

Abstract
Molluscum contagiosum virus (MCV) causes persistent, benign skin neoplasm in children and adults. MCV is refractive to growth in standard tissue culture and there is no relevant animal model of infection. Here we investigated whether another poxvirus (vaccinia virus; VACV) could be used to examine MCV immunoevasion protein properties in vivo. The MCV MC159L or MC160L genes, which encode NF-κB antagonists, were inserted into an attenuated VACV lacking an NF-κB antagonist (vΔA49), creating vMC159 and vMC160. vMC160 slightly increased vΔA49 virulence in the intranasal and intradermal routes of inoculation. vMC159 infection was less virulent than vΔA49 in both inoculation routes. vMC159-infected ear pinnae did not form lesions, but virus replication still occurred. Thus, the lack of lesions was not due to abortive virus replication. This system provides a new approach to examine MCV immunoevasion proteins within the context of a complete and complex immune system.
Keywords: molluscum contagiosum virus, poxvirus, intradermal infection, MC159, MC160, pathogenesis
Molluscum contagiosum virus (MCV) is dermatotropic poxvirus, and is the aetiological agent of molluscum contagiosum (MC) [1]. MCV infections are common and worldwide [2, 3]. MCV infects keratinocytes and infections can persist for months to years [4]. MC neoplasms are small and have little inflammation associated with them [1]. Lesions that spontaneously regress have increased numbers of apoptosing cells, cytotoxic T cells, natural killer cells and type I IFN-expressing plasmacytoid dendritic cells [5]. Thus, it is presumed that one key to MCV persistence lies in MCV modulating the host response.
Very little is known about MCV immune evasion strategies as compared to other viruses. This is because MCV is refractive to growth in standard tissue culture. The sequencing of the MCV genome revealed that MCV encodes at least 40 known or predicted immune evasion molecules [6, 7]. Several of these proteins were characterized by studying them independently of MCV infection [4, 8–11]. However, how these MCV immune evasion molecules play a role in viral pathogenesis remains unknown.
To overcome this technical barrier, we chose to deliver MCV immune evasion proteins (MC159 and MC160) to mice during a related poxvirus (VACV) infection. MC159 and MC160 were examined because each protein inhibits NF-κB activation, yet each uses different mechanisms to antagonize the NF-κB activation pathway [12–15]. We chose VACV because it is the best-studied poxvirus [16], and there are several excellent animal models of VACV infection that allow the impact of viral immune evasion molecules on viral pathogenesis to be studied [17–20]. Additionally, we published that MC159 and MC160 inhibit NF-κB and IRF3 activation in murine cell lines [21]. Further, Randall et al. showed that MC159 interacts with the murine NF-κB essential modulator (NEMO) of the IκB kinase (IKK) complex to inhibit NF-κB activation [15]. We also have unpublished data showing that MC160 induces murine IKK1 degradation, similar to MC160’s effect on human IKK1 [13, 14]. Thus, even though MCV is a human pathogen, it is likely that MC159 and MC160 interact with some of the known equivalent murine-binding partners involved in immune surveillance.
VACV strain vΔA49 was used as the parental virus (Fig. 1a). vΔA49 lacks A49, which is an NF-κB antagonist [22]. We chose this virus because vΔA49 is moderately decreased in virulence [22], and because MC159 and MC160 are reported to inhibit NF-κB activation [12, 14, 15]. Furthermore, MC159 and MC160 each act upstream of A49 [12, 14, 15, 22]. Thus, insertion of MC159 or MC160 into vΔA49 might restore vΔA49 virulence.
Fig. 1.
