

Abstract
Heterocyclic compounds offer a high degree of structural diversity and have proven to be broadly and economically useful as therapeutic agents. Comprehensive research on diverse therapeutic potentials of heterocycles compounds has confirmed their immense significance in the pathophysiology of diseases. Heterocyclic pyrimidine nucleus, which is an essential base component of the genetic material of deoxyribonucleic acid, demonstrated various biological activities. The present review article aims to review the work reported on therapeutic potentials of pyrimidine scaffolds which are valuable for medical applications during new generation.
Keywords: Pyrimidine derivatives, Antimicrobial, Antioxidant, Antimalarial, Anticancer, Anti-inflammatory
Introduction
Pyrimidine is the six membered heterocyclic organic colorless compound containing two nitrogen atoms at 1st and 3rd positions (Fig. 1). The name of the pyrimidine was first applied by Pinner from the combination of two words pyridine and amidine). Pyrimidines(1,3-diazines) and their fused analogues form a large group of heterocyclic compounds. Pyrimidine which is an integral part of DNA and RNA imparts diverse pharmacological properties. The pyrimidine have been isolated from the nucleic acid hydrolyses and much weaker base than pyridine and soluble in water [1]. Pyrimidine and its derivatives have been described with a wide range of biological potential i.e. anticancer [2], antiviral [3], antimicrobial [4], anti-inflammatory [5], analgesic [6], antioxidant [7] and antimalarial [8] etc.
Fig. 1.
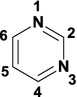
Pyrimidine ring
Biological significance of pyrimidine scaffolds
Antimicrobial activity
The growing health problems demands for a search and synthesis of a new class of antimicrobial molecules which are effective against pathogenic microorganisms. Despite advances in antibacterial and antifungal therapies, many problems remain to be solved for most antimicrobial drugs available. The extensive use of antibiotics has led to the appearance of multidrug resistant microbial pathogens which necessitated the search for new chemical entities for treatment of microbial infections [9].
Anupama et al. synthesized a series of 2,4,6-trisubstituted pyrimidines by reacting chalcone with guanidine hydrochloride. All the synthesized derivatives were confirmed by physicochemical properties and spectral data (IR, NMR and elemental analyses) and screened their in vitro antimicrobial activity against bacterial and fungal strains by cup plate method using Mueller–Hinton agar medium. Among the derivatives tested, compounds, a1, a2 and a3 exhibited promising activity against microbial strains (B. pumilis, B. subtilis, E. coli, P. vulgaris. A. niger and P. crysogenium) and showed activity comparable with standard drugs. Structure activity relationship (SAR) studies indicated that compounds, a1, a2 and a3 having dimethylamino, dichlorophenyl and fluorine substituent on the phenyl ring at 4th position respectively exhibited better antimicrobial activity (Table 1, Fig. 2) [4].
Table 1.
Antimicrobial activity of compounds (a1–a3)
| Compounds | Zone of inhibition (in mm) | |||||
|---|---|---|---|---|---|---|
| Microbial species | ||||||
| B. subtilis | B. pumilis | E. coli | P. vulgaris | A. niger | P. crysogenium | |
| a1 | ||||||
| A | 15 | 12 | 11 | 12 | 11 | 12 |
| B | 20 | 14 | 20 | 18 | 13 | 14 |
| a2 | ||||||
| A | 16 | 13 | 12 | 15 | 16 | 15 |
| B | 20 | 15 | 21 | 21 | 18 | 18 |
| a3 | ||||||
| A | 17 | 14 | 13 | 14 | 15 | 14 |
| B | 20 | 15 | 21 | 20 | 17 | 17 |
| C | – | – | – | – | – | – |
| S | ||||||
| A | 25 | 29 | 26 | 28 | 23 | 24 |
| B | 30 | 31 | 29 | 31 | 28 | 27 |
A: 0.05 ml (50 μg); B: 0.1 ml (100 μg); C: control (DMSO); S: standard (benzyl penicillin for bacterial strains) and fluconazole for fungal strains
Fig. 2.
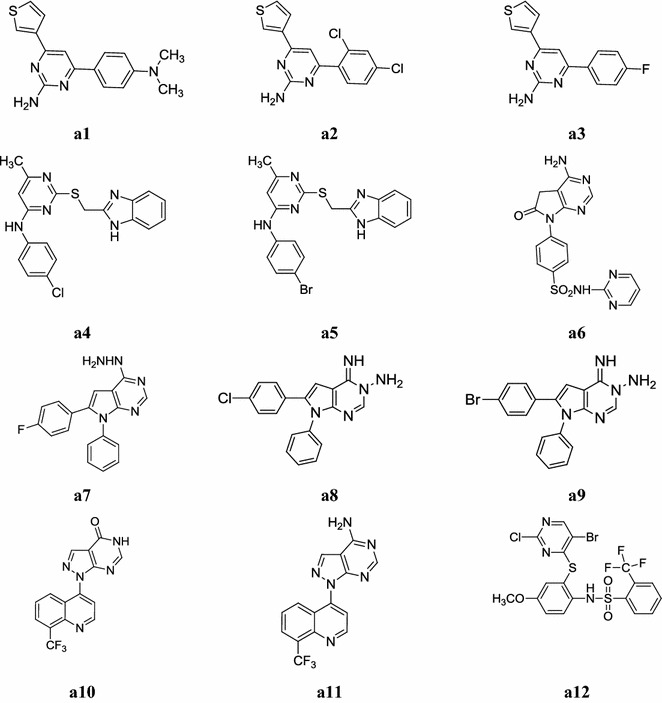
Chemical structure of the most active antimicrobial pyrimidine derivatives (a1–a12)
Chen et al. synthesized a novel series of 4-substituted-2-{[(1H-benzo[d]imidazol-2-yl) methyl]thio}-6-methylpyrimidines from pyrimidine–benzimidazole combination. All the synthesized derivatives were fully characterized by 1H-NMR, 13C-NMR and HRMS study and screened its in vitro antimicrobial activity against Gram-positive bacteria (Staphylococcus aureus, Bacillus subtilis), Gram-negative bacteria (Escherichia coli, Stenotrophomonas maltophilia) and fungi (Candida albicans). The minimum inhibitory concentration (MIC) of the target compounds was determined by broth microdilution method and compared to two commercial antibiotics (levofloxacin and fluconazole). Among the entire synthesized derivatives, compounds, a4 and a5 were found to be the most active antimicrobial agents (Table 2, Fig. 2). Structure activity relationship showed that aromatic amines at pyrimidine ring are beneficial for the antimicrobial activity. Besides, the aniline containing para-substituted groups (especially Cl and Br) is more beneficial for the activity [10].
Table 2.
Antimicrobial activity (MIC = µg/ml) of compounds a4 and a5
| Compounds | Bacterial strains | Fungal strain | |||
|---|---|---|---|---|---|
| Staphylococcus aureus | Bacillus subtilis | Escherichia coli | Stenotrophomonas maltophilia | Candida albicans | |
| a4 | 8 | 128 | 128 | 2 | 64 |
| a5 | 16 | 128 | 128 | 4 | 8 |
| Levofloxacin | 0.5 | 0.25 | 0.125 | 0.25 | – |
| Fluconazole | – | – | – | – | 2 |
El-Gaby et al. developed a new class of pyrrolo[2,3-d]pyrimidines containing sulfonamide moieties and screened its in vitro antifungal activity against four species of fungi viz: Aspergillus ochraceus (Wilhelm), Penicillium chrysogenum (Thom), Aspergillus fleavus (Link) and Candida albicans (Robin) Berkho by disc diffusion technique. Most of the synthesized molecules in this series were found to possess antifungal activity (Table 3, Fig. 2) towards all the microorganisms’ used especially, compound a6 exhibited a remarkable antifungal activity which is comparable to the standard fungicide drug mycostatin [11].
Table 3.
Antifungal activity of synthesized compound a6
| Compound | Zone of inhibition (mm) | |||
|---|---|---|---|---|
| Fungal species | ||||
| A. ochraceus (AUCC-230) | P. chrysogenum (AUCC-530) | A. fleavus (AUCC-164) | C. albicans (AUCC-1720) | |
| a6 | 18 (45%) | 14 (37%) | 16 (42%) | 34 (85%) |
| Mycostatine | 40 (100) | 38 (100%) | 38 (100%) | 40 (100%) |
Hilmy et al. developed a new series of pyrrolo[2,3-d]pyrimidine derivatives. The synthesized compounds were confirmed by IR, NMR, Mass and elemental analysis study and evaluated its antimicrobial activity against bacterial (Staphylococcus aureus, Escherichia coli) and fungal (Candida albicans) organisms was carried out by serial dilution method. All synthesized derivatives showed that good antimicrobial activity, especially, compounds, a7, a8, a9 were exhibited the better antimicrobial activity and compared with the standard drug (ampicillin and fluconazole) (Table 4, Fig. 2) [12].
Table 4.
The MIC (mg/ml) value of the compounds a7, a8 and a9 tested against organisms
| Compounds | Antimicrobial results (MIC = mg/ml) | ||
|---|---|---|---|
| Escherichia coli | Staphylococcus aureus | Candida albicans | |
| a7 | 1.25 | 0.31 | 0.31 |
| a8 | 1.25 | 0.31 | 0.62 |
| a9 | 1.25 | 0.31 | 0.31 |
| Ampicillin | 1.25 | 0.62 | – |
| Fluconazole | – | – | 1.5 |
Holla et al. developed a new class of pyrazolo[3,4-d]pyrimidine derivatives. The synthesized derivatives were analyzed for N content and their structures were confirmed by IR, NMR and Mass spectral data and screened their antibacterial activity against Escherichia coli, Staphylococcus aureus, Pseudomonas aeruginosa and Bacillus subtilis by disk diffusion method and antifungal activity against Aspergillus flavus, Aspergillus fumigates, Candida albicans, Penicillium marneffei and Trichophyton mentagrophytes by serial plate dilution method. All synthesized pyrazolo[3,4-d]pyrimidine derivatives in this series showed that good antimicrobial and fungal activity against bacterial and fungal strains, especially compounds, a10 displayed very good antibacterial activity (Table 5, Fig. 2) and a11 exhibited antifungal activity (Table 6, Fig. 2) [13].
Table 5.
Antibacterial activity data of compound a10
| Compound | Zone of inhibition (mm) of bacterial species | |||
|---|---|---|---|---|
| Escherichia coli | Staphylococcus aureus | Pseudomonas aeruginosa | Bacillus subtilis (recultured) | |
| a10 | 28 | 25 | 24 | 26 |
| Streptomycin | 20 | 21 | 24 | 24 |
Table 6.
Antifungal activity data of prepared compound a11
| Compound | Zone of inhibition (mm) of fungal species | ||
|---|---|---|---|
| Aspergillus flavus | Aspergillus fumigatus | Trichophyton mentagrophytes (recultured) | |
| a11 | 25 | 22 | 24 |
| Fluconazole | 21 | 18 | 19 |
Mallikarjunaswamy et al. synthesized a series of novel 2-(5-bromo-2-chloro-pyrimidin-4-ylsulfanyl)-4-methoxy-phenylamine derivatives by the reaction of 2-(5-bromo-2-chloro-pyrimidin-4-ylsulfanyl)-4-methoxy-phenylamine with various sulfonyl chlorides and its molecular structures were characterized by elemental analyses, FT-IR, 1H-NMR and LC–MS spectral studies and screened in vitro antimicrobial activity against Gram-positive bacteria (Bacillus subtilis, Staphylococcus aureus) and Gram-negative bacteria (Xanthomonas campestris and Escherichia coli) in dimethylformamide by disc diffusion method on nutrient agar medium and antifungal activity against Fusarium oxysporum in dimethylformamide by poisoned food technique. Among them, compound a12 was found to be most potent against fungal strain (Fusarium oxysporum) and bacterial strains (Bacillus subtilis, Staphylococcus aureus, Xanthomonas campestris and Escherichia coli) and compared with standard antimicrobial drugs (Table 7, Fig. 2) [9].
Table 7.
In vitro antibacterial and antifungal activities of compound a12
| Compound | Zone of inhibition in diameter (mm) % inhibition | ||||
|---|---|---|---|---|---|
| Microbial species | |||||
| B. subtilis | S. aureus | X. campestris | E. coli | F. oxysporum | |
| a12 | 33 | 29 | 32 | 33 | 96.9 |
| Bacteriomycin | – | – | 34 | – | – |
| Gentamycin | 35 | 30 | – | 35 | – |
| Nystatin | 100 | ||||
A new series of 1,2,4-triazolo[1,5-a]pyrimidine derivatives bearing 1,3,4-oxadiazole moieties was designed and synthesized by Chen et al. The molecular structures of all new compounds were characterized by spectral means (1H-NMR, Mass and elemental analyses) and evaluated their in vitro antifungal activity against Rhizoctonia solani. In this series, compounds, a13 and a14 displayed the highest antifungal activity against Rhizoctonia solani with EC50 = 3.34 µg/ml and EC50 = 6.57 µg/ml values respectively than the carbendazim (EC50 = 7.62 µg/ml) due to presence of the sec-butyl group (Fig. 3) [14].
Fig. 3.

