

Key Points
Question
Can up-front tumor sequencing replace the prevailing paradigm for universal tumor screening for Lynch syndrome, which includes a sequence of several screening tests?
Findings
This genetic sequencing study of tumor DNA from 419 consecutive patients with colorectal cancer undergoing standard universal tumor screening and germline genetic testing when indicated as part of the multicenter, population-based Ohio Colorectal Cancer Prevention Initiative underwent blinded tumor next-generation sequencing. Tumor sequencing alone had better sensitivity than immunohistochemical staining plus BRAF and microsatellite instability testing plus BRAF and equal specificity to immunohistochemical staining plus BRAF and microsatellite instability testing plus BRAF.
Meaning
Up-front tumor sequencing in colorectal cancer is simpler and has superior sensitivity to current multitest approaches to Lynch syndrome screening, while simultaneously providing critical information for treatment selection.
This genetic sequencing study explores whether up-front tumor sequencing can replace the prevailing paradigm for universal tumor screening for Lynch syndrome, which includes a sequence of several screening tests.
Abstract
Importance
Universal tumor screening for Lynch syndrome (LS) in colorectal cancer (CRC) is recommended and involves up to 6 sequential tests. Somatic gene testing is performed on stage IV CRCs for treatment determination. The diagnostic workup for patients with CRC could be simplified and improved using a single up-front tumor next-generation sequencing test if it has higher sensitivity and specificity than the current screening protocol.
Objective
To determine whether up-front tumor sequencing (TS) could replace the current multiple sequential test approach for universal tumor screening for LS.
Design, Setting, and Participants
Tumor DNA from 419 consecutive CRC cases undergoing standard universal tumor screening and germline genetic testing when indicated as part of the multicenter, population-based Ohio Colorectal Cancer Prevention Initiative from October 2015 through February 2016 (the prospective cohort) and 46 patients with CRC known to have LS due to a germline mutation in a mismatch repair gene from January 2013 through September 2015 (the validation cohort) underwent blinded TS.
Main Outcomes and Measures
Sensitivity of TS compared with microsatellite instability (MSI) testing and immunohistochemical (IHC) staining for the detection of LS.
Results
In the 465 patients, mean age at diagnosis was 59.9 years (range, 20-96 years), and 241 (51.8%) were female. Tumor sequencing identified all 46 known LS cases from the validation cohort and an additional 12 LS cases from the 419-member prospective cohort. Testing with MSI or IHC, followed by BRAF p.V600E testing missed 5 and 6 cases of LS, respectively. Tumor sequencing alone had better sensitivity (100%; 95% CI, 93.8%-100%) than IHC plus BRAF (89.7%; 95% CI, 78.8%-96.1%; P = .04) and MSI plus BRAF (91.4%; 95% CI, 81.0%-97.1%; P = .07). Tumor sequencing had equal specificity (95.3%; 95% CI, 92.6%-97.2%) to IHC plus BRAF (94.6%; 95% CI, 91.9%-96.6%; P > .99) and MSI plus BRAF (94.8%; 95% CI, 92.2%-96.8%; P = .88). Tumor sequencing identified 284 cases with KRAS, NRAS, or BRAF mutations that could affect therapy for stage IV CRC, avoiding another test. Finally, TS identified 8 patients with germline DPYD mutations that confer toxicity to fluorouracil chemotherapy, which could also be useful for treatment selection.
Conclusions and Relevance
Up-front TS in CRC is simpler and has superior sensitivity to current multitest approaches to LS screening, while simultaneously providing critical information for treatment selection.