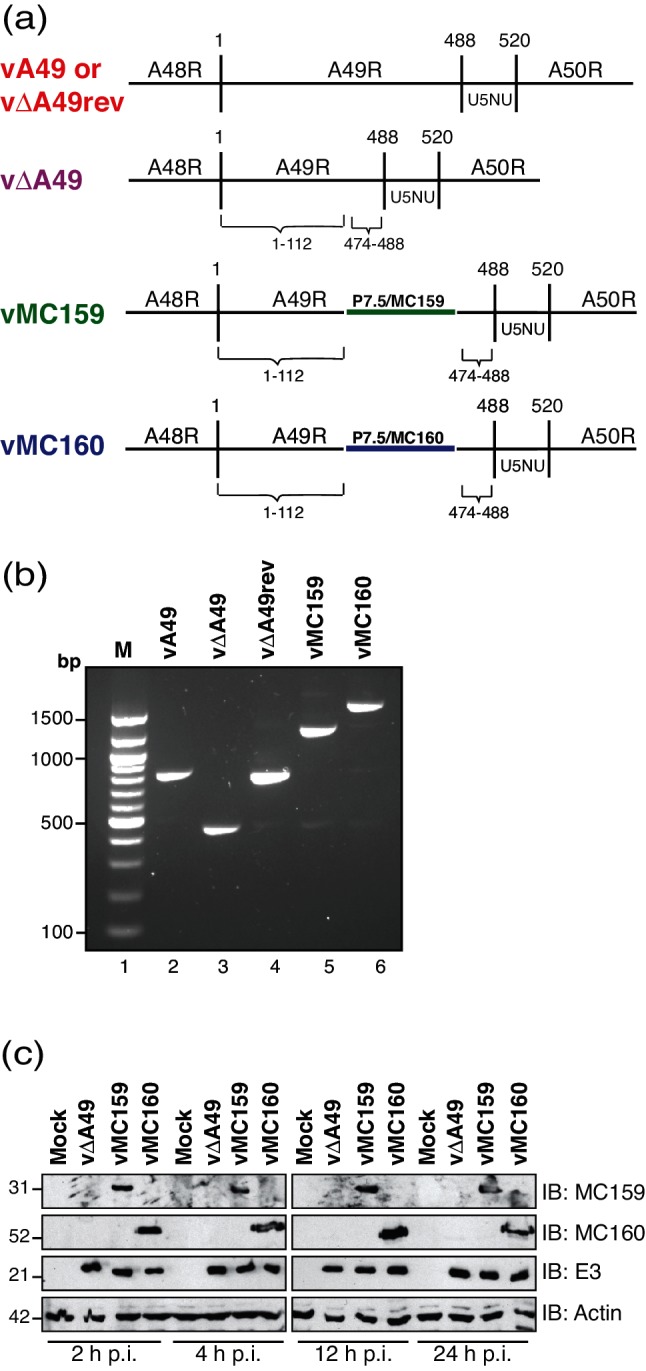
Characterization of vaccinia viruses expressing either MC159 or MC160. (a) A schematic of the viruses used in this study, focusing on the portion of the VACV WR genome containing the A49R gene and portions of the A48R and A50R genes flanking A49R. vΔA49 is a VACV strain in which A49R nucleotides 113–473 are deleted. Either the MCV MC159L or the MC160L genes, each under the control of the VACV p7.5 promoter, were inserted into vΔA49 to create vMC159 or vMC160, respectively. U5NU is the early gene transcription termination signal. (b) BSC40 monolayers were either mock-infected or infected with the indicated viruses (m.o.i.=10). At 24 h p.i., cells were collected and DNA was isolated. The DNA was PCR-amplified using a forward primer specific for A48R and a reverse primer specific for A50R [22]. A portion of each PCR reaction was analysed by gel electrophoresis. Bands were detected by ethidium bromide staining of the gel. (c) BSC40 monolayers were either mock-infected or infected with vΔA49, vMC159 or vMC160 (m.o.i.=10). Cells were lysed at the indicated times and 15 µg of clarified cellular lysates was subjected to immunoblotting for the presence of MCV (MC159 or MC160) or VACV (E3) proteins or cellular β-actin.
The MC159L and MC160L genes were stably inserted into vΔA49, a virus construct that deleted nucleotides 113–473 of the A49R gene [22] (Fig. 1a). The expression of MC159L and MC160L was controlled by the VACV p7.5 promoter to ensure MCV gene expression throughout VACV infection. Of course, the expression profile for these MCV genes in VACV may differ from that of its profile during a natural MCV infection. The creation of vMC159 was described previously [12]. The strategy for constructing vMC160 was similar.
To create vMC160, an MC160L gene under the control of the VACV p7.5 promoter was inserted into the pΔA49MCS plasmid [12]. pΔA49MCS contains a mutated A49R gene lacking nt 113–473 that is flanked by a portion of the A48R and A50R genes [12]. The A48 flank begins at A48R nt 457 and continues through to A49R nt 112 for a 338-bp product [12]. It also possesses multiple cloning sites for insertion of the MC160L gene [12]. To create MC160/pΔA49MCS, the MC160L gene was PCR-amplified from MC160/pCI [13] using the forward primer 5′-GGATCTATAATCATGGCGCACGAGCCA-3′ and the reverse primer 5′- CTAGACTAGTCTAGTAGGAAGCTTTCGTT-3′ to yield a 1212-bp PCR product. The MC160 nucleotides are underlined. The reverse primer for MC160 was engineered in the Spe I restriction enzyme digestion site (italicized). The p7.5 promoter was PCR-amplified from pUC13/gpt/EGFP [22], yielding a 121-bp product. The forward primer was 5′-TTTTATCGATTAAATAATAAATACAATAATTAATTT CTCGT-3′ and the reverse primer was 5′-CTCG TGCGCCATGATTATAGATCCGTCACTG-3′, and this yielded a 121-bp PCR product. The forward primer introduced a ClaI restriction enzyme site (italicized). Next, 50–100 ng of the gel-purified PCR products was joined by SOE and PCR-amplified using the A48R forward and A50R reverse primers described in Biswas et al. [12]. The resultant PCR product (1433 bp) was digested with ClaI and SpeI and inserted into pΔA49MCS that had been digested with ClaI and SpeI and treated with SAP. This plasmid was named MC160/pΔA49MCS.
