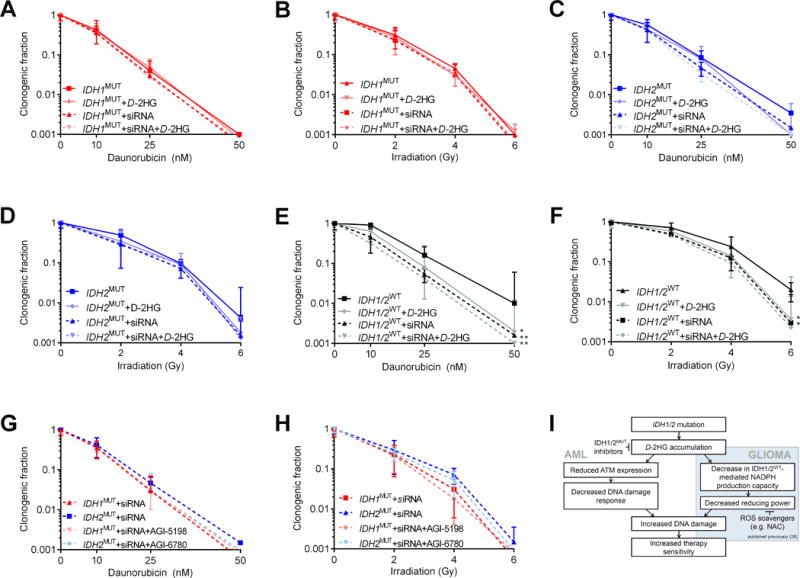
(A-H) Colony-forming assays with IDH1MUT, IDH2MUT or IDH1/2WT AML cells (n=5 for each group) after treatment with 0-50 nM daunorubicin for 48 h or 0-6 Gy IR in the presence or absence of siRNA against ATM and/or 10 mM D2HG and/or 1 μM AGI-5198 or 1 μM AGI-6780. ATM siRNA was controlled for using a negative control siRNA. Data are mean±SD from 3 independent experiments. The clonogenic fraction is the number of colonies counted divided by the number of cells plated and corrected for the plating efficiency. Y-axes are on a logarithmic scale. Significance indicators compare the adjacent group with untreated IDH1/2WT AML cells. P values are indicated as * <.05; ** <.01. (I) Model of IDH1MUT-mediated therapy sensitization in AML cells and glioma cells, based on findings in this study and in a previous study on IDH1MUT in solid tumor cells (shaded part) (30).
IDH1/2MUT decreases NADPH production but does not affect redox states in AML cells
In glioma and colorectal cancer cells, IDH1MUT inhibits IDH1/2WT function, which perturbs redox states and sensitizes these cells to irradiation (30). We interrogated whether or not IDH1/2WT function and redox states could play a role in the therapy sensitization of IDH1/2MUT AML cells. NADP+-dependent IDH1/2 activity was significantly lower in lDH1/2MUT AML cells than in lDH1/2WT and TET2−/− AML cells. However, the impact of the decreased IDH1/2-mediated NADPH production capacity on the total cellular NADPH production capacity in AML cells was limited, because the NADPH production capacity by G6PD was ~4-fold larger than that of IDH1 and IDH2 combined (Figure 6A). IDH1/2MUT were not associated with changes in NAD+-dependent IDH3 activity or NADP+-dependent G6PD activity in AML cells (Figure 6A-B). Pretreatment with an IDH1/2MUT inhibitor for 3 days restored NADP+-dependent IDH1/2 activity in IDH1/2MUT AML cells (Figure 6C). In addition, D2HG administration of 10 mM D2HG (which achieved D2HG levels in IDH1/2WT AML cells similar to untreated IDH1/2MUT AML cells [Figure 1B]) decreased NADP+-dependent IDH1/2 activity in IDH1/2WT cells, which supports a causative role of D2HG accumulation in the suppression of IDH1/2-mediated NADPH production (Figure 6C). In agreement with the modest effects of IDH1/2MUT on the total cellular NADPH production, we observed similar NADP+:NADPH ratios, GSH:GSSG ratios and ROS levels between IDH1/2MUT and IDH1/2WT AML cells under steady-state conditions and after pretreatment with daunorubicin or IR (Figure 6D-F). In TCGA data, the mRNA expression of IDH1, IDH2 and G6PD enzymes was unchanged in IDH1/2MUT versus IDH1/2WT AML, whereas mRNA expression of these enzymes was lower in IDH1/2MUT versus IDH1/2WT glioma (Supplementary Figure S9).