Introduction
Lynch syndrome (LS) affects approximately 3% of all patients with colorectal cancer (CRC), making it the most common hereditary syndrome that predisposes individuals to develop CRC.1,2,3 Individuals with LS are also at increased risk to develop endometrial, ovarian, gastric, and other cancers.4 Identifying LS in patients with CRC and their at-risk relatives allows them to benefit from intensive cancer surveillance,5 chemoprevention,6 and risk-reducing surgical procedures.7
Universal tumor screening for LS is recommended for all patients with CRC at diagnosis,8,9,10 and there are several approaches currently used. Initial screening in tumor tissue can begin with immunohistochemical (IHC) staining for the presence of the mismatch repair (MMR) proteins (MLH1, MSH2, MSH6, PMS2) and/or microsatellite instability (MSI) analysis (Figure 1).1,2,8,11,12 If the tumor is MSI-high and/or has any absent MMR protein, the tumor is considered to have defective mismatch repair (dMMR). The majority of dMMR CRCs (70%) have absence of the MLH1 and PMS2 proteins associated with sporadic MLH1 promoter methylation. Therefore, if the MLH1 protein is absent or if the tumor is MSI-high, analysis for methylation of the MLH1 promoter and/or testing for its surrogate, the somatic BRAF p.V600E mutation, which is found in 69% of methylated cases,12 is performed. If either of these tests have positive results, LS is excluded or highly unlikely. False-negative results are a limitation of universal tumor screening; IHC sensitivity is 83% for MLH1, MSH2, or MSH6 mutations, and MSI sensitivity is 87% for MLH1 or MSH2 mutations and 77% for MSH6 mutations.12 Traditional sequential testing is complex and confusing to patients and clinicians and occurs over a prolonged period, incurring risk for loss to follow-up.
Figure 1. Present Paradigm for Universal Tumor Screening for Lynch Syndrome Among Patients With Colorectal Cancer .
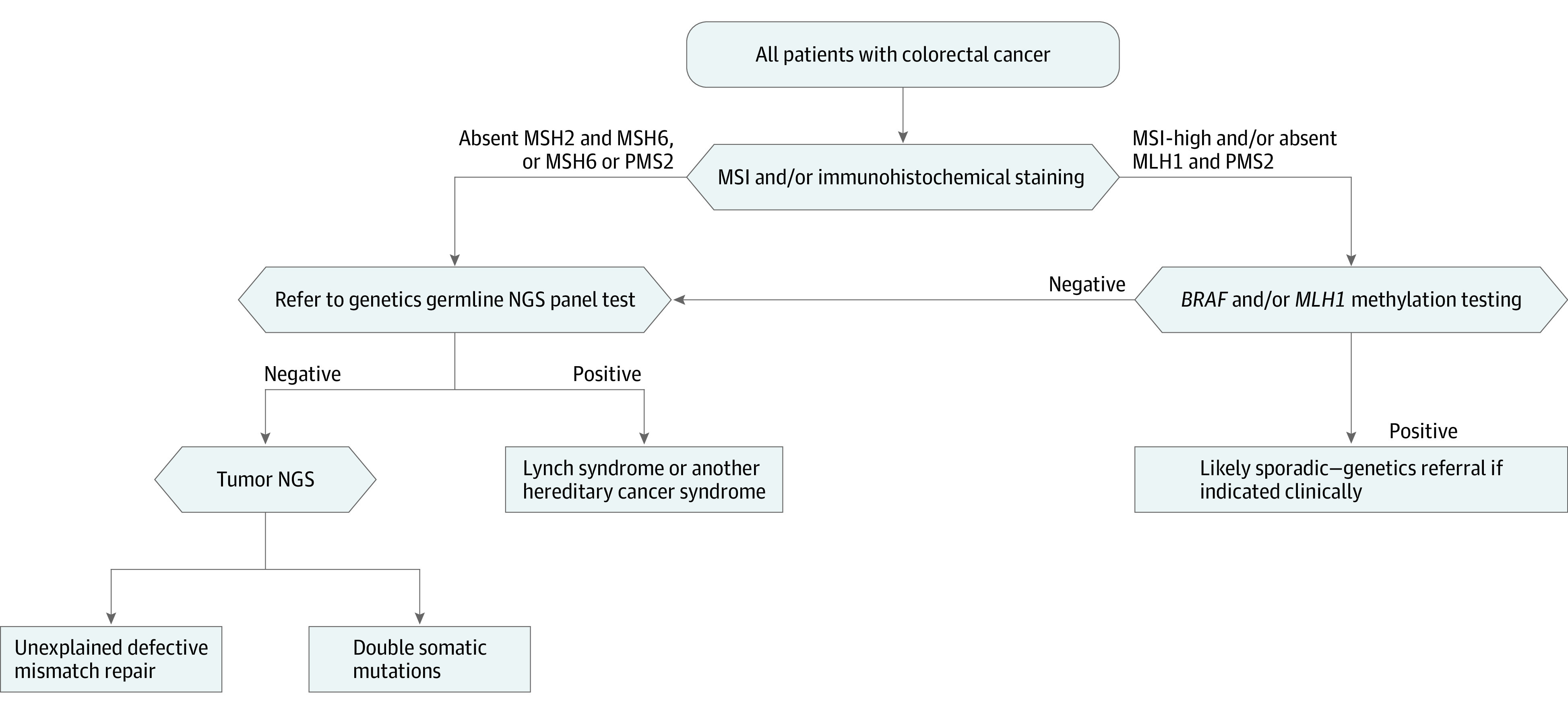
MSI indicates microsatellite instability; MSI-high, microsatellite unstable; NGS, next-generation sequencing.
