

Abstract
Maternal smoking is reported to cause adverse effects on the health of the unborn child, the underlying mechanism for which is thought to involve alterations in DNA methylation. We examined the effects of maternal smoking on DNA methylation in cord blood, in 247 mother–infant pairs in the Sapporo cohort of the Hokkaido Study, using the Infinium HumanMethylation 450K BeadChip. We first identified differentially methylated CpG sites with a false discovery rate (FDR) of <0.05 and the magnitude of DNA methylation changes (|β| >0.02) from the pairwise comparisons of never-smokers (Ne-S), sustained-smokers (Su-S), and stopped-smokers (St-S). Subsequently, secondary comparisons between St-S and Su-S revealed nine common sites that mapped to ACSM3, AHRR, CYP1A1, GFI1, SHANK2, TRIM36, and the intergenic region between ANKRD9 and RCOR1 in Ne-S vs. Su-S, and one common CpG site mapping to EVC2 in Ne-S vs. St-S. Further, we verified these CpG sites and examined neighbouring sites using bisulfite next-generation sequencing, except for AHRR cg21161138. These changes in DNA methylation implicate the effect of smoking cessation. Our findings add to the current knowledge of the association between DNA methylation and maternal smoking and suggest future studies for clarifying this relationship in disease development.
Introduction
The concept of Developmental Origins of Health and Disease (DOHaD) suggests that unfavourable changes in the environment during prenatal and early postnatal periods give rise to health issues later in life, emphasising the importance of environmental exposure during early development. In light of this notion, the mother’s lifestyle, particularly smoking, and its adverse effects on the child’s health have drawn considerable attention. Exposure to tobacco smoke during prenatal period has been found to be directly related to low birth weight and small foetal size1, increased risk of perinatal death2, and many health problems, including asthma, cardiovascular disease, and poor mental health3, via unknown mechanisms. Despite the increasing concerns of smoking on child health, approximately 13% of women in Northern Japan are reported to smoke during pregnancy4. The numbers are similar in USA (12.3%)5 and Europe6, and although this trend is starting to decline, 5%–10% of women still smoke during pregnancy in Scandinavian countries7.
Epigenetics regulates gene expression without altering the DNA sequence and is suggested as a key mechanism underlying the effect of environmental factors on neonatal health. Epigenetics comprises three major mechanisms: DNA methylation, histone modifications, and RNA-based regulations. DNA methylation has been shown to play critical roles in development8. Previous studies have detected alterations in both global DNA methylation and CpG site methylation in different tissues and cell types between smokers and non-smokers. Global hypomethylation has been detected in buccal cells and peripheral blood granulocytes of children exposed to prenatal smoking9,10, but no changes have been observed in the first trimester11. Later, more specific loci and genes related to maternal smoking during pregnancy have been revealed using the Infinium HumanMethylation27 BeadChip12 or the Infinium HumanMethylation 450 K BeadChip (HM450K)13–18, which covers >27,000 and 450,000 methylation sites across the human genome, respectively. Multiple studies using the HM450K assay have shown five genes to be associated with maternal smoking, including Aryl-Hydrocarbon Receptor Repressor (AHRR), Cytochrome P450 Family 1 Subfamily A Member 1 (CYP1A1), Growth Factor Independent 1 Transcriptional Repressor (GFI1), Myosin IG (MYO1G), and Contactin Associated Protein Like 2 (CNTNAP2)13,14,16,17.
Importantly, changes in DNA methylation patterns of some genes, including AHRR, MYO1G, CYP1A1 and CNTNAP2, that occurred in newborns were sustained until childhood or adulthood, whereas those in other genes, such as GFI1, showed a trend of recovery but did not reach the level of the unexposed group15,17–20. Recently, it was shown that alterations in DNA methylation patterns as a result of prenatal tobacco smoke exposure were sustained until the third or fourth decade of life21. These findings demonstrate the long-term effect of prenatal tobacco smoke exposure on DNA methylation, supporting the DOHaD concept.
It should be noted that the HM450K analysis mentioned above were conducted in European or American populations. Although no samples from Asian populations were included, a previous study showed a high divergence in DNA methylation patterns between populations22. Specifically, 32% of the observed sites varied between populations of European origin and African origin; more than 50% of the differentiated sites had a methylation disparity >5%. To our knowledge, there have been neither DNA methylation-based studies nor epigenome-wide association studies (EWAS) have focused on prenatal tobacco smoke exposure in Asian population. Thus, we tested cord blood samples obtained from 247 mother–infant pairs in the Sapporo cohort using HM450K analysis. The objectives of this study were three-fold: (1) to replicate the findings of previous studies on a Japanese population, (2) to gain further insights into the changes in DNA methylation patterns due to smoking cessation during pregnancy and (3) to validate the results at a higher resolution using bisulfite sequencing.
Results
Epigenome-wide analysis of DNA methylation and maternal smoking during pregnancy
Characteristic features of 291 mother–infant pairs are shown in Table 1. No differences were observed among the mothers across the different mother–infant pairs for their maternal age, BMI before pregnancy and parity. Additionally, mothers who never smoked tended to have higher education and were less likely to have been exposed to passive smoking at work place or to have sustained smoking partners. Moreover, there were no differences between sex, gestational age and birth weight of the infants across the different mother–infant pairs.
Table 1.
Characteristics of study population.
| Maternal smoking status | |||||||
|---|---|---|---|---|---|---|---|
| All (n = 291) | Never-smokers (n = 124) | Stopped smokers before pregnancy (n = 44) | Stopped smokers after confirming pregnancy (n = 77) | Sustained smokers during pregnancy (n = 46) | P value (among 4 groups) | P-value (Never-smokers vs sustained-smokers) | |
| Mothers | |||||||
| Maternal Age (years) | 30.1 ± 5.0 | 30.7 ± 4.9 | 31.1 ± 4.0 | 29.0 ± 5.0 | 29.1 ± 5.5 | 0.033* | 0.076 |
| Maternal BMI before pregnancy (kg/m2) | 20.9 ± 2.9 | 20.8 ± 2.9 | 20.7 ± 1.6 | 21.1 ± 3.3 | 20.9 ± 3.2 | 0.835 | 0.865 |
| Parity | |||||||
| 0 | 150 (51.5) | 66 (53.2) | 15 (34.1) | 47 (61.0) | 22 (47.8) | 0.036* | 0.531 |
| ≥1 | 141 (48.5) | 56 (46.8) | 29 (65.9) | 30 (39.0) | 24 (52.2) | ||
| Maternal Educational level (years) | |||||||
| ≤12 | 131 (45.0) | 54 (36.3) | 20 (45.5) | 37 (48.1) | 29 (63.0) | 0.017* | 0.002** |
| >12 | 160 (55.0) | 79 (63.7) | 24 (54.5) | 40 (51.9) | 17 (37.0) | ||
| Partner’s smoking status | |||||||
| Mothers with partners of never smokers | 44 (15.1) | 27 (21.8) | 8 (18.2) | 7 (9.1) | 2 (4.3) | 0.001** | 0.003** |
| Mothers with partners of stopped smoking before pregnancy | 35 (12.0) | 21 (16.9) | 8 (18.2) | 3 (3.9) | 3 (6.5) | ||
| Mothers with partners of stopped smoking after confirming pregnancy | 4 (1.4) | 3 (2.4) | 0 (0.0) | 1 (1.3) | 0 (0.0) | ||
| Mothers with partners of sustained smoking during pregnancy | 207 (71.1) | 73 (58.9) | 28 (63.6) | 66 (85.7) | 40 (87.0) | ||
| Unknown | 1 (0.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.2) | ||
| Infants | |||||||
| Sex | |||||||
| Male | 131 (45.0) | 52 (41.9) | 27 (61.4) | 36 (46.8) | 16 (34.8) | 0.064 | 0.398 |
| Female | 160 (55.0) | 72 (58.1) | 17 (38.6) | 41 (53.2) | 30 (65.2) | ||
| Gestational age (weeks) | 39.4 ± 1.0 | 39.3 ± 1.1 | 39.3 ± 1.2 | 39.5 ± 0.9 | 39.5 ± 1.0 | 0.225 | 0.221 |
| Birth weight (g) | 3129.2 ± 332.7 | 3111.3 ± 324.8 | 3099.9 ± 382.3 | 3176.8 ± 339.3 | 3130.2 ± 292.2 | 0.519 | 0.729 |
n (%) or mean ± standard deviation. ANOVA, independent t-test and chi-squared test.
