

Abstract
Background
Epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae) have an epilithic habitat (rocky slopes) and a parapatric distribution in East Asia, which makes them an ideal model for a more comprehensive understanding of the demographic and divergence history and the influence of climate changes in East Asia. However, the genetic background and resources for these two genera are scarce.
Results
The complete chloroplast (cp) genomes of two Oresitrophe rupifraga and one Mukdenia rossii individuals were reconstructed and comparative analyses were conducted to examine the evolutionary pattern of chloroplast genomes in Saxifragaceae. The cp genomes ranged from 156,738 bp to 156,960 bp in length and had a typical quadripartite structure with a conserved genome arrangement. Comparative analysis revealed the intron of rpl2 has been lost in Heuchera parviflora, Tiarella polyphylla, M. rossii and O. rupifraga but presents in the reference genome of Penthorum chinense. Seven cp hotspot regions (trnH-psbA, trnR-atpA, atpI-rps2, rps2-rpoC2, petN-psbM, rps4-trnT and rpl33-rps18) were identified between Oresitrophe and Mukdenia, while four hotspots (trnQ-psbK, trnR-atpA, trnS-psbZ and rpl33-rps18) were identified within Oresitrophe. In addition, 24 polymorphic cpSSR loci were found between Oresitrophe and Mukdenia. Most importantly, we successfully developed 126 intergeneric polymorphic gSSR markers between Oresitrophe and Mukdenia, as well as 452 intrageneric ones within Oresitrophe. Twelve randomly selected intergeneric gSSRs have shown that these two genera exhibit a significant genetic structure.
Conclusions
In this study, we conducted genome skimming for Oresitrophe rupifraga and Mukdenia rossii. Using these data, we were able to not only assemble their complete chloroplast genomes, but also develop abundant genetic resources (cp hotspots, cpSSRs, polymorphic gSSRs). The genomic patterns and genetic resources presented here will contribute to further studies on population genetics, phylogeny and conservation biology in Saxifragaceae.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4633-x) contains supplementary material, which is available to authorized users.
Keywords: Chloroplast genome, Cp hotspot, East Asia, Population genetics, SSR
Background
Quaternary climatic oscillations accompanied by glacial and inter-glacial cycles have affected the demographic history of many temperate species, shaped their modern distributions [1, 2], and also left a deep footprint on their genetic structure [3, 4]. East Asia did not develop extensive land ice sheets during the the last glacial maximum (LGM) as Europe and eastern North America did [5]. However, the reduced temperatures (mean reduction = 7–10 °C) and increased aridity have still influenced the distribution and evolution of many plant species in China and neighboring areas [6, 7]. Initially, both paleobotanical and modeling results have revealed that temperate forests in the Northern Hemisphere would have retreated southward (below 30 °N and reaching 25 °N) at the LGM and subsequently recolonized the previously uninhabitable northern regions at the warm and wet interglacial [8–10]. However, recent phylogeographic studies of cool-temperate trees in continental East Asia suggested that, during the LGM, cool-temperate deciduous tree species could have persisted within their modern northern range rather than moving to the south [11–13].
Until recently, there were few independent phylogeographic studies of temperate herbs in East Asia to test these two hypotheses regarding how climatic oscillations affected the range distributions. Oresitrophe Bunge and Mukdenia Koidz, which are sister genera in Saxifragaceae [14, 15], are both perennial herbs growing on cliffs or rocks. Oresitrophe is monotypic, with the only species O. rupifraga Bunge occurred in Central and North China [16]; while Mukdenia has two species, M. rossii (Oliv.) Koidz. and M. acanthifolia Nakai, which are distributed from Northeast China to Korean Peninsula [16]. These two sister genera have an epilithic habitat (rocky slopes and ravines) and a parapatric distribution in East Asia, and thus provide an ideal model for a more comprehensive understanding of the demographic and divergence history and the influence of climate changes in East Asia. However, the current studies regarding their genetic background and resources are scarce.
In the last decade, high-throughput sequencing, along with bioinformatic tool development, has provided genomic resources at reasonable prices and schedules [17], with the increasing development of single nucleotide polymorphisms (SNPs) and SSRs in non-model species [18, 19]. In Saxifragaceae, the chloroplast (cp) genome remained relatively unexplored until the release of the only one cp genome of Heuchera parviflora (GenBank accession number: KR478645), and these genomic databases were limited to detect and develop the polymorphic markers. More plastid genomes for Saxifragaceae will soon be published as part of the 1KP project [20].
Chloroplast DNA (cpDNA), which is maternally inherited in most angiosperm, usually have a circular structure ranging from 115 to 165 kb in length and contain two copies of a large inverted repeat (IR) region separated by a large single copy (LSC) region and a small single copy (SSC) region [21]. Chloroplast genomes are more conserved than mitochondrial and nuclear genomes in term of gene content, organization and structure [22], and the nucleotide substitution rate of chloroplast genes is at an intermediate level (higher than mitochondria but lower than nuclear) [23]. Considering its small size, conserved gene content and simple structure, the cp genome has been generally applied for understanding the genome evolution, underlying genome size variations, gene and intron losses at higher taxonomic levels [24, 25]. In addition, the non-recombinant nature of plastid genomes and their (generally) uniparental inheritance, makes plastid data a useful tool to trace demographic history, explore species divergence, hybridization and identify species [26, 27]. Traditional screening of cp DNA regions have been chosen mostly based on their efficacy in related taxa for analysis. However, recent studies related on complete chloroplast genome sequences have allowed a more systematic approach to take into account the mutational dynamics of cp genomes [28]. By this method, cp genomic hotspots in terms of informative regions can be identified for a specific plant genus, tribe or family [29, 30]. The conventional technology of Sanger sequencing was time-consuming, troublesome and difficult for reconstructing complete cp genome [31]. In recent years, with the rapid development of high-throughput sequencing technology, especially like Illumina-based genome skimming, more and more complete cp DNA sequences have been isolated and assembled [25, 32]. Subsequently, this has been proven to be a valid and cost-effective to acquire the complete cp DNA and many assembled cp DNA of non-model species have been obtained for the studies such as differential gene expression, genetic markers development [33] and phylogenomics analysis [34].