To create vMC160, CV-1 cells were infected with vΔA49 and transfected with pMC160, and transient dominant selection was used, as described previously [23]. Recombinant viruses were collected 24 h later, selected in the presence of mycophenolic acid, xanthine and hypoxanthine [23]. This process was repeated three times to isolate recombinant viruses away from parental viruses. Intermediate EcoGPT+ viruses were resolved into vMC160 by plaquing on BSC-1 cells in the absence of drugs.
As shown in Fig. 1(b), the genotype of vMC160 was confirmed by using PCR analysis. Note that the 362-bp amplicon from vΔA49-infected cells increased in size to 1692 bp, reflecting insertion of the 883- and 1333-bp MC160L inserts. Similarly, there was an 883-bp product when PCR-amplifying the region flanking MC159L, as expected. Finally, the 55 kDa MC160 protein expression was detected in infected cells as early as 2 h post-infection (p.i.) and remained detectable at 24 h p.i. using polyclonal antiserum-specific MC160 [24] (Fig. 1c). As previously reported, the 31 kDa MC159 protein was also detected using anti-MC159 antiserum. The VACV E3 protein, an early protein, was detected throughout infection, as predicted [25]. The actin levels were similar in each lane, showing even protein loading.
Intranasal (IN) inoculation allows the examination of VACV virulence; VACV initially infects the lungs and then spreads to the brain and other distal organs [17, 26]. BALB/c mice were inoculated IN with 5×103 p.f.u. of a virus as described [27]. The mice were examined daily and we used a five-point scoring system that determines the extent of illness [28, 29]. This was performed as a blinded study to minimize potential bias. We only analysed the planned comparisons of vΔA49 to either vMC159, vMC160 or vΔA49rev, and set P-values of <0.05 (indicated by one asterisk), P<0.01 (as indicated by two asterisks) and P<0.0001 (as indicated by four asterisks) as the criteria for the statistical significance of these and the remaining assays involving mice.
PBS-inoculated mice showed no signs of illness at any time point. vΔA49rev, which is equivalent to wild-type VACV strain WR, triggered clinical signs of illness, similar to previously reported results [22] (Fig. 2a). vΔA49 infection delayed illness onset, and the clinical scores were consistently lower than those for vΔA49rev-infected mice, as noted previously [22]. These differences were statistically significant on days 7–13 p.i. (Fig. 2a). vMC159 infection appeared to cause a milder disease; the clinical scores from vMC159-infected mice were lower than those from vΔA49-infected mice from days 8–12 p.i., and these differences were statistically significant on days 8, 9 and 11–13 p.i. (Fig. 2a). In comparison to vΔA49 infection, mice infected with vMC160 showed increased signs of illness (Fig. 2a). These differences were statistically significant at days 7 and 8 p.i. (Fig. 2a). These data suggest that the MC159L gene reduced the virulence of vΔA49, while MC160L partially substituted for A49L.
Fig. 2.
