

Abstract
Recent reports indicate that intracellular levels of NAD decline in tissues during chronological aging, and that therapies aim at increasing cellular NAD levels could have beneficial effects in many age-related diseases. The protein CD38 (CLUSTER OF DIFFERENTIATION 38) is a multifunctional enzyme that degrades NAD and modulates cellular NAD homeostasis. At the physiological level, CD38 has been implicated in the regulation of metabolism, and the pathogenesis of multiple conditions including: aging, obesity, diabetes, heart disease, asthma and inflammation. Interestingly, many of these functions are mediated by the CD38 enzymatic activity. In addition, CD38 has also been identified as a cell surface marker in hematologic cancers such as multiple myeloma, and a cytotoxic anti-CD38 antibody has been approved by the FDA for use in this disease. Although this is a remarkable development, killing CD38-positive tumor cells with cytotoxic anti-CD38 antibodies is only one of the potential pharmacological uses of targeting CD38. The present review discusses the biology of the CD38 enzyme and the current state of development of pharmacological tools aim at CD38 and explores how these agents may represent a novel approach to treat human conditions including cancer, metabolic diseases and diseases of aging.
Keywords: CD38, NADase, NAD+, antibodies, small molecules, SIRTUINs, aging, cancer and metabolism
Emerging roles of NAD metabolism in human disease: The promise of “NAD boosting therapies”
Nicotinamide adenine dinucleotide (NAD) is a cofactor in electron transfer during oxidation-reduction reactions, and also plays a crucial role in cell signaling, regulating several pathways from intracellular calcium transients to the epigenetic status of chromatin [1-6]. Thus, NAD is a molecule that provides a link between signaling and metabolism. Importantly, decline in cellular NAD levels has emerged as a potential key player in the pathogenesis of several diseases including age-related conditions (Table 1) [1-32].
Table 1. Conditions where cellular NAD decline or the beneficial effects of NAD “boosting” therapy have been described.
| Conditions | Reference |
|---|---|
| Aging, longevity, healthspan and progeroid syndromes | 5-14, 16 |
| Cataract, genetic macular degeneration | 14, 15 |
| Hearing loss | 16, 17 |
| Obesity and diabetes | 8, 18-20 |
| Non-alcoholic and Alcoholic fat liver disease | 19, 21-23 |
| Kidney and Heart disease | 24, 27 |
| Neurodegeneration-related disease and stroke | 28, 29 |
| Muscular dystrophy and muscular mitochondrial disease | 30, 31 |
| Fetal malformation (Vactrel syndrome) | 32 |
In fact, age-related decline of NAD is of particular interest [1-6]. As the world population ages, the incidence of age-related co-morbidities will continue to increase. It appears that age-related organ dysfunction and age-related diseases are, at least in part, governed by impairment in tissue energy metabolism [1-14]. Understanding the mechanisms that drive these metabolic abnormalities in aging may lead to the development of new therapies for the elderly population. Unfortunately, to date, there are no therapies directly aimed at any of the fundamental biological mechanisms of aging [33]. In animal models, age-related metabolic diseases and decline in healthspan can be mitigated by therapies that restore tissue levels of NAD [1-32], suggesting a role for “NAD-boosting” therapies [1-31].
Experimental “NAD boosting” or “NAD replacement” can be accomplished via administration of NAD precursors such NMN, NR and derivatives of vitamin B3, or via inhibition of NAD-degrading enzymes [1-31]. Therefore, it is important to determine the mechanisms that regulate homeostasis of this nucleotide in vivo. The reactions that consume NAD are mediated by enzymes involved in the non-oxidative roles of NAD [1-4]. Of these enzymes, CD38, PARPs and SARM1 appear to significantly contribute to cellular NAD degradation [1-4, 6, 19, 28, 34-39]. Until recently, the prevailing hypothesis to explain the age-related NAD decline was that activation of DNA-repair enzymes such as PARPs during the aging process would consume and deplete cellular NAD [1-4]. However, we demonstrated that levels and activity of CD38 increase during aging, and that this enzyme is, at least in part, responsible for the age-related NAD decline [6]. For example, genetic ablation of CD38 protects against age-related NAD decline and mitochondrial dysfunction [6]. Due to the emerging role of NAD homeostasis in aging and age-related diseases, our discovery of the role of CD38 in the control of cellular NAD levels opens the possibility that CD38 inhibition may be used as a therapeutic approach to increase cellular NAD levels (Figure 1)[1-4, 6]. Here, we review the biological, biochemical, and pharmacological aspects of CD38, with emphasis on its role in NAD homeostasis.
Figure 1. (key figure). “NAD boosting therapy” via CD38 inhibition in age-related diseases.
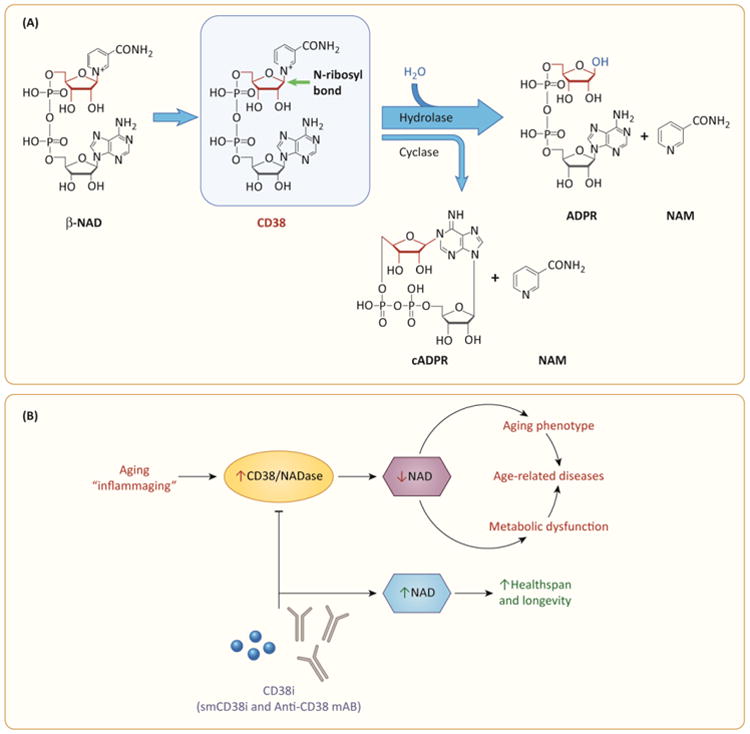
A. CD38 breaks NAD into ADPR/cADPR and NAM. The main site of reaction in the NAD molecule is the terminal ribose, specifically the covalent bond between N1 in nicotinamide and anomeric carbon of the ribose. CD38 breaks the N-glycosyl bond and the main product of the reaction are ADPR and nicotinamide with a very small portion of NAD been converted to cADPR. The figure abbreviations are: ADPR – adenosine ribose cADPR cyclic adenosine ribose NAM – Nicotinamide NAD – nicotinamide adenosine dinucleotide. B. A pro-inflammatory phenotype is observed during the aging process (inflammaging hypothesis). Inflammatory cytokines and endotoxins are potent inducers of CD38 expression, leading to an increase in the tissue NADase activity and a subsequent decline in cellular NAD levels that may play a role in the development of the aging phenotype, decrease resilience, metabolic dysfunction, and the appearance of some age-related diseases. Inhibition of CD38 via small molecule CD38 inhibitors (smCD38i) or non-cytotoxic monoclonal antibodies that inhibit CD38 activity (CD38i mAB) may prevent cellular NAD decline and promote healthspan and successful aging.