Simple sequence repeats (SSRs), also called microsatellites containing repetitive sequences of 1–6 bp in length, have been extensively found in both the coding and non-coding sequences of prokaryotic and eukaryotic genomes [35, 36]. Currently, SSRs are broadly applied in various areas of genetic studies including the evaluation of genetic variation [37], construction of genetic linkage maps [38], population genetics [39] and domestication origin of fruit tree species [40, 41], due to their co-dominant inheritance, high polymorphism, reproducibility and transferability. The traditional methods for screening of the polymorphic SSR (polySSR) markers and their subsequent applicability to genetic researches are extremely time-consuming and labor-intensive. However, the recently increasing availability of genome and transcriptome sequences with the decreasing costs of next generation sequencing provides an excellent opportunity and information resources for large-scale mining this type of molecular markers [42]. In recent years, genomic SSR (gSSR) markers have attracted more attention due to detect higher levels of polymorphism relative to EST-SSRs, because intron or intergenic sequences are more variable than extron sequences [43, 44]. Moreover, a series of bioinformatics tools have been developed for automated SSR discovery and marker development, such as CandiSSR or GMATA, which allowed users to identify putative polySSRs not only from the transcriptome datasets but also from multiple assembled genome sequences of a given species or genus along with several comprehensive assessments [42, 45]. It would help researchers to save significant time on marker-screening experiments.
Here, two individuals of O. rupifraga and one individual of M. rossii were selected for genome skimming. We specifically aimed to: (1) assemble, characterize and compare the cp genomes among representatives of Saxifragaceae in order to gain insights into evolutionary patterns within the family; (2) develop and screen appropriate intergeneric and intrageneric markers (cp hotspot regions, cpSSRs and gSSRs) in Oresitrophe and Mukdenia.
Methods
Plant material, DNA extraction and sequencing
In order to screen polymorphic genomic resources between Oresitrophe and Mukdenia and within Oresitrophe, we selected two individuals of O. rupifraga and one individual of M. rossii with a long geographical distance, which were theoretically assumed to be more genetically different. Fresh young leaves of two O. rupifraga individuals (BJCP: LP161631–1, Muchang, Changping, Beijing, China; HNYD: LP174479–2, Tianmenshan, Zhangjiajie, Hunan, China; Additional file 1: Table S3) and one M. rossii (LP174341–20, Taipinghu, Baishan, Jilin, China; Additional file 1: Table S3) were sampled and dried with silica gel. No specific permissions were required for all the samples which are neither privately owned nor protected and the field study did not involve endangered or protected species. The total DNA was extracted using Plant DNAzol Reagent (LifeFeng, Shanghai) according to the manufacturer’s protocol from approximately 2 mg of the silica-dried leaf tissue. The high molecular weight DNA was sheared (yielding ≤800 bp fragments) and the quality of fragmentation was checked on an Agilent Bioanalyzer 2100 (Agilent Technologies). The short-insert (500 bp) paired-end libraries preparation and sequencing were performed by Beijing Genomics Institute (Shenzhen, China). The three samples were pooled with others and run in a single lane of an Illumina HiSeq 2500 with read length of 150 bp.
Genome assembly and annotation
The raw data was filtered by quality with Phred score < 30 (0.001 probability error) and all remaining high quality sequences were assembled into contigs using the CLC de novo assembler beta 4.06 (CLC Inc., Rarhus, Denmark). The parameters performed in CLC are as follows: deletion and insertion costs of 3, mismatch cost of 2, minimum contig length of 200, bubble size of 98, length fraction and similarity fraction of 0.9. Then, all the contigs were aligned to the reference chloroplast genome (Heuchera parviflora) using BLAST (NCBI BLAST v2.2.31) search. The representative chloroplast sequence contigs were ordered and oriented according to the reference chloroplast genome, and the draft chloroplast genome of O. rupifraga and M. rossii were constructed by connecting overlapping terminal sequences. Finally, clean reads were re-mapped to the draft genome and yielded the complete chloroplast genome sequences.
Initial gene annotation of the three chloroplast genomes was performed through the online program Dual Organellar Genome Annotator [46]. Putative starts, stops, and intron positions were checked according to comparisons with homologous genes of H. parviflora cp genome using Geneious v9.0.5 software (Biomatters, Auckland, New Zealand). The tRNA genes were verified with tRNAscan-SE v1.21 [47] with default settings. The circular gene maps were drawn by the OrganellarGenomeDRAW tool (OGDRAW) following by manual modification [48].
Comparative chloroplast genomic analysis
Multiple complete chloroplast genomes of Saxifragaceae provide an opportunity to compare the sequence variation within the family. Therefore, we included the publicly available chloroplast genome of Heuchera parviflora, and Tiarella polyphylla (the chloroplast genome has been sequenced by us and will be published soon elsewhere), to compare the overall similarities among different chloroplast genomes in Saxifragaceae, using Penthorum chinense (Penthoraceae; JX436155) as the reference based on the results of Dong et al. [24] and Soltis et al. [49]. The sequence identity of the five Saxifragaceae chloroplast genomes was plotted using the mVISTA program with LAGAN mode [50]. The cp DNA rearrangement analyses of five Saxifragaceae chloroplast genomes were performed using Mauve Alignment [51].
Repeat structure and sequence divergence analysis
We determined the four types of repeat sequences, including direct (forward), inverted (palindromic), complement and reverse repeats in the Oresitrophe and Mukdenia chloroplast genomes using the online REPuter software with a minimum repeat size of 30 bp and sequence identity greater than 90% [52]. Chloroplast simple sequence repeats (cpSSRs) were detected using Msatcommander v0.8.2 [53] with a threshold ten, five, five, three, three, and three repeat units for mono-, di-, tri-, tetra-, penta-, and hexanucleotide SSRs, respectively.
Multiple alignments of the three sequenced chloroplast genome sequences in this study were carried out using MAFFT version 7.017 [54]. In order to screen variable characters between Oresitrophe and Mukdenia, the average number of nucleotide differences (K) and total number of mutations (Eta) were determined to analyze nucleotide diversity (Pi) using DnaSP v5.0 [55].
Polymorphic nucleotide SSR development and validation
Firstly, we removed the chloroplast and mitochondria contigs from the assembled sequences using BLAST (NCBI BLAST v2.2.31) search with the sequence of chloroplast and mitochondria genome of H. parviflora (KR478645 & KR559021) as reference. Then, we used CandiSSR [42] to identify candidate polymorphic gSSRs between Oresitrophe and Mukdenia, as well as within Oresitrophe, based on multiple assembled sequences. The parameters performed in CandiSSR are as follows: the flanking sequence length of 100, blast evalue cutoff of 1e-10, blast identity cutoff of 95, blast coverage cutoff of 95. For each target SSRs, primers are automatically designed in the pipeline based on the Primer3 package [56, 57], and global similarities of the primer binding regions is also provided.