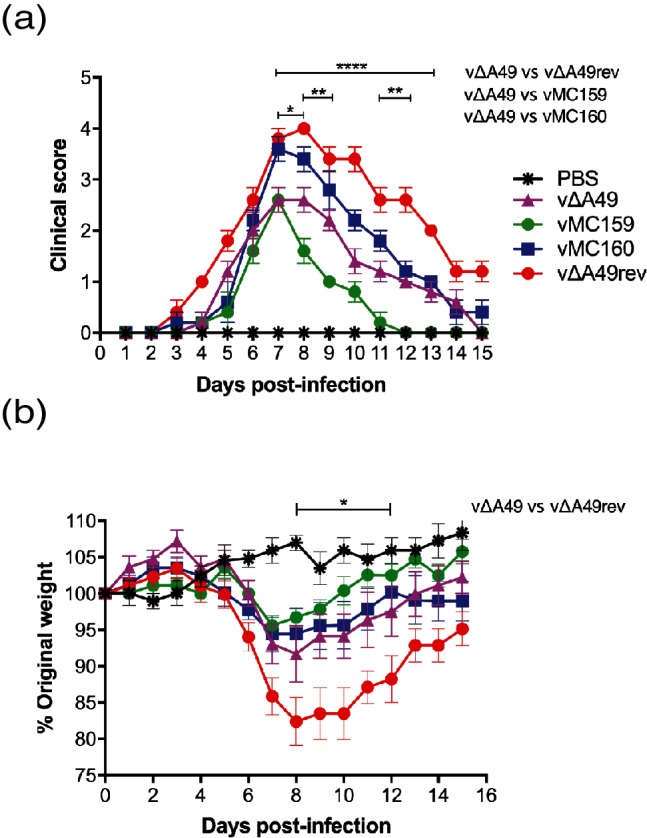
The effect of MCV genes on virus virulence using the intranasal (IN) route of infection. Female BALB/c mice (n=5 per group) were inoculated IN with 5×103 p.f.u. vΔA49, vΔA49rev, vMC159, or vMC160 or PBS. (a) Clinical signs of illness were monitored daily for 15 days and scored from 0 to 5. Clinical scores were expressed as the mean for each group. One-way analysis of variance (ANOVA) was performed followed by Tukey’s multiple comparison test to determine statistical significance. Asterisks indicate the days on which the clinical signs of illness induced were significantly different between the indicated groups. (*P<0.05, **P<0.01 or ****P<0.0001). (b) The mice were weighed daily, and data were expressed as the percentage of weight change from day 0. To determine statistically significant differences between weight change during virus infection, two-way ANOVA, followed by Tukey’s multiple comparison test, was performed. Asterisks indicate the days on which weight changes induced by vΔA49 were significantly different from those induced by vΔA49rev (*P<0.05).
Weight loss is an additional measure of virus virulence for IN inoculations [30]. The same mice as in Fig. 2(a) were also weighed daily, and data were expressed as the percentage of the mean of each individual animal’s weight loss from day 0+/−SEM [27]. The results are shown in Fig. 2(b). We only analysed the planned comparisons of vΔA49 to either vMC159, vMC160 or vΔA49rev. vΔA49rev infection caused weight loss similar to that reported previously [22]. vΔA49-infected mice lost less weight than vΔA49rev-infected mice, as expected [22], and these differences were statistically significant on days 8–12 p.i. (Fig. 2b). vMC159 caused less weight loss than vΔA49 at days 8–15 p.i., although these differences in weight were not statistically significant. vMC160-infected mice also weighed slightly more than vΔA49-infected mice at days 8–12 p.i., but these differences were not statistically significant. These data suggested that MC159 decreased virus virulence to a greater extent than vMC160 in the IN infection model.
The intradermal (ID) inoculation of a mouse ear pinna provides an alternative model to examine VACV virulence [19, 31]. In this case, lesion formation and lesion size is used to quantify virulence [18]. One could argue that ID inoculations most closely represent the location of natural, dermatotropic MCV infections. There also are some parallels in the immune responses to ID VACV infections and MCV infections. For example, type I IFN appears to be important in controlling lesion size in VACV-infected ears [32] and lesion resolution during MCV infections [5].
For ID inoculations, C57BL/6 mice were inoculated ID in the left ear dorsal pinna with 104 p.f.u. of each virus as described in [27]. The infected ears were examined daily for the next 18 days for the presence of lesions, and the results are shown in Fig. 3(a). We only analysed the planned comparisons of vΔA49 to either vMC159, vMC160 or vΔA49rev. For all virus infections, no lesions were visually detected for the first six days p.i., a routine observation [27]. Gross lesions were observed in vΔA49rev-infected mice starting on day 7 p.i., and lesion size increased until day 12, before resolving from days 13–18. vΔA49-induced lesion sizes were slightly smaller than those for vΔA49rev at all times examined, and were only statistically significant on days 10 and 11 p.i. vMC160-associated lesions were similar in size to the lesions produced by vΔA49rev and slightly larger than vΔA49-induced lesions. When comparing lesions from vMC160- versus vΔA49-infected mice, vMC160 lesions were significantly larger on days 7, 8 and 10 p.i., indicating that MC160 may increase virulence to some extent. Surprisingly, vMC159 inoculation did not produce a lesion at any point in time (Fig. 3a, b).
Fig. 3.