Figure 6. IDH1/2MUT AML cells have decreased IDH1/2 activity but similar redox states as IDH1/2WT AML cells.
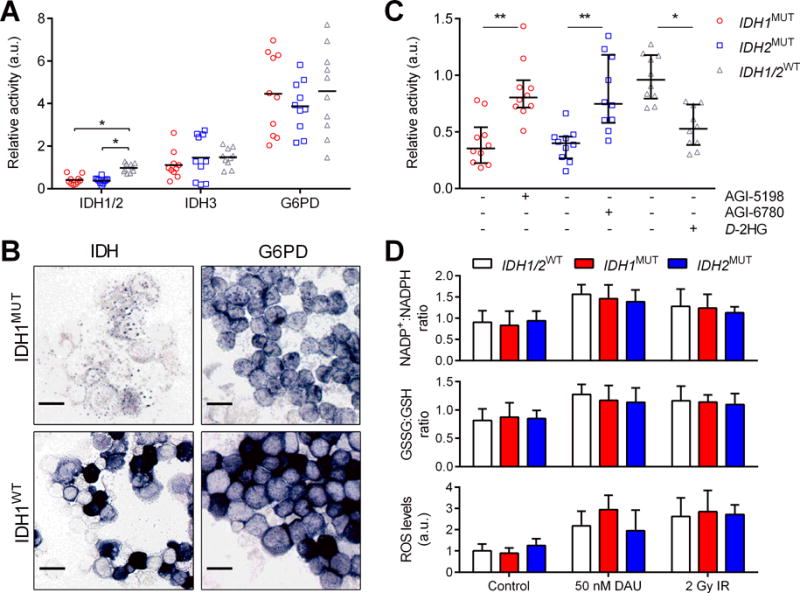
(A) NADP+-dependent IDH1/2 activity, NAD+-dependent IDH3 activity and NADP+-dependent G6PD activity of IDH1MUT, IDH2MUT and IDH1/2WT primary AML cells was determined using image analysis as absorbance of blue formazan produced from nitroBT per cell as a readout of NADPH production. Values were normalized to the NADP+-dependent IDH activity of IDH1/2WT cells and are shown as arbitrary units. (B) Representative photomicrographs of NADP+-dependent IDH and G6PD activity in IDH1MUT and IDH1WT AML cells. Scale bars = 50 μm. (C) NADP+-dependent IDH1/2 activity after pretreatment in the presence or absence of 1 μM AGI-5198, 1 μM AGI-6780 or 10 mM exogenous D2HG; units are arbitrary and relative to IDH1/2WT rates under control conditions. (D) IDH1MUT, IDH2MUT and IDH1/2WT AML cells (n=5 for each group) were pretreated with 0-50 nM daunorubicin (DAU) for 48 h or 0-2 Gy IR and were harvested, prepared, and analyzed, colorimetrically for NADP+:NADPH ratios and fluorometrically for GSH:GSSG ratios and for ROS levels. Data are mean±SD from 3 independent experiments. P values are indicated as * <.05; ** <.01.
Discussion
We found that primary IDH1/2MUT AML cells have reduced DNA damage responses and suppressed expression of ATM. As a consequence, they are sensitized to a PARP inhibitor, daunorubicin or IR and this is negated by pretreatment with an IDH1/2MUT inhibitor, which also restores ATM expression and decreases DNA damage. In mechanistic experiments using siRNA and exogenous D2HG, we obtained further evidence of a cascade wherein D2HG accumulation leads to ATM suppression and decreased DNA damage responses, resulting in increased IDH1/2MUT AML therapy responses. While our results suggest that PARP inhibitors enhance responses of IDH1/2MUT AML to daunorubicin, they also suggest that PARP inhibitors or daunorubicin should not be combined with IDH1/2MUT inhibitors in AML, because IDH1/2MUT inhibitors disrupt the D2HG-ATM-DNA damage cascade. These findings, in combination with earlier findings in IDH1MUT glioma, are summarized in a model shown in Figure 4K.