Chemical structure of the most active antimicrobial pyrimidine derivatives (a13–a21)
A new library of 5-amino-6-(benzo[d]thiazol-2-yl)-2-(2-(substituted benzylidene) hydrazinyl)-7-(4-chlorophenyl)pyrido[2,3-d]pyrimidin-4(3H)-one derivatives was synthesized by Maddila et al. and evaluated its antibacterial activity against Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa and Streptococcus pyogenes and antifungal activity against Aspergillus flavus, Aspergillus fumigatus, Candida albicans, Penicillium marneffei and Mucor by the twofold serial dilution method. Compounds, a15, a16 and a17 showed excellent antibacterial and antifungal activity than the standard drugs ciprofloxacin and clotrimazole respectively (Tables 8, 9, Fig. 3) [15].
Table 8.
Antibacterial activity results of compounds (a15–a17)
| Compounds | Minimum inhibitory concentration (MIC = µg/ml) | ||||
|---|---|---|---|---|---|
| Bacterial species | |||||
| S. aureus | E. coli | K. pneumoniae | P. aeruginosa | S. pyogenes | |
| a15 | 12.5 | 25 | 25 | 25 | 12.5 |
| a16 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 |
| a17 | 25 | 12.5 | 12.5 | 25 | 12.5 |
| Ciprofloxacin | 25 | 25 | 50 | 25 | 12.5 |
Table 9.
Antifungal activity results of compounds (a15–a17)
| Compounds | Minimum inhibitory concentration (MIC = µg/ml) | ||||
|---|---|---|---|---|---|
| Fungal species | |||||
| A. flavus | A. fumigatus | C. albicans | P. marneffei | Mucor | |
| a15 | 12.5 | 12.5 | 25 | 25 | 12.5 |
| a16 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 |
| a17 | 25 | 12.5 | 25 | 12.5 | 25 |
| Clotrimazole | 25 | 25 | 50 | 25 | 50 |
Fellahil et al. synthesized a new series of 5-(1,2-diarylethyl)-2,4,6-trichloro pyrimidines and 2-amino- and 2-(1-piperazinyl)-5-(1,2-diarylethyl)-4,6-dichloro pyrimidines via organozinc reagents and demonstrated its antibacterial activity against human bacterial flora. Biological tests showed that 5-[1-(4-chlorophenyl)-2-phenylethyl]-2,4,6-trichloro pyrimidine derivatives i.e. compounds a18 and a19 were found to be most active against wide range of bacterial flora of the axilla and foot, while 2-(1-piperazinyl)-4,6-dichloro pyrimidine derivatives a20 and a21 displayed a great selectivity against Corynebacterium xerosis and Arcanobacterium haemolyticum of the human axilla (Table 10, Fig. 3) [16].
Table 10.
Pharmacological evaluation (MIC = µg/ml) of the 2-substituted 5-(1,2-diarylethyl)-4,6-dichloropyrimidines
| a18 | a19 | a20 | a21 | |
|---|---|---|---|---|
| Axillary bacterial flora | ||||
| Staphylococcus xylosus | 20 | 100 | 100 | 100 |
| Staphylococcus epidermidis | 100 | 100 | 100 | 75 |
| Staphylococcus haemolyticus | 100 | 100 | 100 | 50 |
| Corynebacterium xerosis | 20 | 30 | 30 | 30 |
| Micrococcus luteus | 20 | 100 | 100 | 100 |
| Arcanobacterium haemolyticum | 10 | 10 | 10 | 10 |
| Foot bacterial flora | ||||
| Staphylococcus epidermidis | > 100 | 100 | 100 | 75 |
| Staphylococcus hominis | 100 | 100 | 100 | 75 |
| Staphylococcus cohnii | 100 | 100 | 100 | 75 |
| Corynebacterium sp. g C | 100 | 100 | 100 | 75 |
| Corynebacterium sp. g B | 30 | 100 | 100 | 50 |
| Corynebacterium sp. g D2 | 30 | 100 | 50 | 50 |
| Micrococcus luteus | 20 | 100 | 100 | 75 |
| Micrococcus sedentarius | 30 | 100 | 100 | 75 |
| Acinetobacter sp. | > 1000 | > 500 | 50 | 30 |
| Moraxella sp. | 300 | 30 | 100 | 50 |
| Alcaligenes sp. | 1000 | > 500 | > 500 | > 500 |
Nagender et al. developed a new series of novel pyrazolo[3,4-b]pyridine and pyrimidine functionalized 1,2,3-triazole derivatives using 6-trifluoro methylpyridine-2(1H) one and screened its antimicrobial activity against i.e. Micrococcus luteus MTCC 2470, Staphylococcus aureus MTCC 96, Staphylococcus aureus MLS-16 MTCC 2940, Bacillus subtilis MTCC 121, Escherichia coli MTCC 739, Pseudomonas aeruginosa MTCC 2453, Klebsiella planticola MTCC 530 and Candida albicans MTCC 3017. In this series, compounds, a22, a23 and a24 were displayed better antimicrobial activity but less than the standard drugs (ciprofloxacin) (Table 11, Fig. 4) [17].
Table 11.
MIC values of the compounds a22, a23 and a24
| Compounds | Minimum inhibitory concentration (µg/ml) | ||||||
|---|---|---|---|---|---|---|---|
| M. luteus | S. aureus | S. aureus | B. subtilis | E. coli | P. aeruginosa | K. planticola | |
| a22 | 7.8 | 15.6 | 15.6 | 15.6 | 7.8 | 7.8 | 15.6 |
| a23 | > 250 | 15.6 | 7.8 | 15.6 | 15.6 | 15.6 | 7.8 |
| a24 | 15.6 | 7.8 | 7.8 | 15.6 | 7.8 | 7.8 | 7.8 |
| Ciprofloxacin | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 | 0.9 |
Fig. 4.
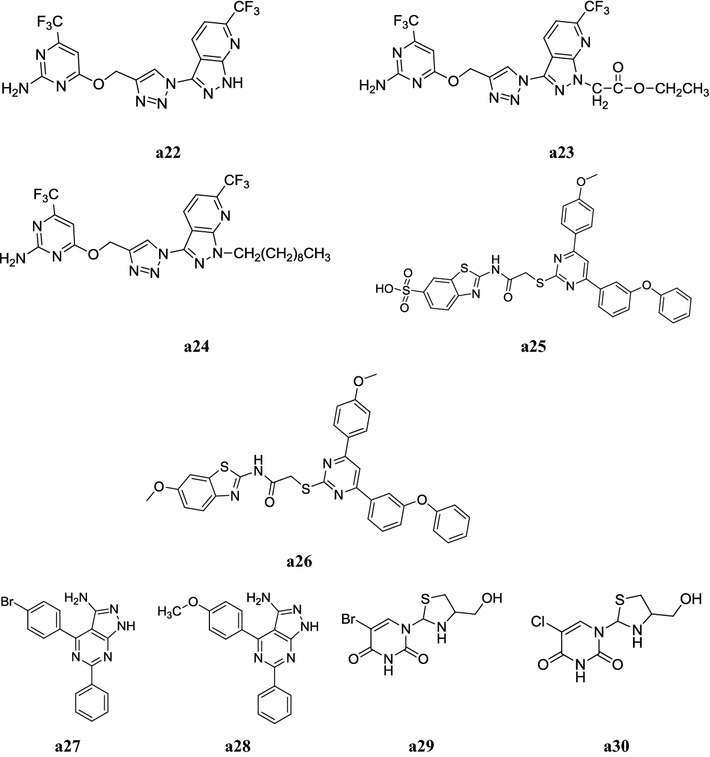
Chemical structure of the most active antimicrobial pyrimidine derivatives (a22–a30)
Patel et al. synthesized a new series of pyrimidine derivatives and demonstrated its antimicrobial activity (Minimum inhibitory concentration) against four different strains, viz two Gram positive bacteria (S. aureus and S. pyogenes) and two Gram negative bacteria and (E. coli and P. aeruginosa) compared it with standard drugs ampicillin, chloramphenicol, ciprofloxacin and norfloxacin and antifungal activities against C. albicans and A. niger using nystatin as standard drug by broth dilution method, compounds, a25 and a26 were showed promising antimicrobial activity (Table 12, Fig. 4) [18].
Table 12.
Antimicrobial activity of compounds a25 and a26
| Compounds | Microbial strains (µg/ml) | |||||
|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. aureus | S. pyogenus | C. albicans | A. niger | |
| a25 | 62.5 | 200 | 100 | 100 | 200 | 250 |
| a26 | 25 | 50 | 100 | 50 | 500 | 250 |
| Chloramphenicol | 50 | 50 | 50 | 50 | – | – |
| Ciprofloxacin | 25 | 25 | 50 | 50 | – | – |
| Norfloxacin | 10 | 10 | 10 | 10 | – | – |
| Nystatin | 100 | 100 | ||||
A new library of pyrazolo[3,4-d]pyrimidine derivatives was synthesized by Rostamizadeh et al. and screened for its antibacterial activity against two Gram-negative strains of bacteria: Pseudomonas aeruginosa and Klebsiella pneumonia and two Gram-positive bacteria: Staphylococcus aureus and Enterococcus raffinosus L. Amongst the tested compounds, compounds a27 and a28 exhibited higher antibacterial activity than the standard drugs (Table 13, Fig. 4) [19].
Table 13.
Antibacterial activity of some novel pyrazolopyrimidine derivatives
| Compounds | MIC (µmol/l) | |
|---|---|---|
| Enterococcus raffinosus | Staphylococcus aureus | |
| a27 | 12.3 | 3.8 |
| a28 | 14.2 | 4.2 |
| Penicillin G | 93.5 | 24.4 |
Sriharsha et al. developed a new series of novel 1,3-thiazolidine pyrimidine derivatives and carried out its antibacterial activity against 14 bacterial strains i.e. Citrobacter sp., Escherichia coli, Klebsiella sp., Proteus mirabilis, Pseudomonas aeruginosa, S. parathyphi A, S. parathyphi B, Salmonella typhi, S. typhimurium, Shigella boydii, Shigella flexneri, Shigella sonnei, Staphylococcus aureus and Streptococcus faecalis. All compounds with free NH group in the pyrimidine moiety showed significant biological activity against all the standard strains used and in that compounds a29 and a30 showed promising activity against 14 human pathogens tested and compared with the ciprofloxacin and bacitracin used as standard drugs (Table 14, Fig. 4) [20].
Table 14.
Antibacterial activity (zone of inhibition = mm) of most active compounds
| S. no | Pathogens | a29 | a30 | Bacitracin | Ciprofloxacin |
|---|---|---|---|---|---|
| 1 | Citrobacter sp. | 37.16 ± 0.15 | 28.66 ± 0.15 | 0.00 ± 0.00 | 19.62 ± 0.18 |
| 2 | Escherichia coli | 36.66 ± 0.15 | 27.83 ± 0.20 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| 3 | Klebsiella sp. | 32.50 ± 0.13 | 25.50 ± 0.27 | 0.00 ± 0.00 | 20.25 ± 0.16 |
| 4 | Proteus mirabilis | 28.66 ± 0.25 | 23.33 ± 0.17 | 0.00 ± 0.00 | 18.25 ± 0.16 |
| 5 | Pseudomonas aeruginosa | 30.66 ± 0.12 | 27.83 ± 0.27 | 0.00 ± 0.00 | 34.25 ± 0.16 |
| 6 | S. parathyphi A | 34.66 ± 0.12 | 24.50 ± 0.12 | 0.00 ± 0.00 | 27.75 ± 0.16 |
| 7 | S. parathyphi B | 32.50 ± 0.13 | 27.83 ± 0.20 | 0.00 ± 0.00 | 27.63 ± 0.18 |
| 8 | Salmonella typhi | 29.50 ± 0.25 | 19.66 ± 0.11 | 0.00 ± 0.00 | 20.25 ± 0.16 |
| 9 | S. typhimurium | 34.66 ± 0.12 | 23.33 ± 0.17 | 0.00 ± 0.00 | 18.75 ± 0.31 |
| 10 | Shigella boydii | 37.50 ± 0.07 | 28.66 ± 0.25 | 0.00 ± 0.00 | 17.75 ± 0.16 |
| 11 | Shigella flexneri | 35.66 ± 0.08 | 25.50 ± 0.27 | 0.00 ± 0.00 | 27.63 ± 0.18 |
| 12 | Shigella sonnei | 32.50 ± 0.13 | 37.50 ± 0.07 | 0.00 ± 0.00 | 21.75 ± 0.16 |
| 13 | Staphylococcus aureus | 37.50 ± 0.07 | 32.50 ± 0.13 | 26.75 ± 0.84 | 18.13 ± 0.48 |
| 14 | Streptococcus faecalis | 38.50 ± 0.12 | 35.66 ± 0.08 | 0.00 ± 0.00 | 0.00 ± 0.00 |
Anticancer activity
Cancer is a multifaceted disease that represents one of the leading causes of mortality in developed countries. Worldwide, one in eight deaths are due to cancer and it is the second most common cause of death in the US, exceeded only by heart disease. Chemotherapy is the mainstay for cancer treatment, the use of available chemotherapeutics is often limited due to undesirable side effects. It is important to identify new molecules and new targets for the treatment of cancer [17].