CD38: from cell surface marker to multifunctional enzyme involved in NAD metabolism
CD38 was first identified by the herculean task of E. L. Reinherz and S. F. Schlossman to characterize and identify cell surface markers with monoclonal antibodies, as part of their pioneer search of molecules for immunophenotyping of T-cells [38-39]. However, beyond a role as structural marker, CD38 is involved in several physiological and pathological conditions [6,19, 34-40]. In particular, CD38 is the main NAD-degrading enzyme in several mammalian tissues [6,19, 34-40]. Some pharmacological approaches to inhibit CD38 take advantage of its role as a cell surface marker when the intention is to target the killing, or modulate CD38-positive cancer and/or immune cells, as in multiple myeloma (MM) and other cancers (Figure 2)[41-50]. On the other hand, modulating CD38 enzymatic activity may serve as a “NAD boosting” therapy for aging and metabolic diseases (Figure 1)[1-4, 6, 19, 36, 39]. Below we will discuss key biological aspects of CD38 and the potential pharmacological tools available to study and target this molecule, including anti-CD38 antibodies and small molecule inhibitors.
Figure 2. CD38 targeted therapy in cancer.
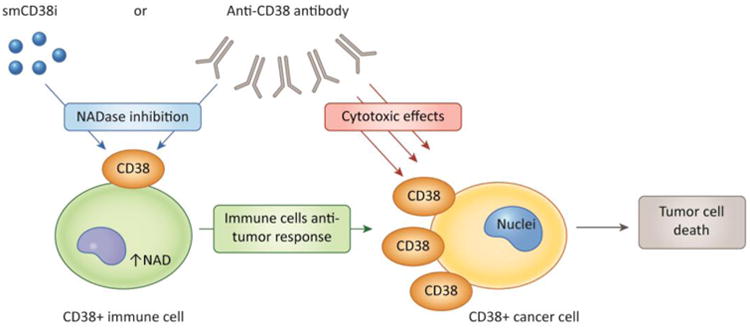
CD38 has at least two potential roles in cancer therapy. First, cytotoxic anti-CD38 antibodies can promote the killing of CD38 positive cancer cells via direct and indirect effects. On the other hand, CD38 expression in immune cells, cancer cells and other cells in the tumor micro environment may cause a decrease in tissue NAD levels that has a negative effect in the immune-response against the tumor. Thus, small molecule CD38 inhibitors (smCD38i), or monoclonal antibodies that inhibit CD38 activity (CD38imAB) can promote an increase in tissue NAD levels and a positive anti-tumor immune response.
The biology of CD38
CD38 is a multifunctional protein involved on cellular and tissue NAD homeostasis and in the generation of the second messengers ADPR and cyclic-ADPR (cADPR) that are involved in intracellular calcium signaling [38-40, 51]. Functional and structural data indicate that the promoter region of the human CD38 gene, located at chromosome 4, is regulated by several factors including NF-κB, R×R, L×R, and STAT [38-39, 52]. Importantly, CD38 expression is induced by inflammatory cytokines, endotoxins, interferon, and some nuclear receptors (Figure 3)[38-39, 52]. Therefore, TNF-α and LPS treatment results in increased expression of CD38/NADase activity and in a subsequent dramatic decrease in cellular NAD levels [38-39, 52]. Understanding the mechanisms regulating CD38 gene expression provides clues to the mechanisms governing the increased expression of CD38. In particular, the pro-inflammatory state observed during the aging process may provide a link between cytokine-induced CD38 expression, age-related tissue NAD decline, and the “inflammaging” theory [1, 53].
Figure 3. The biology of CD38 and topological paradox.