*P < 0.05; **P < 0.01; ***P < 0.001.
Among the 291 mother–infant pairs, 44 mothers had smoked at some point during their life but had stopped smoking before their pregnancy. These 44 mother–infant pairs were excluded from the study, as the duration of their non-smoking periods was unknown. The remaining 247 mother–infant pairs were categorised into three exposure categories: 124 never-smokers (Ne-S), 46 sustained-smokers (Su-S) and 77 stopped-smokers (St-S) who stopped smoking after the confirmation of their pregnancy. To investigate the effect of smoking on DNA methylation patterns, differentially methylated (DM) sites between the children of different groups, i.e. Ne-S vs. Su-S, Ne-S vs. St-S and St-S vs. Su-S were identified and −log 10 p-values of each comparison were plotted (Fig. 1A–C). Data revealed 121, 46 and 99 DM sites in Ne-S vs. Su-S, Ne-S vs. St-S and St-S vs. Su-S comparisons, respectively, with a false discovery rate (FDR) of <0.05 (indicated by data points above the horizontal line in Fig. 1A–C).
Figure 1.

Epigenome-wide association between the smoking status of pregnant mothers and the methylation status of 426,576 CpG sites in cord blood samples. Manhattan plots (left panel) and volcano plots (right panel) showing primary comparisons between never-smokers (Ne-S) vs. sustained-smokers (Su-S) (A and D), stopped-smokers (St-S) vs. Su-S (B and E) and Ne-S vs. St-S (C and F). CpG sites with FDR <0.05 are indicated above the horizontal line in Manhattan plots; different colours represent different chromosomes. CpG sites with FDR >0.5 and |β| >0.2 are located in the striped region in volcano plots.
Next, we inspected DNA methylation change using partial regression coefficient β. Only the DM CpG sites with |β| >0.02 were chosen. Of the 121 DM sites with significant FDR identified in the Ne-S vs. Su-S comparison, 46 met the criteria of β-value and mapped to 27 genes (Fig. 1D and Supplementary Table S1). In the Ne-S vs. St-S comparison, only 15 CpG sites fitted the FDR and β-value requirements (Fig. 1E and Supplementary Table S2), whereas the comparison between St-S vs. Su-S resulted in 64 DM sites fitting the FDR and β-value requirements (Fig. 1F and Supplementary Table S3).
Overlapping differentiated DNA methylation sites in never-smoking and stop-smoking groups compared with smoking group
Taking into account that smoking cessation during pregnancy may prevent adverse effects of smoking on the child’s health, we aimed to identify DM sites associated with maternal smoking that were not altered if the mother quit smoking. To this effect, we compared DM sites identified in the Ne-S vs. Su-S comparison to those identified in the St-S vs. Su-S comparison to identify the DM sites that were common to both comparisons. These DM sites would potentially represent CpG sites that were not affected, if the mother stopped smoking early in the pregnancy. Comparison between 46 Ne-S vs. Su-S DM sites and 64 St-S vs. Su-S DM sites revealed 9 common CpG sites (Fig. 2A). Of these nine CpG sites, eight mapped to six genes, including Acyl-CoA Synthetase Medium Chain Family Member 3 (ACSM3) (cg06478823), AHRR (cg05575921 and cg21161138), CYP1A1 (cg05549655), GFI1 (cg12876356 and cg09662411), SH3 And Multiple Ankyrin Repeat Domains 2 (SHANK2) (cg05780228) and Tripartite Motif Containing 36 (TRIM36) (cg07469926) and one mapped to the intergenic region (IGR) between Ankyrin Repeat Domain 9 (ANKRD9) and REST Corepressor 1 (RCOR1) (cg05150608) (Fig. 2B and Table 2). All nine CpG sites showed similar methylation rates (β-value) in Ne-S and St-S groups, except for a significant difference in cg05575921 of AHRR. Four CpG sites in ACSM3, CYP1A1, TRIM36 and the IGR were found to be hypermethylated, whereas five CpG sites in AHRR, GFI1 and SHANK2 were found to be hypomethylated in the Su-S group (Fig. 2B and Table 2).
Figure 2.

Secondary comparison of differentially methylated CpG sites identified in the Ne-S vs. Su-S comparison relative to those identified in the St-S vs. Su-S comparison. (A) Venn diagram showing 9 CpG sites that were common between the Ne-S vs. Su-S comparison (46 sites) and the St-S vs. Su-S comparison (64 sites). (B) Box plots showing methylation rates (β-value) at 9 common CpG sites.
Table 2.
Details for common CpG sites from secondary comparisons.
| CpG site | Chr.a | Gene region | Ne-S b vs Su-S c | Ne-S vs St-S d | St-S vs Su-S | Mean (SD e ) methylation (ratio) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P-value | FDR f | Beta g | P-value | FDR | Beta | P-value | FDR | Beta | Ne-S | St-S | Su-S | |||
| cg06478823 | 16 | ACSM3 | 2.69E-07 | 7.18E-03 | 0.048 | 0.791 | 0.974 | 0.002 | 2.40E-05 | 0.048 | 0.041 | 0.724 (0.059) | 0.721 (0.064) | 0.766 (0.064) |
| cg05575921 | 5 | AHRR | 9.65E-25 | 4.12E-19 | −0.083 | 0.032 | 0.528 | −0.010 | 5.81E-15 | 2.48E-09 | −0.073 | 0.819 (0.040) | 0.805 (0.038) | 0.737 (0.054) |
| cg21161138 | 5 | AHRR | 8.22E-12 | 1.17E-06 | −0.033 | 0.638 | 0.946 | −0.002 | 9.50E-08 | 0.006 | −0.029 | 0.721 (0.032) | 0.717 (0.026) | 0.690 (0.031) |
| cg05549655 | 15 | CYP1A1 | 2.63E-07 | 7.18E-03 | 0.031 | 0.220 | 0.796 | 0.006 | 9.95E-06 | 0.036 | 0.028 | 0.201 (0.045) | 0.200 (0.040) | 0.242 (0.048) |
| cg12876356 | 1 | GFI1 | 9.67E-08 | 3.44E-03 | −0.063 | 0.913 | 0.990 | 0.001 | 2.03E-06 | 0.021 | −0.058 | 0.752 (0.066) | 0.754 (0.057) | 0.688 (0.087) |
| cg09662411 | 1 | GFI1 | 8.19E-07 | 0.014 | −0.035 | 0.451 | 0.899 | 0.004 | 1.91E-05 | 0.044 | −0.034 | 0.730 (0.041) | 0.734 (0.039) | 0.694 (0.059) |
| cg05150608 | 14 | IGR | 7.49E-06 | 0.036 | 0.030 | 0.945 | 0.994 | 0.000 | 2.56E-05 | 0.048 | 0.034 | 0.417 (0.040) | 0.414 (0.042) | 0.448 (0.053) |
| cg05780228 | 11 | SHANK2 | 8.23E-07 | 0.014 | −0.024 | 0.493 | 0.912 | −0.002 | 1.38E-05 | 0.038 | −0.022 | 0.622 (0.030) | 0.618 (0.025) | 0.601 (0.034) |
| cg07469926 | 5 | TRIM36 | 3.64E-06 | 0.025 | 0.027 | 0.424 | 0.890 | −0.004 | 1.06E-06 | 0.016 | 0.029 | 0.578 (0.044) | 0.570 (0.043) | 0.605 (0.045) |
| cg01290904 | 4 | EVC2 | 5.90E-06 | 0.031 | 0.023 | 2.52E-06 | 0.031 | 0.021 | 3.09E-01 | 0.799981 | 0.005709 | 0.581 (0.042) | 0.593 (0.040) | 0.601 (0.041) |
aChromosome. bNever-smoker. cSustained-smoker. dStopped-smoker. eStandard deviation. fFalse discovery rate. gPartial regression coefficient for the magnitude of the effect on DNA methylation change
Above the line: common CpG sites from comparison between (Ne-S vs Su-S) and (St-S vs Su-S).