Shao et al. synthesized a new derivatives of 2,4,5-trisubstituted pyrimidine CDK inhibitors as potential antitumour agents. The synthesized 2,4,5-trisubstituted pyrimidine derivatives were evaluated for their antitumour activity against a panel of cancer cell lines including colorectal, breast, lung, ovarian, cervical and pancreatic cancer cells. Among the synthesized derivatives, compound b1, possessing appreciable selectivity for CDK9 over other CDKs, is capable of activating caspase 3, reducing the level of Mcl-1 anti-apoptotic protein and inducing cancer cell apoptosis (Table 15, Fig. 5) [21].
Table 15.
Anti-proliferative activity of b1 in human cancer cell lines
| Compound | Human cancer cell lines | ||
|---|---|---|---|
| Origin | Designation | 48 h-MTT GI50 (µM) ± SD | |
| b1 | Colon carcinoma | HCT-116 | 0.79 ± 0.08 |
| Breast carcinoma | MCF-7 | 0.64 ± 0.08 | |
| MDA-MB468 | 1.51 ± 0.34 | ||
| Lung carcinoma | A549 | 2.01 ± 0.55 | |
| Ovarian carcinoma | A2780 | 1.00 ± 0.11 | |
| Cervical carcinoma | HeLa | 0.90 ± 0.07 | |
| Pancreatic carcinoma | Miacapa-2 | 1.25 ± 0.26 | |
Fig. 5.

Chemical structures of the most active anticancer pyrimidine derivatives (b1–b12)
Cocco et al. synthesized a new class of 6-thioxopyrimidine derivatives and its molecular structures were confirmed by IR, NMR and elemental analyses study. The synthesized derivatives were evaluated their in vitro anticancer potential against multiple panels of 60 human cancer cell lines by Sulforhodamine B assay. All synthesized 6-thioxopyrimidine derivatives exhibited good anticancer potential, especially, compound b2 showed the best cytotoxicity (Table 16, Fig. 5) [2].
Table 16.
Anticancer activity results of most active compound b2
| Compound | CNS cancer cell lines | 10−5 M concentration | Ovarian cancer cell lines | 10−5 M concentration |
|---|---|---|---|---|
| b2 | SF-268 | 2.95 | IGROV1 | 7.71 |
| SF-295 | 9.79 | OVCAR-3 | 6.34 | |
| SF-539 | 3.99 | OVCAR-4 | 3.42 | |
| SNB-19 | 5.42 | OVCAR-8 | 4.92 | |
| SNB-57 | 2.49 | – | – | |
| U-251 | 3.58 | – | – |
A new library of sulfonamide derivatives was synthesized and investigated for its in vitro and in vivo antitumor potential by El-Sayed et al. Preliminary biological study revealed that compounds, b3, b4 and b5 showed the highest affinity to DNA and highest percentage increase in lifespan of mice inoculated with Ehrlich ascites cells over 5-flurouracil was taken as standard drug (Table 17, Fig. 5) [22].
Table 17.
In vitro anticancer activity results of active compounds
| Group | Normal | Control (Ehrlich only) | b3 | b4 | b5 | 5-Fluorouracil |
|---|---|---|---|---|---|---|
| % Increase in lifespan over control | 71.43 | 0 | 71.43 | 57.14 | 42.86 | 42.86 |
Two new class of pyrido[2,3-d]pyrimidine and pyrido[2,3-d][1,2,4]triazolo[4,3-a] pyrimidines were synthesized by Fares et al. The molecular structures of synthesized derivatives were confirmed by physicochemical properties and spectral data (IR, NMR, Mass and elemental analyses) and screened for their anticancer activity against human cancer cell lines i.e. PC-3 prostate and A-549 lung. Some of the tested compounds exhibited high growth inhibitory potential against PC-3 cell, among them, compounds, b6 and b7 showed relatively potent antitumor potential (Table 18, Fig. 5) [23].
Table 18.
Anticancer activity results of compounds b6 and b7
| Compounds | Cancer cell lines (IC50 = µM) | |
|---|---|---|
| A-549 | PC-3 | |
| b6 | 3.36 ± 0.39 | 1.54 ± 0.19 |
| b7 | 0.41 ± 0.03 | 0.36 ± 0.02 |
| 5-Fluorouracil | 4.21 ± 0.39 | 12.00 ± 1.15 |
Hu et al. developed a new library of 2,4-diamino-furo[2,3-d]pyrimidine and carried out its in vitro anticancer activity against A459 and SPC-A-1 cancer cell lines. Their structures were confirmed by 1H-NMR, EI-Ms, IR and elemental analysis. Among them, compound b8: ethyl-6-methyl-4-(4-methylpiperazin-1-yl)-2-(phenylamino)furo[2,3-d] pyrimidine-5-carboxylate was found to be most anticancer one against lung cancer cell line (A459 with IC50 0.8 µM) (Fig. 5) [24].
Huang et al. developed a new series of pyrazolo[3,4-d]pyrimidines using 5-aminopyrazoles with formamide in presence of PBr3 as the coupling agent and its chemical structures were characterized by IR, 1H/13C-NMR, Mass, elemental analyses data. The synthesized compounds were screened their in vitro antiproliferative potential by MTT assay against human cancer cell line viz. NCI-H226 (lung carcinoma) and NPC-TW01 (nasopharyngeal carcinoma). From this series, compounds, b9, b10, b11 and b12 possessed better potency against NCI-H226 and NPC-TW01 cancer cells (Table 19, Fig. 5) [25].
Table 19.
Antiproliferative results of active compounds (b9–b12)
| Compounds | Cancer cell lines (GI50 = µM) | |
|---|---|---|
| NCI-H226 | NPC-TW01 | |
| b9 | 18 | 23 |
| b10 | 29 | 30 |
| b11 | 39 | 35 |
| b12 | 37 | 36 |
Song et al. synthesized a new library of fluorinated pyrazolo[3,4-d]pyrimidine derivatives by microwave (MW) irradiation method and evaluated its in vitro antitumor potential against human leukaemia (HL-60) cancer cell line by MTT assay. The preliminary results demonstrated that some of compounds exhibited potent antitumor inhibitory potential than doxorubicin (standard drug), especially compounds, b13 and b14 exhibited higher antitumor activity due to presence of CF group in its molecule structure (Table 20, Fig. 6) [26].
Table 20.
Antitumor potential results of compounds b13 and b14
| Compounds | Human leukaemia (HL-60) cancer cell IC50 = µmol/l |
|---|---|
| b13 | 0.08 |
| b14 | 0.21 |
| Doxorubicin | 0.55 |
Fig. 6.

Chemical structures of the most active anticancer pyrimidine derivatives (b13–b23)
Tangeda and Garlapati, developed new molecules of pyrrolo[2,3-d]pyrimidine and screened its in vitro anticancer activity against HCT116 colon cancer cell line. Especially, compounds, b15 and b16 were found to be most potent ones against HCT116 cell line with IC50 value of 17.61 and 17.60 µM respectively which is comparable with 5-fluorouracil (IC50 = 3.03 µM) (Fig. 6) [27].
Kurumurthy et al. prepared a novel class of alkyltriazole tagged pyrido[2,3-d] pyrimidine derivatives and its molecular structure were confirmed by IR, NMR, Mass and elemental analyses. The synthesized derivatives were evaluated their in vitro anticancer activity against three cancer cell lines i.e. U937 (human leukemic monocytic lymphoma), THP-1 (human acute monocytic leukemia) and Colo205 (human colorectal cancer) using MTT assay. Among the synthesized molecules, compounds b17 and b18 exhibited better anticancer activity than the standard etoposide (Table 21, Fig. 6) [28].
Table 21.
In vitro cytotoxicity of pyrido[2,3-d]pyrimidine derivatives against U937, THP-1 and Colo205 cancer cell lines
| Compounds | IC50 (µg/ml) | ||
|---|---|---|---|
| U937 | THP-1 | Colo205 | |
| b17 | 8.16 ± 0.68 | 16.91 ± 1.42 | 19.25 ± 1.46 |
| b18 | 6.20 ± 0.68 | 11.27 ± 1.67 | 15.01 ± 1.54 |
| Etoposide (positive control) | 17.94 ± 1.19 | 2.16 ± 0.15 | 7.24 ± 1.26 |
Liu et al. synthesized two series of thieno[3,2-d]pyrimidine molecules containing diaryl urea moiety and screened their anticancer potential. The preliminary investigation showed that most compounds displayed good to excellent potency against four tested cancer cell lines compared with GDC-0941 and sorafenib as standard drugs. In particular, the most promising compound b19 showed the most potent antitumor activities with IC50 values of 0.081, 0.058, 0.18 and 0.23 µM against H460, HT-29, MKN-45 and MDA-MB-231 cell lines, respectively (Fig. 6) [29].
Zhu et al. developed a series of 2,6-disubstituted-4-morpholinothieno[3,2-d]pyrimidine molecules and demonstrated its in vitro cytotoxic activity against H460, HT-29, MDA-MB-231, U87MG and H1975 cancer cell lines. Most of the target compounds exhibited moderate to excellent activity to the tested cell lines. The most promising compound b20 is more active than the standard drug (Table 22, Fig. 6) [30].
Table 22.
Cytotoxicity of compound b20
| Compounds | IC50 = (µmol/l) | ||||
|---|---|---|---|---|---|
| H460 | HT29 | MDA-MB-231 | U87MG | H1975 | |
| b20 | 0.84 | 0.23 | 2.52 | 1.80 | 28.82 |
| PAC-1 | 3.57 | 0.97 | 6.11 | ND | ND |
ND not determined
2,4,5-Substituted pyrimidine molecules were prepared and evaluated for their anticancer activity against different human cancer cell lines (A549, Calu-3, H460, SK-BR3, SGC-7901 and HT29) by Xie et al. Among the synthesized molecules, compounds b21 showed good inhibition of several different human cancer cell lines with IC50 values from 0.024 to 0.55 µM (Table 23, Fig. 6) [31].
Table 23.
In vitro anticancer activity of compound b21
| Compound | Human cancer cell lines (IC50 = µM) | |||||
|---|---|---|---|---|---|---|
| A549 | Calu-3 | H460 | SK-BR3 | SGC-7901 | HT29 | |
| b21 | 0.55 | 0.50 | 0.12 | 0.30 | 0.30 | 0.090 |
| Adriamycin | 0.025 | – | – | – | – | 0.018 |
| Docetaxel | – | 0.10 | 0.0097 | – | 0.0084 | – |
| GW572016 | – | – | – | 0.017 | – | – |
Al-Issa, developed a new series of fused pyrimidines and related heterocycles and evaluated its in vitro antitumor activity against human liver cancer cell line (HEPG2). Structures of all synthesized compounds were supported by spectral and elemental analyses. Among the synthesized compounds, compounds b22 and b23 showed significant in vitro antitumor activity (IC50, 17.4, 23.6 µg/ml) (Fig. 6) [32].
Mohareb et al. developed a new class of fused pyran, pyrimidine and thiazole molecules and evaluated its in vitro anticancer potential against cancer cell lines i.e. NUGC- gastric; DLDI-colon; HA22T-liver; HEPG2-liver; HONEI-nasopharyngeal carcinoma; HR-gastric; MCF-breast and WI38-normal fibroblast cells. In this study, compounds, b24 and b25 exhibited more anticancer potential (Table 24, Fig. 7) [33].
Table 24.
Anticancer activity results of b24 and b25
| Compounds | Cytotoxicity (IC50 in nM) | ||||||
|---|---|---|---|---|---|---|---|
| NUGC | DLDI | HA22T | HEPG2 | HONEI | MCF | WI38 | |
| b24 | 180 | 740 | 234 | 837 | 644 | 269 | Na |
| b25 | 40 | 64 | 82 | 328 | 260 | 173 | Na |
| CHS 828 | 25 | 2315 | 2067 | 1245 | 15 | 18 | Na |
Fig. 7.
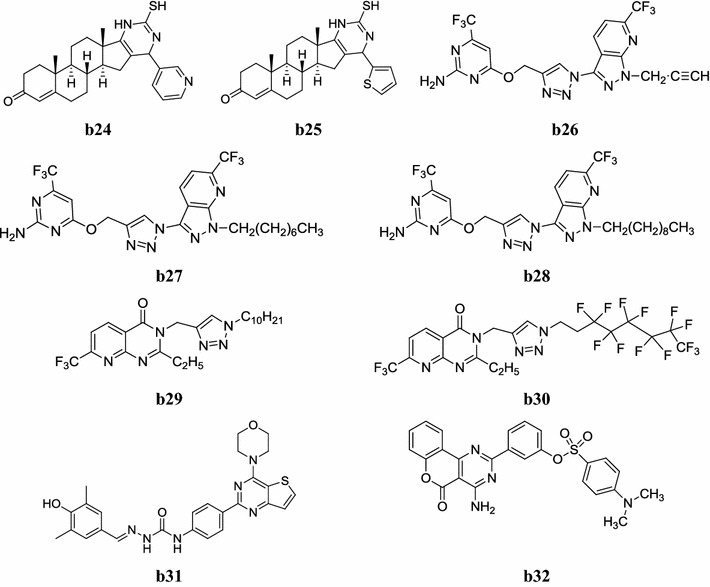
Chemical structures of the most active anticancer pyrimidine derivatives (b24–b32)
A new series of novel pyrazolo[3,4-b]pyridine and pyrimidine functionalized 1,2,3-triazole derivatives were prepared from 6-trifluoro methyl pyridine-2(1H)one by Nagender et al. and screened for its cytotoxicity against four human cancer cell lines such as A549-Lung (CCL-185), MCF7-Breast (HTB-22), DU145-Prostate (HTB-81) and HeLa-Cervical (CCL-2). Among them, compounds, b26, b27 and b28 showed promising cytotoxicity (Table 25, Fig. 7) [17].