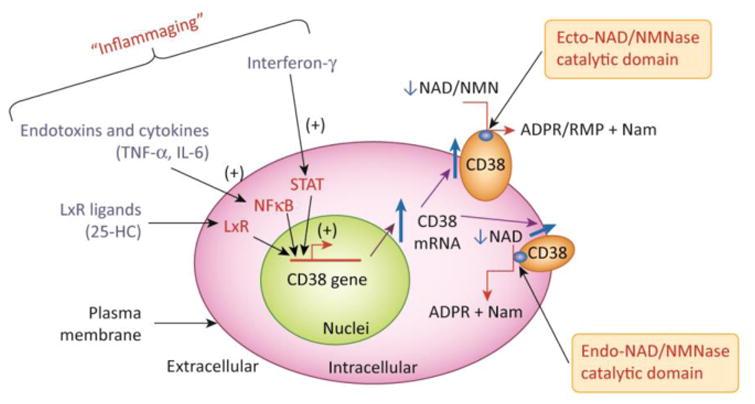
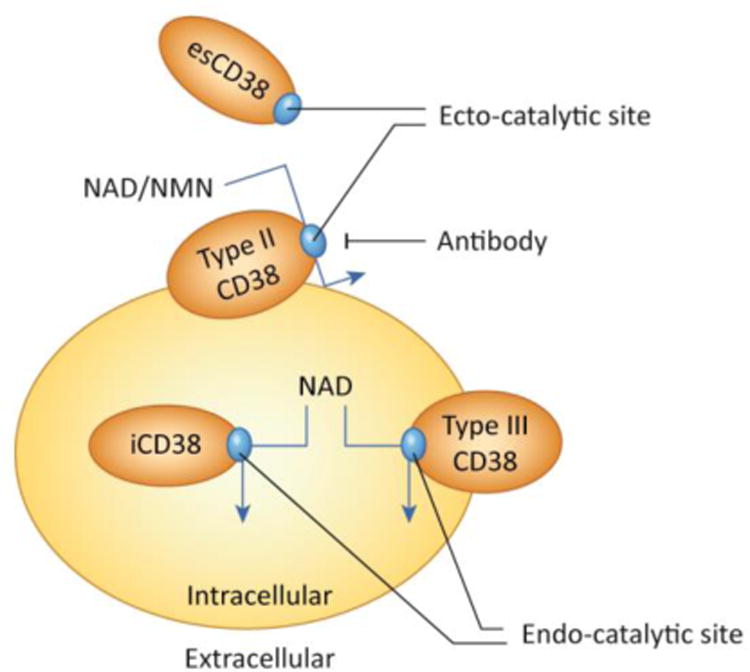
Figure 3A demonstrates the regulation of the CD38 gene expression by inflammatory agents and the link between “inflammaging”, increased CD38 expression, and cellular NAD decline. CD38 expression is activated by LPS, cytokines, interferon, and L×R ligands such as 25-hydroxycholesterolcholesterol (25-HC). In particular, cytokines, interferon and endotoxins have been proposed to play a role on the sterile inflammatory process observed during aging. In B the CD38 topologic paradox dictates that although most NAD is intracellular only a minority of CD38 has its catalytic site facing the inside of the cells. Thus, the most prevalent form of CD38 (type II), that has its catalytic site facing the outside degrades extracellular NAD and NAD precursors, such as NMN, limiting its availability to NAD synthesis in intracellular pools. A secretory soluble form of CD38 with extracellular NADase activity has also been described (esCD38). On the other hand, the type III transmembrane form of CD38 has its catalytic site facing the inside, hence having accessibility to the larger intracellular NAD pools. Minor expression of CD38 has also been described in intracellular membranes such as the nuclear membrane and mitochondrial membrane. The expression of intracellular CD38 may be limited to prevent cellular NAD degradation and cellular metabolic collapse.
A unique characteristic of the CD38 enzyme is its cellular localization. The great majority of CD38 has a type II membrane orientation, with the catalytic site facing the outside of the cell [38-39]. Thus, over 90% of CD38 functions as an ecto-NADase [54]. Since the majority of substrates for NADase- CD38 are expected to be intracellular, the ecto-enzymatic activity of this enzyme represents an intriguing “topological paradox” (Figure 3b). Until recently, it had been difficult to explain this paradox. However, we and others demonstrated that CD38 degrades not only NAD, but also circulating NAD precursors such as NMN and NR, before they can be incorporated inside cells for NAD biosynthetic pathways [6, 55].
CD38 has also been observed in intracellular membranes, such as in the nuclear membrane, mitochondria, and endoplasmic reticulum [38-39]. In addition, a small fraction of CD38 is also expressed as a type III plasma membrane protein with the catalytic site facing the inside of the cell (Figure 3b)[56]. Finally, soluble intra and extracellular forms of CD38 have also been described (Figure 3)[38-39, 57]. The relative roles of the different “anatomical” locations of CD38 in cellular and tissue NAD homeostasis is one of the outstanding key questions in the field.
Catalytic activity and physiological roles of CD38
As discussed above, CD38 is a multifunctional enzyme that uses β-NAD and other β-NAD derivatives, such as β-NADP and β-NMN, as substrates. Interestingly, CD38 does not accept either α-NAD or NADH as substrates. The primary catalytic reaction of CD38 involves the cleavage of a high energy β-glycosidic bond between nicotinamide and the ribose moiety. During catalysis, the removal of the nicotinamide moiety from β-NAD is coupled with the formation of non-covalent enzyme-substrate intermediates such as E-ADPR or the less abundant E-cADPR. These intermediates are stabilized through H-bonds between their ribosyl 2′-, 3′- OH groups and the catalytic residue Glu226, a residue required for the NADase and cyclase activity of the enzyme [38-40, 58-60]. Structural and mutagenesis experiments revealed other important residues in the catalytic site like Glu146, Trp189, Trp125, Ser126, Thr221 and Arg127 [38-40, 59-60]. The intermediates of the reaction are released from the catalytic site forming ADPR or cADPR. In general, the majority of the CD38 NADase catalytic activity will generate nicotinamide, and a nearly stoichiometric amount of ADPR with residual amounts of NAD being converted to cADPR and nicotinamide [34-35, 38-40, 58-60]. These two catalytic activities of the enzyme are classified as glycohydrolase and ADPR-ribosyl cyclase [34-35, 38-40, 51, 58-60]. The amino acid sequence of CD38 has considerable homology with a pure ADP-ribosyl cyclase from Aplysia [38-40, 60]. At least two residues in CD38 contribute to its increased glycohydrolase versus cyclase activity, namely Thr-221 and Glu-146 [58-60]. In fact, mutagenesis of these residues abolishes CD38 NADase activity and increases its cyclase activity [60].
Both ADPR and cADPR have second messenger signaling roles [38-40, 51, 58, 60]. The roles of CD38 as a cyclase and of NAD-derived calcium messengers in physiology and pathology were extensively reviewed by others [38-40, 51, 58, 60]. Importantly, it should be highlighted that when CD38 is catalyzing the degradation of NAD to ADPR, or the cyclization of NAD to cADPR, there is NAD consumption and nicotinamide generation, leading to the crucial role of CD38 in tissue NAD and nicotinamide homeostasis [6, 34-35, 38-40].
Importantly, due to its ecto-enzymatic activity, CD38 not only regulates intracellular and extracellular NAD homeostasis, but also modulates the availability of extracellular NAD and its metabolites [6, 57]. For example, the CD38 ecto-NADase activity plays a role in the extracellular adenosine pathway [61]. In fact, it was reported that extracellular adenosine can be generated from both NAD and ATP degradation via a cascade of events that includes CD38 ecto-NADase, and CD39 ecto-ATPase activity. In turn, adenosine can bind to specific purinergic P1 receptors, eliciting signals in multiple cells including immune-modulatory effects in tumorous microenvironment [61].
Very importantly, CD38 has a crucial role in the regulation of NAD-dependent processes in several cellular compartments. For instance, CD38 regulates the activity of the nuclear and mitochondrial SIRTUINs [6, 34-35]. SIRTUINs are NAD-dependent deacetylases implicated in the regulation of metabolism and several aspects of ageing, healthspan and longevity [2-3]. In particular, we observed that CD38 degrades NAD and decreases the accessibility of NAD to these enzymes [6, 34-35]. It is also possible that generation of nicotinamide by CD38 may regulate SIRTUINs activity, since nicotinamide is an endogenous inhibitor of the SIRTUINs [62-63]. Thus, the potential role of CD38 as SIRTUIN regulator is of major pharmacological relevance.