Twelve developed intergeneric gSSR markers were randomly selected to test the transferability on 32 individuals (four populations) of O. rupifraga and 15 individuals (two populations) of M. rossii. Standard PCR amplifications were performed following the conditions below: 94 °C for 1 min; 28 cycles of 94 °C for 30 s, 50–59 °C for 30 s, and 72 °C for 30 s; a final extension at 72 °C for 5 min. Amplification products were checked on 2% agarose gel stained with GeneGreen Nucleic Acid dye (TIANGEN, Beijing, China). Reaction products were subsequently run on an ABI PRISM 3730xl Genetic Analyzer (Applied Biosystems). Genotypes were scored by using the software GeneMarker v2.2.0 (SoftGenetics, LLC, State College, PA, USA). Genetic diversity parameters, including the number of alleles, observed and expected heterozygosity, and polymorphism information content, were estimated using CERVUS v3.0 [58]. Deviations from Hardy-Weinberg equilibrium were tested through GENEPOP v4.2 [59]. SSR genotypes’ assignment to different clusters was tested with STRUCTURE v2.3.3 [60], using 10 replicates of an admixture model allowing for correlated allele frequencies with K ranging from 1 to 10, a burn-in period of 100,000 iterations and a post-burn-in period of 1,000,000 iterations, following recommendations by Gilbert et al,. [61].
Results
Genome organization and features
We generated a total of 18,694,896, 15,247,794 and 14,404,890 paired-end (150 bp) clean reads for O. rupifraga-BJCP, O. rupifraga-HNYD and M. rossii, respectively. The de novo assembly generated 352,393 contigs with an N50 length of 346 bp and a total length of 21.69 Mb for O. rupifraga-BJCP, 382,827 contigs with an N50 length of 460 bp and a total length of 18.46 Mb for O. rupifraga-HNYD, and 352,181 contigs with an N50 length of 397 bp and a total length of 13.64 Mb for M. rossii. Each draft chloroplast genome was generated from a combined product of initial contigs (O. rupifraga-BJCP: contigs 76, 98, 136, 412 and 1913; O. rupifraga-HNYD: contigs 16, 70 and 131; M. rossii: contigs 4, 11 and 12), with no gaps and no Ns.
The complete chloroplast genomes of the three samples ranged narrowly from 156,738 bp in O. rupifraga-HNYD to 156,960 bp in M. rossii (Fig. 1, Table 1). All three chloroplast genomes shared the common feature of comprising two copies of IR (25,507–25,519 bp) separated by the LSC (87,496–87,604 bp) and SSC (18,222–18,342 bp) regions. The overall GC content was 37.80% for O. rupifraga and 37.70% for M. rossii, whereas the GC content in the LSC, SSC and IR regions were 35.70–35.80, 32.00–32.20 and 43.20%, respectively (Table 1). The chloroplast genome sequences were deposited in GenBank (accession numbers: MF774190 for O. rupifraga-BJCP, MG470845 for O. rupifraga-HNYD, and MG470844 for M. rossii).
Fig. 1.
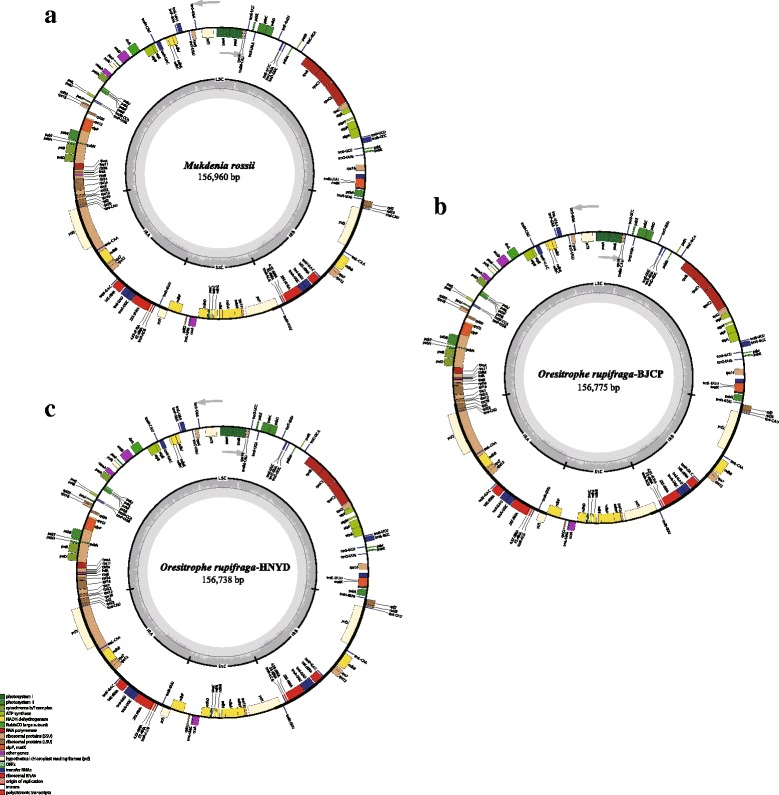
Chloroplast genome maps of Mukdenia and Oresitrophe: (a) M. rossii, (b) O. rupifraga-BJCP and (C) O. rupifraga-HNYD. Genes inside the circle are transcribed clockwise, genes outside are transcribed counter-clockwise. The light gray inner circle corresponds to the AT content, the dark gray to the GC content. Genes belonging to different functional groups are shown in different colors
Table 1.