The effect of MCV genes on virus virulence using the intradermal (ID) route of inoculation. C57BL/6 mice (n=5 per group) were infected ID with 104 p.f.u. vΔA49, vΔA49rev, vMC159, or vMC160 in the left ear pinna. Lesion size was expressed as the mean for the group +/−SEM. (a) The sizes of the resulting lesions were measured daily for 18 days. The lesion size was measured by using a 0.01 mm digital caliper. The data are expressed as the means of lesion sizes±SEM. Statistical significance was determined by two-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison test. The asterisks indicate the days on which the lesion size caused was statistically significantly between indicated groups (*P<0.05 or ****P<0.0001). (b) Representative images of inoculated ear pinnae at 10 days p.i. vΔA49-infected mice (left panel) or vMC159-infected mice (right panel). (c, d) At the indicated days p.i., ears were collected, homogenized and lysed, and the viral titres of the lysates were determined by plaque assay. Each symbol represents the virus titre from an individual animal, and the mean titre is indicated by a line. The data are expressed as the mean titre of virus (p.f.u.) per gram of tissue (d) and as the total p.f.u. per ear (c). Statistical significance was determined by the Kruskal–Wallis test. The asterisks indicate data points at which the titres of the viruses were statistically significantly different from the others (*P<0.05).
The most striking results observed were those for the ID model of infection, and that there was a complete lack of lesion formation during vMC159 infection. Only two other VACV strains have been reported to not cause lesion formation in skin: vΔA36R and NYVAC [18, 33]. vΔA36R and NYVAC cannot spread from cell to cell efficiently in vitro [18, 33, 34]. It is thought that ΔA36R or NYVAC do not cause lesions because they spread less efficiently to neighbouring cells in vivo, resulting in an abortive infection process.
One question was why vMC159 would cause no lesions. vMC159 and vMC160 each replicated to the same levels as vΔA49 using either one-step or multi-step growth curve assays in mouse embryo fibroblasts (MEFs) (data not shown). Also, vMC159- and vMC160-formed plaques were similar to those for vΔA49 and vA49 in MEFs and BSC40 cellular monolayers (data not shown). Thus, it was unlikely that vMC159 spreads less efficiently for the reasons suspected for vΔA36R and NYVAC. Another possibility was that MC159 and MC160 may not interact with the murine homologues of their binding partners. This is also unlikely because MC159 interacts with murine NEMO [15] and MC160 induces degradation of murine IKK1 (data not shown). Next, the viral titres were quantified at 3, 5 and 11 days p.i. [27]. The maximum VACV titres were detected at day 5 p.i. [19], and we chose these time points to detect virus replication prior to and after maximal replication. vMC160 titres were not examined because the vMC160-induced ear lesions were similar to those for vΔA49 and vΔA49rev.
The data in Fig. 3 are shown as both p.f.u. per ear (panel c) and p.f.u. per gram of tissue (panel d). The starting inoculum was 104 p.f.u. in each ear pinna [27]. The data showed that all of the viruses replicated because the virus titres were higher than the initial inoculum (104 p.f.u.) on days 3 and 5 p.i. The vMC159 titres were lower than those of the wild-type (vA49rev) or parental (vΔA49) viruses at all times tested. The vΔA49 and vΔA49rev titres increased to approximately 1.3×106 by day 3 p.i. (Fig. 3c). In contrast, the vMC159 titres were 1.2×105 p.f.u. at day 3 p.i. All of the viruses continued to replicate during the next 48 h because the titres increased between 3 and 5 days p.i. By day 11 p.i., virus replication had waned, as indicated by the decrease in the virus titres. Note that the decrease in the virus titres from day 5 to day 11 p.i. implies that there is indeed immune-mediated clearance of virus, but this occurred without accompanying inflammation.
One could argue that no lesions arise because the vMC159 titres are lower than those of vΔA49 in ear pinnae. Indeed, it is unclear what virus titre threshold is needed for lesion formation. Tscharke et al. showed that inoculation with as little as 102 p.f.u. of VACV induces ear lesion formation [18]. The vMC159 titres greatly exceed that amount at days 3 and 5 p.i. This suggests that vMC159 decouples replication from lesion formation. Interestingly, both ΔA36R and NYVAC elicit protective immune responses (e.g. antibody production) [18, 35]. Thus, it is tempting to ask whether vMC159 also retains its immunogenicity and, if so, whether vMC159 would be useful for the vaccine field.