Below the line: common CpG sites from comparison between (Ne-S vs Su-S) and (Ne-S vs St-S).
On the other hand, to identify the CpG sites whose methylation pattern was altered even after the mother stopped smoking, we compared the 46 CpG sites identified in the Ne-S vs. Su-S with the 15 sites identified in the Ne-S vs. St-S comparison. Data revealed only one common CpG site (cg01290904). This CpG site mapped to the EvC Ciliary Complex Subunit 2 (EVC2) gene and was hypermethylated in both the Su-S and St-S groups compared with the Ne-S group (Fig. 3 and Table 2).
Figure 3.
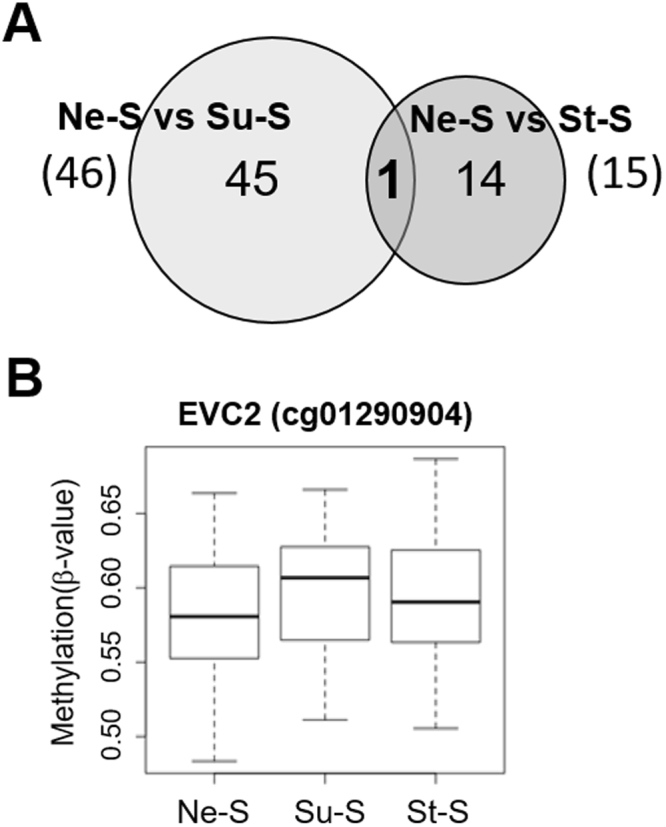
Secondary comparison of differentially methylated CpG sites identified in the Ne-S vs. Su-S comparison relative to those identified in the Ne-S vs. St-S comparison. (A) Venn diagram showing one CpG site that was common between the Ne-S vs. Su-S comparison (46 sites) and the Ne-S vs. St-S comparison (15 sites). (B) Box plots showing methylation rates (β-value) at one common CpG site.
Verification of DNA methylation analysis using bisulfite sequencing
Despite being widely used in the field of epigenetics for large-scale DNA methylation profiling, HM450K and analysis of tandem HM450K have their limitations, e.g., restriction to predesigned CpG sites or complications in data normalisation and analysis23. Moreover, some of the selected CpG sites from our secondary comparison fall within the list of suspected low-quality probes provided by Naeem et al.24, namely ACSM3 cg06478823, AHRR cg21161138, and SHANK2 cg05780228 (Supplementary Tables S1, S2, and S3). Thus, we further verified our findings using next-generation sequencing (NGS) technique following bisulfite conversion. Because of their locations in GC-rich regions, suitable primers could not be designed for cg12876356 and cg09662411 located in GFI1, Therefore, GFI1 was excluded from verification experiments.
Due to some outliers in the NGS result for AHRR cg21161138, SHANK2 cg05780228, and EVC2 cg01290904 (black arrows in Supplementary Fig. S1), we chose to perform analysis both including and excluding outliers. Our NGS analysis showed that seven out of eight CpG sites exhibited similar DNA methylation profiles to that of HM450K, with the exception of cg21161138 within the AHRR gene, which showed no changes in methylation rates between the Ne-S, Su-S and St-S groups (Fig. 4, bottom panel in each target, Table 3, and Supplementary Fig. S1). The statistical significance in NGS analysis remained the same for all sites after discounting outliers, except for cg012090904 within the EVC2 gene, which fell slightly outside the range of significance (p = 0.0526) after discounting outliers (Table 3). Correlation analysis revealed a moderate to strong positive correlation (ρ > 0.6) between DNA methylation ratio at the seven CpG sites determined by HM450K analysis and by NGS analysis, excluding AHRR cg21161138 (ρ = 0.347 and ρ = 0.331 after and before discounting outlier, respectively) (Fig. 5 and Supplementary Fig. S2).
Figure 4.

Next-generation sequencing (NGS) analysis of eight CpG sites from secondary comparisons after excluding outliners in AHRR cg21161138, SHANK2 cg05780228, EVC2 cg01290904, and 2 sites associated with GFI1 due to technical issue. The upper part of each panel represents the length and schematic position in each sequence. Numbers represent the CpG sites; the same sites in the HumanMethylation450K array (HM450K) are marked in red. DNA methylation ratios from NGS analysis of the same CpG sites in HM450K are shown in box plots in the lower part of each panel.
Table 3.