Summary of three chloroplast genomes sequenced in this study
| Category | O. rupifraga-BJCP | O. rupifraga-HNYD | Mukdenia rossii |
|---|---|---|---|
| Total cp DNA size (bp) | 156,775 | 156,738 | 156,960 |
| Length of large single copy (LSC) region (bp) | 87,515 | 87,496 | 87,604 |
| Length of inverted repeat (IR) region (bp) | 25,519 | 25,509 | 25,507 |
| Length of small single copy (SSC) region (bp) | 18,222 | 18,224 | 8342 |
| Coding size (bp) | 90,954 | 90,954 | 90,954 |
| Intron size (bp) | 20,301 | 20,285 | 20,290 |
| Spacer size (bp) | 45,520 | 45,499 | 45,716 |
| Total GC content (%) | 37.80 | 37.80 | 37.70 |
| GC content of LSC (%) | 35.70 | 35.80 | 35.70 |
| GC content of IR (%) | 43.20 | 43.20 | 43.20 |
| GC content of SSC (%) | 32.20 | 32.20 | 32.00 |
| Total number of genes | 113 | 113 | 113 |
| Number of protein encoding genes | 79 | 79 | 79 |
| Number of tRNA genes | 30 | 30 | 30 |
| Number of rRNA genes | 4 | 4 | 4 |
| Number of genes duplicated in IR | 18 | 18 | 18 |
The three chloroplast genomes encoded an identical set of 131 genes, of which 113 were unique and 18 were duplicated in the IR regions (Tables 1 and 2). The 113 unique genes contained 79 protein-coding genes, 30 tRNA genes, and four rRNA genes. Coding regions, including protein-coding genes, tRNA genes, and rRNA genes, account for 57.95–58.03% of the whole genome, and the remaining regions were non-coding sequences, including inter-genic spacers and introns. Among the 113 unique genes, 14 contain one intron (six tRNA genes and eight protein-coding genes) and three (rps12, clpP, and ycf3) contain two introns. The 5′-end exon of the rps12 gene is located in the LSC region, and the intron and 3′-end exon of the gene are situated in the IR region.
Table 2.
Genes contained in chloroplast genomes (113 genes in total)
| Category | Group of gene | Name of gene | |||
|---|---|---|---|---|---|
| Self-replication | Ribosomal RNA genes | rrn4.5a | rrn5a | rrn16a | rrn23a |
| Transfer RNA genes |
trnA-UGCa* trnF-GAA trnH-GUG trnL-CAAa trnN-GUUa trnR-UCU trnT-GGU trnW-CCA |
trnC-GCA trnfM-CAU trnI-CAUa trnL-UAA* trnP-UGG trnS-GCU trnT-UGU trnY-GUA |
trnD-GUC trnG-GCC* trnI-GAUa* trnL-UAG trnQ-UUG trnS-GGA trnV-GACa |
trnE-UUC trnG-UCC trnK-UUU* trnM-CAU trnR-ACGa trnS-UGA trnV-UAC* |
|
| Small subunit of ribosome |
rps2 rps8 rps15 |
rps3 rps11 rps16* |
rps4 rps12a,b** rps18 |
rps7a rps14 rps19 |
|
| Large subunit of ribosome |
rpl2a rpl22 rpl36 |
rpl14 rpl23a |
rpl16* rpl32 |
rpl20 rpl33 |
|
| RNA polymerase subunits | rpoA | rpoB | rpoC1* | rpoC2 | |
| Photosynthesis | Subunits of photosystem I |
psaA psaJ |
psaB ycf3** |
psaC | psaI |
| Subunits of photosystem II |
psbA psbE psbJ psbN |
psbB psbF psbK psbT |
psbC psbH psbL psbZ |
psbD psbI psbM |
|
| Subunits of cytochrome |
petA petL |
petB* petN |
petD* | petG | |
| Subunits of ATP synthase |
atpA atpH |
atpB atpI |
atpE | atpF* | |
| Large subunit of Rubisco | rbcL | ||||
| Subunits of NADH Dehydrogenase |
ndhA* ndhE ndhI |
ndhBa* ndhF ndhJ |
ndhC ndhG ndhK |
ndhD ndhH |
|
| Other gene | Translational initiation factor | infA | |||
| Maturase | matK | ||||
| Envelope membrane protein | cemA | ||||
| Subunit of acetyl-CoA | accD | ||||
| C-type cytochrome synthesis gene |
ccsA | ||||
| Protease | clpP** | ||||
| Unknown function | Conserved open reading frames | ycf1a (part) | ycf2a | ycf4 | |
aTwo gene copies in IRs; b gene divided into two independent transcription units; one and two asterisks indicate one- and two-intron containing genes, respectively
Genome comparison of Saxifragaceae
The five Saxifragaceae chloroplast genomes were relatively conserved (Fig. 2), and no rearrangement occurred in gene organization after verification (Fig. 3), but differences were still found in terms of genome size, intron losses, and IR expansion and contraction. In addition, the IR region is more conserved in these species than the LSC and SSC regions, which is consistent with other angiosperms [25, 62].
Fig. 2.

Visualization of alignment of the five Saxifragaceae chloroplast genome sequences, with Penthorum chinense as the reference. The horizontal axis indicates the coordinates within the chloroplast genome. The vertical scale indicates the percentage of identity, ranging from 50 to 100%. Genome regions are color coded as protein coding, intron, mRNA, and conserved non-coding sequences (CNS)
Fig. 3.
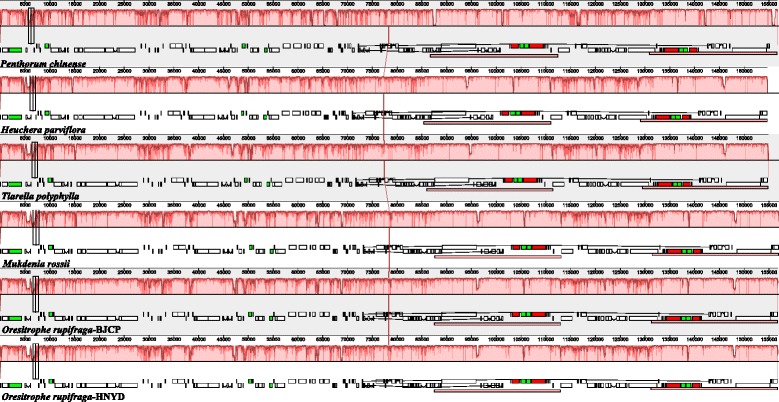
MAUVE alignment of five Saxifragaceae chloroplast genomes. The Penthorum chinense genome is shown at top as the reference. Within each of the alignment, local collinear blocks are represented by blocks of the same color connected by lines
Genome size
In terms of the chloroplast genome size observed among the representative Saxifragaceae species, M. rossii and O. rupifraga exhibited the similar genome size comparing with the reference genome with ranging from 156,690 bp to 156,960 bp, while H. parviflora and T. polyphylla had the smaller chloroplast genome comparing with the others (154,696 bp for H. parviflora and 154.850 bp for T. polyphylla, respectively; Fig. 4).
