

Key Points
Question
How can we interpret the variants identified in titin and distinguish the pathogenic from the benign?
Findings
In this case series, 504 patients with skeletal muscle disorders were screened with a targeted resequencing approach. An evaluation of titin gene variants that combined genetic, clinical, and imaging data with messenger RNA and/or protein studies identified 9 patients with a titinopathy and 4 patients with possible titinopathy.
Meaning
The clinical interpretation of titin gene variants is challenging and requires comprehensive analyses. A specific workflow for the clinical interpretation of genetic findings in titin is suggested.
Abstract
Importance
Mutations in the titin gene (TTN) cause a wide spectrum of genetic diseases. The interpretation of the numerous rare variants identified in TTN is a difficult challenge given its large size.
Objective
To identify genetic variants in titin in a cohort of patients with muscle disorders.
Design, Setting, and Participants
In this case series, 9 patients with titinopathy and 4 other patients with possibly disease-causing variants in TTN were identified. Titin mutations were detected through targeted resequencing performed on DNA from 504 patients with muscular dystrophy, congenital myopathy, or other skeletal muscle disorders. Patients were enrolled from 10 clinical centers in April 2012 to December 2013. All of them had not received a diagnosis after undergoing an extensive investigation, including Sanger sequencing of candidate genes. The data analysis was performed between September 2013 and January 2017. Sequencing data were analyzed using an internal custom bioinformatics pipeline.
Main Outcomes and Measures
The identification of novel mutations in the TTN gene and novel patients with titinopathy. We performed an evaluation of putative causative variants in the TTN gene, combining genetic, clinical, and imaging data with messenger RNA and/or protein studies.
Results
Of the 9 novel patients with titinopathy, 5 (55.5%) were men and the mean (SD) age at onset was 25 (15.8) years (range, 0-46 years). Of the 4 other patients (3 men and 1 woman) with possibly disease-causing TTN variants, 2 (50%) had a congenital myopathy and 2 (50%) had a slowly progressive distal myopathy with onset in the second decade. Most of the identified mutations were previously unreported. However, all the variants, even the already described mutations, require careful clinical and molecular evaluation of probands and relatives. Heterozygous truncating variants or unique missense changes are not sufficient to make a diagnosis of titinopathy.
Conclusions and Relevance
The interpretation of TTN variants often requires further analyses, including a comprehensive evaluation of the clinical phenotype (deep phenotyping) as well as messenger RNA and protein studies. We propose a specific workflow for the clinical interpretation of genetic findings in titin.
This case series identifies genetic variants associated with titinopathy in patients with muscle disorders and describes a workflow for a more straightforward and reproducible interpretation of the clinical meaning of titin variants.
Introduction
The TTN gene encodes titin, a giant sarcomeric protein, spanning from the Z-disc to the M-band.1 Titin plays crucial functional and structural roles in the sarcomere.2 Mutations in the TTN gene cause several different muscle disorders, cardiomyopathies, and combinations of these.3,4
The skeletal muscle diseases caused by TTN mutations include a wide spectrum of disorders.5 The late-onset autosomal dominant tibial muscular dystrophy (TMD) is caused by mutations in the last exon (364), which cause a posttranslational pathological cleavage of a larger portion of the C-terminal titin protein.6,7,8
Young- or early-adult–onset recessive distal titinopathy is due to either 2 mutations in the last 2 exons (363–364), or 1 mutation in these exons and a truncating mutation on the other allele.9 Similarly, 2 C-terminal mutations or 1 C-terminal mutation along with a truncating variant in trans cause an early-onset recessive limb-girdle muscular dystrophy 2J.10,11,12
Other congenital or early-onset recessive titinopathies comprise disorders with heterogeneous clinical and histological features: congenital centronuclear myopathy,13,14 early-onset myopathy with fatal cardiomyopathy,15 multiminicore disease with heart disease,16 and childhood-juvenile–onset Emery-Dreifuss–like myopathy phenotype without cardiomyopathy.17 Hereditary myopathy with early respiratory failure (HMERF) represents an increasingly identified, completely different adult-onset myopathy, mainly because of dominant mutations in exon 344.18
Many additional TTN-related muscular phenotypes are emerging as a consequence of next-generation sequencing (NGS) screening in patients with myopathy.5 For instance, adult-onset proximal lower limb weakness without ankle dorsiflexion weakness has been described in 2 unrelated patients who had a TMD-causing mutation combined with a second missense mutation.9,19 Recently, a novel TTN homozygous truncating mutation was found in a patient with arthrogryposis multiplex congenita and severe axial hypotonia as a form of congenital amyoplasia without cardiac involvement.20 The mutation occurs within an exon, which seems to be expressed only in the fetal skeletal isoform.20
Next-generation sequencing is rapidly being implemented into routine clinical practice, improving the diagnostic rate for patients with neuromuscular diseases.21,22,23 Almost all NGS screenings reveal many rare and private titin variants and their clinical interpretation is particularly challenging.5,19,24,25,26 By using MotorPlex (Agilent Technologies), a targeted NGS panel, we screened TTN and the other muscle disease genes in 504 patients with skeletal muscle disorders.25,26 Here, we describe the approach used for the NGS data interpretation and we propose a workflow for a more straightforward and reproducible interpretation of the clinical meaning of titin variants.