Germline DNA testing of the MMR genes is performed for patients with dMMR without MLH1 promoter methylation, and a diagnosis of LS is confirmed by the presence of a germline mutation. However, only 25% to 67% of patients with a dMMR CRC are found to have a germline mutation in an MMR gene.1,2 In the past, when a germline mutation was not found, patients were left with unexplained MMR deficiency and typically treated as if they had LS without a detectable germline mutation. However, up to 68% of nonmethylated dMMR cases without a germline MMR mutation have acquired 2 somatic mutations in the MMR gene corresponding to the protein absence on IHC, resulting in dMMR in the tumor.13,14,15 These “double somatic” patients and their relatives do not have LS and do not need to follow the intensive LS cancer surveillance recommendations.
In addition to identifying patients more likely to have LS, universal tumor screening identifies patients with MSI-high tumors who may respond well to US Food and Drug Administration–approved immunotherapy.16 For treatment purposes, the National Comprehensive Cancer Network guidelines recommend that stage IV CRC tumors undergo targeted mutation testing of the KRAS, NRAS, and BRAF genes to help guide treatment.17,18,19
While it is known that tumor sequencing (TS) can be used to determine the MSI status of tumors,20,21,22,23 it is not known whether it can be used more specifically to identify patients with LS. The primary objective of this study was to compare the performance of up-front TS (Figure 2) with the current universal tumor screening for LS approaches (Figure 1). The secondary aim of this study was to evaluate TS for detection of other variants that might alter cancer therapy.
Figure 2. Proposed Universal Tumor Screening Pathway Using Tumor Sequencing for All Patients With Colorectal Cancer.

NGS indicates next-generation sequencing.
Methods
Participants
The Ohio Colorectal Cancer Prevention Initiative (OCCPI) was created with the aim of decreasing CRC incidence in Ohio by identifying patients with hereditary predisposition, increasing colonoscopy adherence for first-degree relatives of patients with CRC, and encouraging future research through the creation of a biorepository. Patients were enrolled at 51 participating Ohio hospitals. Institutional review board approval was obtained by the individual hospitals, community oncology programs, or by ceding review to the Ohio State University Institutional Review Board. Written informed consent was obtained.
All CRC cases (n = 419) undergoing testing as part of the OCCPI study between October 2015 and February 2016 were chosen for this project. Cases were coded so all individuals involved in the TS diagnostic process were blinded to previous tumor screening and germline test results. An additional validation set of 46 cases known to have germline MMR gene mutations were coded and mixed with the initial set to ensure that there were enough mutation-positive cases to test the performance of TS in the identification of germline MMR mutations. These 46 cases included all germline MMR gene mutation carriers identified from January 2013 through September 2015 in the OCCPI study for which there was leftover tumor DNA after all prior standard tests had been completed (MSI and, when necessary, MLH1 methylation). The demographic characteristics of these 465 patients are presented in Table 1.
Table 1. Demographic Characteristics of 465 Patients With Colorectal Cancer (CRC).