Pharmacological approaches to target/modulate CD38
The identification of CD38 as a key enzyme involved in NAD metabolism, cell signaling, and cancer suggests its potential as a target for multiple conditions. Based on the therapeutic goals, CD38 can be targeted using different pharmacological approaches such as cytotoxic antibodies, enzymatic-blocking antibodies, and with reversible or irreversible small molecule inhibitors.
Anti-CD38 antibodies: a success story in multiple myeloma and future perspectives in NAD replacement therapy
CD38 is highly and uniformly expressed at the cell surface of MM cells, and at lower levels in normal lymphoid and myeloid cells, and in some tissues of non-hematopoietic origin [38, 41-49]. This spur the development of several anti-CD38 antibodies for MM [41-49]. In fact, monoclonal antibodies (mAbs) against CD38 are highly efficacious in the treatment of MM [41-49]. Currently, four monoclonal antibodies are in clinical trials for the treatment of CD38+ malignancies [41-49]. The most advanced is Daratumumab (Janssen Biotech) which received FDA approval for MM in 2015 [41-42]. Three other CD38 binding antibodies are in clinical trials: Isatuximab (Sanofi-Aventis), MOR202 (Morphosys), and TAK079 (Takeda) (Table 2) [42, 48-49, 64]. Direct in vitro comparisons of these mAbs showed comparable antibody-dependent cell-mediated toxicity (ADCC) and binding affinities, but remarkable differences in the ability to induce direct apoptosis, to induce complement-mediated cytotoxicity (CDC), to inhibit enzymatic activities, and to induce antibody-dependent cell-mediated phagocytosis (ADCP)(Figure 2 and Table 12)[41-42]. In addition, it has been demonstrated that these anti-CD38 mAbs also have a potential in vivo immune modulatory effects on the tumor microenvironment such as enhancing effector T-cell function and inhibiting suppressive T-reg activity (42). In light of the fact that these three anti-CD38 show similar safety and efficacy profiles, it is hypothesized that ADCC is the main mechanism of action of these antibodies in MM [41-42]. However, as discussed above, CD38 is a multifunctional membrane enzyme and regulates a variety of NAD-dependent cellular processes. Although these antibodies were selected on the basis of cytolysis, it is possible that some of their therapeutic effects may be mediated by inhibition of the NADase activity and subsequent NAD boosting effects. In particular, immune modulatory effects of anti-CD38 antibodies during cancer therapy may be at least in part related to the decrease in CD38 NADase activity. Very recently, Chatterjee et al. showed that the CD38-NADase-NAD+ axis plays an important role in the immune response of T cells in a preclinical model of melanoma [50]. These studies indicate that high levels of NAD+, negatively regulated by CD38, preserve T cell function against tumors cells, raising the possibility that inhibition of CD38 may work synergistically with blockade of PD-1/PD-L1 pathway in immune therapy for cancer. These findings suggest that combined therapy may lead to superior tumor responses. To date, isatuximab is the only clinically relevant anti-CD38 antibody shown to inhibit the catalytic activity of the enzyme (Table 2). Anti-CD38 antibodies that specifically inhibit CD38 NADase activity without cytotoxic effects may become an important tool for the boosting NAD, immune modulation, and for use in age-related diseases. These new antibodies may pave the way for the development of highly specific CD38 NADase inhibitors aimed at “NAD boosting” therapy in the near future. However, to date there is no in vivo evidence that therapeutic anti-CD38 mAb exert their effects via inhibition of the CD38 NADase activity. In fact, if the anti-tumor effects of inhibiting CD38 can be at least in part mediated by inhibition of its CD38 NADase activity is not known.
Table 2.
Pharmacological tools for targeting CD38.
| Compound Name | Mechanism of Action | IC50 or Ki | In vivo/ex vivo data | Refs | ||
|---|---|---|---|---|---|---|
| NAD Analogous | Covalent Inhibitors | Ara-F-NAD | Competitive Inhibition of NADase activity | 169 nM | No | 66, 69 |
| Ara-F-NMN | Competitive Inhibition of NADase activity | 69 nM | no | 69 | ||
| Ara-F-NMN phosphoester/C48 | Competitive Inhibition of NADase activity | NA | yes | 70 | ||
| Non-covalent inhibitors | Carba-NAD | Competitive Inhibition of NADase activity | 100 μM | no | 67, 68 | |
| Pseudo-Carba-NAD | Competitive Inhibition of NADase activity | 186 μM | no | 67 | ||
| Flavonoids | Apigenin | Competitive Inhibition of NADase activity | 15 μM | Yes | 59, 73 | |
| Luteolinidin | Competitive Inhibition of NADase activity | 6.0 μM 11.4 μM | Yes | 71, 73 | ||
| Kuromanin | Competitive Inhibition of NADase activity | 6.3 μM | Yes | 59, 71, 77 | ||
| Rhein/K-Rhein | Uncompetitive Inhibition of NADase activity | 1.24 μM 0.84 μM | Yes | 59, 75, 76 | ||
| 4-amino-Quinolines | 78c | Competitive Inhibition of NADase activity | 7.3 nM | Yes | 78 | |
| 1ah | Competitive Inhibition of NADase activity | 510 nM | Yes | 79 | ||
| 1ai | Competitive Inhibition of NADase activity | 150 nM | Yes | 79 | ||
| Antibodies (IgG mAB) | Isatuximab | Allosteric Inhibition of NADase activity | NA | Yes | 42, 49 | |
| Daratumumab | Cytotoxic Effect/Clearance of CD38+ cells | NA | Yes | 41, 42, 43 | ||
| TAK-079 | Cytotoxic Effect/Clearance of CD38+ cells | NA | Yes | 64 | ||
| MOR-202 | Cytotoxic Effect/Clearance of CD38+ cells | NA | Yes | 42 | ||
Monoclonal antibodies have direct and indirect cytotoxic effects. Direct in vitro comparisons of these mAbs showed comparable antibody-dependent cell-mediated toxicity (ADCC) and binding affinities, but remarkable differences in the ability to induce direct apoptosis, to induce complement-mediated cytotoxicity (CDC), and to induce antibody-dependent cell-mediated phagocytosis (ADCP). Inhibition of enzymatic activities of CD38 has been reported only with Isatuximab.