Fig. 4.
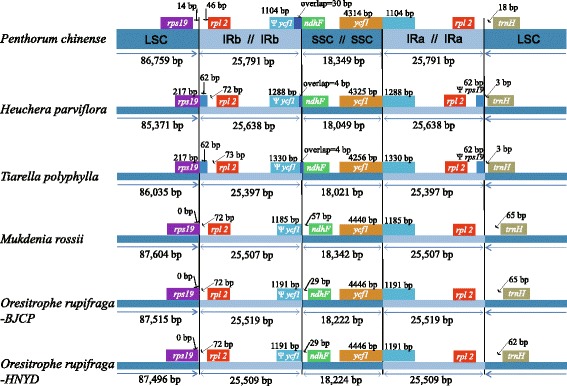
Comparison of the borders of large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) regions among the five Saxifragaceae chloroplast genomes, with the Penthorum chinense genome is shown at top as the reference. The location of two parts of inverted repeat region (IRA and IRB) was referred to Fig. 1
Intron loss
The rps16 intron has been lost from the reference genome of Penthorum chinense, although it is present in H. parviflora, T. polyphylla, M. rossii and O. rupifraga. On the contrary, the rpl2 gene in the chloroplast genomes of H. parviflora, T. polyphylla, M. rossii and O. rupifraga have lost their only intron except for P. chinense.
IR expansion and contraction
The expansion and contraction of the border regions between the two IR regions and the single-copy regions will cause the genome size differences among plant lineages. Therefore, we compared the exact IR border positions and their adjacent genes between the five Saxifragaceae chloroplast genomes and the reference genome (Fig. 4). The genes ycf1-ndhF and rps19-rpl2-trnH were located in the junctions of SSC/IR and LSC/IR regions. The ycf1 gene spanned the SSC/IRA region and the pseudogene fragment of ψycf1 varies from 1104 to 1330 bp. The ndhF gene is separated from ψycf1 by spacers with 29 bp in O. rupifraga and 57 bp in M. rossii respectively, but shares some nucleotides (from 4 to 30 bp) in other three species. The rps19 gene in H. parviflora and T. polyphylla crossed the LSC/IRB region with 62 bp located at the IRB region, and does not extend to the IRB region in P. chinense, M. rossii and O. rupifraga. The rpl2 gene is separated from the LSC/IRB border by a spacer varies from 46 to 135 bp, as well as the trnH gene is separated from the IRA/LSC border by a spacer varies from 3 to 65 bp.
Repetitive sequences and hotspot regions in cp genomes
In the current study, the type, distribution and presence of microsatellites were studied between the cp genomes of O. rupifraga and M. rossii. A total of 58 perfect microsatellites were identified in the O. rupifraga-BJCP cp genome. Among them, 44 were located in the LSC region, whereas 8 and 6 were found in the IR and SSC regions, respectively. Moreover, 6 SSRs were found in the protein-coding regions, 6 were in the introns and 46 were in intergenic spacers of the O. rupifraga-BJCP cp genome (Fig. 5a). The distribution and type of microsatellites of other two genomes (O. rupifraga-HNYD and M. rossii) is shown in supplementary Additional file 2: Figure S1. Among these SSRs, 43 are mononucleotides, 11 are dinucleotides, and 4 are tetranucleotides, tri-, penta-, and hexanucleotides are not found in the cp genomes of O. rupifraga and M. rossii (Fig. 5b).
Fig. 5.
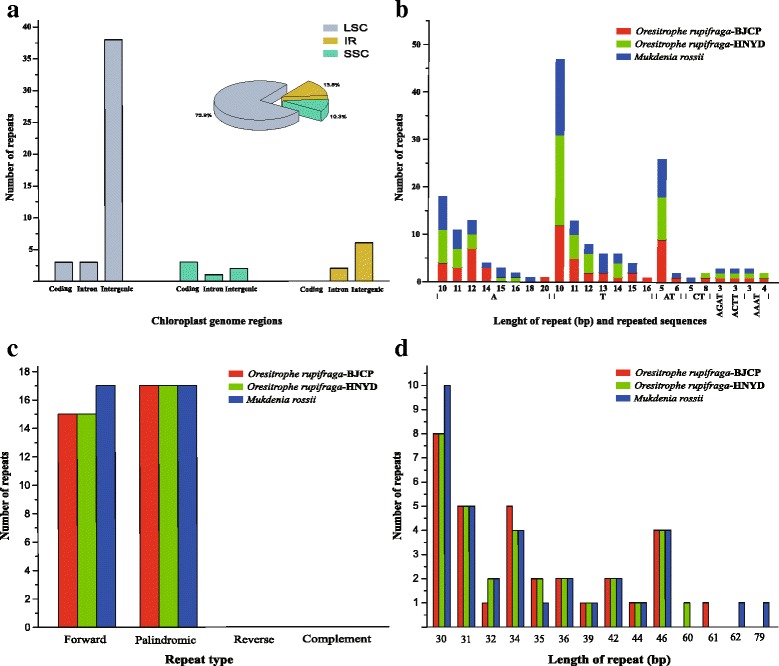
The distribution, type, and presence of simple sequence repeats (SSRs) and analysis of repeated sequences in the cp genome of Oresitrophe rupifraga and Mukdenia rossii: (a) Presence of SSRs in the different region of O. rupifraga-BJCP cp genome, (b) Presence of polymers in the cp genome of O. rupifraga and M. rossii, (c) Frequency of repeat types, (d) Frequency of repeats by length
In the chloroplast genome of O. rupifraga and M. rossii, 32 and 34 pairs of repeats (30 bp or longer) were detected using the program REPuter (Kurtz and Schleiermacher, 1999). Among these repeat sequences, 15 and 17 are forward repeats in O. rupifraga and M. rossii respectively, and the rest of 17 are palindromic repeats in all the three chloroplast genomes (Fig. 5c). In addition, 30–46 bp long repeats occurred in the three chloroplast genomes, as well as 60 bp, 61 bp, 62 and 79 bp long repeats are only detected in O. rupifraga-HNYD, O. rupifraga-BJCP and M. rossii respectively (Fig. 5d).