Our results corroborate other studies showing that compared to IDH1/2WT counterparts, IDH1(/2)MUT human glioma, human colorectal cancer and murine HSCs are sensitized to treatment with daunorubicin, IR or PARP inhibitors due to ATM suppression and increased DNA damage levels (21,22,29,30). Mechanistic studies have provided evidence that IDH1/2 mutations decrease ATM expression by increasing methylation of the repressive histone mark H3K9 that may rely on inhibition of the histone demethylases KDM4A and/or KDM4B by D2HG (21,22). TET2 is a major downstream target of D2HG in IDH1/2MUT AML (45), but ATM downregulation was not observed in TET2−/− mice (22), nor was homologous recombination significantly impaired after treatment of U2OS DR-GFP cells with an siRNA against TET2 (21). This is supported by the finding that restoration of TET2 function sensitizes, rather than protects, TET2+/− AML cells to PARP inhibitors (46) and it may also be supported by our finding that ATM mRNA and protein expression were not different in TET2−/− AML cells compared to TET2+/+ AML cells.
Several mechanistic results from IDH1/2MUT AML cells in the present study contrast our earlier findings in IDH1MUT glioma and colon carcinoma cells, where pretreatment with an IDH1MUT inhibitor or the ROS scavenger NAC for 3 days achieved radioprotection due to restored NADPH production and ROS detoxification (30). In IDH1/2MUT AML cells, such protection against cytotoxic therapy required incubation with an IDH1/2MUT inhibitor for 7 days and was not achieved by using NAC. Similarly to the profound metabolic effects of IDH1/2MUT in glioma (41,47), IDH1/2MUT reduced IDH1/2-mediated NADPH production in IDH1/2MUT primary AML cells. However, this did not affect therapy responses in AML cells wherein IDH1/2 provides <20% of the cell’s NADPH; in contrast, in glioma, IDH1/2 provides ~two-thirds of the cell’s capacity to produce NADPH (48). IDH1/2MUT were associated with decreased mRNA expression of IDH1/2 and G6PD in glioma but not in AML, suggesting that IDH1/2MUT may alter the metabolism of AML cells to a lesser extent than that of glioma. Relative to glioma and colon carcinoma cells, slower protection of IDH1/2MUT AML cells by IDH1/2MUT inhibitors is likely due to it being mediated by a different mechanism that involves slow epigenetic alterations needed to suppress ATM expression. Reversing redox states in glioma and colon carcinoma cells is likely to be much faster. Theoretically, the increased DNA damage in IDH1/2MUT AML cells can be explained by differences in cell doubling times between IDH1/2MUT and IDH1/2WT cells (49) and IDH1MUT inhibitors are reported to affect cell cycle duration (50). However, we found no differences in doubling time between IDH1/2MUT and IDH1/2WT AML cells and increased ɣH2AX+ foci argue against cell cycle perturbations as being responsible for our results.
Patients with IDH1/2MUT glioma have longer survival times than IDH1WT counterparts (2,51,52), probably by virtue of improved responses to chemotherapy and IR (30,53). Although our data suggest that IDH1/2MUT AML cells are sensitive to chemotherapy and IR, there is no difference between the survival of patients with IDH1/2MUT AML or IDH1/2WT AML (4,54). This discrepancy between our results and the data from observational studies might be inherent to limitations of our in vitro data, such as a relatively small sample size, the inclusion of AML with ancestral IDH1/2MUT only or different behaviour of IDH1/2MUT AML cells in vitro and in vivo. The latter issue would be interesting to investigate in a translational clinical trial wherein the therapy response of IDH1/2MUT AML patients (in vivo) and their primary samples (in vitro) are compared. Alternatively, the aforementioned discrepancy may be inherent to limitations of observational studies. Of note, high-dose IR is nowadays rarely used in the treatment of AML and only 30-40% of elderly AML patients (aged ≥65 years) are reported to receive any type of chemotherapy, of which not all receive intensive treatment regimens such as daunorubicin (55,56). Indeed, examining data from the NCI’s Surveillance, Epidemiology and End Results (SEER) program we found that only ~60% of AML patients of all ages receive any type of chemotherapy (see online supplements). As a possible explanation for the absence of survival differences between patients with IDH1/2MUT AML or IDH1/2WT AML, low use of daunorubicin and IR may prevent the putative predictive effects of IDH1/2MUT to materialize into a significant prognostic association in retrospective studies.