It is not yet clear how MC159 suppresses lesion formation, and this is a direction for future studies. It is appreciated that VACV lesions are due, in part, to immunopathology because smaller lesions are associated with decreases in the expression of multiple cytokines and chemokines [27, 36]. Thus, MC159 may either directly or indirectly prevent the expression of these host cell proteins to halt pro-inflammatory processes. In this case, the ability of MC159 to inhibit NF-κB and IRF3 activation may be relevant. MC159 inhibits apoptosis, while MC160 does not [24, 37]. Another speculation, then, is that this anti-apoptotic property of MC159 affords virus attenuation, perhaps by allowing virus-infected cells to survive for prolonged time periods. Interestingly, a mutant VACV lacking an apoptosis antagonist (ΔB13R) has an increased lesion size as compared to wild-type VACV [18], showing an instance where inhibition of apoptosis diminishes lesion formation.
This is the first report that examines MCV immune antagonists in the context of an animal infection. MC159 and MC160 are members of the FLIP family of proteins [4]. They each possess tandem death effector domains (DEDs) that share 43 % similarity. MC159 and MC160 also share some biological features, including inhibition of NF-κB and IRF3 [4]. Despite their similarity, MC159 and MC160 likely have distinct roles during MCV infection in vivo, as indicated by the data here. VACV itself expresses at least 10 different NF-κB inhibitory proteins [20], indicating that control of this pathway is critical for the survival of VACV in vivo. Perhaps MCV also expresses MC159 and MC160, along with the two other known MCV NF-κB antagonists (MC005, MC132), for similar reasons when confronting a complex, multi-faceted anti-viral immune response [10, 11].
To date, there is no cell culture system or laboratory animal model to study MCV replication and pathogenesis. Researchers have used creative approaches to extrapolate the biological importance of MC159 as an immune evasion molecule. This includes the development of transgenic mouse strains that express MC159 [38, 39] or using murine cytomegalovirus to express MC159 [40]. Our system uses VACV during ID mouse infections, and is perhaps the model that is closest to mimicking MCV infection at this current time. One could argue that the addition of MC159 to vΔA49 resulted in a disease that mimics MC because, like MC, there is little inflammation.
The current study demonstrates the biological effects of two well-characterized MCV immune evasion proteins in a newly created system. MCV encodes at least 40 other known or predicted immune evasion molecules [6, 7]. Thus, studies of MCV immune evasion molecules provide a rich opportunity to identify novel aspects of virus–host interactions during persistent infections. The surrogate system described here allows these types of studies to be performed to better understand MCV pathogenesis and persistent virus infections.
Funding information
This work was supported by grants from the University of Illinois Campus Research Board and the National Institutes of Health (AI117105). G. L.S. is a Wellcome Trust Principal Research Fellow.
Acknowledgements
The authors thank Dr Ariana Bravo Cruz, Lauren Gates and Melissa Ryerson for advice and review.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
The work described was undertaken with ethical approval from the University of Illinois IACUC.
Footnotes
Abbreviations: DED, death effector domain; FLIP, FLICE-like inhibitory protein; ID, intradermal; IFN, interferon; IKK, inhibitor of NF-κB (IκB) kinase complex; IKK1, IKK1 subunit; IN, intranasal; IRF3, interferon regulatory transcription factor 3; MC, molluscum contagiosum; MCV, molluscum contagiosum virus; NEMO, NF-κB essential modulator; NF-κB, nuclear factor kappa B; VACV, vaccinia virus; WR, Western Reserve.
References
- 1.Damon IK. Poxviruses. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013. pp. 2160–2184. (editors) [Google Scholar]
- 2.Hay RJ, Johns NE, Williams HC, Bolliger IW, Dellavalle RP, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol. 2014;134:1527–1534. doi: 10.1038/jid.2013.446. [DOI] [PubMed] [Google Scholar]
- 3.Gottlieb SL, Myskowski PL. Molluscum contagiosum. Int J Dermatol. 1994;33:453–461. doi: 10.1111/j.1365-4362.1994.tb02853.x. [DOI] [PubMed] [Google Scholar]