The coding genes, non-coding regions and intron regions were comparing within Oresitrophe and between Oresitrophe and Mukdenia divergence hotspots. We generated 72 loci (20 coding genes, 40 inter-genic spacers, and 12 intron regions) within Oresitrophe and 116 loci (47 coding genes, 53 inter-genic spacers, and 16 intron regions) between Oresitrophe and Mukdenia with more than 200 bp in length and the nucleotide variability (Pi) values calculated with the DnaSP v5.0 software. Among the values received from comparative analysis, we found it is ranged from 0.0004 (ndhB gene) to 0.0259 (trnR-atpA region) between Oresitrophe and Mukdenia (Fig. 6a) and from 0.002 (ycf2 gene) to 0.0174 (rpl33-rps18 region) within Oresitrophe (Fig. 6b), and the IR region is much more conserved than the LSC and SSC regions. Seven of these variable loci (Pi > 0.009) including trnH-psbA, trnR-atpA, atpI-rps2, rps2-rpoC2, petN-psbM, rps4-trnT and rpl33-rps18, as well as four variable loci (Pi > 0.006) including trnQ-psbK, trnR-atpA, trnS-psbZ and rpl33-rps18, showed high levels of intergeneric and intrageneric variation.
Fig. 6.
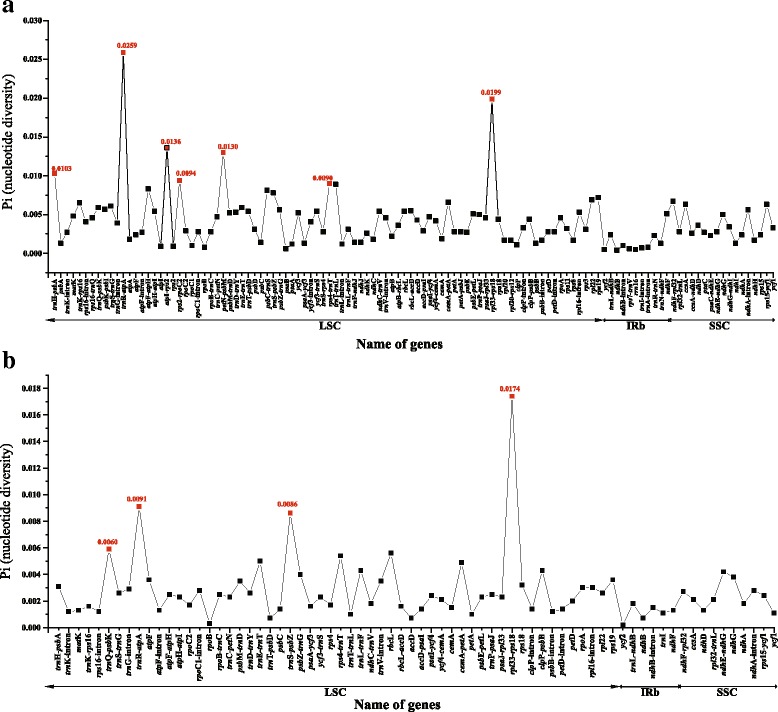
Comparative analysis of the nucleotide variability (Pi) values between Mukdenia rossii and Oresitrophe rupifraga (a), and within O. rupifraga (b)
Polymorphic genomic SSRs development and validation
A total of 242 candidate polymorphic gSSRs were identified in both Oresitrophe and Mukdenia. After screening by similarity < 90% (27) and no available primers designed (89), we obtained 126 polymorphic gSSRs with the standard deviation ranged from 0.47 to 4.00 between the two genera (Fig. 7a, Additional file 3: Table S1). Among them, di-, tri-and tetranucleotides account for 77.0%, 22.2% and 0.79%, respectively. In addition, we also detected 691 candidate polymorphic gSSRs within Oresitrophe, after removing the loci with the similarity < 90% (31) and no available primers designed (208), we received 452 polymorphic gSSRs with the standard deviation ranged from 0.50 to 5.50, and di-, tri-, tetra- and hexanucleotides account for 78.10%, 19.90%, 1.77% and 0.22%, respectively (Fig. 7b, Additional file 4: Table S2).
Fig. 7.
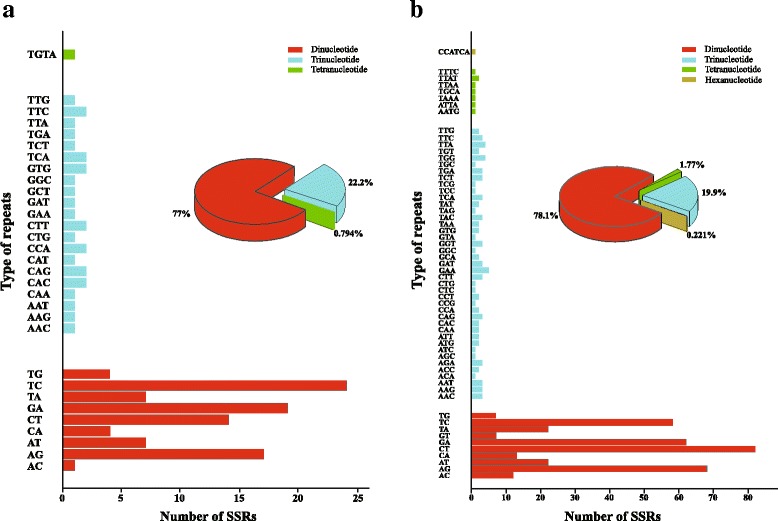
The distribution of polymorphic genomic simple sequence repeats (gSSRs) between Mukdenia rossii and Oresitrophe rupifraga (a), and within O. rupifraga (b)
To test the transferability of the developed markers, we selected twelve pairs of candidate polySSRs primers (Additional file 3: Table S1) and six populations (Additional file 1: Table S3) including four populations for O. rupifraga and two populations for M. rossii to detect the effectiveness of primer amplification and to preliminarily assess the genetic variation. Genetic diversity parameters were calculated for two species (Table 3). The polymorphism information content ranged from 0.030 to 0.778, the number of alleles ranged from 2 to 11, and the observed heterozygosity and expected heterozygosity varied from 0.031 to 1.000 and 0.031 to 0.825, respectively. No significant deviation from Hardy-Weinberg equilibrium (P < 0.001) was observed for the selected 12 loci except OR242 and OR41 in O. rupifraga group, which might be caused by wahlund effect, inbreeding, null alleles and sampling effect.
Table 3.