| Characteristic | Value (N = 465) |
|---|---|
| Age at CRC diagnosis, mean (range), y | 59.9 (20-96) |
| Age at diagnosis, No. (%), y | |
| <50 | 104 (22.4) |
| ≥50 | 361 (78.6) |
| CRC site, No. (%) | |
| Right colon | 184 (39.6) |
| Left colon | 148 (31.8) |
| Transverse colon | 35 (7.5) |
| Rectum | 86 (18.5) |
| Not specified | 12 (2.6) |
| Pathologic stage, No. (%) | |
| I | 84 (18.1) |
| II | 122 (26.2) |
| III | 184 (39.6) |
| IV | 29 (6.2) |
| Unknown | 46 (9.9) |
| Sex, No. (%) | |
| Male | 224 (48.2) |
| Female | 241 (51.8) |
| Self-reported race, No. (%) | |
| White | 414 (89.0) |
| African American or black | 38 (8.2) |
| Asian | 4 (0.9) |
| Other | 9 (1.9) |
| Hispanic, No. (%) | |
| Yes | 5 (1.1) |
| No | 460 (98.9) |
Traditional Universal Tumor Screening
The standard universal tumor screening tests and germline mutation testing panels used in the OCCPI study have been described.24 Microsatellite instability testing was completed using the Promega MSI Analysis System with instability in at least 2 of 5 markers required for MSI-high and instability in 1 of 5 markers considered MSI-low. Immunohistochemistry for the MMR proteins was performed either (1) by the hospital that performed the original pathology review using all 4 MMR stains or (2) by the central study pathologist (W.L.F.) using the 2-stain method as previously described to reduce IHC costs.25 Staining for all 4 MMR proteins was undertaken if MSI analysis could not be performed or if the MSI and IHC results were discordant. Mismatch repair proteins staining more than 5% of cancer cells were considered “present”; 1% to 5% staining was “equivocal” and we deferred to the MSI result to determine whether germline genetic testing was necessary; and less than 1% was considered “absent.” MLH1 promoter methylation was assessed at 4 CpG sites between −209 and −188 using pyrosequencing26 when tumors were MSI-high and/or absent MLH1 and PMS2 proteins on IHC. The mean percent of methylation detected at the 4 CpG sites was used to classify tumors as methylated (≥10%) or unmethylated (<10%).
A total of 197 of the 419 prospective cases underwent germline genetic testing. In accordance with current practice, any patients with CRC found to have a dMMR tumor (MSI-high and/or IHC showing lack of expression of ≥1 MMR protein) without MLH1 promoter methylation underwent germline genetic testing (n = 26). In addition, as part of the OCCPI research study, any patient with CRC with a MMR proficient tumor who received a diagnosis before age 50 years (n = 75), had a first-degree relative with CRC or endrometrial cancer (n = 82), or had synchronous or metachronous CRC or endrometrial cancer tumors underwent germline genetic testing (n = 19). Germline genetic testing for this study included a next-generation sequencing (NGS) panel of cancer susceptibility genes including all of the LS genes as described previously.24
Tumor Next-Generation Sequencing
Tumor sequencing was performed using University of Washington (UW)-OncoPlex in the Clinical Laboratory Improvement Amendments–certified laboratory setting (http://web.labmed.washington.edu/tests/genetics/UW-OncoPlex) as previously described.19,27 UW-OncoPlex is a clinically validated targeted NGS deep sequencing panel that sequences to 500 × mean depth all exons, introns, and flanking regions of MLH1, MSH2, and MSH6, and all exons of PMS2 in addition to assessing BRAF, KRAS, NRAS, and MSI status among other genes (full description of test in the eAppendix in Supplement 1).19 Analysis and interpretation of sequencing results was performed by an expert molecular pathologist (C.C.P.) who was blinded to all clinical and molecular data, and a second pass was performed by an independent molecular pathologist (B.H.S.).
Analysis
The definition of a positive screening test result in the current screening pathway is any case that was MSI-high and/or had abnormal IHC with negative BRAF V600E test results. The definition of a positive screening test result in the proposed TS pathway is any case that had mutations identified in at least 1 of the MMR genes at a variant allele fraction that was consistent with the possibility of a germline mutation with MSI and BRAF V600E status taken into account. A true positive result was any case with a germline MMR gene mutation confirmed by germline genetic testing. False-positive results included double somatic MMR mutation cases and methylated cases that were missed with BRAF testing. Sensitivity and specificity were calculated using standard methods on the combined results from all completed tests in both the prospective and validation cohorts. Positive predictive values (PPVs) and negative predictive values (NPVs) were calculated using population prevalence from the prospective cohort only because an accurate prevalence in the tested population is important for this calculation. Statistical comparisons of sensitivity and specificity results were performed in R using the McNemar test (mcnemar.test function) with continuity correction.
Results
Complete data for all 465 cases from this study can be found in the eWorksheet in Supplement 2.