- 4.Shisler JL. Immune evasion strategies of molluscum contagiosum virus. Adv Virus Res. 2015;92:201–252. doi: 10.1016/bs.aivir.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Vermi W, Fisogni S, Salogni L, Schärer L, Kutzner H, et al. Spontaneous regression of highly immunogenic molluscum contagiosum virus (MCV)-induced skin lesions is associated with plasmacytoid dendritic cells and IFN-DC infiltration. J Invest Dermatol. 2011;131:426–434. doi: 10.1038/jid.2010.256. [DOI] [PubMed] [Google Scholar]
- 6.Senkevich TG, Bugert JJ, Sisler JR, Koonin EV, Darai G, et al. Genome sequence of a human tumorigenic poxvirus: prediction of specific host response-evasion genes. Science. 1996;273:813–816. doi: 10.1126/science.273.5276.813. [DOI] [PubMed] [Google Scholar]
- 7.Senkevich TG, Koonin EV, Bugert JJ, Darai G, Moss B. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology. 1997;233:19–42. doi: 10.1006/viro.1997.8607. [DOI] [PubMed] [Google Scholar]
- 8.Chen X, Anstey AV, Bugert JJ. Molluscum contagiosum virus infection. Lancet Infect Dis. 2013;13:877–888. doi: 10.1016/S1473-3099(13)70109-9. Research Support, Non-U.S. Gov't Review. [DOI] [PubMed] [Google Scholar]
- 9.Coutu J, Ryerson MR, Bugert J, Brian Nichols D. The molluscum contagiosum virus protein MC163 localizes to the mitochondria and dampens mitochondrial mediated apoptotic responses. Virology. 2017;505:91–101. doi: 10.1016/j.virol.2017.02.017. [DOI] [PubMed] [Google Scholar]
- 10.Brady G, Haas DA, Farrell PJ, Pichlmair A, Bowie AG. Molluscum contagiosum virus protein MC005 inhibits NF-κB activation by targeting NEMO-regulated IκB kinase activation. J Virol. 2017;91:e00545-17. doi: 10.1128/JVI.00545-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brady G, Haas DA, Farrell PJ, Pichlmair A, Bowie AG. Poxvirus protein MC132 from molluscum contagiosum virus inhibits NF-B activation by targeting p65 for degradation. J Virol. 2015;89:8406–8415. doi: 10.1128/JVI.00799-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas S, Shisler JL. Molluscum contagiosum virus MC159 abrogates cIAP1-NEMO interactions and inhibits NEMO polyubiquitination. J Virol. 2017;91:e00276-17. doi: 10.1128/JVI.00276-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichols DB, Shisler JL. The MC160 protein expressed by the dermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosis factor alpha-induced NF-κB activation via inhibition of I kappa kinase complex formation. J Virol. 2006;80:578–586. doi: 10.1128/JVI.80.2.578-586.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nichols DB, Shisler JL. Poxvirus MC160 protein utilizes multiple mechanisms to inhibit NF-κB activation mediated via components of the tumor necrosis factor receptor 1 signal transduction pathway. J Virol. 2009;83:3162–3174. doi: 10.1128/JVI.02009-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Randall CM, Jokela JA, Shisler JL. The MC159 protein from the molluscum contagiosum poxvirus inhibits NF-κB activation by interacting with the IκB kinase complex. J Immunol. 2012;188:2371–2379. doi: 10.4049/jimmunol.1100136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moss B. Poxviridae. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2013. [Google Scholar]
- 17.Reading PC, Smith GL. A kinetic analysis of immune mediators in the lungs of mice infected with vaccinia virus and comparison with intradermal infection. J Gen Virol. 2003;84:1973–1983. doi: 10.1099/vir.0.19285-0. [DOI] [PubMed] [Google Scholar]
- 18.Tscharke DC, Reading PC, Smith GL. Dermal infection with vaccinia virus reveals roles for virus proteins not seen using other inoculation routes. J Gen Virol. 2002;83:1977–1986. doi: 10.1099/0022-1317-83-8-1977. [DOI] [PubMed] [Google Scholar]
- 19.Tscharke DC, Smith GL. A model for vaccinia virus pathogenesis and immunity based on intradermal injection of mouse ear pinnae. J Gen Virol. 1999;80:2751–2755. doi: 10.1099/0022-1317-80-10-2751. [DOI] [PubMed] [Google Scholar]