Verification result for selected CpG sites using Next Generation Sequencing.
| Gene region | CpG site a | P-value | Mean (SD e ) methylation (ratio) | ||||
|---|---|---|---|---|---|---|---|
| Ne-S b vs Su-S c | Ne-S vs St-S d | St-S vs Su-S | Ne-S | St-S | Su-S | ||
| ACSM3 | cg59 (cg06478823) | 0.032* | 0.728 | 0.033* | 0.700 (0.062) | 0.690 (0.078) | 0.722 (0.080) |
| AHRR | cg26 | 1.17E-10** | 0.258 | 1.55E-08** | 0.644 (0.052) | 0.636 (0.049) | 0.564 (0.068) |
| cg28 | 6.64E-12** | 0.205 | 6.24E-09** | 0.640 (0.054) | 0.630 (0.050) | 0.549 (0.067) | |
| cg51 (cg05575921) | 1.05E-11** | 0.625 | 9.76E-11** | 0.615 (0.060) | 0.609 (0.050) | 0.519 (0.072) | |
| cg71 | 3.73E-12** | 0.587 | 1.48E-10** | 0.738 (0.064) | 0.733 (0.057) | 0.611 (0.097) | |
| cg96 | 1.33E-10** | 0.388 | 6.18E-08** | 0.679 (0.058) | 0.670 (0.057) | 0.579 (0.082) | |
| AHRR | cg26 | 0.964 (0.984) | 0.768 (0.826) | 0.980 (0.980) | 0.854 (0.034) | 0.851 (0.037) | 0.854 (0.035) |
| cg56 | 0.007**(0.656) | 0.827 (0.892) | 0.042* (0.429) | 0.785 (0.150) | 0.814 (0.111) | 0.768 (0.150) | |
| cg79 (cg21161138) | 0.064 (0.064) | 0.669 (0.576) | 0.280 (0.350) | 0.767 (0.040) | 0.755 (0.063) | 0.749 (0.046) | |
| CYP1A1 | cg26 (cg05549655) | 8.47E-04** | 0.941 | 7.06E-04** | 0.210 (0.061) | 0.202 (0.053) | 0.245 (0.061) |
| cg31 | 0.008** | 0.986 | 0.005** | 0.242 (0.066) | 0.236 (0.054) | 0.274 (0.060) | |
| cg42 | 0.009** | 0.312 | 3.67E-04** | 0.480 (0.081) | 0.461 (0.073) | 0.518 (0.071) | |
| cg49 | 0.166 | 0.288 | 0.007** | 0.470 (0.070) | 0.453 (0.065) | 0.491 (0.070) | |
| cg57 | 8.93E-4** (0.002**) | 0.781 (0.781) | 7.07E-5** (2.19E-04**) | 0.566 (0.077) | 0.558 (0.064) | 0.604 (0.069) | |
| IGR | cg26 | 0.72 (0.849) | 0.600 (0.600) | 0.303 (0.422) | 0.371 (0.076) | 0.367 (0.070) | 0.374 (0.087) |
| cg75 (cg05150608) | 0.024* | 0.736 | 0.010* | 0.368 (0.066) | 0.365 (0.071) | 0.403 (0.080) | |
| cg77 | 0.044* | 0.707 | 0.041* | 0.376 (0.078) | 0.372 (0.085) | 0.415 (0.110) | |
| SHANK2 | cg39 (cg05780228) | 2.50E-04** (1.98E-04**) | 0.287 (0.246) | 0.034* (0.034*) | 0.590 (0.042) | 0.579 (0.040) | 0.558 (0.046) |
| cg67 | 0.09 (0.120) | 0.986 (0.964) | 0.163 (0.163) | 0.937 (0.018) | 0.938 (0.013) | 0.932 (0.017) | |
| cg74 | 0.025* | 0.793 | 0.022* | 0.848 (0.027) | 0.849 (0.028) | 0.837 (0.027) | |
| TRIM36 | cg26 | 0.003** | 0.777 | 0.003** | 0.549 (0.085) | 0.538 (0.088) | 0.602 (0.098) |
| cg38 | 0.031* | 0.562 | 0.009** | 0.569 (0.082) | 0.556 (0.087) | 0.606 (0.088) | |
| cg40 (cg07469926) | 0.005** | 0.678 | 0.002** | 0.563 (0.057) | 0.551 (0.065) | 0.595 (0.063) | |
| EVC2 | cg34 (cg01290904) | 0.007** (0.007)** | 0.0526 (0.042*) | 0.429 (0.527) | 0.637 (0.056) | 0.658 (0.088) | 0.667 (0.054) |
aCpG sites are numbered according to their position on the sequencing products, overlapped CpG sites with Human Methylation 450K array are indicated in parentheses. bNever-smoker. cSustained-smoker. dStopped-smoker. eStandard deviation.
Steel-Dwass tests. *P < 0.05; **P < 0.01, P value from analysis before removal of outliers are indicated in parentheses.
Figure 5.

Correlation between HumanMethylation450K array (HM450K) and next generation sequencing (NGS) data analysis for the DNA methylation status of eight CpG sites after removing outliers. Values of Spearmen correlation coefficient (ρ) are indicated.
Because NGS provides single base-resolution, we were able to examine adjacent CpG sites falling within the region spanned by the primers (Fig. 4, upper panel in each target). Of the 16 neighbouring CpG sites, methylation patterns of 13 sites were consistent with those determined using HM450K for Su-S and St-S groups (Table 3 and Supplementary Fig. S3). Notably, multiple CpG sites near AHRR cg05575921, CYP1A1 cg05549655 and TRIM36 cg07469926 were altered by smoking and were not changed if smoking was stopped in early pregnancy (Table 3 and Supplementary Fig. S3A,S3C and S3F), suggesting that these regions are highly susceptible to smoking.
Discussion
In the current study, we conducted an EWAS using HM450K analysis in a Japanese population from the Sapporo cohort of the Hokkaido Study to address the current interest in DNA methylation alteration due to prenatal tobacco smoke exposure. Considering the beneficial effects of quitting smoking early in the pregnancy, we identified nine CpG sites located in six genes and an intergenic region, whose DNA methylation patterns were altered by smoking but recovered after early smoking cessation. However, the methylation pattern of CpG sites in EVC2 (cg01290904) was observed even after the cessation of smoking. We authenticated the findings of HM450K analysis using bisulfite sequencing, with the exclusion of CpG sites in GFI1 due to technical issues. We found a relatively strong correlation between the two approaches for the methylation status of CpG sites, except for the sites in AHRR (cg21161138).
In a primary comparison between each smoking group, CpG sites that passed both criteria, including epigenome-wide significance level (FDR <0.05) and effect size (|β| >0.02), were selected. We chose a difference of 2% to narrow down the CpG sites based on a previous publication, which defined the mean methylation change for both hyper- and hypomethylation as 2%16. Moreover, a recent review summarised that most environmental exposure studies have reported changes in DNA methylation from 2–10% and called for focusing on small magnitude alterations25. In the EWAS comparison between Ne-S and Su-S groups, we replicated the CpG sites that were consistent with previous studies, particularly the hypomethylated sites in AHRR and GFI1 and the hypermethylated sites in CYP1A1 and MYO1G13,14,16,17. Among the 64 DM CpG sites, cg05575921 associated with AHRR was found to be consistently replicated across studies assessing DNA methylation in cord blood or newborn blood6,13,14,17. In detail, all CpG sites identified in GFI1, CYP1A1 and MYO1G in this study were also reported in previous studies; AHRR cg21161138 was reported by Joubert et al.13, Kupers et al.14 and Markunas et al.16 but not by Richmond et al.17. Another AHRR-associated CpG site, cg17924476, although not reported in studies using cord blood or newborn blood, was detected in adolescents exposed to maternal smoking15. Moreover, multiple DM CpG sites, including two in MARCH3, two in FLYWCH1 and three in ACSM3 were not identified in previous studies, which suggests these to be novel targets affected by prenatal tobacco smoke exposure and warrants further studies for validation. We did not detect any variation consistent with the genome-wide significant criteria in CTCNAP2 cg25949550, a CpG site that was agreeable with previous studies (data not shown). This may be explained by the differences between populations, as our study was performed in an Asian population when others were conducted mostly in Caucasian populations. Moreover, evidence suggests that genetic background and genetic variants effect differences in DNA methylation at CpG sites associated with maternal smoking19,26,27. Although we did not investigate genetic variants in our study, we attempted to search for single-nucleotide polymorphism (SNP) sites in CTCNAP2 and several novel genes that may affect the reported CpG sites from the Infinium HD Methylation SNP List provided by Illumina. We then compared SNP sites between Japanese and Caucasian populations using the 1000 Genomes Browser (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/). However, we detected a higher frequency of SNP sites that may affect the methylation rate at cg05150608 only in the intergenic region between ANKRD9 and RCOR1 (Supplementary Table S4). Nevertheless, a study comparing European and African populations found that genetic variances may explain as much as half of the diversity in DNA methylation between populations; the remaining variance is thought to be due to complex gene-gene and gene-environment interactions22.