Microsatellite Instability
Using the Promega polymerase chain reaction (PCR) panel, MSI-high was detected in 76 of 419 (18.1%) of the unselected CRC cases undergoing standard tumor screening for LS. All 76 tumors were also found to be MSI-high based on MSI testing by the mSINGS method with TS (sensitivity, 100%; 95% CI, 95.3%-100%).21 Among the 341 tumors classified as microsatellite stable (MSS) or MSI-low using PCR-based MSI analysis, TS findings were concordant for 340 cases (specificity, 99.7%; 95% CI, 98.4%-100%). The 1 discrepant tumor was classified as MSS by the PCR panel and MSI-high by TS. Another case was classified as MSI-low by the PCR panel but MSS by TS; this was considered concordant because MSI-low cases are treated like MSS cases for the purposes of LS screening. Two tumors that were classified as MSS by the PCR panel failed MSI testing by TS due to low DNA quality. The PPV of TS to determine MSI was 98.7% (95% CI, 91.5%-99.8%) and the NPV was 100%. This compares favorably to the ability of IHC to detect MSI-high cases (Table 2).
Table 2. Analytic Validity of Tumor Sequencing for the Detection of Microsatellite Instability (MSI) and Lynch Syndrome (LS) Compared With MSI and Immunohistochemical (IHC) Staining Followed by BRAF Testing.
| Parameter | Tumor Sequencing Including BRAF | MSI + BRAF | IHC + BRAF |
|---|---|---|---|
| MSI-high detection, % (95% CI) | |||
| Sensitivity | 100 (95.3-100) | 100 [Reference] | 98.3 (94.0-99.8) |
| Specificity | 99.7 (98.4-100) | 100 [Reference] | 99.7 (98.4-100) |
| PPVa | 98.7 (91.5-99.8) | 100 [Reference] | 99.2 (94.3-99.9) |
| NPVa | 100 | 100 [Reference] | 99.4 (97.8-99.9) |
| LS mutation detection, % (95% CI) | |||
| Sensitivity | 100 (93.8-100) | 91.4 (81.0-97.1) | 89.7 (78.8-96.1) |
| Specificity | 95.3 (92.6-97.2) | 94.8 (92.2-96.8) | 94.6 (91.9-96.6) |
| PPVa | 40 (29.8-51.1) | 34.4 (25.0-45.1) | 33.3 (24.3-43.7) |
| NPVa | 100 (99.1-100) | 99.7 (98.3-100) | 99.7 (98.3-100) |
| LS cases missed, No. | 0 | 5 | 6 |
Abbreviations: NPV, negative predictive value; PPV, positive predictive value.
Calculated using only the prospective cohort because disease prevalence can affect these measures.
MLH1 Promoter Methylation
On the basis of pyrosequencing, 50 of 76 (66%) of MSI-high cases were MLH1 methylated and 26 of 76 (34%) were unmethylated. Tumor sequencing used the included BRAF V600E mutation as a surrogate for MLH1 methylation in MSI-high tumors.12,28 Because approximately 68% of tumors with MLH1 methylation have the BRAF V600E mutation, some methylated cases will be missed using this approach.12 In this manner, TS correctly identified 42 of 50 (84% sensitivity) tumors with MLH1 promoter methylation and 26 of 26 MSI-high tumors without MLH1 methylation (100% specificity). The PPV was 100%, and the NPV was 63.6%.
LS Germline Gene Mutations
Twelve patients were found to have LS (eTable 1 in Supplement 1) (2 MLH1, 5 MSH2, 1 MSH6, 4 PMS2) in the prospective group, and TS correctly identified all 12 (100%). Current screening methods would have missed 1 patient with a PMS2 mutation (8%) because although the tumor had absent MLH1 and PMS2 proteins and was MSI-high, the MLH1 promoter was methylated. As a result, this case would have been considered a sporadic tumor and no genetic counseling or testing would have been recommended.
Validation Set of Germline MMR Gene Mutations
Tumor DNA samples from 46 patients known to have pathogenic germline mutations in MMR genes were analyzed along with the prospective cases (eTable 1 in Supplement 1) (11 MLH1, 20 MSH2, 6 MSH6, 9 PMS2). Tumor sequencing correctly identified 46 of 46 (100%) pathogenic germline MMR gene mutations in this validation set.