Methods
Patients
We recruited 504 European patients from 10 clinical centers, mainly adults (mean [SD] age of recruitment, 39.04 [19.09] years) with skeletal muscle disorders. The patients had not received diagnoses despite extensive diagnostic investigations performed according to the observed phenotype. All the patients or their legal guardians provided written informed consent. The study, approved by the ethics committee of the Universitá della Campania “Luigi Vanvitelli,” was performed in accordance with the Declaration of Helsinki.
Molecular Analysis
Genomic DNA was extracted from the peripheral blood by phenol/chloroform purification. Next-generation sequencing libraries were prepared using MotorPlex, as previously described.25,26 An in-house pipeline25,26,27 was used to analyze the raw data.
We focused on patients with previously reported TTN mutations or with at least a single TTN truncating variant. Missense variants were explicitly studied in a single large recessive family only (family X). Further possible causative variants in genes other than titin were ruled out by a segregation analysis.
Titin gene mutated exons were amplified by polymerase chain reaction using M13-tailed primers. M13 primers were used to perform Sanger sequencing using an ABI PRISM 3130XL Genetic Analyzer (Applied Biosystems).
Direct-zol RNA MiniPrep Kit (Zymo Research) was used to extract RNA from muscle biopsies. Complementary DNA (cDNA) synthesis was performed using RevertAid H Minus Reverse Transcriptase (Thermo Scientific). Reverse-transcription polymerase chain reactions were performed using primers designed with Primer3 software and a DreamTaq DNA Polymerase (Thermo Scientific). Copy number variant analysis was performed using a custom array-comparative genomic hybridization, MotorChip28 (Agilent Technologies). Sequence variants in TTN are described according to the coding DNA reference sequence (LRG_391t1), covering transcript variant-IC (NM_001267550.1).
An in silico analysis of missense variants and the prediction of their deleterious effects were performed by homology modeling in DeepView/Swiss-PdbViewer, version 4.1.0 (GlaxoSmithKline R&D and Swiss Institute of Bioinformatics)29 using the most similar structures available in the Protein Data Bank for each titin domain. For titin domains A168 to A170, the crystal structure is available (2NZI).30 The models were visualized using POV-Ray, version 3.7 (Persistence of Vision Raytracer Pty Ltd); (http://www.povray.org).
Muscle Imaging, Histological Studies, and Western Blot Analysis
Muscle magnetic resonance imaging of the lower limbs using 1.5-T magnetic resonance scanners (Siemens and Philips)31 and histological and histochemical examinations in muscle biopsies followed standard procedures.32 Western blotting (WB) of muscle biopsy samples was performed according to standard methods.9 Two previously described in-house–generated antibodies (rabbit polyclonal antibody M10-111 and mouse monoclonal antibody 11-4-39) were used to detect the titin M10 domain, followed by horseradish peroxidase–conjugated secondary antibodies (Dako) and enhanced chemilumescent detection using the Pierce SuperSignal West Femto substrate (Thermo Fisher).9
Results
In an extensive study of 504 mainly adult, patients who had not received a genetic diagnosis and were presenting with clinical signs of muscular dystrophy, congenital myopathy, or other skeletal muscle disorders, we identified 9 novel patients (1.8%) with titinopathy and 4 patients (0.8%) with very likely disease-causing TTN mutations. The clinical details of each patient are summarized in Table 1 and described in the eAppendix in the Supplement.
Table 1. Clinical Summary of Index Patients.
| Variant | Patient No./Sex/Onset, Age/Current, Age | First Symptom | Muscle Weakness | CK | Muscle Involvement (MRI) | Biopsy Findings | Cardiac Impairment | Genotype (LRG_391) | Titinopathy |
|---|---|---|---|---|---|---|---|---|---|
| Previously reported variants | I/M/early 40s /late 50s | Weakness and toe-walking | Prox UL, prox and dist LL | NA | GM, QD, BF, PSM | Connective increase, necrotic fibers | None | (c.95134T>C p[.Cys31712Arg]) | Confirmed |
| II/M/mid-40s/ mid-50s | Myalgia and exercise intolerance | Prox and dist LL | 2-3X | NA | Cytopl. bodies | None | (c.95134T>C p.[Cys31712Arg]) | ||
| III/F/early 30s/ early 40s | Walking difficulties | Dist > prox LL | N | TA, EDL, SOL | Atrophic fibers, FTD | None | (c.107840T>A p.[Ile35947Asn]) + (c.67519C>T p.[Gln22507*]) | ||