Small molecule CD38 inhibitors
To date, over 200 compounds are listed as CD38 inhibitors in the literature [40, 54, 58-60, 65-78]. Regarding chemical structures, CD38 inhibitors can be classified as NAD-analogs, flavonoids and heterocycles compounds [40, 54, 60, 65-78]. Based on mechanisms of action, they can be pooled into two groups: covalent and non-covalent inhibitors (Table 2). Covalent inhibitors form a bond in the active site at Glu226. Even though an excess of nicotinamide can recover the enzymatic activity, under physiological conditions the rate of dissociation is expected to be very slow [3]. On the other hand, non-covalent inhibitors bind to amino acid resides in the active site of the enzyme through weaker interactions, like hydrogen and hydrophobic bonds [1-4].
Mechanistic/structural-based NAD analogs CD38 inhibitors
This class of CD38 inhibitors was developed via modification of the nicotinamide ribose in the molecule of NAD and NMN [58-59, 66-67]. These inhibitors were designed based on the knowledge of the mechanisms of catalysis and crystal structure of CD38 and are considered mechanistic/structural-based inhibitors. These molecules include the carba-NAD and the ara-NAD analogs [54, 59, 66-67]. Carba-NAD and pseudo carba-NAD are non-covalent inhibitors of CD38, and have low affinity for the enzyme (Table 2). On the contrary, arabinose-derivatives are covalent inhibitors of CD38, and their potency is further increased when the 2-hydroxyl group was replaced by a fluoride (Table 2 and Figure 4). Figure 4 shows the modifications that led to these analogs. Likewise, modification of the nicotinamide ribose in NMN and NR led to even more potent covalent and non-covalent inhibitors of CD38 (Figure 4 and Table 2).
Figure 4. The chemical structure of the different classes of smCD38i.
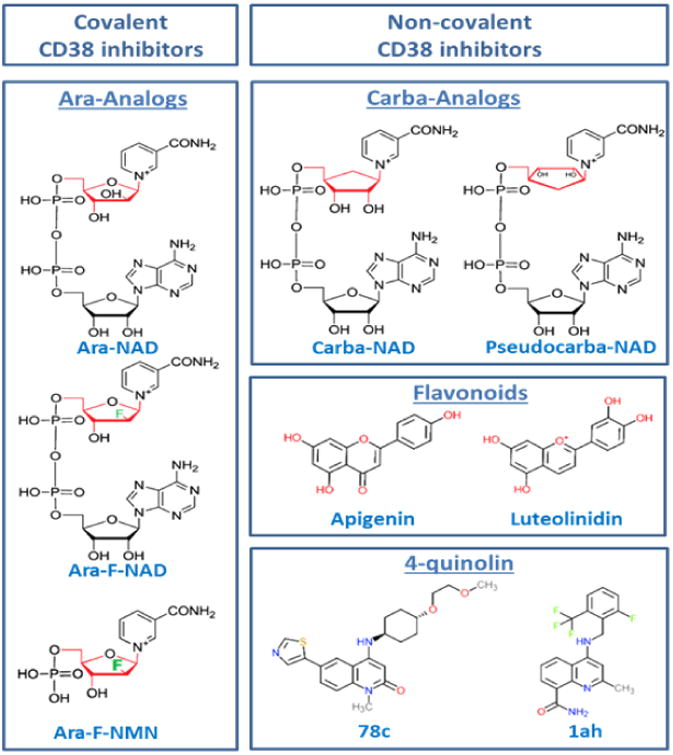
The overall mechanism of action is the inhibition of NAD breakdown. To date no inhibitors can selectively block the glycohydrolase vs the cyclase activity of CD38. NAD analogs can be considered covalent and non-covalent inhibitors. Ara-NAD analogs are covalent competitive inhibitors. Carba-analogs are non-covalent competitive inhibitors. Most flavonoids and small molecule inhibitors of CD38 are non-covalent competitive inhibitors. RHein is the only uncompetitive inhibitor in the flavonoid class.
The therapeutic potential of these NAD analog inhibitors may be limited due to the inhibitory effect they may have in several other NAD dependent enzymes, which raises concerns about their specificity. Thus, these “non-specific” effects may limit their in vivo applicability. However, it is possible that in the future some covalent inhibitors may serve as CD38 modifiers in therapies aimed at ex vivo optimization of immune cells therapy for cancer [50]. For example, based on recent published data, one could envision that treatment of ex vivo hybrid Th1/17 cells with these inhibitors could increase their tumor suppressive effects [50].
Flavonoids
Several natural compounds were reported to inhibit the catalytic activity of CD38 [40, 71-76]. These include flavonoids such as apigenin, quercetin, and leteolinidin (Figure 4 and Table 2). The beneficial effects of flavonoid CD38 inhibitors were reported in animal models of obesity, heart ischemia, kidney injury, viral infection, and cancer [71-77]. Mechanistically, inhibition of CD38 in vivo via apigenin leads to an increase in cellular NAD levels and activation of NAD-dependent enzymes, (e.g.SIRTUINs) [72]. Most flavonoids are competitive antagonists of CD38 and 4,5-dihydroxyanthraquinone-2-carboxylic acid (RHein) is the only uncompetitive inhibitor reported to date (Table 2)[75-76]. Unfortunately, flavonoids are far from being specific inhibitors of CD38, showing several “off-target” effects, and also have poor oral pharmacokinetic profile [1,2, 9-13]. On the other hand, some of these compounds were described as safe for human use, supporting druggability of CD38.