Table 25.
In vitro cytotoxicity of most active compounds
| Compounds | IC50 values (in µM) | |||
|---|---|---|---|---|
| A549 | MCF7 | DU145 | HeLa | |
| b26 | 4.1 ± 0.12 | – | 4.7 ± 0.18 | – |
| b27 | 5.7 ± 0.22 | 24.7 ± 0.16 | 6.3 ± 0.21 | 22.7 ± 0.11 |
| b28 | 4.2 ± 0.31 | 37.2 ± 0.31 | 5.8 ± 0.14 | 34.3 ± 0.32 |
| 5-Fluorouracil | 1.3 ± 0.11 | 1.4 ± 0.09 | 1.5 ± 0.12 | 1.3 ± 0.14 |
Kumar et al. developed a new library of triazole/isoxazole functionalized 7-(trifluoromethyl)pyrido[2,3-d]pyrimidine derivatives and screened their anticancer activity against four human cancer cell lines using nocodazole as standard. Compounds b29 and b30 showed highest activity against PANC-1 (pancreatic cancer) and A549 (lung cancer) cell lines respectively (Table 26, Fig. 7) [34].
Table 26.
Anticancer activity of triazole/isoxazole functionalized pyridopyrimidine derivatives
| Compounds | GI50 values in µM | |||
|---|---|---|---|---|
| MDA MB-231 | PANC1 | A549 | HeLa | |
| b29 | 2.21 ± 0.08 | 0.02 ± 0.01 | 0.86 ± 0.03 | 0.81 ± 0.02 |
| b30 | 2.83 ± 0.05 | 0.73 ± 0.01 | 0.03 ± 0.01 | 0.93 ± 0.03 |
| Nocodazole | 0.042 ± 0.001 | 0.029 ± 0.003 | 0.08 ± 0.001 | 0.063 ± 0.002 |
A new class of novel thieno[3,2-d]pyrimidine derivatives was synthesized by Liu et al. and studied for its anticancer potential against selected cancer cell lines viz: H460, HT-29, MKN-45 and MDA-MB-231. Most of compounds displayed good to excellent potency against four tested cancer cell lines as compared with GDC-0941 and sorafenib.
In this study, compound b31 was found to be most active anticancer one (Table 27, Fig. 7) [35].
Table 27.
Cytotoxicity of compound b31
| Compound | IC50 (µmol/l) ± SD | |||
|---|---|---|---|---|
| H460 | HT-29 | MKN-45 | MDA-MB-231 | |
| b31 | 0.057 ± 0.011 | 0.039 ± 0.008 | 0.25 ± 0.019 | 0.23 ± 0.020 |
| GDC-0941 | 0.87 ± 0.20 | 0.86 ± 0.081 | 0.60 ± 0.12 | 0.28 ± 0.06 |
| Sorafenib | 2.19 ± 0.11 | 3.61 ± 0.36 | 2.32 ± 0.35 | 0.94 ± 0.13 |
Lv et al. synthesized a new series of 2-phenylpyrimidine coumarin derivatives and evaluated its in vitro antiproliferative activity against CNE2, KB and Cal27 cancer cell lines. The results showed that most of the derivatives had a favorable effect on resisting tumor cell proliferation, among them, compound b32 exhibited the best antiproliferative activity and comparable to the standard drug (Table 28, Fig. 7) [36].
Table 28.
In vitro anticancer activity of the synthesized compound
| Compound | IC50 (µM) | ||
|---|---|---|---|
| CNE2 | KB | Cal27 | |
| b32 | 1.92 ± 0.13 | 3.72 ± 0.54 | 1.97 ± 0.51 |
| Doxorubicin | 2.12 ± 0.56 | 3.04 ± 0.87 | 1.56 ± 0.64 |
Antiviral activity
Antiviral nucleoside compounds inhibit viral genome replication by acting as mimetics of the natural nucleosides. Nucleoside analogues (NAs) can either act as chain terminators after being incorporated into growing DNA/RNA strands and/or inhibit the viral polymerase function by competition with the natural nucleoside 50-triphosphate substrate [3].
A new library of 4H,6H-[1,2,5]oxadiazolo[3,4-d]pyrimidine-5,7-dione 1-oxide nucleoside was synthesized by Xu et al. and screened for its in vitro anti-vesicular stomatitis virus (VSV) activity in Wish cell. All the synthesized derivatives showed obvious anti-VSV potential whereas, compound c1 with ribofuranoside enhanced the anti-VSV potential by approximately 10–18 times compared to didanosine and acyclovir (standard drugs), respectively (Table 29, Fig. 8) [37].
Table 29.
Experimental antiviral results of compound c1
| Compound | Toxicity for wish cells and antivirus effect (TC0 µmol/l) | ED50 | Model 1 | Model 2 |
|---|---|---|---|---|
| c1 | 2095 | 78 | 148 | 100 |
| Acyclovir | 3414 | 1411 | – | – |
| Didanosine | 2646 | 792 | – | – |
Fig. 8.

Chemical structures of the most active antiviral pyrimidine derivatives (c1–c10)
Hockova et al. synthesized a new series of 2,4-diamino-5-cyano-6[2-(phosphono methoxy)ethoxy]pyrimidine derivatives and evaluated its antiviral activity. The 5-cyano and 5-formyl derivatives (c2–c4) showed pronounced antiretroviral activity, comparable to that of the reference drugs adefovir and tenofovir (Table 30, Fig. 8) [38].
Table 30.
Antiviral activity results of test compounds (c2–c4) in cell culture
| Compounds | ECa50 (µmol/ml) | CCb50 | ||
|---|---|---|---|---|
| HIV (IIIB) | HIV-2 (ROD) | MSV | (µmol/ml) (CEM) | |
| c2 | 0.011 | 0.0045 | 0.0095 | ≥ 0.3 |
| c3 | 0.0045 | 0.0027 | 0.021 | ≥ 0.3 |
| c4 | 0.080 | 0.050 | – | ≥ 0.2 |
| Adefovir | 0.0033 | 0.0066 | 0.0022 | 0.056 |
| Tenofovir | 0.0012 | 0.0014 | 0.0046 | 0.14 |
a50% effective concentration; b 50% cytostatic concentration
Tian et al. developed a novel library of 5,7-disubstituted pyrazolo[1,5-a]pyrimidine molecules and carried out its anti-HIV potential. From the series, compound c5: 4-(7-(mesityloxy)-4,5-dihydropyrazolo[1,5-a]pyrimidin-5-ylamino)benzonitrile was found to be the most active one (Fig. 8) with an EC50 = 0.07 µM against wild-type HIV-1 and very high selectivity index (SI, 3999) than the reference drugs (nevirapine and delavirdine) [39].
A new class of novel acyclic nucleosides in the 5-alkynyl and 6-alkylfuro[2,3-d] pyrimidines was synthesized by Amblard et al. and screened for its antiviral activity against human immunodeficiency virus (HIV), herpes simplex virus (HSV-1). Compounds, c6 and c7 exhibited moderate antiviral activity (Table 31, Fig. 8) [40].
Table 31.
Antiviral activity results (µM) of compounds c6 and c7
| Compounds | Anti-HIV-1 activity in PBMCs | HSV-1 plaque reduction assay | ||
|---|---|---|---|---|
| EC50 | EC90 | EC50 | EC90 | |
| c6 | 2.7 | 19.8 | 6.3 | 16.4 |
| c7 | 4.9 | 13.07 | 4.8 | 46.2 |
| AZTa | 0.016 | 0.20 | > 10 | > 10 |
| Acyclovira | > 100 | > 100 | 0.11 | 0.69 |
A series of pyrazole and fused pyrazolo pyrimidines was synthesized by Rashad et al. and studied for their antiviral activity against hepatitis-A virus (HAV) and herpes simplex virus type-1 (HSV-1). The substituted pyrazole and fused pyrazolopyrimidine derivatives, c8 and c9 revealed higher anti-HSV-1 activity at concentration of 10 µg/105 cells and antiviral results are compared with amantadine and acyclovir (Fig. 8) [41].
Sari et al. developed a new library of dihydropyrimidine α,γ-diketobutanoic acid molecules and screened its antiviral potential. Among the series, compound c10 ((Z)-ethyl-4-benzyl-1-(4-(3-hydroxy-4-isopropoxy-4-oxobut-2-enoyl)benzyl)-6-methyl-2-oxo-1,2-dihydro pyrimidine-5-carboxylate) was found to be most active anti-HIV agent (Table 32, Fig. 8) [42].
Table 32.
Antiviral activity results of compound c10
| Compound | EC50 (µM) |
|---|---|
| c10 | 17.2 |
| AZT | 0.0074 |
Antimalarial activity
Malaria is the most serious and widespread parasitic disease because of its prevalence, virulence and drug resistance, having an overwhelming impact on public health in developing regions of the world. Plasmodium falciparum is the main cause of severe clinical malaria and death. Endemic mapping indicates that P. falciparum and P. vivax account for 95% of the malarial infections [43]. According to a WHO report, malaria accounted for 207 million cases and an estimated 627,000 deaths worldwide in 2013 [8].
Kumar et al. synthesized a new series of 4-aminoquinoline-pyrimidine hybrids and evaluated its antimalarial potential. Several compounds showed promising in vitro antimalarial activity against both CQ sensitive and CQ-resistant strains with high selectivity index. The in vitro evaluation of these hybrids against D6 and W2 strains of P. falciparum depicted the antimalarial activity in the nanomolar range. Also, these hybrids exhibited high selectivity indices and low toxicity against the tested cell lines. Compounds (d1, d2 and d3) (Fig. 9) exhibited very potent antimalarial activity with IC50 = 0.033, 0.019 and 0.028 µM respectively which were comparable to the standard drug chloroquine (IC50 = 0.035 µM) against CQ-sensitive strain [8].
Fig. 9.
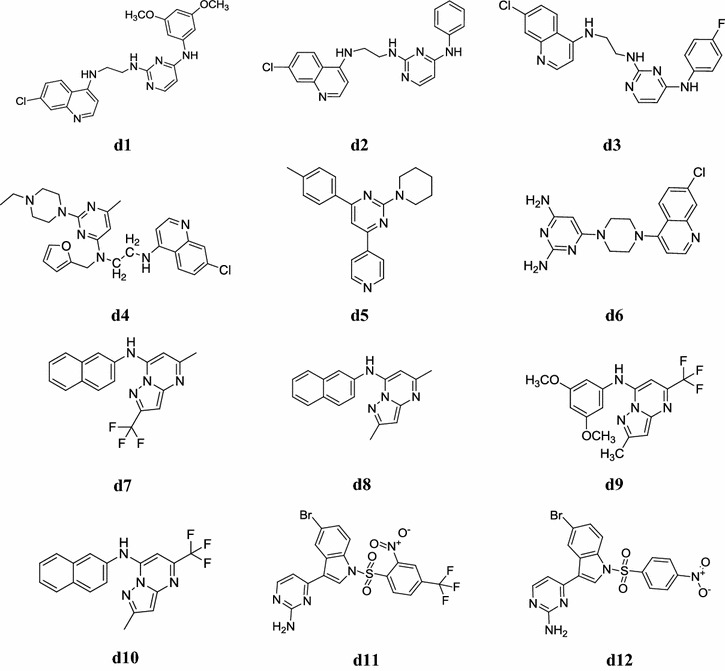
Chemical structures of the most active antimalarial pyrimidine derivatives (d1–d12)
Maurya et al. developed a new series of novel N-substituted 4-aminoquinoline-pyrimidine hybrids via simple and economic route and evaluated its antimalarial activity. Most compounds showed potent antimalarial activity against both CQ-sensitive and CQ-resistant strains with high selectivity index. All the compounds were found to be non-toxic to the mammalian cell lines. The most active compound d4 was analyzed for heme binding activity using UV spectrophotometer. Compound d4 was found to interact with heme and a complex formation between compound d4 and heme in a 1:1 stoichiometry ratio was determined using job plots. The interaction of these hybrids was also investigated by the molecular docking studies in the binding site of wild type Pf-DHFR-TS and quadruple mutant Pf-DHFR-TS (Table 33, Fig. 9) [44].
Table 33.
In vitro antimalarial activity of AQ-furfural-2-carbaldehyde-pyrimidine hybrids
| Compound | P. falciparum D6 | P. falciparum W2 | VERO cells | Resistance index | ||
|---|---|---|---|---|---|---|
| IC50 (µM) | (SI) | IC50 (µM) | (SI) | |||
| d4 | 0.038 ± 0.000 | > 263.15 | 0.040 ± 0.001 | > 250.0 | NC | 1.05 |
| Chloroquine | 0.011 ± 0.004 | > 909.09 | 0.317 ± 0.051 | > 31.54 | NC | 28.81 |
| Pyrimethamine | 0.009 ± 0.003 | > 1111.1 | NA | – | NC | – |
| Artemisinin | 0.045 ± 0.001 | > 222.22 | 0.023 ± 0.001 | 434.78 | NC | 0.511 |
Agarwal et al. developed a new series of 2,4,6-trisubstituted-pyrimidines and evaluated its in vitro antimalarial activity against Plasmodium falciparum. All the synthesized compounds showed good antimalarial activity against Plasmodium falciparum whereas, compound d5 exhibited higher antimalarial activity than pyrimethamine used as standard drug (Table 34, Fig. 9) [43].