In summary, this study is the first to show that IDH1/2MUT AML is vulnerable for PARP inhibition as monotherapy, but especially when combined with daunorubicin treatment. IDH1/2MUT inhibitors protect IDH1/2MUT AML cells against PARP inhibitors, daunorubicin or IR, which suggests that combinations of IDH1/2MUT inhibitors and DNA-damaging agents should be avoided. Our data are crucial to the rational design and analysis of clinical trials with IDH1/2MUT inhibitors, especially for clinical trials that investigate combinations of IDH1/2MUT inhibitors with conventional chemotherapy for AML (e.g. ClinicalTrials.gov NCT02632708). Instead, our results show that exploiting impaired DNA repair in IDH1/2MUT AML cells using a PARP inhibitor, ideally combined with a DNA-damaging agent, may be a better strategy for the treatment of IDH1/2MUT AML. The investigation of PARP inhibitor monotherapy in clinical trials in AML patients with somatic IDH1/2MUT is warranted.
Supplementary Material
Summary statement.
IDH1/2 mutations affect ~20% of patients with acute myeloid leukemia (AML). In the present study, we describe that IDH1/2MUT caused increased levels of DNA damage in primary AML cells and that this phenomenon could be therapeutically exploited using by therapies that induce or augment DNA damage, such as daunorubicin chemotherapy, irradiation or the PARP inhibitors olaparib and talazoparib. Combined treatment with a PARP inhibitor and daunorubicin had an additive effect on the killing of IDH1/2MUT primary AML cells. On the other hand, pharmacological inhibition of IDH1/2MUT decreased the therapeutic responses of IDH1/2MUT primary AML cells to daunorubicin, irradiation, olaparib and talazoparib. Collectively, these results advocate in favor of clinical trials of PARP inhibitors either or not in combination with daunorubicin in IDH1/2MUT AML.
Acknowledgments
Funding: This work was supported by AMC PhD Scholarship (R.J.M), the Dutch Cancer Society (KWF; UVA 2014-6839 and AMC2016.1-10460, to R.J.M., M.K., F.E.B. and C.J.F.v.N), the National Institutes of Health (Bethesda, MD; NIH) grants R01HL118281, R01HL123904, R01HL132071, R35HL135795, a grant from the AA & MDS International Foundation (Rockville, MD), the Robert Duggan Charitable Fund (Cleveland, OH, all to J.P.M.), a Scott Hamilton CARES grant (Cleveland, OH; H.M.), a grant from the AA & MDS International Foundation (H.M.).
Footnotes
Conflict of Interest disclosure: Dr. Maciejewski has received honoraria, has performed consultancy and has served as a speaker on behalf of Celgene. Dr. Sekeres has served on an advisory committee of Celgene. Dr. Carraway has received research funding and has served as a speaker on behalf of Celgene, and has served on an advisory committee of Novartis. These companies have IDH1/2-mutant inhibitors in development.
Authorship contribution: R.J.M., C.J.F.v.N. and J.P.M. designed the research; R.J.M., B.P., H.M. and C.H., performed the research; R.J.M and T.R. analyzed the data; F.E.B., M.X., J.W.W, H.E., S.M. and M.S. supervised the research, R.J.M. and J.P.M. wrote the paper, all authors read and approved the paper.