CD38 inhibitors derived from 4-amino-quinoline
The availability of data from crystallography of CD38, and a better understanding of its mechanism of catalysis, paved the road for the design of new high affinity and more specific CD38 inhibitors [78-79]. These small molecule candidates are non-covalent reversible inhibitors of the enzyme. Two studies using high throughput screening discovered active compounds with IC50 in the nanomolar range [78-79]. The compounds are heterocycles derivatives of 4-amino-quinoline [78-79]. The three lead compounds, 78c (IC50 7.3 nM), 1ai (IC50 46nM), and Iah (IC50 115nM), increased NAD levels in muscle and liver of diet-induced obese mice [78-79]. They have improved pharmacokinetic profile for oral administration and acceptable specificity, chemical, and biological stability compared to NAD analogous and flavonoids. Mechanistically, they may interact with other amino acid residues in the catalytic site, rather than Glu226. For 78c, the most important interaction in the active site appears to be a hydrophobic interaction of C4 group and Trp176 and the hydrogen bond between the 2-ketoresidue and the side chain amide of Asp183 [78]. For the other derivatives, hydrogen bonds between the carboxylate side chain of both Glu146 and Asp155 and face-to-face π-π stacking with Trp189 appear to be more important [78-79]. As previously discussed, mutagenesis studies revealed that these residues are important for the NADase activity of human CD38, and changes in these residues led to the abolishment of the glycohydrolase activity and to an increase in the cyclase activity. So, the blockade of the interaction of endogenous substrates with these residues appears to be the molecular mechanism of inhibition by these drugs [78-79]. It is possible that these inhibitors may also have selective inhibitory effects on the CD38 NADase over its cyclase activity. Thus, these compounds may become powerful tools to understand the importance of the different catalytic activities of CD38 in a multiplicity of conditions like asthma, cancer, metabolic syndrome and aging. Because these drugs increase NAD levels in many tissues in vivo, and our studies show that CD38 is the main NADase regulating NAD levels during aging, these compounds can also improve and advance “NAD boosting” therapy for aging-related diseases.
Concluding Remarks
The development of non-cytotoxic anti-CD38 antibodies and small molecules targeting the NADase enzymatic activity of CD38 may present as an important advancement in therapies for cancers and age-related diseases, but many important questions remain about CD38 biology and potential therapeutic uses (see outstanding questions). One of these questions concerns the CD38 “topological paradox” and its role in intercellular NAD homeostasis. Of particular interest, is there a specific role for CD38 sub-cellular localization on the dynamics of intracellular NAD compartmentalization? For example, does CD38 regulates preferentially NAD in specific locations such as the mitochondrial or nuclei? This is a very important question that could potentially be approached experimentally by using CD38 constructs targeted for different sub-cellular distributions and the newly developed NAD biosensors [80-81]. Another important question in the biology of NAD and CD38 is related with the potential role of the CD38 gene homologous BST-1 in organismal NAD homeostasis. Finally, from the pharmacological point of view it is necessary to determine the therapeutic role of CD38/NADase inhibitors and other NAD “boosting” therapies for diseases of aging and metabolic syndrome. In this regard, a very crucial question is what are the potential unwanted side effects of CD38 inhibitors? Based on the role of CD38 in the immune system and behavior these may include impaired immune response to infections and neurological abnormalities [40, 50-52, 82-83]. These side effects vary between different types of CD38 inhibitors due to different mechanisms of inhibition, effect on the cyclase vs NADase activity of CD38, tissue distribution, and pharmacokinetic profiles. For example, the low blood brain barrier penetrability of antibodies and some small molecule inhibitors may mitigate the theoretical CNS side effects of CD38 inhibitors. Future pre-clinical and clinical studies will bring light to the potential transformative role of CD38 inhibitors in human diseases.
Outstanding Questions Box.
Is CD38 ecto-NADase activity the main contributor to NAD homeostasis in vivo?
How does CD38, a mainly ecto-NADase, regulate the homeostasis of NAD inside of the cell?
How do the different orientations and subcellular distribution of CD38 contribute to the compartmentalization of NAD homeostasis?
Does a CD38 positive cell regulate the availability of NAD precursors to a CD38 negative cell in vivo?
Does BST-1, a protein homologous to CD38, play a role in organismal NAD homeostasis?
Will NAD-boosting therapies be an effective way to treat or prevent age-related diseases and metabolic syndrome?
Does the combination of CD38 inhibitors with NAD precursors present an advantage in NAD boosting therapy for age-related diseases?
What will be the unwanted side effects of inhibiting CD38 NADase activity?
Highlights.
Changes in NAD metabolism play an important role in the aging process and the pathogenesis of several diseases.
“NAD boosting” therapy can promote increases in longevity and healthspan in animals models of aging, accelerated aging and age-related diseases.
CD38 is one of the main NAD degrading enzymes in mammalian tissues, and plays a key role in age-related NAD decline.
CD38 is a druggable target in the therapy of human cancers.
Inhibition of CD38 with small molecules or monoclonal antibodies can decrease NADase activity and promote boosting in cellular NAD.
Acknowledgments
The work in Dr. Chini's laboratory is supported in part by grants from the Ted Nash Long Life Foundation, the Glenn foundation for medical research via the Paul F. Glenn Laboratories for Senescence at the Mayo Clinic, National Institutes of Health (NIH) grants from the National Institute of Aging (NIA, grant AG-26094), and the NCI via the Mayo Clinic pancreatic cancer SPORE program (CA 102701-14P2). Dr. Chini has a patent on CD38 inhibitors, and is a consultant for Teneobio. Dr. Wim van Schooten is the Chief Scientific Officer for Teneobio a company interested in the development of therapeutic antibodies.