Considering that the negative effects of prenatal tobacco smoke exposure can be diminished if the mother quits smoking in the first trimester of pregnancy, it is vital to determine the critical window of exposure. We conducted a secondary comparison to identify CpG sites that overlapped across the different primary comparisons, with the aim to focus on CpG sites that may be specific to the St-S group. Five of the nine CpG sites identified for unaltered DNA methylation patterns if the mothers stopped smoking early during pregnancy, namely AHRR cg05575921, AHRR cg21161138, CYP1A1 cg05549655, GFI1 cg12876356 and GFI1 cg09662411 were previously reported, as discussed above. Except for AHRR cg21161138, which showed a weak correlation with HM450K and no significant difference in methylation between groups in NGS, we successfully verified the changes in DNA methylation at AHRR cg05575921 and CYP1A1 cg05549655. Two CpG sites in GFI1 gene were not verified by NGS due to technical issues. Novakovic et al. performed a detailed investigation on AHRR methylation and found no significant differences between children from women who never smoked and those who stopped smoking after confirmation of their pregnancy19. Similarly, Joubert et al. reported that alterations in DNA methylation occurred in the foetus only if women smoked past the 18th week of pregnancy28. The authors further indicated that changes in DNA methylation patterns result from maternal smoking, and not from paternal smoking before pregnancy, or from an epigenetic inheritance of smoking effects from the grandmother to the mother. Richmond et al. found no differences at AHRR cg05575921 and CYP1A1 cg05549655 between cord blood samples that were unexposed or exposed only during the first trimester17. The authors also reported the same finding in GFI1, although the CpG site identified in their study (cg09935388) was different from those identified in this study (cg12876356 and cg09662411). Our findings at AHRR cg05575921 were consistent with previously published data19, where hypomethylated state of CpG sites neighbouring cg05575921 was observed in the cord blood samples of mothers in the Su-S and Ne-S but not among those of mothers in the St-S. In adult smokers, stopping or reducing smoking was related to the recovery of DNA methylation status at AHRR in peripheral blood29. A very recent study examined AHRR and CYP1A1 methylation and mRNA expression using first trimester foetal livers and placentas; significant changes in AHRR methylation were only observed in placentas from female foetus30. However, significant increases in the mRNA of AHRR in livers and of CYP1A1 in placentas were detected, which suggests that smoking may have an early effect on gene expression before inducing noticeable alterations in DNA methylation30. Thus, the question regarding of the duration of maternal smoking exposure required to make remarkable and sustained changes in the DNA methylation level is still unclear and warrants further investigations. In total, together with other studies, our results further support AHRR as an exemplary gene for the interaction between environment and DNA methylation.
Amidst the reported genes associated with smoking, in particular maternal smoking, AHRR and CYP1A1 were thoroughly investigated. Both AHRR and CYP1A1 function downstream of AHR; while AHRR represses AHR, CYP1A1 was activated by AHR19. Consistent with previous studies, we found a hypomethylated status of AHRR and a hypermethylated status of the CYP1A1 promoter13–19. Moreover, it has been shown that the hypomethylation at AHRR cg05575921 is correlated with an increase in AHRR expression31. Notably, alterations in the DNA methylation status of both AHRR and GFI1 in cord blood contributes to low birth weight of children of mothers in the Su-S group14,32. Specifically, hypomethylation cg05575921 was found to associate with low birth weight33. As discussed in Tian et al., AHRR cg05575921 hypomethylation was correlated with an increase in AHRR expression and several effects, including repression of oxygen transport-related embryonic globin genes, inhibition of cell growth and proliferation, and suppression of angiogenesis. Although genetic polymorphisms in CYP1A1 have been implicated to affect birth weight and birth size1, the contribution of CYP1A1 hypermethylation in cord blood has not been fully investigated. However, we did not observe significant change in birth weights between Ne-S, Su-S, and St-S. We can only assume that our relatively small sample size contributed to the absence of a low birth weight phenotype. In addition to being recognised as a risk factor for lower birth weight and birth size in newborns, intra utero tobacco smoke exposure has been shown to increase the risk of neurodevelopmental disorders, particularly autism spectrum disorder and attention deficit hyperactivity disorder (ADHD)34. This notion was recently addressed in a study investigating the association between DNA methylation in the peripheral blood of ADHD children and maternal smoking during pregnancy32. Despite the small size of the population studied, DNA methylation at AHRR and GFI1, but not at CYP1A1, were shown to be related to the severity of conduct disorder and ADHD in children of smoking mothers. Furthermore, maternal smoking has been shown to induce hypomethylation at GFI1 in the peripheral blood of children suffering from sudden infant death syndrome (SIDS) compared with healthy children35, suggesting a complex triad relationship among intra utero tobacco smoke exposure, GFI1 methylation status and SIDS.
In addition to the previously known CpG sites, we identified four novel CpG sites, including ACSM3 cg0647882, TRIM36 cg07469926, and cg05150608 in the intergenic region between ANKRD9 and RCOR1, which represented a hypermethylated status in the Su-S group, and SHANK2 cg05780228, representing a hypomethylated status in the Su-S group. NGS analysis revealed similar changes in DNA methylation of the adjacent CpG sites in TRIM36 and SHANK2, suggesting that these sites are susceptible to maternal smoking. A recent meta-analysis suggested maternal smoking during pregnancy as a high-risk factor for neural tube defects36. TRIM36 is expressed in the developing brain and is thought to be indispensable for somite formation37; lack of TRIM36 due to a homozygous mutation has been reported to result in autosomal recessive anencephaly38. Recently, TRIM36 hypermethylation has been linked to a cell-transformation effect of Benzo[a]pyrene, a polycyclic aromatic hydrocarbon found in tobacco smoke, in bronchial epithelial cells39. Hypermethylation status in TRIM36 was also identified in tumours of lung, breast, liver and ovarian cancer39. Although no study has focused on SHANK2 hypomethylation, mutation and copy number variation of SHANK2, encoding a member of synaptic scaffolding protein in the SHANK family was reported in patients with autism spectrum disorder and mental retardation40. ACSM3, formerly known as an acyl-CoA synthetase medium chain family member 3, is associated with metabolic syndrome41. Recently, down-regulation of ACSM3 has been implicated in the poor outcome of patients with hepatocellular carcinoma42. Like SHANK2, little is known about the epigenetic modification of ACSM3. The function of the CpG site, cg05150608, identified in the noncoding region between the ANKRD9 and RCOR1 genes is unknown. Taken together, our findings indicate changes in DNA methylation patterns of target genes, implicating prenatal tobacco smoke exposure as a risk factor for cancer, congenital abnormalities and neurodevelopmental disorders in the unborn child. However, our observations were limited to alterations in DNA methylation of cord blood, which may not represent methylation status in the foetal brain or other organs. Further studies are awaited to reinforce the relationship between tobacco smoke exposure and disease development as well as the role of novel target genes in this process.