References
- 1.Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49(2):204–12. doi: 10.1038/ng.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molenaar RJ, Radivoyevitch T, Maciejewski JP, van Noorden CJ, Bleeker FE. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim Biophys Acta. 2014;1846(2):326–41. doi: 10.1016/j.bbcan.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Tefferi A, Lasho TL, Abdel-Wahab O, Guglielmelli P, Patel J, Caramazza D, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24(7):1302–9. doi: 10.1038/leu.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molenaar RJ, Thota S, Nagata Y, Patel B, Clemente M, Przychodzen B, et al. Clinical and biological implications of ancestral and non-ancestral IDH1 and IDH2 mutations in myeloid neoplasms. Leukemia. 2015;29(11):2134–42. doi: 10.1038/leu.2015.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–7. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–8. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–83. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C, Liu Y, Lu C, Cross JR, Morris JPT, Shroff AS, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. GenesDev. 2013;27(18):1974–85. doi: 10.1101/gad.226613.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kats LM, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014;14(3):329–41. doi: 10.1016/j.stem.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mylonas E, Janin M, Bawa O, Opolon P, David M, Quivoron C, et al. Isocitrate dehydrogenase (IDH)2 R140Q mutation induces myeloid and lymphoid neoplasms in mice. Leukemia. 2014;28(6):1343–6. doi: 10.1038/leu.2014.18. [DOI] [PubMed] [Google Scholar]
- 12.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–30. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130(6):732–41. doi: 10.1182/blood-2017-04-779447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–31. doi: 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullard A. Cancer metabolism pipeline breaks new ground. Nat Rev Drug Discov. 2016;15(11):735–7. doi: 10.1038/nrd.2016.223. [DOI] [PubMed] [Google Scholar]
- 16.Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJ. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene. 2017 doi: 10.1038/s41388-017-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang P, Wu J, Ma S, Zhang L, Yao J, Hoadley KA, et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015;13(11):2353–61. doi: 10.1016/j.celrep.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen F, Bian K, Tang Q, Fedeles BI, Singh V, Humulock ZT, et al. Oncometabolites d- and l-2-Hydroxyglutarate Inhibit the AlkB Family DNA Repair Enzymes under Physiological Conditions. Chem Res Toxicol. 2017;30(4):1102–10. doi: 10.1021/acs.chemrestox.7b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31(8):1865–78. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young LC, McDonald DW, Hendzel MJ. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. J Biol Chem. 2013;288(29):21376–88. doi: 10.1074/jbc.M113.491514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9(375) doi: 10.1126/scitranslmed.aal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell. 2016;30(2):337–48. doi: 10.1016/j.ccell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197–210. doi: 10.1038/nrm3546. [DOI] [PubMed] [Google Scholar]
- 24.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373(18):1697–708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, et al. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J Exp Clin Cancer Res. 2013;32:95. doi: 10.1186/1756-9966-32-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C, Jette N, Moussienko D, Bebb DG, Lees-Miller SP. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl Oncol. 2017;10(2):190–6. doi: 10.1016/j.tranon.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitt A, Knittel G, Welcker D, Yang TP, George J, Nowak M, et al. ATM Deficiency Is Associated with Sensitivity to PARP1- and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res. 2017;77(11):3040–56. doi: 10.1158/0008-5472.CAN-16-3398. [DOI] [PubMed] [Google Scholar]
- 28.Williamson CT, Muzik H, Turhan AG, Zamo A, O’Connor MJ, Bebb DG, et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol Cancer Ther. 2010;9(2):347–57. doi: 10.1158/1535-7163.MCT-09-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su YT, et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017;77(7):1709–18. doi: 10.1158/0008-5472.CAN-16-2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molenaar RJ, Botman D, Smits MA, Hira VV, van Lith SA, Stap J, et al. Radioprotection of IDH1-Mutated Cancer Cells by the IDH1-Mutant Inhibitor AGI-5198. Cancer Res. 2015;75(22):4790–802. doi: 10.1158/0008-5472.CAN-14-3603. [DOI] [PubMed] [Google Scholar]