Glossary
- NADase
An enzymatic activity that leads to the degradation of NAD unrespect of the products
- NAD glycohydrolase
The break of the glycosidic bond between nicotinamide and ribose of NAD. This reaction leads to the degradation of NAD and formation of the products adenosine diphosphate-ribose (ADPR) and nicotinamide
- ADP-rybosyl cyclase
A reaction where NAD is converted to its cyclic analog cyclic-adenosine-diphosphate ribose (cADPR) and nicotinamide
- cADPR and ADPR
cADPR and ADPR are second messengers involved in calcium signaling
- SIRTUINs
NAD-dependent deacetylases involve don the control of metabolism, healthspan and aging. In mammals there are seven SIRTUINS (SIRT1-7)
- NAD precursors
NAD precursors are molecules that can be used by cells and tissues to make NAD. They include nicotinamide and vitamin B3 derivatives such as nicotinamide riboside (NR) and nicotinamide mono nucleotide (NMN)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chini CCS, et al. NAD and the aging process: Role in life, death and everything in between. Mol Cell Endocrinol. 2017;455:62–74. doi: 10.1016/j.mce.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 3.Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schultz MD, Sinclair DA. Why NAD+ Declines during Aging: It's Destroyed. Cell Metab. 2016;23(6):965–966. doi: 10.1016/j.cmet.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes AP, et al. Declining NAD[+] induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell= 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.J Camacho-Pereira J, et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016;23:1127–1139. doi: 10.1016/j.cmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mouchiroud L, et al. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshino J. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14(4):528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Picciotto NE, et al. Seals. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016;15(3):522–530. doi: 10.1111/acel.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H, et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352(6292):1436–43. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- 11.North BJ, et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J. 2014;33(13):1438–53. doi: 10.15252/embj.201386907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mills KF, et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016;24(6):795–806. doi: 10.1016/j.cmet.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheibye-Knudsen M, et al. A high-fat diet and NAD[+] activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014;420(5):840–55. doi: 10.1016/j.cmet.2014.10.005. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams PA, et al. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017;355(6326):756–760. doi: 10.1126/science.aal0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koenekoop RK, et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012;44(9):1035–1039. doi: 10.1038/ng.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Someya S, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown KD, et al. Activation of SIRT3 by the NAD+ precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014;20(6):1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cantó C, et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbosa MT, et al. The enzyme CD38 [a NAD glycohydrolase, EC 3.2.2.5] is necessary for the development of diet-induced obesity. FASEB J. 2007;21:3629–3639. doi: 10.1096/fj.07-8290com. [DOI] [PubMed] [Google Scholar]
- 20.Trammell SAJ, et al. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep. 2016;6:26933. doi: 10.1038/srep26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gariani K, et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology. 2016;63:1190–1204. doi: 10.1002/hep.28245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.French SW. Chronic alcohol binging injures the liver and other organs by reducing NAD+ levels required for sirtuin's deacetylase activity. Exp Mol Pathol. 2016;100(2):303–306. doi: 10.1016/j.yexmp.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Tummala KS. Inhibition of de novo NAD[+] synthesis by oncogenic URI causes liver tumorigenesis through DNA damage. Cancer Cell. 2014;26(6):826–839. doi: 10.1016/j.ccell.2014.10.002. 2014. [DOI] [PubMed] [Google Scholar]
- 24.Guan Y, et al. Nicotinamide Mononucleotide, an NAD+ Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J Am Soc Nephrol. 2017;28(8):2337–2352. doi: 10.1681/ASN.2016040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran MT, et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531:528–532. doi: 10.1038/nature17184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boslett J, et al. Endothelial Cells Highly Express CD38 with Activation by Hypoxia/Reoxygenation Depleting NAD[P]H. Am J Physiol Cell Physiol. 2017 doi: 10.1152/ajpcell.00139.2017. doi:ajpcell.00139.2017. Pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y. Exogenous NAD[+] administration significantly protects against myocardial ischemia/reperfusion injury in rat model. Am J Transl Res. 2016;8(8):3342–3350. [PMC free article] [PubMed] [Google Scholar]
- 28.Gerdts J, et al. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science. 2015;348:453–457. doi: 10.1126/science.1258366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park JH, et al. Nicotinamide mononucleotide inhibits post-ischemic NAD[+] degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol Dis. 2016;95:102–110. doi: 10.1016/j.nbd.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryu D, et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med. 2016;8(361):361ra139. doi: 10.1126/scitranslmed.aaf5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerutti R, et al. NAD+-Dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014;19:1042–1049. doi: 10.1016/j.cmet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi H, et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N Engl J Med. 2017;377(6):544–552. doi: 10.1056/NEJMoa1616361. [DOI] [PubMed] [Google Scholar]
- 33.Longo VD, et al. Interventions to slow aging in humans: are we ready? Aging Cell. 14:497–510F. doi: 10.1111/acel.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aksoy P, et al. Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem Biophys Res Commun. 2006;349:353–359. doi: 10.1016/j.bbrc.2006.08.066. [DOI] [PubMed] [Google Scholar]
- 35.P Aksoy P, et al. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun. 2006;345:1386–1392. doi: 10.1016/j.bbrc.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 36.Chiang SH, et al. Genetic ablation of CD38 protects against western diet-induced exercise intolerance and metabolic inflexibility. PLoS One. 2015;10:e0134927. doi: 10.1371/journal.pone.0134927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu Y, et al. Overexpression of CD38 decreases cellular NAD levels and alters the expression of proteins involved in energy metabolism and antioxidant defense. J Proteome Res. 2014;13:786–795. doi: 10.1021/pr4010597. [DOI] [PubMed] [Google Scholar]
- 38.Malavasi S, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 39.Chini EN. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr Pharm Des. 2009;15:57–63. doi: 10.2174/138161209787185788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deshpande DA, et al. CD38 in the Pathogenesis of Allergic Airway Disease: Potential Therapeutic Targets. Pharmacology & therapeutics. 2017:172, 116–26. doi: 10.1016/j.pharmthera.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lokhorst HM, et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N Engl J Med. 2015;373(13):1207–19. doi: 10.1056/NEJMoa1506348. [DOI] [PubMed] [Google Scholar]