The genetic parameters (per locus) in Oresitrophe rupifraga and Mukdenia rossii
| Locus | Oresitrophe rupifraga (N = 32) | Mukdenia rossii (N = 15) | ||||||
|---|---|---|---|---|---|---|---|---|
| A | H O | H E | PICa | A | H O | H E | PICa | |
| OR133 | 8 | 0.594 | 0.800 | 0.754 | 6 | 0.467 | 0.761 | 0.696 |
| OR242 | 2 | 1.000 | 0.508 | 0.375*** | 2 | 0.867 | 0.517 | 0.375 |
| OR9 | 5 | 0.375 | 0.328 | 0.300 | 7 | 0.733 | 0.818 | 0.763 |
| OR103 | 2 | 0.563 | 0.411 | 0.323 | 3 | 0.933 | 0.549 | 0.421 |
| OR107 | 6 | 0.469 | 0.643 | 0.566 | 8 | 0.800 | 0.807 | 0.749 |
| OR127 | 3 | 0.094 | 0.272 | 0.240 | 5 | 0.733 | 0.634 | 0.553 |
| OR148 | 8 | 0.563 | 0.743 | 0.691 | 7 | 0.800 | 0.825 | 0.772 |
| OR179 | 11 | 0.750 | 0.813 | 0.778 | 7 | 0.933 | 0.752 | 0.691 |
| OR212 | 4 | 0.188 | 0.345 | 0.307 | 3 | 0.467 | 0.559 | 0.466 |
| OR224 | 7 | 0.344 | 0.568 | 0.534 | 4 | 0.600 | 0.522 | 0.458 |
| OR41 | 7 | 0.344 | 0.675 | 0.624*** | 5 | 0.600 | 0.641 | 0.580 |
| OR131 | 2 | 0.031 | 0.031 | 0.030 | 4 | 0.400 | 0.531 | 0.475 |
Note: A = number of alleles per locus; HE = expected heterozygosity; HO = observed heterozygosity; N = number of individuals sampled; PIC = polymorphism information content
aSignificant deviations from Hardy-Weinberg equilibrium at *P < 0.05, **P < 0.01, ***P < 0.001, respectively
In the STRUCTURE analysis, the true number of clusters K in the data were difficult to determine following Falush et al. [63], due to ln P(D) increased progressively as K increased (Additional file 5: Figure S2). The ΔK statistic of Evanno et al. [64], however, permitted detection of a rate change in ln P(D) corresponding to K = 2. At K = 2, all the six populations were separated into two clusters according to the different species (Fig. 8a). Moreover, for K = 3, we found that four O. rupifraga populations were further separated into two clusters, with HBQL, TJLX and BJCP assigned into one cluster, and HNYD into the second cluster (Fig. 8b).
Fig. 8.
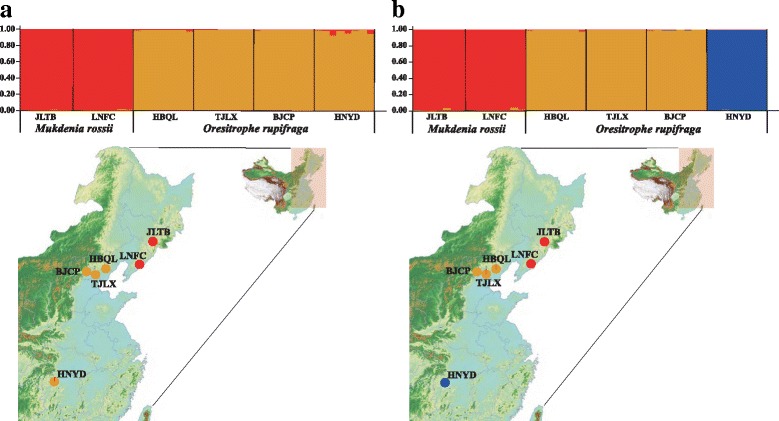
The probability of membership and geographical distribution of gene pools in Mukdenia rossii and Oresitrophe rupifraga, detected by STRUCTURE analysis: K = 2 (a) and K = 3 (b). Each vertical bar represents one individual (N = 47), with populations arranged by collection site from Northeast to Central China
Discussion
Chloroplast genome organization of Oresitrophe and Mukdenia and genome evolution in Saxifragaceae
The availability of plastid genome sequences for most major lineages of angiosperms has increased rapidly with next generation sequencing (NGS) methods development during the past decade. These data have provided many new insights into angiosperm phylogenetic relationships [25, 65], genomic rearrangements [66, 67], and genome-wide patterns and rates of nucleotide substitutions [68, 69]. In Saxifragaceae, the chloroplast genomes remained relatively limited, with only one species (Heuchera parviflora) was sequenced. In this study, we assembled and annotated three complete chloroplast genomes including two of Oresitrophe rupifraga and one of Mukdenia rossii. By comparing cp genomes in Saxifragaceae, we were able to gain s insights into evolutionary patterns of the family.
Comparative analyses of three chloroplast genomes sequenced in this study also showed highly conserved structures and genes. The size of O. rupifraga-BJCP, O. rupifraga-HNYD, and M. rossii ranged narrowly from 156,738 bp to 156,960 bp with sharing the common feature of comprising two copies of IR separated by the LSC and SSC regions. Most angiosperms commonly encode 74 protein-coding genes, while an additional five are present in only some species [70]. However, the three cp genomes contained 79 protein-coding genes, 30 tRNA genes, and four rRNA genes, which is similar to Heuchera parviflora and Penthorum chinense. This might have been because the genome shares its gene contents with the Saxifragaceae family.
After comparing the cp genomes between four Saxifragaceae species and the reference, we found the gene content and genome structure were relatively conserved, and no rearrangement occurred in gene organization, but some differences were detected in terms of intron losses and IR expansion and contraction. Two genes, rpl2 and rps16, presented the intron loss phenomenon. The rpl2 intron loss has been reported in some Saxifragaceae genera, such as Saxifraga and Heuchera [71]. This phenomenon was subsequently confirmed in Heuchera sanguinea (HQ664603), but was absent in H. micrantha (EF207446) and Saxifraga stolonifera (EF207457). In this study, the rpl2 intron is lost in all four representative species, suggesting that intron loss in the rpl2 gene is not occasional in Saxifragaceae. The rps16 gene has lost its only intron in the chloroplast genome of Penthorum chinense, but present in Oresitrophe rupifraga, Mukdenia rossii, Heuchera parviflora and Tiarella polyphylla, which is similar to those of the other published Saxifragales species [24]. Previously studies have reported the rps16 intron loss is also detected in Trachelium (Campanulaceae) [67] and Pelargonium (Geraniaceae) [72], we still deduced this phenomenon is unusual in normal angiospermous chloroplast genomes because the genome of Trachelium and Pelargonium have been extensively restructured. Moreover, the ycf15 gene, which displays a small open reading frame (ORF) with potential function in tobacco [73], was pseudogenized in all five representatives of Saxifragaceae. The infA gene, which functions as a translation initiation factor [74] with loss of it having independently experienced multiple times during the evolution of land plants [70], appears in all of the species in this study. Thus, we inferred that the pseudogenization of ycf15 and attendant of infA are ancestral condition in Saxifragaceae.