| Biallelic PTVs | IV/M/child/ early 30s | Difficulty in running and toe-walking | Dist UL, prox, and dist LL | 10X | AM, BF, SM, GST | Rimmed vacuoles, FTD | DCM | (c.107377 + 1G>A) + (c.65157T>A p.[Tyr21719*]) |
|
| V/M/infant/ early 30s | Hypotonia | Prox UL, prox, and dist LL | N | GM, QD, RF, BF, TA, SOL, GST | FTD, internal nuclei | None | (c.35828dupA p.[Glu11945Argfs*6]) + (c.25063 + 1G>A) | ||
| VI/F/early 30s/ early 50s | Walking difficulties | Dist LL | 2X | NA | FTD, cores | Ventricular hypokinesia and reduced ejection fraction | (c.79072delG p.[Val26358Phefs*4]) + (c.107635C>T p.[Gln35879*]) | ||
| VIIa/F/early 30s/ early 50s | Walking difficulties | Dist LL | NA | TA, SOL, GST | FTD, autophagic vacuoles | NA | (c.27966T>A p.[Cys9322*])+(c.107397dupC p.[Arg35800Glnfs*10]) | ||
| VIIb/F/early 30s/ early 60s | Walking difficulties | Dist LL | NA | NA | FTD, autophagicvacuoles | Paroxysmal sinus tachycardias and frequent ventricular extrasystoles | (c.27966T>A p.[Cys9322*]) + (c.107397dupC p.[Arg35800GInfs*10]) | ||
| Monoallelic PTV | VIII/M/preteen/ late 40s | Difficulty in running | Pectoralis and LL | 2-3X | BF, SM, ST, GM, IP and TA | FTD, internal nuclei | None | (c.107397dupC p.[Arg35800Glnfs*10]) + ? | |
| IXa/M/infant/ teenage | Hypotonia | Prox and dist UL and LL | N | NA | FTD, minicores | NA | (c.79683dupA p.[Arg26562Thrfs*12]) + (c.94015A>G p.[Thr31339Ala]; c.18970A>C p.[Thr6324Pro]) | Possible | |
| IXb/F/infant/ teenage | Hypotonia | Prox and dist UL and LL | N | NA | NA | NA | (c.79683dupA p.[Arg26562Thrfs*12]) + (c.94015A>G p.[Thr31339Ala]; c.18970A>C p.[Thr6324Pro]) | ||
| Missense variants | Xa/M/teenage/ early 50s | Difficulty in running | Dist > prox LL | N | TA and SOL | FTD, autophagic vacuoles | None | (c.100585T>C p.[Trp33529Arg]) + (c.98390A>G p.[Asn32797Ser]) | |
| Xb/M/teenage/ early 40s | Difficulty in running | Dist>Prox LL | N | TA and SOL | FTD, autophagicvacuoles | None | (c.100585T>C p.[Trp33529Arg]) + (c.98390A>G p.[Asn32797Ser]) |
Abbreviations: AM, adductor magnus; BF, biceps femoris; CK, creatine kinase; Cytopl bodies, cytoplasmic bodies; DCM, dilated cardiomyopathy; Dist, distal; EDL, extensor digitorum longus; F, female; FTD, fiber-type disproportion; GM, gluteus maximus; GST, gastrocnemius; IP, iliopsoas; LL, lower limbs; M, male; MRI, magnetic resonance imaging; NA, not available; Prox, proximal; PSM, paraspinal muscles; PTV, protein truncating variant; QD, quadriceps; RF, rectus femoris; SM, semimembranosus; SOL, soleus; ST, semitendinosus; TA, tibialis anterior; UL, upper limbs.
Patients With Previously Described Mutations
We identified disease-associated mutations in the TTN gene in 3 patients (0.6%). The most common mutation responsible for the HMERF phenotype (p.Cys31712Arg in exon 344)18 was identified in 2 cases (I and II).
Patient I was a man in his late 50s with no family history for neuromuscular disorders. He presented with a progressive distal weakness in the lower limbs (onset at 40 years) and a restrictive respiratory insufficiency due to respiratory muscle weakness.
Patient II was a man in his mid-50s presenting with a distal myopathy (onset in his mid-40s with myalgia and exercise intolerance). Although pulmonary function test results were only minimally impaired, muscle biopsy results revealed typical histopathological features seen in HMERF, including cytoplasmic bodies and rimmed vacuoles.
A previously reported TMD mutation (p.Ile35947Asn)33 was identified in compound heterozygosity with a nonsense mutation in a Belgian woman in her early 40s (patient III). This patient has been described elsewhere.34 In brief, she shows an earlier onset (at 30 years) and a more severe phenotype compared with previously reported patients with TMD who carried the same missense variant in heterozygosity.33
Patients With Biallelic Protein Truncating Variants
In 4 patients (0.8%), protein truncating variants (PTVs) were identified on both alleles. In a man in his early 30s with healthy parents and siblings (patient IV), we found a splice site variant (c.107377 + 1G>A in intron 362) on the maternal allele and a nonsense variant (p.Tyr21719* in exon 312) on the paternal allele. Since childhood, the patient had shown mildly progressive generalized muscular weakness. He received a diagnosis of dilated cardiomyopathy without arrhythmias in his late teens. Messenger RNA analyses confirmed the splicing effect of the intronic variant (eFigure in the Supplement). Western blotting analyses showed a reduced intensity of small C-terminal titin protein fragments and the presence of an additional band due to the splicing defect (Figure 1).
Figure 1. Western Blot for C-Terminal Titin Fragments.