Comparison of TS With MSI and IHC
The diagnostic performance of the TS, MSI, and IHC assays to predict MMR gene mutations was evaluated in both cohorts combined (Table 2) (n = 464 for MSI and IHC and n = 439 for TS omitting 25 cases that failed TS due to low tumor DNA quality or quantity). One dMMR tumor (MSI-high, IHC absent MSH2 and MSH6) was removed from analysis because it had no germline mutation and no evidence of double somatic MMR gene mutations, so it was unclear whether to count this case as a true- or false-positive result.
Overall, TS (which includes BRAF testing) successfully identified all LS cases (58 of 58), whereas MSI followed by BRAF testing failed to identify 5 cases of LS and IHC followed by BRAF testing failed to identify 6 cases of LS. Three hundred sixty-three cases had a negative screening test result and 76 cases had a positive screening test result based on TS. Of the 76 cases with a positive screening test result, 58 had LS and 18 were considered false-positive results (including 9 cases with double somatic MMR mutations and 9 with MLH1 methylation). Therefore, the sensitivity of TS for the detection of LS was 100% (95% CI, 93.8%-100%) and specificity was 95.3% (95% CI, 92.6%-97.2%).
The PPV and NPV for all of the LS screening tests were calculated using only the prospective cohort because disease prevalence can affect these measures. The PPV of TS was 40% (12 of 30; 95% CI, 29.8%-51.1%) and NPV was 100% (363 of 363; 95% CI, 99.1%-100%).
For comparison in the same cohort, the sensitivity of MSI followed by BRAF testing for the detection of LS was 91.4% (95% CI, 81.0%-97.1%) and specificity was 94.8% (95% CI, 92.2%-96.8%). The PPV was 34.4% (11 of 32; 95% CI, 25.0%-45.1%) and NPV was 99.7% (385 of 386; 95% CI, 98.3%-100%). The sensitivity of IHC followed by BRAF testing for the detection of LS was 89.7% (95% CI, 78.8%-96.1%) and the specificity was 94.6% (95% CI, 91.9%-96.6%). The PPV was 33.3% (11 of 33; 95% CI, 24.3%-43.7%) and NPV was 99.7% (383 of 384; 95% CI, 98.3%-100%).
The sensitivity of TS (which includes BRAF testing) was significantly higher than the sensitivity of IHC followed by BRAF (P = .04) and was higher (but not statistically significantly) than the sensitivity of MSI followed by BRAF (P = .07). Tumor sequencing also had slightly better specificity (95.3%; 95% CI, 92.6%-97.2%) than IHC plus BRAF testing (94.6%) and MSI plus BRAF testing (94.8%).
Germline and Somatic Mutations With Potential to Affect Treatment
Tumor sequencing as the sole molecular test for CRC must include testing both to aid in LS detection and for standard-of-care treatment purposes. Current National Comprehensive Cancer Network guidelines (v2.2017) recommend tumor KRAS, NRAS, and BRAF testing in all stage IV CRC.29 While only 6.2% of this cohort had stage IV disease, generally approximately 25% of CRCs are diagnosed at stage IV and require this testing. In addition, many patients with earlier-stage disease will progress to stage IV, at which time they could benefit from having this information from their primary tumor. In total, 380 of the 465 (81.7%) CRC tumors had at least 1 somatic mutation that could have therapeutic implications; 283 cases had either a KRAS (n = 190), NRAS (n = 22), or BRAF (n = 71) mutation. In addition, 8 patients (8 of 465 [1.7%]) were found to have known pathogenic germline mutations in DPYD, placing them at risk for severe reactions to fluorouracil chemotherapy.30,31 Having this knowledge at the time of diagnosis could prevent severe, potentially life-threatening adverse reactions to fluorouracil in these patients.
Discussion
Here we show that up-front TS could effectively replace all current standard-of-care tests performed on CRC tumors, including those used for universal tumor screening for LS and those used for treatment purposes, as long as there is enough tumor available for this testing. The sensitivity of TS for LS screening is superior to the current standard-of-care approach, while simultaneously providing additional data that may be useful in guiding treatment. Tumor sequencing can also simplify the complex and time-consuming multistep algorithm to screen for LS (Figures 1 and 2).