- 20.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, et al. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol. 2013;94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 21.Randall CM, Biswas S, Selen CV, Shisler JL. Inhibition of interferon gene activation by death-effector domain-containing proteins from the molluscum contagiosum virus. Proc Natl Acad Sci USA. 2014;111:E265. doi: 10.1073/pnas.1314569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mansur DS, Maluquer de Motes C, Unterholzner L, Sumner RP, Ferguson BJ, et al. Poxvirus targeting of E3 ligase β-TrCP by molecular mimicry: a mechanism to inhibit NF-κB activation and promote immune evasion and virulence. PLoS Pathog. 2013;9:e1003183. doi: 10.1371/journal.ppat.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falkner FG, Moss B. Transient dominant selection of recombinant vaccinia viruses. J Virol. 1990;64:3108–3111. doi: 10.1128/jvi.64.6.3108-3111.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shisler JL, Moss B. Molluscum contagiosum virus inhibitors of apoptosis: the MC159 v-FLIP protein blocks Fas-induced activation of procaspases and degradation of the related MC160 protein. Virology. 2001;282:14–25. doi: 10.1006/viro.2001.0834. [DOI] [PubMed] [Google Scholar]
- 25.Weaver JR, Shamim M, Alexander E, Davies DH, Felgner PL, et al. The identification and characterization of a monoclonal antibody to the vaccinia virus E3 protein. Virus Res. 2007;130:269–274. doi: 10.1016/j.virusres.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alcamí A, Smith GL. A mechanism for the inhibition of fever by a virus. Proc Natl Acad Sci USA. 1996;93:11029–11034. doi: 10.1073/pnas.93.20.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bravo Cruz AG, Han A, Roy EJ, Guzmán AB, Miller RJ, et al. Deletion of the K1L gene results in a vaccinia virus that is less pathogenic due to muted innate immune responses, yet still elicits protective immunity. J Virol. 2017;91:e00542-17. doi: 10.1128/JVI.00542-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berhanu A, King DS, Mosier S, Jordan R, Jones KF, et al. ST-246 inhibits in vivo poxvirus dissemination, virus shedding, and systemic disease manifestation. Antimicrob Agents Chemother. 2009;53:4999–5009. doi: 10.1128/AAC.00678-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alcamí A, Smith GL. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell. 1992;71:153–167. doi: 10.1016/0092-8674(92)90274-G. [DOI] [PubMed] [Google Scholar]
- 30.Williamson JD, Reith RW, Jeffrey LJ, Arrand JR, Mackett M. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J Gen Virol. 1990;71:2761–2767. doi: 10.1099/0022-1317-71-11-2761. [DOI] [PubMed] [Google Scholar]
- 31.Lin LC, Smith SA, Tscharke DC. An intradermal model for vaccinia virus pathogenesis in mice. Methods Mol Biol. 2012;890:147–159. doi: 10.1007/978-1-61779-876-4_9. [DOI] [PubMed] [Google Scholar]
- 32.Fischer MA, Davies ML, Reider IE, Heipertz EL, Epler MR, et al. CD11b+, Ly6G+ cells produce type I interferon and exhibit tissue protective properties following peripheral virus infection. PLoS Pathog. 2011;7:e1002374. doi: 10.1371/journal.ppat.1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet JC, et al. NYVAC: a highly attenuated strain of vaccinia virus. Virology. 1992;188:217–232. doi: 10.1016/0042-6822(92)90752-B. [DOI] [PubMed] [Google Scholar]
- 34.Parkinson JE, Smith GL. Vaccinia virus gene A36R encodes a M(r) 43-50 K protein on the surface of extracellular enveloped virus. Virology. 1994;204:376–390. doi: 10.1006/viro.1994.1542. [DOI] [PubMed] [Google Scholar]
- 35.Tartaglia J, Cox WI, Pincus S, Paoletti E. Safety and immunogenicity of recombinants based on the genetically-engineered vaccinia strain, NYVAC. Dev Biol Stand. 1994;82:125–129. [PubMed] [Google Scholar]
- 36.Stanford MM, McFadden G, Karupiah G, Chaudhri G. Immunopathogenesis of poxvirus infections: forecasting the impending storm. Immunol Cell Biol. 2007;85:93–102. doi: 10.1038/sj.icb.7100033. [DOI] [PubMed] [Google Scholar]
- 37.Shisler JL, Senkevich TG, Berry MJ, Moss B. Ultraviolet-induced cell death blocked by a selenoprotein from a human dermatotropic poxvirus. Science. 1998;279:102–105. doi: 10.1126/science.279.5347.102. [DOI] [PubMed] [Google Scholar]
- 38.Woelfel M, Bixby J, Brehm MA, Chan FK. Transgenic expression of the viral FLIP MC159 causes lpr/gld-like lymphoproliferation and autoimmunity. J Immunol. 2006;177:3814–3820. doi: 10.4049/jimmunol.177.6.3814. [DOI] [PubMed] [Google Scholar]
- 39.Wu Z, Roberts M, Porter M, Walker F, Wherry EJ, et al. Viral FLIP impairs survival of activated T cells and generation of CD8+ T cell memory. J Immunol. 2004;172:6313–6323. doi: 10.4049/jimmunol.172.10.6313. [DOI] [PubMed] [Google Scholar]
- 40.Hüttmann J, Krause E, Schommartz T, Brune W. Functional comparison of molluscum contagiosum virus vFLIP MC159 with murine cytomegalovirus M36/vICA and M45/vIRA proteins. J Virol. 2015;90:2895–2905. doi: 10.1128/JVI.02729-15. [DOI] [PMC free article] [PubMed] [Google Scholar]