Western blotting using 2 different antibodies (M10-1 and 11-4-3) against the titin C-terminal M10 domain. A, Patient VIII with a single identified protein truncating variant shows a severe reduction of titin C-terminal fractions of all sizes; patient IV presents a reduced amount of the small (<20 kDa) titin fragments, and additionally the presence of a truncated fragment (arrowheads) resulting from the aberrant splicing due to the splice site mutation in intron 362. B, Patient Xa with missense mutations showed a normal titin C-terminal pattern, while patient IXa with a single protein truncating variant and 2 missense variants showed a reduction of the small (<20 kDa) titin C-terminal fragments in particular. The myosin heavy chain (MyHC) serves as the loading control. Ctrl indicates control; LGMD2J, limb-girdle muscular dystrophy 2J; TMD, tibial muscular dystrophy.
A 34-year-old Belgian patient (patient V) with an unremarkable family history harbored the p.Glu11945Argfs*6 variant in exon 164 and the c.25063 + 1G>A variant in intron 87 in compound heterozygosity. Since childhood, the patient had shown a slowly progressive generalized muscular weakness and gait abnormalities with frequent falling. In silico predictions confirmed that c.25063 + 1G>A would result in a splicing defect. Further messenger RNA and WB analyses were not performed because of the unavailability of muscle tissue.
Two further biallelic PTVs were identified in patient VI (p.Val26358Phefs*4 and p.Gln35879*, the latter recently reported as a Balkan–Middle East founder mutation34,35) and in 2 siblings (cases VIIa and VIIb) (p.Cys9322* and p.Arg35800Glnfs*10).
Patient VI was a woman in her mid-50s presenting in her early 30s with frequent tripping. Her family history was unremarkable. The disease worsened and the patient has required a cane to walk for the last 5 years. Echocardiography results in her early 50s showed mild left ventricular hypokinesia and a mildly reduced ejection fraction (43%).
The index case of family VII was a woman in her early 50s (VIIa), with onset in adulthood (in her early 30s) characterized by walking difficulty and distal lower limb muscle weakness. No signs of respiratory or cardiac involvement were detected at a recent follow-up (2016). Patient VIIb, a sibling, showed similar clinical and histological features.
Patients With a Single Heterozygous Protein Truncating Variant
In 1 additional patient (patient VIII) harboring the heterozygous p.Arg35800Glnfs*10 in exon 363, protein analysis results showed a severe reduction with the C-terminal titin antibodies, indicating truncating mutations on both alleles as previously reported in patients with limb-girdle muscular dystrophy 2J.9 No additional variants were detected by MotorPlex, and MotorChip did not reveal any copy number variants. The patient had presented with difficulties in running and Achilles’ tendon contractures since the preteen years. The weakness in the lower extremities worsened in the early 30s. No signs of cardiomyopathy were detected on heart ultrasonography.
In family IX, the proband was a teenage boy who presented with hypotonia and congenital torticollis at birth. He had delayed motor milestones, reaching independent walking after the toddler years. He was referred to the neuromuscular unit as a child because of a proximal and distal weakness. The patient, as well as his similarly affected sibling, harbored a single-nucleotide duplication (p.Arg26562Thrfs*12) on the maternal allele. In addition, 2 missense variants were identified on the paternal allele. In particular, a c.18970A>C causing a substitution of a threonine with a proline at position 6324 was identified. The mutated amino acid is located on the external surface of a β strand in an Ig-domain in the I-band region, probably affecting the stability (Figure 2A). The second detected variant was a c.94015A>G leading to a substitution of a threonine at position 31339 with an alanine in an Fn3 domain (A-band portion of titin). The amino acid substitution may affect the interaction with ligands in this region (Figure 2B). A comparative genomic hybridization array excluded the presence of copy number variants in the proband and WB results showed a reduced intensity of C-terminal M10 fragments (Figure 1B).
Figure 2. Positions of TTN Missense Variants Identified in Families IX and X on a Structural Model.