Table 34.
Antimalarial in vitro activity against P. falciparum
| Compound | MIC (µg/ml) |
|---|---|
| d5 | 0.25 |
| Pyrimethamine | 10 |
Pretorius et al. synthesized a new library of quinoline–pyrimidine hybrids and evaluated its in vitro antimalarial activity against the D10 and Dd2 strains of Plasmodium falciparum. The compounds were all active against both strains. However, hybrid (d6, Fig. 9) featuring piperazine linker stood as the most active of all. It was found as potent as CQ and PM against the D10 strain and possessed a moderately superior potency over CQ against the Dd2 strain (IC50: 0.157 vs 0.417 µM) and also displayed activity comparable to that of the equimolar fixed combination of CQ and PM against both strains [45].
Azeredo et al. synthesized a new series of 7-aryl aminopyrazolo[1,5-a]pyrimidine derivatives with different combinations of substituent’s at positions 2-,5- and 7- of the pyrazolo[1,5-a]pyrimidine ring. The compounds were tested against Plasmodium falciparum, as antimalarials in mice with P. berghei and as inhibitors of PfDHODH. From this series, compounds, d7, d8, d9 and d10 were found to be the most active ones (Table 35, Fig. 9) [46].
Table 35.
In vitro antimalarial activity results of active compounds
| Compounds | (%) Activity PfDHODH | IC50 against PfDHODH (µM) |
|---|---|---|
| d7 | 67.474 ± 0.002 | 6 ± 1 |
| d8 | 41 ± 3 | 4 ± 1 |
| d9 | 77 ± 1 | – |
| d10 | 60 ± 3 | 0.16 ± 0.01 |
A series of N-aryl and heteroaryl sulfonamide derivatives of meridianins were prepared by Yadav et al. and screened for its antimalarial activity against D6 and W2 strains of Plasmodium falciparum. Especially, compounds, d11 and d12 displayed promising antiplasmodial activity and comparable to the standard drugs (Table 36, Fig. 9) [47].
Table 36.
In vitro antimalarial activity of N-aryl and heteroaryl sulfonamide derivatives
| Compounds | P. falciparum (IC50 in µM (µg/ml)) | |||
|---|---|---|---|---|
| P. falciparum (D6) | P. falciparum (W2) | |||
| IC50 | SI | IC50 | SI | |
| d11 | 4.86 (2.3) | > 10.8 | 6.39 (3.02) | > 8.2 |
| d12 | 2.56 (1.38) | > 18 | 3.41 (1.84) | > 13.5 |
| Artemisinin | < 0.09 (< 0.03) | – | < 0.09 (< 0.03) | – |
| Chloroquine | < 0.08 (< 0.03) | – | 0.72 (0.23) | – |
Anti-inflammatory activity
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely used therapeutics, primarily for the treatment of pain, rheumatic arthritis and various types of inflammatory conditions. However, their use is mainly restricted by their well known and serious adverse gastrointestinal side effects such as gastroduodenal erosions, ulcerations and nephrotoxicity [6].
Tozkoparan et al. synthesized a new class of 2-benzylidene-7-methyl-3-oxo-5-(substituted phenyl)-2,3-dihydro-5H-thiazolo[3,2-a]pyrimidine-6-carboxylic acid methyl esters and evaluated its anti-inflammatory activity by carrageenan induced edema test using indomethacin as reference drug. Test results revealed that compounds, e1, e2, e3, e4 exerted moderate anti-inflammatory activity at the 100 mg/kg dose level compared with indomethacin (Table 37, Fig. 10) [5].
Table 37.
Anti-inflammatory activity in percentage (%) of synthesized compounds (e1–e4)
| Compounds | Anti-inflammatory activity (%)a |
|---|---|
| e1 | 41 |
| e2 | 38 |
| e3 | 16 |
| e4 | 28 |
| Indomethacin | 32 |
a 100 mg/kg p.o. (n = 6)
Fig. 10.

Chemical structures of the most active anti-inflammatory pyrimidine derivatives (e1–e15)
Two new series of thieno[2′,3′:4,5]pyrimido[1,2-b][1,2,4]triazines and thieno[2,3-d][1,2,4]triazolo[1,5-a]pyrimidines were synthesized by Ashour et al. and evaluated for their anti-inflammatory and analgesic activity using diclofenac as reference drug. In general, the thieno[2,3-d][1,2,4]triazolo[1,5-a]pyrimidine derivatives exhibited better anti-inflammatory activity than the thieno[2′,3′5′:4,5]pyrimido[1,2-b][1,2,4]triazines. The thienotriazolo pyrimidine derivatives, e5, e6 and e7 (Fig. 10) were proved to display distinctive anti-inflammatory activity at the acute and sub acute models as well as good analgesic profile with a delayed onset of action. The anti-inflammatory screening results are presented in Tables 38 and 39 [6].
Table 38.
Anti-inflammatory activity of compounds (e5–e7) in formal in induced rat paw edema bioassay (sub-acute inflammatory model)
| Compounds | Volume of edema (ml)a | ||
|---|---|---|---|
| 0 | 1st day | 8th day | |
| e5 | 0.31 ± 0.01 | 0.51 ± 0.03b (44)c | 0.68 ± 0.02b (31) |
| e6 | 0.35 ± 0.02 | 0.54 ± 0.01b (47) | 0.67 ± 0.02b (40) |
| e7 | 0.33 ± 0.02 | 0.15 ± 0.01b (50) | 0.67 ± 0.02b (37) |
| Control | 0.32 ± 0.01 | 0.68 ± 0.01 | 0.86 ± 0.03 |
| Diclofenac | 0.32 ± 0.02 | 0.52 ± 0.02b (44) | 0.64 ± 0.02b (40) |
aValues are expressed as mean ± S.E. (Number of animals N = 5 rats)
bSignificantly different compared to corresponding control P ≤ 0.05
cBetween parentheses (percentage anti-inflammatory activity %)
Table 39.
Anti-inflammatory activity of the fused thienopyrimidines in formalin-induced rat paw edema bioassay (acute inflammatory model)
| Compounds | Volume of edema (ml)a | ED50 (mg/kg) | |||
|---|---|---|---|---|---|
| 0 | 1 h | 2 h | 4 h | ||
| e5 | 0.31 ± 0.01 | 0.44 ± 0.02b (38)c | 0.49 ± 0.01b (43) | 0.52 ± 0.02b (52) | 23.45d |
| e6 | 0.35 ± 0.02 | 0.46 ± 0.01b (47) | 0.50 ± 0.01b (53) | 0.54 ± 0.02b (56) | 28.15 |
| e7 | 0.33 ± 0.02 | 0.46 ± 0.01b (42) | 0.53 ± 0.01b (37) | 0.59 ± 0.02b (40) | 26.12 |
| Control | 0.32 ± 0.01 | 0.55 ± 0.01 | 0.64 ± 0.02 | 0.76 ± 0.01 | – |
| Diclofenac | 0.32 ± 0.02 | 0.45 ± 0.01b (38) | 0.50 ± 0.02b (43) | 0.53 ± 0.02b (52) | 25.13 |
aValues are expressed as mean ± SE (number of animals N = 5 rats)
bSignificantly different compared to corresponding control P ≤ 0.05
cBetween parentheses (percentage anti-inflammatory activity %)
dED50 is the effective dose calculated after 2 h
Yejella and Atla, synthesized a new series of 2,4,6-trisubstituted pyrimidines and screened its in vivo anti-inflammatory activity by carrageenan induced rat paw edema model. Compounds, e8: 2-amino-4-(4-aminophenyl)-6-(2,4-dichlorophenyl)pyrimidine and e9: 2-amino-4-(4-aminophenyl)-6-(3-bromophenyl)pyrimidine were found to be the most potent anti-inflammatory agents compared with ibuprofen (Table 40, Fig. 10) [48].
Table 40.
Anti-inflammatory activity of pyrimidine derivatives
| Comp. | Percent inhibition ± SEM at various time intervals | |||||
|---|---|---|---|---|---|---|
| 0.5 h | 1.0 h | 2.0 h | 3.0 h | 4.0 h | 6.0 h | |
| e8 | 15.22 ± 0.68* | 50.45 ± 1.23* | 87.23 ± 2.61* | 62.51 ± 2.33* | 56.94 ± 1.79 | 48.39 ± 2.65 |
| e9 | 18.26 ± 0.68* | 49.35 ± 1.41* | 86.99 ± 2.62* | 62.13 ± 2.25* | 53.32 ± 2.01 | 42.11 ± 2.75 |
| Ibuprofen | 20.26 ± 0.90* | 53.95 ± 0.97* | 97.09 ± 2.86* | 79.97 ± 2.38* | 67.93 ± 2.22* | 58.02 ± 1.87* |
All values are represented as mean ± SEM (n = 6). *P < 0.01 compared to saline control group. One-way ANOVA, Dunnett’s t test. Dosage: Ibuprofen-10 mg/kg and test compounds-10 mg/kg body weight by orally
Zhou et al. synthesized a new series of imidazo[1,2-a]pyrimidine derivatives and screened its anti-inflammatory potential with selective cyclooxygenase-2 (COX-2) inhibitors. In this series, compound e10 exhibited potent activity (63.8%) than ibuprofen (44.3%). The human whole blood assay still revealed that e10 (Fig. 10) has selective COX-2 inhibition (IC50 = 13 µmol/l) which is 13 times more potent than its inhibitory activity to COX-1 (IC50 = 170 µmol/l) and swollen inhibition 63.8%. The results indicated that imidazo[1,2-a] pyrimidine compounds keep moderate anti-inflammatory activity as compared to ibuprofen (standard drug) [49].
Gondkar et al. prepared a new class of substituted 1,2,3,4-tetrahydropyrimidine and screened its in vitro anti-inflammatory activity by inhibition of protein denaturation method using diclofenac (standard drug). The results revealed that almost all the tested compounds showed potent anti-inflammatory potential. All synthesized derivatives were tested their in vitro anti-inflammatory activity using inhibition of albumin denaturation technique compared to standard diclofenac. Derivatives, e11, e12, e13, e14 and e15 (Fig. 10) showed significant in vitro anti-inflammatory activity with % inhibition of albumin denaturation 98, 97, 90, 94, and 96% respectively [50].
Keche et al. developed a new series of novel 4-(3-(trifluoromethyl)phenylamino-6-(4-(3-arylureiodo/arylthioureido/arylsulfonamido)-pyrimidine derivatives by the sequential Suzuki cross coupling and screened for their anti-inflammatory activity. Among all the synthesized derivatives, compounds, e16, e17, e18, e19, e20 and e21 were found to have moderate to potent anti-inflammatory activity and compared to dexamethasone used as reference drug (Table 41, Fig. 11) [51].
Table 41.
Anti-inflammatory activity of novel pyrimidine derivatives
| Compounds | % Inhibition at 10 µM NF-α | IL-6 |
|---|---|---|
| e16 | 78 | 96 |
| e17 | 71 | 90 |
| e18 | 61 | 80 |
| e19 | 68 | 82 |
| e20 | 50 | 62 |
| Dexamethasone | 72 | 86 |
Fig. 11.

Chemical structures of the most active anti-inflammatory pyrimidine derivatives (e16–e24)
Mohamed et al. synthesized a new library of thio containing pyrrolo[2,3-d]pyrimidine derivatives and carried out its in vitro anti-inflammatory potential using the carrageenan-induced rat paw oedema assay. The potency and duration of action was compared to ibuprofen was taken as standard drug. From tested compounds, compounds e21, e22 and e23 showed best anti-inflammatory activity (Table 42, Fig. 11) [52].
Table 42.
In vivo anti-inflammatory activity
| Compounds | Oedema induced by carrageenan (% oedema % inhibition relative to control) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 h | 2 h | 3 h | 4 h | |||||
| Swel | % inh | Swel | % inh | Swel | % inh | Swel | % inh | |
| e21 | 0.206 | 10.43 | 0.101 | 61.15 | 0.142c | 73.9 | 0.132b | 79.04 |
| e22 | 0.196 | 14.78 | 0.182 | 30 | 0.022c | 95.58 | 0.282 | 67.43 |
| e23 | 0.216 | 6.08 | 0.012b | 95.38 | 0.024c | 95.95 | 0.202a | 76.82 |
| Ibuprofen | 0.216 | 6.08 | 0.14 | 45 | 0.214b | 60.66 | 0.192a | 69.52 |
As indicated: a P < 0.05; b P < 0.01; c P < 0.001
Sondhi et al. synthesized new derivatives of pyrimidine and screened their anti inflammatory activity carried out using carrageenin-induced paw oedema assay. All compounds exhibited good activity whereas, compound e24 was found to be most active one comparable to the standard drug ibuprofen (Table 43, Fig. 11) [53].
Table 43.