- 31.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–6. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 32.Fernandez SL, Russell DW, Hurlin PJ. Development of human gene reporter cell lines using rAAV mediated homologous recombination. Biol Procedures Online. 2007;9:84–90. doi: 10.1251/bpo136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan F, Wingo TS, Zhao Z, Gao R, Makishima H, Qu G, et al. Tet2 loss leads to hypermutagenicity in haematopoietic stem/progenitor cells. Nat Commun. 2017;8:15102. doi: 10.1038/ncomms15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chieco P, Jonker A, De Boer BA, Ruijter JM, Van Noorden CJ. Image cytometry: protocols for 2D and 3D quantification in microscopic images. Progr Histochem Cytochem. 2013;47(4):211–333. doi: 10.1016/j.proghi.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Molenaar RJ, Khurshed M, Hira VV, van Noorden CJ. Metabolic Mapping: Quantitative Enzyme Cytochemistry and Histochemistry to Determine the Activity of Dehydrogenases in Cells and Tissues. J Vis Exp. 2017:e56843. doi: 10.3791/56843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Struys EA, Jansen EE, Verhoeven NM, Jakobs C. Measurement of urinary D- and L-2-hydroxyglutarate enantiomers by stable-isotope-dilution liquid chromatography-tandem mass spectrometry after derivatization with diacetyl-L-tartaric anhydride. Clin Chem. 2004;50(8):1391–5. doi: 10.1373/clinchem.2004.033399. [DOI] [PubMed] [Google Scholar]
- 37.Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia. 2003;17(12):2318–57. doi: 10.1038/sj.leu.2403135. [DOI] [PubMed] [Google Scholar]
- 38.Beillard E, Pallisgaard N, van der Velden VH, Bi W, Dee R, van der Schoot E, et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR) - a Europe against cancer program. Leukemia. 2003;17(12):2474–86. doi: 10.1038/sj.leu.2403136. [DOI] [PubMed] [Google Scholar]
- 39.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khurshed M, Molenaar RJ, Lenting K, Leenders WP, van Noorden CJF. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget. 2017;8(30):49165–77. doi: 10.18632/oncotarget.17106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward PS, Lu C, Cross JR, Abdel-Wahab O, Levine RL, Schwartz GK, et al. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J Biol Chem. 2013;288(6):3804–15. doi: 10.1074/jbc.M112.435495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 44.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315–9. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 45.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell. 2017;170(6):1079–95 e20. doi: 10.1016/j.cell.2017.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Molenaar RJ, Coelen RJ, Khurshed M, Roos E, Caan MW, van Linde ME, et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1-mutated or IDH2-mutated solid tumours. BMJ Open. 2017;7(6):e014961. doi: 10.1136/bmjopen-2016-014961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010;119(4):487–94. doi: 10.1007/s00401-010-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74(12):3317–31. doi: 10.1158/0008-5472.CAN-14-0772-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li L, Paz AC, Wilky BA, Johnson B, Galoian K, Rosenberg A, et al. Treatment with a Small Molecule Mutant IDH1 Inhibitor Suppresses Tumorigenic Activity and Decreases Production of the Oncometabolite 2-Hydroxyglutarate in Human Chondrosarcoma Cells. PLoS One. 2015;10(9):e0133813. doi: 10.1371/journal.pone.0133813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molenaar RJ, Verbaan D, Lamba S, Zanon C, Jeuken JW, Boots-Sprenger SH, et al. The combination of IDH1 mutations and MGMT methylation status predicts survival in glioblastoma better than either IDH1 or MGMT alone. Neuro Oncol. 2014;16(9):1263–73. doi: 10.1093/neuonc/nou005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32(8):783–90. doi: 10.1200/JCO.2013.49.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu Q, Li Y, Lv N, Jing Y, Xu Y, Li Y, et al. Correlation Between Isocitrate Dehydrogenase Gene Aberrations and Prognosis of Patients with Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. Clin Cancer Res. 2017;23(15):4511–22. doi: 10.1158/1078-0432.CCR-16-2628. [DOI] [PubMed] [Google Scholar]
- 55.Medeiros BC, Satram-Hoang S, Hurst D, Hoang KQ, Momin F, Reyes C. Big data analysis of treatment patterns and outcomes among elderly acute myeloid leukemia patients in the United States. Annals Hematol. 2015;94(7):1127–38. doi: 10.1007/s00277-015-2351-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Menzin J, Lang K, Earle CC, Kerney D, Mallick R. The outcomes and costs of acute myeloid leukemia among the elderly. Arch Intern Med. 2002;162(14):1597–603. doi: 10.1001/archinte.162.14.1597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.