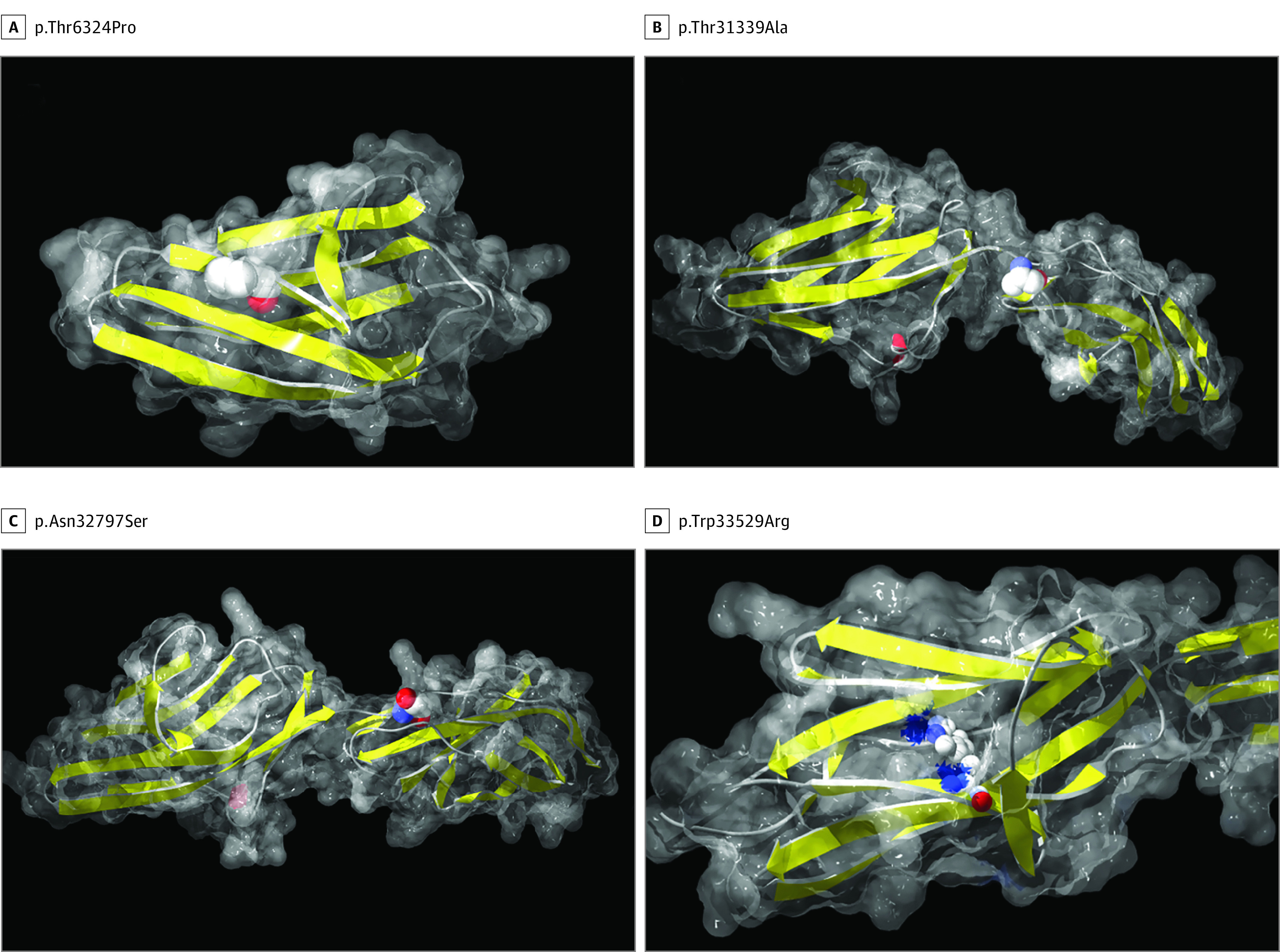
All images were made in DeepView/Swiss-PdbViewer, version 4.1.0 (GlaxoSmithKline R&D and Swiss Institute of Bioinformatics). The computed molecular surface is semitransparent gray and the secondary structure is shown with yellow β strands and red α helices. The mutated residue is shown as CPK. A, Position of p.Thr6324Pro using the most similar structure available in the Protein Data Bank (3B43). The mutated residue is located in a β strand. The mutation to proline will induce steric restrictions most probably causing a reduced stability and a structural disruption. B, p.Thr31339Ala modeled using the structure 2NZI of titin domains A168-A170. The change from threonine to alanine is predicted in a loop and will probably not interfere with the structure. However, the hydroxyl group on the sidechain of threonine allows for hydrogen bonding with other molecules. The amino acid substitution may alter interactions with TTN ligands in this specific region. C, Position of p.Asn32797Ser using the structure 2NZI. The mutated amino acid, one of the first residues in the domain, is on the surface of the model and it seems not to cause any important structural change. It will probably affect the binding to the interactors of this domain. D, Position of p.Trp33529Arg using the structure 2JBO. The tryptophan residue p.Trp33529 is almost totally buried in the hydrophobic core of the protein. The change to a positively charged arginine will probably be detrimental for the structural stability and will lead to an unfolding of this domain.
Patients With Rare Missense Variants
Because rare missense variants were found in most analyzed patients, we focused on a single recessive family (family X) in whom 2 rare variants segregated with the observed phenotype.
The 2 patients were siblings (mid-40s and mid-50s, respectively) and showed a slowly progressive distal myopathy with onset in the second decade. At the latest neurological examination, the patients walked with a waddling gait and bilateral steppage. Their serum creatine kinase levels were normal. No heart or respiratory involvement was observed. Both siblings harbored 2 compound heterozygous missense variants: p.Asn32797Ser and p.Trp33529Arg. The former is located in an Fn3 domain in the A-band portion of titin, and in silico studies predicted that the amino acid is located on the external surface of the domain, possibly affecting the binding to the interactors. The latter variant is in an Ig-domain, which is located just before Ser/Thr kinase domain (TK). The amino acid change probably affects the folding of the domain (Figure 2). No further clearly or potentially damaging variants were detected by MotorPlex (not even in additional causative or candidate genes) and MotorChip studies did not reveal any causative deletion or duplication. A segregation study confirmed that none of the 3 unaffected siblings were compound heterozygous for these TTN missense variants. Western blotting results revealed a normal C-terminal titin pattern, as expected (Figure 1).
Discussion
Because of its size, many rare or private variants are usually identified in the titin gene by NGS analyses.5 The correct interpretation of these variants is a critical challenge for making a diagnosis for patients affected by neuromuscular disorders.5 Although mainly truncating mutations have been identified in patients with titinopathy, missense variants may similarly have a crucial role, as also suggested by our data (Figure 3).