Secondary comparison between the CpG sites identified in the Ne-S vs. Su-S comparison and those identified in the St-S vs. Su-S comparison pointed out EVC2 cg01290904 as the only common CpG site, which was verified by NGS analysis, suggesting that this CpG site was affected by short-term tobacco smoke exposure in the first trimester. Mutation in EVC2 causes congenital skeletal dysplasia, resulting in very short stature, known as Ellis-van Creveld syndrome43. Recent studies on Evc2 mutant mice further implicate the importance of EVC2 in the normal development of teeth and cranial bone44,45. Notably, children born to actively or passively smoking mothers show higher risk of hypodontia46. Moreover, a slightly increased risk of oral clefts has been reported in children born to actively smoking mothers47. These findings suggest a striking association between maternal smoking and EVC2. However, we could not find any previous study linking EVC2 to early maternal smoking. Future studies focusing on EVC2-related outcomes and smoking exposure during the first trimester are needed to validate whether EVC2 links maternal smoking with congenital anomalies in children.
The HM450K array allows access to >450,000 CpG sites across the genome that span 99% of the RefSeq genes and 96% of the CpG islands. Thus, HM450K array is a top choice for EWAS study. HM450K utilises the detection of DM CpG sites, providing insights into the affected genes. However, it is limited to the CpG sites included in the array and does not provide information on the neighbouring sites. Moreover, it is thought that the results of HM450K analysis may vary between different arrays (batch effect) or may not reflect bona fide DNA methylation due to problematic probes and intricacies in data normalisation and/or data analysis23. Therefore, with the availability of NGS in our laboratory, we chose to perform NGS analysis of DM CpG sites detected by HM450K. Using this approach helped us accomplish two objectives: to verify the changes in DNA methylation status of selected CpG sites identified using HM450K array and to assess the neighbouring CpG sites contained within the range of primers. The relatively strong correlation between results obtained with the two methods, together with the high-throughput capacity and single nucleotide resolution of NGS, favours the application of NGS for assessing DNA methylation of target gene(s) in large cohort studies. Moreover, with the increasing number of studies on DNA methylation as a biomarker for smoking29,48, especially maternal smoking, NGS may serve as a high-throughput and reliable approach for translational studies.
To the best of our knowledge, this is the first study to report the association of cord blood DNA methylation and prenatal cigarette smoking exposure in a Japanese population. Our study has several strengths. Firstly, all participants were native Japanese, living in Sapporo city and the surrounding areas, which were presumably homogenous in term of environment, culture, and daily life. Thus, the study population limited the effect of other environmental factors and was suitable for investigation of the interaction between a specific factor (prenatal smoking exposure) and the epigenome. Secondly, previously reported CpG sites were replicated in our studies, demonstrating the robustness of HM450K. Moreover, to overcome the drawbacks of HM450K, we applied bisulfite-NGS to validate our observations. Thirdly, by focusing on the windows of exposure with the exclusion of mothers who had stopped smoking before pregnancy, we demonstrated the effect of smoking on DNA methylation in the cord blood even if the mother stopped smoking as soon as the pregnancy was confirmed.
Although data were carefully analysed and verified, we are aware of the lack of a method of quantitative analysis of maternal smoking and the possible impact this may have on the results. In this study, mother–infant pairs were categorised based on self-reported information, without measuring any biomarkers for smoking (e.g. cotinine level). This approach might result in misclassification of maternal smoking groups. Nevertheless, the inaccuracy was presumed to be small, as a previous report on a Japanese population showed a high agreement between self-reported information and cotinine measurement4. Another limitation of this study might be that the effect of second-hand smoking was not accounted for in our analysis, which may lead to false unexposed status. However, this underestimation in smoking exposure would reduce the significance between Ne-S or St-S and Su-S groups. Thus, the CpG sites reported in this study are likely not overstated. Lastly, our results had shortcomings: replication in a second cohort and for the relatively small population studied, especially for Su-S and St-S groups (n = 46 and 77, respectively), which may reduce statistical strength and contribute to the absence of phenotypes, for instance, low birth weight, as discussed above. Therefore, the novel CpG sites should be interpreted with caution and must be validated in future cohort studies.
In conclusion, we reported novel CpG sites and verified previously established sites associated with maternal smoking and cessation of maternal smoking upon the confirmation of pregnancy. These sites were linked with adverse outcomes of intra utero tobacco smoke exposure, including recently reported effects on neurodevelopment disorders and congenital defects. However, the triad relation among maternal smoking, DNA methylation alterations and certain diseases remain unclear. To address this issue, future studies on the effects of maternal smoking on DNA methylation and disease development using large-scale cohorts are needed.
Methods
Study population
We performed a prospective study using Sapporo birth cohort from previously described Hokkaido Study on Environment and Children’s Health49,50. In this study, 514 women, who had enrolled at 23–35 weeks of gestation and delivered at the Sapporo Toho Hospital between 2002 and 2005 in Japan, agreed to participate. All participants were native Japanese and residents of Sapporo City or surrounding areas. Participants completed a questionnaire that asked for basic information, including their dietary habits, exposure to chemical compounds in their daily life, smoking history, alcohol consumption, caffeine intake, family income and educational levels of themselves and their partners. Of the 514 participants, 295 cord blood samples were obtained. Genomic DNA of sufficient quantity and quality was obtained from 292 participants.
Genome-wide DNA methylation analysis
Umbilical cord blood samples were taken immediately after birth and stored at −80 °C. Genomic DNA was extracted from cord blood using a Maxwell® 16 DNA Purification Kit (Promega, Madison, WI, USA). Genome-wide DNA methylation analysis was performed using the Infinium HumanMethylation 450 BeadChip (HM450K) (Illumina, San Diego, CA, USA) following the manufacturer’s instructions. All analyses were performed by G&G SCIENCE CO., LTD. (Matsukawa, Fukushima, Japan).
Methylation data were pre-processed using the R/Bioconductor package minfi51. From the 292 mother–infant pairs, one pair was rejected on the basis of poor quality control (QC) parameters. After QC, signal intensities were normalised using functional normalisation52. CpG probes containing single nucleotide pair-affected probes53, with a detection P-value of >0.05, sex chromosomes were removed54. As a result, 426,576 CpG probes were included in the working set. Inter-slide differences were adjusted using ComBat55. Methylation values of each CpG site were represented as β-values ranging from 0 to 1 and calculated from signal intensities prior to statistical analysis56.
Data analysis
The 291 mother–infant pairs were categorised into three exposure classes based on the smoking history of mothers: 124 Ne-S, 46 Su-S, 77 St-S, who stopped smoking after the confirmation of their pregnancy; 44 St-S were excluded from this study because of the unknown duration of non-smoking periods. Robust linear regression analyses57 and empirical Bayesian methods58 were applied to determine the associations of β-value at each CpG site, adjusted for maternal age, infant sex, maternal education and surrogate variables. In addition, cell type fraction (CD4+ T cells, CD8+ T cells, granulocytes, monocytes, B cell and nucleated red blood cells) for each subject were calculated using reference-free cell mixture adjustments59. Statistical analyses were performed using minfi, SVA, limma and RefFreeEWAS packages in R ver. 3.2.3 and Bioconductor ver. 3.2. Differential methylated sites were identified between Ne-S and Su-S, Ne-S and St-S and St-S and Su-S. To correct for multiple testing, P-values were adjusted for FDR based on the Benjamini and Hochberg method to obtain q-values. Partial regression coefficient (β) for the magnitude of the effect on DNA methylation change and selected CpGs with an FDR of <0.05 and |β| >0.02. On specific genes, statistical significance of differences in methylation values between groups was determined using Steel–Dwass test; differences with P-value of <0.05 were considered statistically significant. Associations between DNA methylation ratios at the seven CpG sites obtained from HM450K analysis and from NGS analysis were tested using Spearman’s correlation coeffcients (ρ).