- 42.van de Donk N, et al. CD38 antibodies in multiple myeloma: back to the future. Blood. 2017 doi: 10.1182/blood-2017-06-740944. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 43.de Weers M, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840–8. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 44.Horenstein AL, et al. NAD[+]-Metabolizing Ectoenzymes in Remodeling Tumor-Host Interactions: The Human Myeloma Model. Cells. 2015;4(3):520–37. doi: 10.3390/cells4030520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krejcik J, et al. Monocytes and granulocytes reduce CD38 expression levels on myeloma cells in patients treated with daratumumab. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-17-2027. 2017. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng X, et al. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin Cancer Res. 2017;23(15):4290–4300. doi: 10.1158/1078-0432.CCR-16-3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nijhof IS, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood. 2016;128(7):959–70. doi: 10.1182/blood-2016-03-703439. [DOI] [PubMed] [Google Scholar]
- 48.Deckert J, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20(17):4574–83. doi: 10.1158/1078-0432.CCR-14-0695. [DOI] [PubMed] [Google Scholar]
- 49.Martin T, et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;129(25):3294–3303. doi: 10.1182/blood-2016-09-740787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chatterjee S, et al. CD38-NAD+Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2017 doi: 10.1016/j.cmet.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee HC, Aarhus R. ADP-ribosyl cyclase: an enzyme that cyclizes NAD+ into a calcium-mobilizing metabolite. Cell Regul. 1991;3:203–209. doi: 10.1091/mbc.2.3.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matalonga J, et al. The Nuclear Receptor LXR Limits Bacterial Infection of Host Macrophages through a Mechanism that Impacts Cellular NAD Metabolism. Cell Rep. 2017;18(5):1241–1255. doi: 10.1016/j.celrep.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 53.Franceschi C, Campisi J. Chronic inflammation [inflammaging] and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69:S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 54.Shrimp JH, et al. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J Am Chem Soc. 2014;136(15):5656–5663. doi: 10.1021/ja411046j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grozio A. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J Biol Chem. 2013;288:25938–25949. doi: 10.1074/jbc.M113.470435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu J, et al. Cytosolic interaction of type III human CD38 with CIB1 modulates cellular cyclic ADP-ribose levels. Proc Natl Acad Sci. 2017 doi: 10.1073/pnas.1703718114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zielinska W, et al. Metabolism of cyclic ADPribose: zinc is an endogenous modulator of the cyclase/NAD glycohydrolase ratio of a CD38-like enzyme from human seminal fluid. Life Sci. 2004;74:1781–1790. doi: 10.1016/j.lfs.2003.08.033. [DOI] [PubMed] [Google Scholar]
- 58.Sauve AA. A Covalent Intermediate in CD38 Is Responsible for ADP-Ribosylation and Cyclization Reactions. J Am Chem Soc. 2000;122(33):7855–59. [Google Scholar]
- 59.Zhang S, et al. Comparative Analysis of Pharmacophore Features and Quantitative Structure-Activity Relationships for CD38 Covalent and Non-Covalent Inhibitors. Chem Biol and Drug Design. 2015;86(6):1411–24. doi: 10.1111/cbdd.12606. [DOI] [PubMed] [Google Scholar]
- 60.Liu Q, et al. Structural basis for enzymatic evolution from a dedicated ADP-ribosyl cyclase to a multifunctional NAD hydrolase. J Biol Chem. 2009;284(40):27637–27645. doi: 10.1074/jbc.M109.031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horenstein AL, et al. Adenosine Generated in the Bone Marrow Niche Through a CD38-Mediated Pathway Correlates with Progression of Human Myeloma. Mol Med. 2016 doi: 10.2119/molmed.2016.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krauset D, et al. Nicotinamide N-methyltransferase knockdown protects against iet-induced obesity. Nature. 2014;508:258–262. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Avalos JL, et al. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD[+] cosubstrate specificity of a Sir2 enzyme. Mol Cell. 2005;17:855–868. doi: 10.1016/j.molcel.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 64.Smithson G, et al. CD38+ cell depletion with TAK-079 reduces arthritis in a cynomolgus collagen-induced arthritis (CIA) model. J Immunol. 2017;198(1 Supplement):127.17. [Google Scholar]
- 65.Liu Z, et al. Studies on CD38 Inhibitors and Their Application to cADPR-Mediated Ca2+; Signaling. Messenger. 2013;2(1):19–32. [Google Scholar]
- 66.Muller-Steffner HM, et al. Slow-Binding Inhibition of NAD+ Glycohydrolase by Arabino Analogues of β-NAD+ J of Biol Chem. 1992;267(14):9606–11. [PubMed] [Google Scholar]
- 67.Slama JT, Simmons AM. Carbanicotinamide Adenine Dinucleotide: Synthesis and Enzymological Properties of a Carbocyclic Analogue of Oxidized Nicotinamide Adenine Dinucleotide. Biochemistry. 1988;27(1):183–93. doi: 10.1021/bi00401a028. [DOI] [PubMed] [Google Scholar]
- 68.Wall K, et al. Inhibition of the Intrinsic NAD+ Glycohydrolase Activity of CD38 by Carbocyclic NAD Analogues. The Bioch J. 1998;335:631–636. doi: 10.1042/bj3350631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sauve AA, Schramm VL. Mechanism-Based Inhibitors of CD38: A Mammalian Cyclic ADP-Ribose Synthetase. Biochemistry. 2002;41(26):8455–63. doi: 10.1021/bi0258795. [DOI] [PubMed] [Google Scholar]
- 70.Kwong AKY, et al. Catalysis-Based Inhibitors of the Calcium Signaling Function of CD38. Biochemistry. 2012;51(1):555–64. doi: 10.1021/bi201509f. [DOI] [PubMed] [Google Scholar]
- 71.Kellenberger E, et al. Flavonoids as Inhibitors of Human CD38. Bioorg and Med Chem Lett. 2011;21(13):3939–3942. doi: 10.1016/j.bmcl.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 72.Escande C, et al. Flavonoid Apigenin Is an Inhibitor of the NAD+ase CD38: Implications for Cellular NAD+ Metabolism, Protein Acetylation, and Treatment of Metabolic Syndrome. Diabetes. 2013;62(4):1084–93. doi: 10.2337/db12-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boslett James, et al. Luteolinidin Protects the Postischemic Heart through CD38 Inhibition with Preservation of NAD[P][H] J Pharmacol Exp Ther. 2017;361(1):99–108. doi: 10.1124/jpet.116.239459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shu B, et al. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-kB signaling suppression. Cell Signal. 2017 doi: 10.1016/j.cellsig.2017.10.014. [DOI] [PubMed] [Google Scholar]
- 75.Blacher E, et al. Inhibition of Glioma Progression by a Newly Discovered CD38 Inhibitor. Int J Cancer. 2015;136(6):1422–33. doi: 10.1002/ijc.29095. [DOI] [PubMed] [Google Scholar]
- 76.Blacher E, et al. “Targeting CD38 in the Tumor Microenvironment; a Novel Approach to Treat Glioma. Cancer Cell & Microenv. 2015;2:e486. 1–6. [Google Scholar]
- 77.Schiavoni I, et al. CD38 modulates respiratory syncytial virus-driven pro-inflammatory processes in human monocyte-derived dendritic cells. Immunology. 2017 doi: 10.1111/imm.12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haffner CD, et al. Discovery, Synthesis, and Biological Evaluation of Thiazoloquin[az]olin[on]es as Potent CD38 Inhibitors. J Med Chem. 2015;58(8):3548–71. doi: 10.1021/jm502009h. [DOI] [PubMed] [Google Scholar]
- 79.Becherer JD, et al. Discovery of 4-Amino-8-Quinoline Carboxamides as Novel, Submicromolar Inhibitors of NAD-Hydrolyzing Enzyme CD38. J Med Chem. 2015;58(17):7021–7056. doi: 10.1021/acs.jmedchem.5b00992. [DOI] [PubMed] [Google Scholar]
- 80.Cambronne XA, et al. Biosensor reveals multiple sources for mitochondrial NAD+ Science. 2016;352(6292):1474–1477. doi: 10.1126/science.aad5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao Y, et al. SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metab. 2015;21(5):777–789. doi: 10.1016/j.cmet.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Partida-Sanchez DA, et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med. 2001;7:1209–1216. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- 83.Lopatina O, et al. The roles of oxytocin and CD38 in social or parental behaviors. Front Neurosci. 2012;6:182. doi: 10.3389/fnins.2012.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]