Anti-inflammatory of compound e24
| Compound | Dose mg/kg po | Anti-inflammatory activity % |
|---|---|---|
| e24 | 100 | 65 |
| Ibuprofen | 100 | 66.8 |
Antioxidant activity
Oxidative stress seems to play a significant role in various human diseases, including cancers. Antioxidant compounds are the agents that neutralize free radicals, which scavenge reactive oxygen species, may have potent value in preventing the onset and propagation of oxidative diseases such as neurovascular, cardiovascular diseases. Pyrimidine and its derivatives have recently attracted the attention of medicinal chemists in exploring their potential as antioxidant agents [1].
Bhalgat et al. developed a new class of novel pyrimidines and its triazole fused derivatives and investigated its in vitro antioxidant by various methods as scavenging of hydrogen peroxide, scavenging of nitric oxide radical and lipid per oxidation inhibitory activity. Compounds, f1 showed good antioxidant activity as compared to standard by scavenging of nitric oxide radical and hydrogen peroxide, while f2 showed most potent antioxidant activity by scavenging of nitric oxide (Table 44, Fig. 12) [7].
Table 44.
Antioxidant activity (IC-50 values) of compounds f1 and f2
| Compound | IC-50 (mean ± SD)a (µg/ml) | ||
|---|---|---|---|
| Scavenging of nitric oxide radical | Scavenging of hydrogen peroxide | Lipid peroxidation inhibitory activity | |
| f1 | 51 ± 0.058 | 41 ± 0.087 | 40 ± 0.121 |
| f2 | 47 ± 0.052 | 52 ± 0.279 | 43 ± 0.333 |
| Standard | 56 ± 0.087 | 38 ± 0.121 | 26 ± 0.333 |
aAverage of three determination
Fig. 12.

Chemical structures of the most active antioxidant pyrimidine derivatives (f1–f10)
Kotaiah et al. synthesized new molecules of novel 1,2,4-triazolo[3,4-b][1,3,4]thiadiazol-6-yl)selenopheno[2,3-d]pyrimidines with substituted anilines and benzoic acid. The antioxidant activity of the synthesized compounds was evaluated by DPPH, NO and H2O2 radical scavenging methods. In this series, compounds, f3, f4 and f5 showed promising antioxidant activity compared to standard drug (Table 45, Fig. 12) [54].
Table 45.
Antioxidant activity of most compounds
| Compounds | Scavenging activity (IC50 µg/ml) | ||
|---|---|---|---|
| DPPH | NO | H2O2 | |
| f3 | 11.02 ± 0.27 | 13.72 ± 1.26 | 15.38 ± 0.96 |
| f4 | 10.41 ± 0.23 | 12.74 ± 0.18 | 17.08 ± 0.12 |
| f5 | 9.46 ± 0.91 | 8.20 ± 1.60 | 12.54 ± 1.17 |
| AA | 12.27 ± 0.86 | 14.62 ± 0.97 | 15.24 ± 0.44 |
| BHT | 16.53 ± 1.74 | 19.06 ± 1.04 | 17.82 ± 0.28 |
Lower IC50 values indicate higher radical scavenging activity
AA ascorbic acid, BHT butylated hydroxy toluene
Mohana et al. reported a new series of pyrimidine derivatives and evaluated its antioxidant activity by DPPH method. The structures of all the new compounds are established on the basis of FT-IR, 1H-NMR and Mass spectral data. All the compounds showed DPPH radical scavenging activity, whereas, compounds, f6, f7 and f8 exhibited best radical scavengers due to presence of electron donating methoxy group at different position (ortho, meta and para) (Table 46, Fig. 12) [55].
Table 46.
DPPH radical scavenging activity of the tested compounds
| Compounds | Scavenging effect (%) | ||
|---|---|---|---|
| Concentration of the tested compounds (µg/ml) | |||
| 100 | 150 | 200 | |
| f6 | 51.1 | 60.8 | 68.1 |
| f7 | 35.2 | 46.3 | 52.1 |
| f8 | 32.2 | 43.4 | 54.8 |
| Ascorbic acid | 73.0 | 85.3 | 98.2 |
Quiroga et al. developed a new library of 5-aryl-4-oxo-3,4,5,8-tetrahydropyrido[2,3-d] pyrimidine-7-carboxylic acids and carried out their antioxidant activity by DPPH (1,1-diphenyl-2-picryl-hydrazyl) radical scavenging assay. Compounds f9 and f10 showed antioxidant properties and compared to standard drugs (Table 47, Fig. 12) [56].
Table 47.
Free radical scavenging (FRS50) for the tested pyrido[2,3-d]pyrimidines (f9 and f10)
| Compounds | FRS50 (µg/ml) | |
|---|---|---|
| Mean | %RSD | |
| f9 | 367 | 10 |
| f10 | 472 | 10 |
| Asc. acid | 1.1 | 12 |
| Quercetin | 3.4 | 7 |
Antileishmanial activity
Leishmaniasis, a vector-borne parasitic disease, is a major cause of concern in developing countries. The disease is caused by more than 20 species of protozoan Leishmania and transmitted by the bite of female phlebotomine sand flies. Leishmaniasis has traditionally been classified into three major clinical forms: visceral leishmaniasis (VL), cutaneous leishmaniasis (CL) and mucocutaneous leishmaniasis (MCL) which differs in immunopathologies and degree of morbidity and mortality. VL caused by Leishmania donovani is the most severe form of leishmaniasis and is usually fatal in the absence of treatment. Most of the first line drugs available for the treatment of leishmaniasis such as sodium stibogluconate, meglumine antimoniate, pentamidine etc. cause serious side effects and toxicity [57].
A new series of substituted aryl pyrimidine derivatives was synthesized by Suryawanshi et al. and evaluated for its in vitro antileishmanial potential against intracellular amastigotes of Leishmania donovani using reporter gene luciferase assay. All synthesized compounds showed promising IC50 values ranging from 0.5 to 12.9 µM. Selectivity indices (S.I.) of all these compounds are far better than sodium stibogluconate (SSG) and miltefosine used as standard drugs. On the basis of good selectivity indices compounds were further screened their in vivo antileishmanial activity against L. donovani/hamster model. Compounds, g1, g2 and g3 showed good inhibition (Table 48, Fig. 13) of parasitic multiplication that is 88.4, 78.1 and 78.2%, respectively at a daily dose of 50 mg/kg × 5 days, when administered intraperitoneally [57].
Table 48.
In vitro and in vivo antileishmanial activity and cytotoxicity results of synthetic pyrimidine derivatives
| Compounds | In vitro assessment | Selectivity index CC50/IC50 |
In vivo activity (dose—50 mg/kg × 5 days, ipb) % Inhibition ± SD |
|
|---|---|---|---|---|
| IC50 (µM) | CC50 (µM) | |||
| g1 | 2.0 ± 0.1 | 375.9 ± 5.1 | 188 | 88.4 ± 10.6 |
| g2 | 0.5 ± 0.1 | 57.8 ± 5.9 | 116 | 78.1 ± 17.7 |
| g3 | 2.7 ± 0.5 | 345.4 ± 19.6 | 128 | 78.2 ± 4.4 |
| SSGa | 59.8 ± 7.5 | > 400 ± 0 | > 7 | 88.5 ± 4.4 |
| Miltefosinec | 12.5 ± 0.9 | 54.7 ± 6.9 | 4 | 98.1 ± 1.0 |
IC50 and CC50 values are the mean ± SD of two independent experiments
The selectivity index is defined as the ratio of CC50 on vero cells to IC50 on L. donovani intramacrophagic amastigotes
aSSG = sodium stibogluconate (40 mg/kg × 5 days, ip)
bip = intraperitonial; c Miltefosine (30 mg/kg × 5 days, po) used as a reference drugs
Fig. 13.

Chemical structures of the most active antileishmanial pyrimidine derivatives (g1–g7)
Pandey et al. synthesized some novel terpenyl pyrimidine from α/β-ionone keteneacetals and screened their in vivo leishmanicidal activity against amastigote stage of Leishmania donovani was determined in Golden hamsters (Mesocricotus aurctus) infected with HOM/IN/80/DD8 strain of L. donovani. The compounds, g4, g5, g6 and g7 showed promising in vivo antileishmanial activity (Table 49, Fig. 13) [58].
Table 49.
Antileishmanial activity of compounds against amastigotes of Leishmania donovani in hamsters
| Compounds | Dose (mg/kg) | In vivo inhibition (%) | |
|---|---|---|---|
| Day-7 | Day-28 | ||
| g4 | 50 | 66 | – |
| g5 | 50 | 22 | 63 |
| g6 | 50 | 64 | – |
| g7 | 50 | 64 | – |
Miscellaneous activities
A new series of strobilurin-pyrimidine derivatives was synthesized by Chai et al. The synthesized compounds were evaluated for their acaricidal activity. Preliminary bioassays demonstrated that compounds, h1 and h2 exhibited significant control against Tetranychus cinnabarinus (Boisd.) at 0.625 mg/l, and their acaricidal potencies were higher than pyriminostrobin in a green house. Compounds, h1 and h2 (Fig. 14) were chosen as candidates for extensive greenhouse bioassays on larvae and eggs of T. cinnabarinus. Both of them showed potency consistent with pyriminostrobin against larvae and weaker potency than pyriminostrobin against eggs, as shown in Table 50 [59].
Fig. 14.

Chemical structures of the most active pyrimidine derivatives (h1–h15)
Table 50.
Acaricidal activity of h1 and h2 against T. cinnabarinus
| Compounds | T. cinnabarinus | (% mortality at given concentration mg/l) | ||
|---|---|---|---|---|
| 10 | 25 | 0.625 | ||
| h1 | Larvae | 100 | 98 | 77 |
| Eggs | 100 | 70 | 25 | |
| h2 | Larvae | 100 | 100 | 100 |
| Eggs | 75 | 20 | 10 | |
| Pyriminostrobin | Larvae | 100 | 100 | 96 |
| Eggs | 100 | 100 | 20 | |
Amin et al. synthesized a new series of novel coumarin–pyrimidine hybrids and evaluated its vasorelaxant activity against nor-adrenaline-induced spasm on thoracic rat aorta rings and compared to prazocin (reference drug). From the series, compounds, h3: (6-(4,6-dimethylpyrimidin-2-ylamino)-2H-chromen-2-one) and h4: (6-(diethylamino)-5-isocyano-2-(2-oxo-2H-chromen-6-ylamino)pyrimidin-4(3H)-one) were found to be most prospective vasorelaxant agent with IC50 = 0.411 and IC50 = 0.421 mM respectively when compared with reference drug prazocin (IC50 = 0.487 mM). The chemical structure depicted in Fig. 14 [60].
Duan et al. designd and synthesized a new series of S(−)-2-(4-chlorophenyl)-N-(5,7-di- substituted-2H-[1,2,4]-thiadiazolo[2,3-a]pyrimidin-2-ylidene)-3-methylbutanamide derivatives. The synthesized compounds were evaluated for their herbicidal activity against three monocotyledon weeds and two dicotyledon weeds i.e. Echinochloa crusgallis L., Sorghum bicolort, Digitaria sanguinalis (L.) scop Chenopodium serotinum (L.) and Amaranthus retroflexus L., respectively. Compounds h5 and h6 showed the highest inhibitory activity against root and stalk of Amaranthus retroflexus L. in higher concentration (1.0 × 10−4 µg/ml), while compounds h7 and h8 showed good activity against root of Echinochloa crusgallis L. and stalk of Chenopodium serotinum L., respectively (Table 51, Fig. 14). The chiral target compounds showed improved herbicidal activity to some extent over their racemic counterparts against a variety of tested weeds, which might be contributed by the introduction of chiral active unit [61].
Table 51.
The inhibition percentage of the target compounds against various weeds
| Compounds | Concentration (ppm) | Echinochloa crusgallis L. | Chenopodium serotinum L. | Amaranthus retroflexus L. | |||
|---|---|---|---|---|---|---|---|
| Stalk | Root | Stalk | Root | Stalk | Root | ||
| h5 | 50 | 50 | 30 | 50 | 50 | 50 | 50 |
| 100 | 80 | 30 | 80 | 85 | 95 | 100 | |
| h6 | 50 | 10 | 70 | 80 | 80 | 90 | 85 |
| 100 | 30 | 90 | 85 | 90 | 100 | 100 | |
| h7 | 50 | 60 | 80 | 10 | 10 | 0 | 0 |
| 100 | 70 | 100 | 20 | 10 | 20 | 30 | |
| h8 | 50 | 30 | 70 | 70 | 50 | 60 | 50 |
| 100 | 40 | 80 | 100 | 80 | 95 | 90 | |
Katiyar et al. developed a new series of trisubstituted pyrimidine derivatives and evaluated its in vitro topoisomerase II inhibitory activity against filarial parasite Setaria cervi. Compounds (h9–h15) have shown 60–80% inhibition at 40 and 20 µg/ml concentrations. Structure activity relationship of most active compounds have given clear indication that amino group and 4-aminophenyl group at position-2 are very crucial in exerting topoisomerase II inhibitory activity against filarial parasite Setaria cervi than standard antifilarial drug (DEC) and enzyme topoisomerase II inhibitors (novobiocin, nalidixic acid) (Table 52, Fig. 14) [62].
Table 52.