Bisulfite next-generation sequencing
DNA was subjected to bisulfite conversion and amplified using FastStart Taq DNA Polymerase (Roche, Basel, Schweiz). PCR primers for bisulfite PCR were designed using MethPrimer (http://www.urogene.org/methprimer/) (Supplementary Table S5). For NGS, amplicon libraries were generated using an Ion Plus Fragment Library Kit (ThermoFisher Scientific, MA, USA) and Ion Xpress Barcode Adaptors Kit (ThermoFisher Scientific). Following Agencourt AMPure XP purification (Beckman Coulter, CA, USA), individual libraries were amplified using quantitative real-time PCR, diluted and pooled in equimolar ratios. The libraries were then processed with an Ion Chef System using an Ion PG Hi-Q Chef Kit (ThermoFisher Scientific). Sequencing was performed using an Ion PGM Hi-Q Sequencing Kit (ThermoFisher Scientific) and 850 flows on an Ion 318 Chip Kit v2 (ThermoFisher Scientific), according to the manufacturer’s protocol. After sequencing, single processing and base calling were performed using Torrent Suite 5.0.2 (ThermoFisher Scientific). Methylation analysis was performed using MethylationAnalysis_Amplicon plug-in v1.3 (ThermoFisher Scientific). Extreme outliers in methylation data can have significant influences on results. Therefore, outliers were removed using the Tukey method60. Comparison of methylation ratio among the different smoking groups was performed using Steel–Dwass test. Differences with P-value of <0.05 were considered statistically significant.
Ethics
This study was conducted with informed consent of all subjects in writing. All procedures involving human subjects were approved by the University of Yamanashi, the Hokkaido University Graduate School of Medicine and the Hokkaido University Center for Environmental and Health Science and were performed in accordance with relevant guidelines and regulations.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on request.
Electronic supplementary material
Acknowledgements
We thank all the participants and stuff of Sapporo Toho Hospital. This study was financially supported by the Japanese Ministry of Health, Labour and Welfare (H29-Kagaku Ippan-002) and the Environment Research and Technology Development Fund (5-1454) from the Ministry of the Environment, Japan.
Author Contributions
K.M. designed and implemented the data analysis, performed experimental work, interpreted results and participated in writing the manuscript. T.K. and R.K. conceived and designed the study and interpreted results. A.K. contributed to data analysis, statistical analysis and interpreted results. K.M. and A.K. contributed equally to this work. Z.Y. contributed to data analysis and interpreted results. R.M., Sachiko K., Sumitaka K., C.M. and A.A. collected data and contributed to data analysis. N.Q.V.T. wrote the manuscript and participated in performing experimental work. All authors read and approved the final version of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Kunio Miyake and Akio Kawaguchi contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-23772-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sasaki S, et al. Maternal smoking during pregnancy and genetic polymorphisms in the Ah receptor, CYP1A1 and GSTM1 affect infant birth size in Japanese subjects. Molecular human reproduction. 2006;12:77–83. doi: 10.1093/molehr/gal013. [DOI] [PubMed] [Google Scholar]
- 2.Pineles BL, Hsu S, Park E, Samet JM. Systematic Review and Meta-Analyses of Perinatal Death and Maternal Exposure to Tobacco Smoke During Pregnancy. American journal of epidemiology. 2016;184:87–97. doi: 10.1093/aje/kwv301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holbrook BD. The effects of nicotine on human fetal development. Birth defects research. Part C, Embryo today: reviews. 2016;108:181–192. doi: 10.1002/bdrc.21128. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki S, Braimoh TS, Yila TA, Yoshioka E, Kishi R. Self-reported tobacco smoke exposure and plasma cotinine levels during pregnancy–a validation study in Northern Japan. The Science of the total environment. 2011;412–413:114–118. doi: 10.1016/j.scitotenv.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 5.Tong VT, et al. Trends in smoking before, during, and after pregnancy–Pregnancy Risk Assessment Monitoring System, United States, 40 sites, 2000-2010. Morbidity and mortality weekly report. Surveillance summaries. 2013;62:1–19. [PubMed] [Google Scholar]
- 6.Joubert BR, et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. American journal of human genetics. 2016;98:680–696. doi: 10.1016/j.ajhg.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ekblad M, Gissler M, Korkeila J, Lehtonen L. Trends and risk groups for smoking during pregnancy in Finland and other Nordic countries. European journal of public health. 2014;24:544–551. doi: 10.1093/eurpub/ckt128. [DOI] [PubMed] [Google Scholar]
- 8.Geiman TM, Muegge K. DNA methylation in early development. Molecular reproduction and development. 2010;77:105–113. doi: 10.1002/mrd.21118. [DOI] [PubMed] [Google Scholar]
- 9.Breton CV, et al. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. American journal of respiratory and critical care medicine. 2009;180:462–467. doi: 10.1164/rccm.200901-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flom JD, et al. Prenatal smoke exposure and genomic DNA methylation in a multiethnic birth cohort. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2011;20:2518–2523. doi: 10.1158/1055-9965.EPI-11-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fa S, et al. Assessment of global DNA methylation in the first trimester fetal tissues exposed to maternal cigarette smoking. Clinical epigenetics. 2016;8:128. doi: 10.1186/s13148-016-0296-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suter M, et al. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics. 2011;6:1284–1294. doi: 10.4161/epi.6.11.17819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joubert BR, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environmental health perspectives. 2012;120:1425–1431. doi: 10.1289/ehp.1205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kupers LK, et al. DNA methylation mediates the effect of maternal smoking during pregnancy on birthweight of the offspring. International journal of epidemiology. 2015;44:1224–1237. doi: 10.1093/ije/dyv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KW, et al. Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environmental health perspectives. 2015;123:193–199. doi: 10.1289/ehp.1408614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markunas CA, et al. Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environmental health perspectives. 2014;122:1147–1153. doi: 10.1289/ehp.1307892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richmond RC, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC) Human molecular genetics. 2015;24:2201–2217. doi: 10.1093/hmg/ddu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rzehak P, et al. Maternal Smoking during Pregnancy and DNA-Methylation in Children at Age 5.5 Years: Epigenome-Wide-Analysis in the European Childhood Obesity Project (CHOP)-Study. PloS one. 2016;11:e0155554. doi: 10.1371/journal.pone.0155554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Novakovic B, et al. Postnatal stability, tissue, and time specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics. 2014;9:377–386. doi: 10.4161/epi.27248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladd-Acosta C, et al. Presence of an epigenetic signature of prenatal cigarette smoke exposure in childhood. Environmental research. 2016;144:139–148. doi: 10.1016/j.envres.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tehranifar, P. et al. Maternal cigarette smoking during pregnancy and offspring DNA methylation in midlife. Epigenetics 0, 10.1080/15592294.2017.1325065 (2017). [DOI] [PMC free article] [PubMed]