Limitations
While unlikely, it is possible that some cases of LS were missed by both the reference standard and index tests in this study, which would result in an overestimate of the sensitivity and specificity of all tests.
A limitation of TS is that it has not been optimized to detect all possible germline mutations. For example, it is unlikely that TS would be able to detect mutations in the pseudogene region (exons 12-15) of PMS2 for which mutation detection is particularly challenging.32 While TS may not be able to detect all large rearrangements in cancer susceptibility genes including EPCAM deletions,33 it did detect 10 large deletions in this cohort. Cases like these would presumably be MSI-high without a BRAF mutation, which would be suspicious for LS even if no somatic MMR gene mutations were identified and the patient should still be referred for cancer genetic testing. Like MSI and IHC, TS is only a screening test. Some patients with normal TS results would still need to be referred to cancer genetics if their personal and/or family history of cancer is concerning for a possible hereditary cancer syndrome. In addition, all patients found to have putative germline mutations on TS will need to confirm the mutation with germline sequencing; however, this may be done using the less expensive single-mutation test rather than a full gene sequence or NGS panel. It should be noted that an advantage of TS is the ability to help identify double somatic MMR mutations as were found in 14 of 419 (3.3%) of the prospective cohort. These patients would have had to have TS testing after having MSI or IHC, BRAF, or MLH1 methylation testing and germline sequencing because their dMMR tumor would have been left unexplained when no germline mutation was found.
Another limitation to implementing TS as a single molecular test for all patients with CRC is the possibility of a longer turnaround time compared with MSI or IHC; however, the turnaround time for TS is currently a median of 2 weeks and it requires less time overall by eliminating multiple follow-up tests in a subset of cases. Up-front TS may also create the need for new workflows as some hospitals will need to shift to using reference laboratories instead of performing tests locally; however, it should ultimately streamline the molecular workup of CRC. Another barrier for TS is insufficient or poor-quality DNA from formalin-fixed paraffin-embedded tissue (5.4% in this series). It is possible that we could have salvaged some of these cases by getting recuts from the blocks. Insufficient tumor DNA is a known problem for MSI testing as well, with 14.2% (474 of 3346) of CRC cases having insufficient tumor to perform MSI testing in the OCCPI study and these would also have been insufficient for TS. Immunohistochemistry could be used as a backup test for cases with insufficient tumor material for MSI or TS because it requires far less material. Ultimately, the use of 1 test requiring tumor for TS instead of potentially more than 4 tests requiring tumor will leave more tissue available for clinical trials or other needs. Finally, cost-effectiveness will need to be considered; however, it is anticipated that the costs of TS will decrease substantially as has been seen for germline NGS.
Tumor sequencing could also be optimized to search for putative germline mutations in other cancer susceptibility genes. While this was not the aim of the present study, we did identify 36 additional cases with potential germline mutations in other cancer susceptibility genes. Seventeen of these cases had germline NGS as part of the OCCPI study and 8 of these mutations (47%) were confirmed to be present in the germline (eTable 2 in Supplement 1).
Conclusions
In summary, our data support that up-front TS is both simpler and analytically superior to current LS screening tests and could replace all current molecular testing for patients with CRC.