Figure 3. Schematic Representation of Mutations Identified and Algorithm for the Clinical Interpretation of Genetic Findings in Titin.

A, Schematic visualization of truncating (circle) and missense (triangle) variants identified in TTN gene in this study. B, Workflow for interpreting titin variants. Previously reported, disease-causing mutations in the TTN gene easily address the diagnosis toward a titinopathy. Identifying 2 truncating variants on both the alleles results in a diagnosis of titinopathy. A single heterozygous protein truncating variant is not sufficient for a diagnosis of titinopathy. Missense variants can lead to a diagnosis of titinopathy only when sufficient evidence supporting their pathogenicity is obtained.
For the interpretation of genetic findings in TTN, we have developed a workflow (Figure 3) based on 3 categories of sequence variants: (1) previously reported mutations, (2) truncating variants, and (3) missense changes and on deep phenotyping (ie, a comprehensive and precise evaluation of phenotypic abnormalities in which each component of the clinical phenotype is observed and described).36 Although the limited number of patients with titinopathy described so far has hampered the identification of specific and unique hallmarks for each TTN-related disease, significant key points have been reported (Table 2). The integration of structured clinical data with genetic variations is crucial for a correct evaluation of TTN findings, as detailed below.
Table 2. Features of Titin-Related Skeletal Musical Disorders.
| Dominant Inheritance | Recessive Inheritance | ||||
|---|---|---|---|---|---|
| Disease | |||||
| HMERF | TMD | LGMD2J | Adult-onset distal myopathy | Proximal adult-onset rimmed vacuolar myopathy | Congenital or early onset titinopathies |
| Genetics | |||||
| Heterozygous exon 344 mutations | Heterozygous exon 364 mutations | Homozygosity (mainly exon 364 mutations)a Compound heterozygosity (mainly exon 364 + truncating mutations)a |
Compound heterozygosity (exon 364 + missense mutations) | Compound heterozygosity (mainly truncating variants) | |
| Key Point(s) | |||||
| Respiratory muscle weakness; “necklace” cytoplasmic bodies and myofibrillar changes in muscle biopsies |
Degeneration of the muscles of the anterior lower legs starting in the anterior tibial muscle | Slowly progressive fatty degeneration (preserved muscles can be found in young patients) | Severe degeneration of tibialis anterior and soleus (unusual in TMD) | Prominent quadriceps and soleus involvement (sparing of tibial muscle); peculiar rimmed vacuoles in their muscle biopsies | Cardiac involvement often seen; sparing of extraocular muscles; early degeneration of semitendinosus, soleus, and peroneal; late involvement of the rectus femoris and vasti |
The clinical phenotype depends on the specific exon 364 mutation.
Previously Reported Mutations
If previously reported disease-causing mutations are identified, they may easily address the diagnosis of a titinopathy; however, segregation studies and a deep phenotyping are mandatory for a correct genotype-phenotype correlation and for proper genetic counselling. In the presence of a previously reported HMERF variant (eg, p.Cys31712Arg), a respiratory involvement and/or the presence of cytoplasmic bodies and myofibrillar changes (seen in patient I and II, respectively) confirm the diagnosis of titinopathy.5,18
The clinical interpretation of mutations in exon 364, previously associated with TMD (like the p.Ile35947Asn in patient III), is more complex. These mutations cause either a dominant, mild, and late-onset distal leg phenotype, or recessive phenotypes.7,8,9,11 Muscle imaging is mandatory and often very informative (Table 2).
Protein Truncating Variants
Identifying 2 truncating variants in trans results in a diagnosis of titinopathy, which may be corroborated by a WB showing the absence or a severe reduction of the C-terminal protein (patient IV or previously reported patients9,34). However, a complete molecular characterization of variants affecting the canonical or noncanonical splice sites by cDNA or protein studies is suggested.
Biallelic truncating mutations have been so far associated with a wide range of phenotypes, showing heterogeneous clinical and histological features. However, a primary cardiac involvement is often seen and peculiar imaging findings seem to characterize congenital or early onset titinopathies.
Clinically evaluating single heterozygous truncating variants is more complex (Figure 3). Increasing evidence is indicating that titin truncating variants cause recessive skeletal muscle disorders.9,15,16,34 In the presence of monoallelic PTVs, we suggest performing a WB analysis that represents the most valuable and potentially conclusive test, as it is the only available tool able to predict the presence of further elusive truncating variants in trans (as seen in patient VIII and in a previously reported patient9).
Why are there elusive variants in TTN? First, the huge size of the TTN gene and its complex structure, due to a 10-kb triplicate region where 9 exons are repeated 3 times, may hamper an exhaustive gene analysis by NGS, resulting in low-covered or noncovered regions and thus in unidentified mutations. Second, additional elusive mutations may be deep intronic or structural variants. Currently available bioinformatics tools37 combined with customized comparative genomic hybridization arrays28,38 should be used to assess the presence of large deletions or duplications39 in unsolved cases. Western blotting is an effective strategy, although with well-recognized limitations. Inframe deletions, the skipping of inframe exons or truncating variants in exons not expressed in the adult muscles, and small size variations would still not be recognizable by a titin Western blot.