Topoisomerase II inhibitory activity against filarial parasite Setaria cervi
| Compounds | % Inhibition at different concentrations | |||
|---|---|---|---|---|
| 40 µg/ml | 20 µg/ml | 10 µg/ml | 5 µg/ml | |
| h9 | 60 | 60 | – | – |
| h10 | 60 | 60 | – | – |
| h11 | 80 | 80 | 80 | – |
| h12 | 80 | 80 | 80 | 60 |
| h13 | 80 | 80 | 80 | 60 |
| h14 | 80 | 80 | 80 | 25 |
| h15 | 70 | 70 | 70 | 40 |
| DEC (antifilarial) | 45 | 10 | – | – |
| Novobiocin (topo II inhibitor) | 80 | 20 | 10 | – |
| Nalidixic acid (topo II inhibitor) | 80 | 40 | 20 | – |
A new class of 2,4,6-trisubstituted bis-pyrimidines was synthesized by Parveen et al. and screened for its in vitro antiamoebic activity against HM1:IMSS strain of Entamoeba histolytica and toxicological studies on PC12-rat pheochoromocytoma cell line.
Bis-pyrimidine having methyl-substituent exhibited higher antiamoebic activity than the reference drug metronidazole (IC50 = 1.9 µM). Compound h16: 1,3-bis(2-(piperidin-1-yl)-6-(p-tolyl)pyrimidin-4-yl)benzene was found most active (IC50 = 0.10 µM) and least toxic among all the synthesized compounds (Table 53, Fig. 15) [63].
Table 53.
Antiamoebic activity and toxicity profile of compound h16
| Compound | Antiamoebic activity | Toxicity profile | ||
|---|---|---|---|---|
| (IC50 = µM) | SD (±) | (IC50 = µM) | Safety index | |
| h16 | 0.10 | 0.014 | > 100 | > 1000 |
| Metronidazole | 9 | 0.020 | > 100 | > 52.63 |
SD standard deviation
Fig. 15.
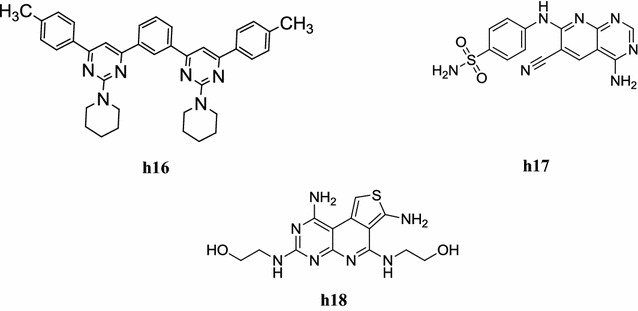
Chemical structures of the most active pyrimidine derivatives (h16–h18)
A new class of pyrido[2,3-d]pyrimidine derivatives was designed and synthesized by Ibrahim and Ismail. The pyrido[2,3-d]pyrimidine derivatives were evaluated for their in vitro anti-proliferative activity against A431a, SNU638b, HCT116 and inhibition of CDK2-Cyclin A, CDK4/Cyclin D and EGFR enzyme. In this class, the anti-proliferative and CDK2-Cyclin A inhibitory activity of compounds, h17 and h18 (Fig. 15) was significantly more active than roscovotine (as standard drug) with IC50 values of 0.3 and 0.09 µM respectively [64].
Conclusion
In conclusion, the biological potentials i.e. antimicrobial, anticancer, antiviral, anti-inflammatory, analgesic, antioxidant and antimalarial of pyrimidine derivatives are summarized. Pyrimidine is the important heterocyclic compound as they are being an essential constituent of cells and large number of marketed drugs. The biological activities of the pyrimidine derivatives indicated the maneuverability and versatility, which offer the medicinal chemist a continued interest in the pyrimidine skeleton in medicinal field.
Authors’ contributions
Authors BN and SK have designed and prepared the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Present in manuscript.
Ethics approval and consent to participate
Not applicable.
Funding
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sanjiv Kumar, Email: sanjiv.pharmchem@gmail.com.
Balasubramanian Narasimhan, Email: naru2000us@yahoo.com.
References
- 1.Rani J, Kumar S, Saini M, Mundlia J, Verma PK. Biological potential of pyrimidine derivatives in a new era. Res Chem Intermed. 2016;42:6777–6804. doi: 10.1007/s11164-016-2525-8. [DOI] [Google Scholar]
- 2.Cocco MT, Congiu C, Onnis V, Piras R. Synthesis and antitumor evaluation of 6-thioxo-, 6-oxo- and 2,4-dioxopyrimidine derivatives. Farmaco. 2001;56:741–748. doi: 10.1016/S0014-827X(01)01123-5. [DOI] [PubMed] [Google Scholar]
- 3.Meneghesso S, Vanderlinden E, Stevaert A, McGuigan C, Balzarini J, Naesens L. Synthesis and biological evaluation of pyrimidine nucleoside monophosphate prodrugs targeted against influenza virus. Antivir Res. 2012;94:35–43. doi: 10.1016/j.antiviral.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Anupama B, Dinda SC, Prasad YR, Rao AV. Synthesis and antimicrobial activity of some new 2,4,6-trisubstituted pyrimidines. Int J Res Pharm Chem. 2012;2(2):231–236. [Google Scholar]
- 5.Tozkoparan B, Ertan M, Kelicen P, Demirdamar R. Synthesis and anti-inflammatory activities of some thiazolo[3,2-a]pyrimidine derivatives. Farmaco. 1999;54:588–593. doi: 10.1016/S0014-827X(99)00068-3. [DOI] [PubMed] [Google Scholar]
- 6.Ashour HM, Shaaban OG, Rizk OH, El-Ashmawy IM. Synthesis and biological evaluation of thieno [2′,3′:4,5]pyrimido[1,2-b][1,2,4]triazines and thieno[2,3-d] [1,2,4]triazolo[1,5-a]pyrimidines as anti-inflammatory and analgesic agents. Eur J Med Chem. 2013;62:341–351. doi: 10.1016/j.ejmech.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Bhalgat CM, Ali MI, Ramesh B, Ramu G. Novel pyrimidine and its triazole fused derivatives: synthesis and investigation of antioxidant and anti-inflammatory activity. Arab J Chem. 2014;7:986–993. doi: 10.1016/j.arabjc.2010.12.021. [DOI] [Google Scholar]
- 8.Kumar D, Khan SI, Tekwani BL, Diwan PP, Rawat S. 4-Aminoquinoline–pyrimidine hybrids: synthesis, antimalarial activity, heme binding and docking studies. Eur J Med Chem. 2015;89:490–502. doi: 10.1016/j.ejmech.2014.10.061. [DOI] [PubMed] [Google Scholar]
- 9.Mallikarjunaswamy C, Mallesha L, Bhadregowda DG, Pinto O. Studies on synthesis of pyrimidine derivatives and their antimicrobial activity. Arab J Chem. 2014;7:986–993. doi: 10.1016/j.arabjc.2010.12.021. [DOI] [Google Scholar]
- 10.Chen PJ, Yang A, Gu YF, Zhang XS, Shao KP, Xue DQ, He P, Jiang TF, Zhang QR, Liu HM. Synthesis, in vitro antimicrobial and cytotoxic activities of novel pyrimidine–benzimidazol combinations. Bioorg Med Chem Lett. 2014;24:2741–2743. doi: 10.1016/j.bmcl.2014.04.037. [DOI] [PubMed] [Google Scholar]
- 11.El-Gaby MSA, Gaber AM, Atalla AA, Al-Wahab KAA. Novel synthesis and antifungal activity of pyrrole and pyrrolo[2,3-d]pyrimidine derivatives containing sulfonamide moieties. Farmaco. 2002;57:613–617. doi: 10.1016/S0014-827X(01)01178-8. [DOI] [PubMed] [Google Scholar]
- 12.Hilmy KMH, Khalifa MMA, Hawata MAA, Keshk RMA, El-Torgman AA. Synthesis of new pyrrolo[2,3-d]pyrimidine derivatives as antibacterial and antifungal agents. Eur J Med Chem. 2010;45:5243–5250. doi: 10.1016/j.ejmech.2010.08.043. [DOI] [PubMed] [Google Scholar]
- 13.Holla BS, Mahalinga M, Karthikeyan MS, Akberalib PM, Shettyc NS. Synthesis of some novel pyrazolo[3,4-d]pyrimidine derivatives as potential antimicrobial agents. Bioorg Med Chem. 2006;14:2040–2047. doi: 10.1016/j.bmc.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 14.Chen Q, Zhu X-L, Jiang L-L, Liu Z-M, Yang G-F. Synthesis, antifungal activity and CoMFA analysis of novel 1,2,4-triazolo[1,5-a]pyrimidine derivatives. Eur J Med Chem. 2008;43:595–603. doi: 10.1016/j.ejmech.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 15.Maddila S, Gorle S, Seshadri N, Lavanya P, Jonnalagadda SB. Synthesis, antibacterial and antifungal activity of novel benzothiazole pyrimidine derivatives. Arab J Chem. 2016;9:681–687. doi: 10.1016/j.arabjc.2013.04.003. [DOI] [Google Scholar]
- 16.Fellahil Y, Duboisz P, Mandin D, Ombetta-Goka JE, Guenzet J, Chaumont JP, Frangin Y. Synthesis and antibacterial activity of 2-substituted 5-(1,2-diarylethyl)-4,6-dichloropyrimidine derivatives. Eur J Med Chem. 1995;30:633–639. doi: 10.1016/0223-5234(96)88279-1. [DOI] [Google Scholar]
- 17.Nagender P, Reddy GM, Kumar RN, Poornachandra Y, Kumar CG, Narsaiah B. Synthesis, cytotoxicity, antimicrobial and anti-biofilm activities of novel pyrazolo[3,4-b] pyridine and pyrimidine functionalized 1,2,3-triazole derivatives. Bioorg Med Chem Lett. 2014;24:2905–2908. doi: 10.1016/j.bmcl.2014.04.084. [DOI] [PubMed] [Google Scholar]
- 18.Patel YM, Mehta KM, Patel KC. Studies on synthesis characterization and antimicrobial activity of pyrimidine based derivatives. Int J Chemtech Res. 2011;3(4):1734–1739. [Google Scholar]
- 19.Rostamizadeh S, Nojavan M, Aryan R, Sadeghian H, Davoodnejad M. A novel and efficient synthesis of pyrazolo[3,4-d]pyrimidine derivatives and the study of their anti-bacterial activity. Chin Chem Lett. 2013;24:629–632. doi: 10.1016/j.cclet.2013.04.035. [DOI] [Google Scholar]
- 20.Sriharsha SN, Satish S, Shashikanth S, Raveesha KA. Design, synthesis and antibacterial activity of novel 1,3-thiazolidine pyrimidine nucleoside analogues. Bioorg Med Chem. 2006;14:7476–7481. doi: 10.1016/j.bmc.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Shao H, Shi S, Foley DW, Lam F, Abbas AY, Liu X, Huang S, Jiang X, Baharin N, Fischer PM, Wang S. Synthesis, structure-activity relationship and biological evaluation of 2,4,5-trisubstituted pyrimidine CDK inhibitors as potential anti-tumour agents. Eur J Med Chem. 2013;70:447–455. doi: 10.1016/j.ejmech.2013.08.052. [DOI] [PubMed] [Google Scholar]
- 22.El-Sayed NS, El-Bendary ER, Saadia M, El-Ashry SM, El-Kerdawy MM. Synthesis and antitumor activity of new sulfonamide derivatives of thiadiazole [3,2-a] pyrimidines. Eur J Med Chem. 2011;46:3714–3720. doi: 10.1016/j.ejmech.2011.05.037. [DOI] [PubMed] [Google Scholar]
- 23.Fares M, Abou-Seri SM, Abdel-Aziz HA, Abbas SES, Youssef MM, Eladwy RA. Synthesis and antitumor activity of pyrido[2,3-d]pyrimidine and pyrido[2,3-d] [1,2,4]triazolo[4,3-a]pyrimidine derivatives that induce apoptosis through G1 cell-cycle arrest. Eur J Med Chem. 2014;83:155–166. doi: 10.1016/j.ejmech.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 24.Hu Y-G, Wang Y, Du S-M, Chen X-B, Ding M-W. Efficient synthesis and biological evaluation of some 2,4-diamino-furo[2,3-d]pyrimidine derivatives. Bioorg Med Chem Lett. 2010;20:6188–6190. doi: 10.1016/j.bmcl.2010.08.122. [DOI] [PubMed] [Google Scholar]
- 25.Huang YY, Wang LY, Chang CH, Kuo YH, Kaneko K, Takayama H, Kimura M, Juang SH, Wong FF. One-pot synthesis and antiproliferative evaluation of pyrazolo[3,4-d]pyrimidine derivatives. Tetrahedron. 2012;68:9658–9664. doi: 10.1016/j.tet.2012.09.054. [DOI] [Google Scholar]
- 26.Song XJ, Shao Y, Gao X. Microwave-assisted synthesis of some novel fluorinated pyrazolo[3,4-d]pyrimidine derivatives containing 1,3,4-thiadiazole as potential antitumor agents. Chin Chem Lett. 2011;22:1036–1038. doi: 10.1016/j.cclet.2011.05.012. [DOI] [Google Scholar]
- 27.Tangeda SJ, Garlapati A. Synthesis of new pyrrolo[2,3-d]pyrimidine derivatives and evaluation of their activities against human colon cancer cell lines. Eur J Med Chem. 2010;45:1453–1458. doi: 10.1016/j.ejmech.2009.12.050. [DOI] [PubMed] [Google Scholar]