eAppendix. Description of the UW Oncoplex Test Including a List of the Genes Included
eTable 1. Germline Mutations in Mismatch Repair Genes Detected by Tumor Sequencing
eTable 2. Non-MMR Pathogenic Potentially Germline Cancer Gene Mutations Identified in the Study
eWorksheet. Overall Results for All Cases
References
- 1.Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783-5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352(18):1851-1860. [DOI] [PubMed] [Google Scholar]
- 3.Yurgelun MB. Germline testing for individuals with pancreatic cancer: the benefits and challenges to casting a wider net. J Clin Oncol. 2017;35(30):3375-3377. [DOI] [PubMed] [Google Scholar]
- 4.Bonadona V, Bonaïti B, Olschwang S, et al. ; French Cancer Genetics Network . Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305(22):2304-2310. [DOI] [PubMed] [Google Scholar]
- 5.Järvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118(5):829-834. [DOI] [PubMed] [Google Scholar]
- 6.Burn J, Gerdes AM, Macrae F, et al. ; CAPP2 Investigators . Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378(9809):2081-2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354(3):261-269. [DOI] [PubMed] [Google Scholar]
- 8.Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group . Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11(1):35-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multisociety Task Force on Colorectal Cancer. Am J Gastroenterol. 2014;109(8):1159-1179. [DOI] [PubMed] [Google Scholar]
- 10.Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW; American College of Gastroenterology . ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hampel H, de la Chapelle A. How do we approach the goal of identifying everybody with Lynch syndrome? Fam Cancer. 2013;12(2):313-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. 2009;11(1):42-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haraldsdottir S, Hampel H, Tomsic J, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147(6):1308-1316.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology. 2014;146(3):643-646.e8. [DOI] [PubMed] [Google Scholar]
- 15.Sourrouille I, Coulet F, Lefevre JH, et al. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam Cancer. 2013;12(1):27-33. [DOI] [PubMed] [Google Scholar]
- 16.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509-2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Haene N, Le Mercier M, De Nève N, et al. Clinical validation of targeted next generation sequencing for colon and lung cancers. PLoS One. 2015;10(9):e0138245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J, Wu H, Wang L, et al. Validation of targeted next-generation sequencing for RAS mutation detection in FFPE colorectal cancer tissues: comparison with Sanger sequencing and ARMS-Scorpion real-time PCR. BMJ Open. 2016;6(1):e009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pritchard CC, Salipante SJ, Koehler K, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16(1):56-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kautto EA, Bonneville R, Miya J, et al. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget. 2017;8(5):7452-7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC. Microsatellite instability detection by next generation sequencing. Clin Chem. 2014;60(9):1192-1199. [DOI] [PubMed] [Google Scholar]
- 22.Stadler ZK, Battaglin F, Middha S, et al. Reliable detection of mismatch repair deficiency in colorectal cancers using mutational load in next-generation sequencing panels. J Clin Oncol. 2016;34(18):2141-2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowak JA, Yurgelun MB, Bruce JL, et al. Detection of mismatch repair deficiency and microsatellite instability in colorectal adenocarcinoma by targeted next-generation sequencing. J Mol Diagn. 2017;19(1):84-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pearlman R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3(4):464-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shia J, Tang LH, Vakiani E, et al. Immunohistochemistry as first-line screening for detecting colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome: a 2-antibody panel may be as predictive as a 4-antibody panel. Am J Surg Pathol. 2009;33(11):1639-1645. [DOI] [PubMed] [Google Scholar]
- 26.Newton K, Jorgensen NM, Wallace AJ, et al. Tumour MLH1 promoter region methylation testing is an effective prescreen for Lynch syndrome (HNPCC). J Med Genet. 2014;51(12):789-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen SA, Turner EH, Beightol MB, et al. Frequent PIK3CA mutations in colorectal and endometrial tumors with 2 or more somatic mutations in mismatch repair genes. Gastroenterology. 2016;151(3):440-447.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adar T, Rodgers LH, Shannon KM, et al. A tailored approach to BRAF and MLH1 methylation testing in a universal screening program for Lynch syndrome. Mod Pathol. 2017;30(3):440-447. [DOI] [PubMed] [Google Scholar]
- 29.NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Genetic/Familial High-Risk Assessment: Breast and Ovarian (version 1.2017). 2017. National Comprehensive Cancer Network, Inc. https://www.nccn.org/store/login/login.aspx?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed June 30, 2017.
- 30.Milano G, Etienne MC, Pierrefite V, Barberi-Heyob M, Deporte-Fety R, Renée N. Dihydropyrimidine dehydrogenase deficiency and fluorouracil-related toxicity. Br J Cancer. 1999;79(3-4):627-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Kuilenburg AB. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur J Cancer. 2004;40(7):939-950. [DOI] [PubMed] [Google Scholar]
- 32.Clendenning M, Hampel H, LaJeunesse J, et al. Long-range PCR facilitates the identification of PMS2-specific mutations. Hum Mutat. 2006;27(5):490-495. [DOI] [PubMed] [Google Scholar]
- 33.Niessen RC, Hofstra RM, Westers H, et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009;48(8):737-744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix. Description of the UW Oncoplex Test Including a List of the Genes Included
eTable 1. Germline Mutations in Mismatch Repair Genes Detected by Tumor Sequencing
eTable 2. Non-MMR Pathogenic Potentially Germline Cancer Gene Mutations Identified in the Study
eWorksheet. Overall Results for All Cases