Several recent studies suggest that heterozygous titin truncating variants cause dominant dilated cardiomyopathy.40,41 However, a positional effect and an incomplete and age-dependent penetrance (probably related to other genetic or environmental factors) may explain the lack of any cardiac symptoms in some individuals with mono or biallelic PTVs (eg, patient V and VIII).41 A systematic follow-up to evaluate the cardiac status of such individuals, as well as their asymptomatic relatives who carry truncating variants, is highly recommended.
Missense Variants
The clinical significance of missense variants in TTN represents a major issue related to NGS investigation in the field of neuromuscular disorders.5 A WB analysis is not effective in the presence of missense variants, as demonstrated in cases IX and X. The evaluation of TTN missense variants should reflect the current genetic guidelines.42 A segregation analysis and/or in silico predictions can only suggest a pathogenic or a noncausative effect of a missense variant.42
In the presence of monoallelic truncating variants, as well as of missense variants, the possible causative effect of mutations in genes other than titin has to be ruled out and the presence of the aforementioned key clinical points has to be assessed by deep phenotyping.
However, the definitive proof of pathogenicity for missense variants can only be established by functional tests, segregation studies in very large families, and/or identifying unrelated patients or families with the same mutations. The interpretation of TTN missense variants may also benefit from the establishment of clinical and research consortia able to combine cohorts of patients into larger groups.43
Limitations
Our study has limitations. First, we enrolled, in a multicenter study, patients with clinically and genetically heterogenous conditions and specific clinical studies (magnetic resonance imaging or cardiac tests) were unavailable or not performed for some patients.
Second, we report missense variants with an unconfirmed causative role (cases IX and X). Although further studies are needed to attribute causality to missense changes, reporting possible causative variants is an effective strategy to improve consistency in the interpretation of molecular findings in titin. Ultimately, the proposed workflow is meant for interpreting titin variants in a mendelian disorder. The possible role of titin variants as modifiers or within a digenic or multigenic disease is not discussed here.
Conclusions
An increasing number of rare, ultrarare, and private variants in the titin gene is detected in any sequencing approach, and NGS has dramatically expanded the spectrum of skeletal muscle disorders associated with causative mutations in TTN.5 Our workflow results in a greater understanding and more consistent interpretation of titin variants by neurologists, pediatricians, and geneticists less familiar with the titin gene and titinopathies.
eAppendix. Clinical Data
eFigure. Muscle cDNA Analysis in Patient IV Confirms that the Variant c.107377+1G>A Causes a Misplicing
References
- 1.Bang ML, Centner T, Fornoff F, et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89(11):1065-1072. [DOI] [PubMed] [Google Scholar]
- 2.Krüger M, Kötter S. Titin, a central mediator for hypertrophic signaling, exercise-induced mechanosignaling and skeletal muscle remodeling. Front Physiol. 2016;7:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerull B. The rapidly evolving role of titin in cardiac physiology and cardiomyopathy. Can J Cardiol. 2015;31(11):1351-1359. [DOI] [PubMed] [Google Scholar]
- 4.Chauveau C, Rowell J, Ferreiro A. A rising titan: TTN review and mutation update. Hum Mutat. 2014;35(9):1046-1059. [DOI] [PubMed] [Google Scholar]
- 5.Savarese M, Sarparanta J, Vihola A, Udd B, Hackman P. Increasing role of titin mutations in neuromuscular disorders. J Neuromuscul Dis. 2016;3(3):293-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charton K, Sarparanta J, Vihola A, et al. CAPN3-mediated processing of C-terminal titin replaced by pathological cleavage in titinopathy. Hum Mol Genet. 2015;24(13):3718-3731. [DOI] [PubMed] [Google Scholar]
- 7.Udd B, Partanen J, Halonen P, et al. Tibial muscular dystrophy. late adult-onset distal myopathy in 66 Finnish patients. Arch Neurol. 1993;50(6):604-608. [DOI] [PubMed] [Google Scholar]
- 8.Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71(3):492-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evilä A, Vihola A, Sarparanta J, et al. Atypical phenotypes in titinopathies explained by second titin mutations. Ann Neurol. 2014;75(2):230-240. [DOI] [PubMed] [Google Scholar]
- 10.Udd B, Kääriänen H, Somer H. Muscular dystrophy with separate clinical phenotypes in a large family. Muscle Nerve. 1991;14(11):1050-1058. [DOI] [PubMed] [Google Scholar]
- 11.Hackman P, Marchand S, Sarparanta J, et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul Disord. 2008;18(12):922-928. [DOI] [PubMed] [Google Scholar]
- 12.Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. 2014;33(1):1-12. [PMC free article] [PubMed] [Google Scholar]
- 13.Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. 2013;81(14):1205-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fattori F, Maggi L, Bruno C, et al. Centronuclear myopathies: genotype-phenotype correlation and frequency of defined genetic forms in an Italian cohort. J Neurol. 2015;262(7):1728-1740. [DOI] [PubMed] [Google Scholar]