- 22.Fraser HB, Lam LL, Neumann SM, Kobor MS. Population-specificity of human DNA methylation. Genome Biol. 2012;13:R8. doi: 10.1186/gb-2012-13-2-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dedeurwaerder S, et al. A comprehensive overview of Infinium HumanMethylation450 data processing. Briefings in bioinformatics. 2014;15:929–941. doi: 10.1093/bib/bbt054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naeem H, et al. Reducing the risk of false discovery enabling identification of biologically significant genome-wide methylation status using the HumanMethylation450 array. BMC Genomics. 2014;15:51. doi: 10.1186/1471-2164-15-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breton CV, et al. Small-Magnitude Effect Sizes in Epigenetic End Points are Important in Children’s Environmental Health Studies: The Children’s Environmental Health and Disease Prevention Research Center’s Epigenetics Working Group. Environmental health perspectives. 2017;125:511–526. doi: 10.1289/EHP595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonseth S, et al. Genetic contribution to variation in DNA methylation at maternal smoking-sensitive loci in exposed neonates. Epigenetics. 2016;11:664–673. doi: 10.1080/15592294.2016.1209614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dogan MV, Beach SRH, Philibert RA. Genetically contextual effects of smoking on genome wide DNA methylation. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2017;174:595–607. doi: 10.1002/ajmg.b.32565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joubert BR, et al. Maternal smoking and DNA methylation in newborns: in utero effect or epigenetic inheritance? Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2014;23:1007–1017. doi: 10.1158/1055-9965.EPI-13-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Philibert R, et al. Reversion of AHRR Demethylation Is a Quantitative Biomarker of Smoking Cessation. Frontiers in psychiatry. 2016;7:55. doi: 10.3389/fpsyt.2016.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fa S, et al. Changes in first trimester fetal CYP1A1 and AHRR DNA methylation and mRNA expression in response to exposure to maternal cigarette smoking. Environmental toxicology and pharmacology. 2018;57:19–27. doi: 10.1016/j.etap.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 31.Reynolds LM, et al. DNA Methylation of the Aryl Hydrocarbon Receptor Repressor Associations With Cigarette Smoking and Subclinical Atherosclerosis. Circulation. Cardiovascular genetics. 2015;8:707–716. doi: 10.1161/CIRCGENETICS.115.001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sengupta SM, Smith AK, Grizenko N, Joober R. Locus-specific DNA methylation changes and phenotypic variability in children with attention-deficit hyperactivity disorder. Psychiatry research. 2017;256:298–304. doi: 10.1016/j.psychres.2017.06.048. [DOI] [PubMed] [Google Scholar]
- 33.Tian FY, et al. Tissue differences in DNA methylation changes at AHRR in full term low birth weight in maternal blood, placenta and cord blood in Chinese. Placenta. 2017;52:49–57. doi: 10.1016/j.placenta.2017.02.017. [DOI] [PubMed] [Google Scholar]
- 34.Tran NQV, Miyake K. Neurodevelopmental Disorders and Environmental Toxicants: Epigenetics as an Underlying Mechanism. International journal of genomics. 2017;2017:7526592. doi: 10.1155/2017/7526592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwender K, et al. Sudden infant death syndrome: exposure to cigarette smoke leads to hypomethylation upstream of the growth factor independent 1 (GFI1) gene promoter. Forensic science, medicine, and pathology. 2016;12:399–406. doi: 10.1007/s12024-016-9812-y. [DOI] [PubMed] [Google Scholar]
- 36.Meng, X., Sun, Y., Duan, W. & Jia, C. Meta-analysis of the association of maternal smoking and passive smoking during pregnancy with neural tube defects. International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics, 10.1002/ijgo.12334 (2017). [DOI] [PubMed]
- 37.Yoshigai E, Kawamura S, Kuhara S, Tashiro K. Trim36/Haprin plays a critical role in the arrangement of somites during Xenopus embryogenesis. Biochemical and biophysical research communications. 2009;378:428–432. doi: 10.1016/j.bbrc.2008.11.069. [DOI] [PubMed] [Google Scholar]
- 38.Singh N, et al. A homozygous mutation in TRIM36 causes autosomal recessive anencephaly in an Indian family. Human molecular genetics. 2017;26:1104–1114. doi: 10.1093/hmg/ddx295. [DOI] [PubMed] [Google Scholar]
- 39.He Z, et al. TRIM36 hypermethylation is involved in polycyclic aromatic hydrocarbons-induced cell transformation. Environmental pollution. 2017;225:93–103. doi: 10.1016/j.envpol.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 40.Berkel S, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nature genetics. 2010;42:489–491. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- 41.Iwai N, et al. Association Between <strong> <em> SAH </em></strong>, an Acyl-CoA Synthetase Gene, and Hypertriglyceridemia, Obesity, and Hypertension. Circulation. 2002;105:41–47. doi: 10.1161/hc0102.101780. [DOI] [PubMed] [Google Scholar]
- 42.Gopal R, Selvarasu K, Pandian PP, Ganesan K. Integrative transcriptome analysis of liver cancer profiles identifies upstream regulators and clinical significance of ACSM3 gene expression. Cellular oncology. 2017;40:219–233. doi: 10.1007/s13402-017-0321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galdzicka M, et al. A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Molecular genetics and metabolism. 2002;77:291–295. doi: 10.1016/S1096-7192(02)00178-6. [DOI] [PubMed] [Google Scholar]
- 44.Kwon, E. K. et al. The Role of Ellis-Van Creveld 2(EVC2) in Mice During Cranial Bone Development. Anatomical record, 10.1002/ar.23692 (2017). [DOI] [PMC free article] [PubMed]
- 45.Zhang H, et al. Loss of Function of Evc2 in Dental Mesenchyme Leads to Hypomorphic Enamel. Journal of dental research. 2017;96:421–429. doi: 10.1177/0022034516683674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Ani AH, Antoun JS, Thomson WM, Merriman TR, Farella M. Maternal Smoking during Pregnancy Is Associated with Offspring Hypodontia. Journal of dental research. 2017;96:1014–1019. doi: 10.1177/0022034517711156. [DOI] [PubMed] [Google Scholar]
- 47.Xuan Z, et al. Maternal active smoking and risk of oral clefts: a meta-analysis. Oral surgery, oral medicine, oral pathology and oral radiology. 2016;122:680–690. doi: 10.1016/j.oooo.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 48.Reese SE, et al. DNA Methylation Score as a Biomarker in Newborns for Sustained Maternal Smoking during Pregnancy. Environmental health perspectives. 2017;125:760–766. doi: 10.1289/EHP333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kishi R, et al. Cohort profile: the Hokkaido study on environment and children’s health in Japan. International journal of epidemiology. 2011;40:611–618. doi: 10.1093/ije/dyq071. [DOI] [PubMed] [Google Scholar]
- 50.Kishi R, et al. Ten years of progress in the Hokkaido birth cohort study on environment and children’s health: cohort profile–updated 2013. Environ Health Prev Med. 2013;18:429–450. doi: 10.1007/s12199-013-0357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aryee MJ, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fortin JP, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen YA, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Touleimat N, Tost J. Complete pipeline for Infinium((R)) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4:325–341. doi: 10.2217/epi.12.21. [DOI] [PubMed] [Google Scholar]
- 55.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bibikova M, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 57.Fox, J. & Weisberg, S. Robust Regression in R: An Appendix to An R Companion to Applied Regression. 2nd edn, (Sage, 2011).
- 58.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 59.Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;30:1431–9. doi: 10.1093/bioinformatics/btu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharp QC, et al. Maternal alcohol consumption and offspring DNA methylation: findings from six general population-based birth cohorts. Epigenomics. 2018;10:27–42. doi: 10.2217/epi-2017-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analysed during the current study are available from the corresponding author on request.