- 28.Kurumurthy C, Rao PS, Swamy BV, Kumar GS, Rao PS, Narsaiah B, Velatooru LR, Pamanji R, Rao JV. Synthesis of novel alkyltriazole tagged pyrido[2,3-d] pyrimidine derivatives and their anticancer activity. Eur J Med Chem. 2011;46:3462–3468. doi: 10.1016/j.ejmech.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 29.Liu Z, Wang Y, Lin H, Zuo D, Wang L, Zhao Y, Gong P. Design, synthesis and biological evaluation of novel thieno[3,2-d]pyrimidine derivatives containing diaryl urea moiety as potent antitumor agents. Eur J Med Chem. 2014;85:215–227. doi: 10.1016/j.ejmech.2014.07.099. [DOI] [PubMed] [Google Scholar]
- 30.Zhu WF, Zhai X, Li S, Cao YY, Gong P, Liu YJ. Synthesis and cytotoxic activity of novel 2,6-disubstituted-4-morpholinothieno[3,2-d]pyrimidines as potent anti-tumor agents. Chin Chem Lett. 2012;23:703–706. doi: 10.1016/j.cclet.2012.04.012. [DOI] [Google Scholar]
- 31.Xie F, Zhao H, Zhao L, Lou L, Hu Y. Synthesis and biological evaluation of novel 2,4,5-substituted pyrimidine derivatives for anticancer activity. Bioorg Med Chem Lett. 2009;19:275–278. doi: 10.1016/j.bmcl.2008.09.067. [DOI] [PubMed] [Google Scholar]
- 32.Al-Issa SA. Synthesis and anticancer activity of some fused pyrimidines and related heterocycles. Saudi Pharm J. 2013;21:305–316. doi: 10.1016/j.jsps.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohareb RM, Abbas NS, Abdelaziz MA. Heterocyclic ring extension of androstenedione: synthesis and cytotoxicity of fused pyran, pyrimidine and thiazole derivatives. Steroids. 2014;86:45–55. doi: 10.1016/j.steroids.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Kumar RN, Dev GJ, Ravikumar N, Swaroop DK, Debanjan B, Bharath G, Narsaiah B, Jain SN, Rao AG. Synthesis of novel triazole/isoxazole functionalized-(trifluoromethyl)pyrido[2,3-d]pyrimidine derivatives as promising anticancer and antibacterial agents. Bioorg Med Chem Lett. 2016;26:2927–2930. doi: 10.1016/j.bmcl.2016.04.038. [DOI] [PubMed] [Google Scholar]
- 35.Liu Z, Wu S, Wang Y, Li R, Wang J, Wang L, Zhao Y, Gong P. Design, synthesis and biological evaluation of novel thieno[3,2-d]pyrimidine derivatives possessing diaryl semicarbazone scaffolds as potent antitumor agents. Eur J Med Chem. 2014;87:782–793. doi: 10.1016/j.ejmech.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 36.Lv N, Sun M, Liu C, Li J. Design and synthesis of 2-phenylpyrimidine coumarin derivatives as anticancer agents. Bioorg Med Chem Lett. 2017;27:4578–4581. doi: 10.1016/j.bmcl.2017.08.044. [DOI] [PubMed] [Google Scholar]
- 37.Xu X, Wang J, Yao Q. Synthesis and quantitative structure–activity relationship (QSAR) analysis of some novel oxadiazolo[3,4-d]pyrimidine nucleosides derivatives as antiviral agents. Bioorg Med Chem Lett. 2015;25:241–244. doi: 10.1016/j.bmcl.2014.11.065. [DOI] [PubMed] [Google Scholar]
- 38.Hockov D, Holy A, Masojidkov M, Andrei G, Snoeck R, Clercq ED, Balzariniv J. Synthesis and antiviral activity of 2,4-diamino-5-cyano-6-[2-(phosphonomethoxy) ethoxy]pyrimidine and related compounds. Bioorg Med Chem. 2004;12:3197–3202. doi: 10.1016/j.bmc.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Tian Y, Du D, Rai D, Wang L, Liu H, Zhan P, Clercq ED, Pannecouque C, Liu X. Fused heterocyclic compounds bearing bridgehead nitrogen as potent HIV-1 NNRTIs. Part 1: design, synthesis and biological evaluation of novel 5,7-disubstituted pyrazolo[1,5-a]pyrimidine derivatives. Bioorg Med Chem. 2014;22:2052–2059. doi: 10.1016/j.bmc.2014.02.029. [DOI] [PubMed] [Google Scholar]
- 40.Amblard F, Aucagne V, Guenot P, Schinazi RF, Agrofoglio LA. Synthesis and antiviral activity of novel acyclic nucleosides in the 5-alkynyl- and 6-alkylfuro[2,3-d] pyrimidine series. Bioorg Med Chem. 2005;13:1239–1248. doi: 10.1016/j.bmc.2004.11.057. [DOI] [PubMed] [Google Scholar]
- 41.Rashad AE, Hegab MI, Abdel-Megeid RE, Micky JA, Abdel-Megeid FME. Synthesis and antiviral evaluation of some new pyrazole and fused pyrazolopyrimidine derivatives. Bioorg Med Chem. 2008;16:7102–7106. doi: 10.1016/j.bmc.2008.06.054. [DOI] [PubMed] [Google Scholar]
- 42.Sari O, Roy V, Metifiot M, Marchand C, Pommier Y, Bourg S, Bonnet P, Schinazi RF, Agrofoglio L. Synthesis of dihydropyrimidine α, γ-diketobutanoic acid derivatives targeting HIV integrase. Eur J Med Chem. 2015;104:127–138. doi: 10.1016/j.ejmech.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agarwal A, Srivastava K, Puri SK, Chauhan PMS. Synthesis of 2,4,6-trisubstituted pyrimidines as antimalarial agents. Bioorg Med Chem. 2005;13:4645–4650. doi: 10.1016/j.bmc.2005.04.061. [DOI] [PubMed] [Google Scholar]
- 44.Maurya SS, Khan SI, Bahuguna A, Kumar D, Rawat DS. Synthesis, antimalarial activity, heme binding and docking studies of N-substituted 4-aminoquinoline–pyrimidine molecular hybrids. Eur J Med Chem. 2017;129:175–185. doi: 10.1016/j.ejmech.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 45.Pretorius SI, Breytenbach WJ, de Kock C, Smith PJ, N’Da DD. Synthesis, characterization and antimalarial activity of quinoline–pyrimidine hybrids. Bioorg Med Chem. 2013;21:269–277. doi: 10.1016/j.bmc.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 46.Azeredo LFSP, Coutinho JP, Jabor VAP, Feliciano PR, Nonato MC, Kaiser CR, Menezes CMS, Hammes ASO, Caffarena ER, Hoelz LVB, de Souza NB, Pereira GAN, Ceravolo IP, Krettli AU, Boechat N. Evaluation of 7-arylaminopyrazolo[1,5-a]pyrimidines as anti-Plasmodium falciparum, antimalarial and Pf-dihydroorotate dehydrogenase inhibitors. Eur J Med Chem. 2017;126:72–83. doi: 10.1016/j.ejmech.2016.09.073. [DOI] [PubMed] [Google Scholar]
- 47.Yadav RR, Khan SI, Singh S, Khan IA, Vishwakarma RA, Bharate SB. Synthesis, antimalarial and antitubercular activities of meridianin derivatives. Eur J Med Chem. 2015;98:160–169. doi: 10.1016/j.ejmech.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 48.Yejella RP, Atla SR. A study of anti-inflammatory and analgesic activity of new 2,4,6-trisubstituted pyrimidines. Chem Pharm Bull. 2011;59(9):1079–1082. doi: 10.1248/cpb.59.1079. [DOI] [PubMed] [Google Scholar]
- 49.Zhou JP, Ding YW, Zhang HB, Xu L, Dai Y. Synthesis and anti-inflammatory activity of imidazo[1,2-a]pyrimidine derivatives. Chin Chem Lett. 2008;19:669–672. doi: 10.1016/j.cclet.2008.04.020. [DOI] [Google Scholar]
- 50.Gondkar AS, Deshmukh VK, Chaudhari SR. Synthesis, characterization and in vitro anti-inflammatory activity of some substituted 1,2,3,4-tetrahydropyrimidine derivatives. Drug Invent Today. 2013;5:175–181. doi: 10.1016/j.dit.2013.04.004. [DOI] [Google Scholar]
- 51.Keche AP, Hatnapure GD, Tale RH, Rodge AH, Birajdar SS, Kamble VM. A novel pyrimidine derivatives with aryl urea, thiourea and sulfonamide moieties: synthesis, anti-inflammatory and antimicrobial evaluation. Bioorg Med Chem Lett. 2012;22:3445–3448. doi: 10.1016/j.bmcl.2012.03.092. [DOI] [PubMed] [Google Scholar]
- 52.Mohamed MS, Kamel R, Fatahala SS. Synthesis and biological evaluation of some thio containing pyrrolo[2,3-d]pyrimidine derivatives for their anti-inflammatory and anti-microbial activities. Eur J Med Chem. 2010;45:2994–3004. doi: 10.1016/j.ejmech.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 53.Sondhi SM, Jain S, Dinodia M, Shukla R, Raghubir R. One pot synthesis of pyrimidine and bispyrimidine derivatives and their evaluation for anti-inflammatory and analgesic activities. Bioorg Med Chem. 2007;15:3334–3344. doi: 10.1016/j.bmc.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 54.Kotaiah Y, Nagaraju K, Harikrishna N, Rao CV, Yamini L, Vijjulatha M. Synthesis, docking and evaluation of antioxidant and antimicrobial activities of novel 1,2,4-triazolo[3,4-b][1,3,4]thiadiazol-6-yl)selenopheno[2,3-d]pyrimidines. Eur J Med Chem. 2014;75:195–202. doi: 10.1016/j.ejmech.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 55.Mohana KN, Kumar BNP, Mallesha L. Synthesis and biological activity of some pyrimidine derivatives. Drug Invent Today. 2013;5:216–222. doi: 10.1016/j.dit.2013.08.004. [DOI] [Google Scholar]
- 56.Quiroga J, Romo PE, Ortiz A, Isaza JH, Insuasty B, Abonia R, Nogueras M, Cobo J. Synthesis, structures, electrochemical studies and antioxidant activity of 5-aryl-4-oxo-3,4,5,8-tetrahydropyrido[2,3-d]pyrimidine-7-carboxylic acids. J Biomol Struct. 2016;1120:294–301. [Google Scholar]
- 57.Suryawanshi SN, Kumar S, Shivahare R, Pandey S, Gupta S, Tiwari A. Design, synthesis and biological evaluation of aryl pyrimidine derivatives as potential leishmanicidal agents. Bioorg Med Chem Lett. 2013;23:5235–5238. doi: 10.1016/j.bmcl.2013.06.060. [DOI] [PubMed] [Google Scholar]
- 58.Pandey S, Suryawanshi SN, Gupta S, Srivastava VML. Synthesis and antileishmanial profile of some novel terpenyl pyrimidines. Eur J Med Chem. 2004;39:969–973. doi: 10.1016/j.ejmech.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 59.Chai B-S, Liu C-L, Li H-C, Liu S-W, Xu Y, Song Y-Q, Chang J-B. Synthesis and acaricidal activity of strobilurin-pyrimidine derivatives. Chin Chem Lett. 2014;25:137–140. doi: 10.1016/j.cclet.2013.10.006. [DOI] [Google Scholar]
- 60.Amin KM, Awadalla FM, Eissa AAM, Abou-Seri AM, Hassan GS. Design, synthesis and vasorelaxant evaluation of novel coumarin–pyrimidine hybrids. Bioorg Med Chem. 2011;19:6087–6097. doi: 10.1016/j.bmc.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 61.Duan LP, Zhao QF, Zhang HB. Novel 2H-[1,2,4]thiadiazolo[2,3-a]pyrimidine derivatives bearing chiral S(−)-2-(4-chlorophenyl)-3-methylbutyric acid moiety: design, synthesis and herbicidal activity. Arab J Chem. 2010;3:225–228. doi: 10.1016/j.arabjc.2010.06.004. [DOI] [Google Scholar]
- 62.Katiyar SB, Bansal I, Saxena JK, Chauhan PMS. Syntheses of 2,4,6-trisubstituted pyrimidine derivatives as a new class of antifilarial topoisomerase II inhibitors. Bioorg Med Chem Lett. 2005;15:47–50. doi: 10.1016/j.bmcl.2004.10.046. [DOI] [PubMed] [Google Scholar]
- 63.Parveen H, Hayat F, Mukhtar S, Salahuddin A, Khan A, Islam F, Azam A. Synthesis, characterization and biological evaluation of novel 2,4,6-trisubstituted bis-pyrimidine derivatives. Eur J Med Chem. 2011;46:4669–4675. doi: 10.1016/j.ejmech.2011.05.055. [DOI] [PubMed] [Google Scholar]
- 64.Ibrahim DA, Ismail NSM. Design, synthesis and biological study of novel pyrido[2,3-d]pyrimidine as anti-proliferative CDK2 inhibitors. Eur J Med Chem. 2011;46:5825–5832. doi: 10.1016/j.ejmech.2011.09.041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Present in manuscript.