- 15.Carmignac V, Salih MA, Quijano-Roy S, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol. 2007;61(4):340-351. [DOI] [PubMed] [Google Scholar]
- 16.Chauveau C, Bonnemann CG, Julien C, et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet. 2014;23(4):980-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Cid R, Ben Yaou R, Roudaut C, et al. A new titinopathy: childhood-juvenile onset Emery-Dreifuss–like phenotype without cardiomyopathy. Neurology. 2015;85(24):2126-2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmio J, Evilä A, Chapon F, et al. Hereditary myopathy with early respiratory failure: occurrence in various populations. J Neurol Neurosurg Psychiatry. 2014;85(3):345-353. [DOI] [PubMed] [Google Scholar]
- 19.Evilä A, Arumilli M, Udd B, Hackman P. Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul Disord. 2016;26(1):7-15. [DOI] [PubMed] [Google Scholar]
- 20.Fernández-Marmiesse A, Carrascosa-Romero MC, Alfaro Ponce B, et al. Homozygous truncating mutation in prenatally expressed skeletal isoform of TTN gene results in arthrogryposis multiplex congenita and myopathy without cardiac involvement. Neuromuscul Disord. 2017;27(2):188-192. [DOI] [PubMed] [Google Scholar]
- 21.Thompson R, Straub V. Limb-girdle muscular dystrophies—international collaborations for translational research. Nat Rev Neurol. 2016;12(5):294-309. [DOI] [PubMed] [Google Scholar]
- 22.Nigro V, Savarese M. Next-generation sequencing approaches for the diagnosis of skeletal muscle disorders. Curr Opin Neurol. 2016;29(5):621-627. [DOI] [PubMed] [Google Scholar]
- 23.Helman G, Bonkowsky JL, Vanderver A. Neurologist comfort in the use of next-generation sequencing diagnostics: current state and future prospects. JAMA Neurol. 2016;73(6):621-622. [DOI] [PubMed] [Google Scholar]
- 24.Vasli N, Böhm J, Le Gras S, et al. Next-generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathol. 2012;124(2):273-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savarese M, Di Fruscio G, Torella A, et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology. 2016;87(1):71-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savarese M, Di Fruscio G, Mutarelli M, et al. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol Commun. 2014;2:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mutarelli M, Marwah V, Rispoli R, et al. A community-based resource for automatic exome variant-calling and annotation in Mendelian disorders. BMC Genomics. 2014;15(suppl 3):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piluso G, Dionisi M, Del Vecchio Blanco F, et al. Motor chip: a comparative genomic hybridization microarray for copy-number mutations in 245 neuromuscular disorders. Clin Chem. 2011;57(11):1584-1596. [DOI] [PubMed] [Google Scholar]
- 29.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714-2723. [DOI] [PubMed] [Google Scholar]
- 30.Mrosek M, Labeit D, Witt S, et al. Molecular determinants for the recruitment of the ubiquitin-ligase MuRF-1 onto M-line titin. FASEB J. 2007;21(7):1383-1392. [DOI] [PubMed] [Google Scholar]
- 31.Tasca G, Ricci E, Monforte M, et al. Muscle imaging findings in GNE myopathy. J Neurol. 2012;259(7):1358-1365. [DOI] [PubMed] [Google Scholar]
- 32.Dubowitz VSC, Oldfors A. Muscle Biopsy: A Practical Approach. 4th ed. Philadelphia, PA. Saunders Ltd; 2013. [Google Scholar]
- 33.Van den Bergh PYK, Bouquiaux O, Verellen C, et al. Tibial muscular dystrophy in a Belgian family. Ann Neurol. 2003;54(2):248-251. [DOI] [PubMed] [Google Scholar]
- 34.Evilä A, Palmio J, Vihola A, et al. Targeted next-generation sequencing reveals novel TTN mutations causing recessive distal titinopathy. Mol Neurobiol. 2016;54(9):7212-7223. [DOI] [PubMed] [Google Scholar]
- 35.Perić S, Glumac JN, Töpf A, et al. A novel recessive TTN founder variant is a common cause of distal myopathy in the Serbian population. Eur J Hum Genet. 2017;25(5):572-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson PN. Deep phenotyping for precision medicine. Hum Mutat. 2012;33(5):777-780. [DOI] [PubMed] [Google Scholar]
- 37.Tattini L, D’Aurizio R, Magi A. Detection of genomic structural variants from next-generation sequencing data. Front Bioeng Biotechnol. 2015;3:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savarese M, Piluso G, Orteschi D, et al. Enhancer chip: detecting human copy number variations in regulatory elements. PLoS One. 2012;7(12):e52264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giugliano T, Fanin M, Savarese M, Piluso G, Angelini C, Nigro V. Identification of an intragenic deletion in the SGCB gene through a re-evaluation of negative next-generation sequencing results. Neuromuscul Disord. 2016;26(6):367-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schafer S, de Marvao A, Adami E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49(1):46-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hackman P, Udd B, Bönnemann CG, Ferreiro A; Titinopathy Database Consortium . 219th ENMC International Workshop Titinopathies International database of titin mutations and phenotypes, Heemskerk, The Netherlands, 29 April-1 May 2016. Neuromuscul Disord. 2017;27(4):396-407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix. Clinical Data
eFigure. Muscle cDNA Analysis in Patient IV Confirms that the Variant c.107377+1G>A Causes a Misplicing