The border regions of LSC/IRB, IRB/SSC, SSC/IRA, and IRA/LSC represent highly variable regions with many nucleotide changes in cp genomes of closely related species [75]. Therefore, we compared the exact IR border positions and their adjacent genes among the five Saxifragaceae chloroplast genomes and the reference genome. The result showed that T. polyphylla and H. parviflora have relatively similar boundary characteristics with the rps19 gene locating at the junction of LSC/IRB region of cp genome and the ndhF gene sharing some nucleotides with the ycf1 pseudogene. Whereas M. rossii and O. rupifraga presented similar boundary characteristics with the rps19 gene does not extending to the IRB region and the ndhF gene does not sharing any nucleotides with the ycf1 pseudogene. The reference genome of P. chinense showed a relatively independent boundary feature comparing with the Saxifragaceae species. In Saxifragaceae, we deduced that the species with closer phylogenetic relationship will have more similar boundary feature. However, due to limited species were sampled, we need more chloroplast genome sequences to test our hypothesis in the future.
Molecular markers development using genome skimming
Oresitrophe and Mukdenia provide an ideal model for a more comprehensive understanding of the divergence history and the influence of climate changes on lithophytes in Northeast China and adjacent regions. However, no genetic background and resources are available for these two genera. By analyzing genome skimming data of Oresitrophe and Mukdenia, here we developed abundant genetic resources, including cp hotspot regions, cpSSRs and polymorphic gSSRs.
Mutation events in the chloroplast genome are usually clustered in “hotspots”, and these mutational dynamics created highly variable regions dispersed throughout the chloroplast genomes [76, 77]. We identified seven regions including trnH-psbA, trnR-atpA, atpI-rps2, rps2-rpoC2, petN-psbM, rps4-trnT and rpl33-rps18 between Oresitrophe and Mukdenia, as well as four highly variable regions including trnQ-psbK, trnR-atpA, trnS-psbZ and rpl33-rps18 within Oresitrophe, which enabled the development of novel cp markers for genetic studies in these two genera. As our results showed, all of them occurred in the LSC region but not in SSC or IR regions. Among these regions, the highly variable regions trnH-psbA, atpI-rps2, petN-psbM and rpl33-rps18 have been reported in seed plants before [25, 78–81]. The hotspot regions will provide important genetic information for the subsequent studies on phylogeography and divergence history of Oresitrophe and Mukdenia.
Chloroplast simple sequence repeats (cpSSRs) markers, which possess unique and important characteristics such as non-recombination, haploidy, uniparental inheritance and a low nucleotide substitution rate, are excellent tool in population genetics [82]. Particularly, the chloroplast genome holds ancient genetic patterns and can therefore provide unique insight into evolutionary processes [83], and cpSSR loci are generally distributed throughout noncoding regions with higher sequence variations than coding regions [84]. Moreover, the cpSSR markers developed based on a species are frequently universal to amplify homologous loci across related taxa [85]. Thus, cpSSR markers can be used to reveal population genetic variation and phylogeographic patterns [86, 87]. In this study, the type, distribution and presence of cpSSRs were detected between the chloroplast genomes of O. rupifraga and M. rossii. We received a lot of 58, 61 and 61 perfect cpSSR loci in O. rupifraga-BJCP, O. rupifraga-HNYD and Mukdenia rossii, respectively. After comparative analysis, 24 polymorphic cpSSR loci were developed between Oresitrophe and Mukdenia (Additional file 6: Table S4), which will contribute to further researches relating on population genetic and phylogeography of these two genera.
With the application of the NGS technologies, genomic resources have greatly increased in the last decade [88]. Recently, the increasing of available whole-genome or transcriptome sequences has provided considerable resources for SSR mining and SSR marker applications for research and genetic improvements [89]. A series of bioinformatics tools for SSRs have also been developed, such as MISA [90], SSR Primer [91], and SSR Locator [92]. However, these tools have not yet integrated a computational solution for systematic assessment of SSR polymorphic status, thus the detected SSRs still require manual screening for the polymorphy [45]. CandiSSR is a new pipeline to detect candidate polymorphic SSRs not only from the transcriptome datasets but also from multiple assembled genome sequences [42].
In this study, we employed genome skimming data not only to complete the plastid genome assembly of O. rupifraga and M. rossii, but also to identify appropriate intergeneric and intrageneric polymorphic gSSRs using CandiSSR. Some of these markers may have wide utility in Saxifragaceae, a family that with other Saxifragales has provided a useful well-sampled model for the study of niche evolution and ecological diversification [93]. We developed 126 and 452 intergeneric and intrageneric polySSR markers between Oresitrophe and Mukdenia and within Oresitrophe. Twelve pairs of candidate gSSR primers were selected to test their transferability following Qi et al. [94], primer transferability was detected using 2% agarose gels, and amplification was considered successful when one clear distinct band was visible in the expected size range. In total, 100% of the developed microsatellite markers we selected could be successfully amplified in two populations of M. rossii and four populations of O. rupifraga. Genetic diversity parameters initially indicated M. rossii (HE = 0.66) and O. rupifraga (HE = 0.51) have a pattern of moderate genetic diversity, and the genetic diversity observed in M. rossii is very similar to the average HE of 0.65 for outcrossing plant species from other microsatellite studies [39, 95]. STRUCTURE analysis separated the six populations into two clusters according to the different species at K = 2, and O. rupifraga populations were further assigned to two distinct clusters at K = 3, preliminarily showing that the two close genera have relatively significant geographical structure. In the near future, we will expand our sampling of Oresitrophe and Mukdenia to study the population genetic structure and phylogeography of these two genera.
Conclusions
In present study, we conducted genome skimming for Oresitrophe and Mukdenia. Using these data, we assembled their complete chloroplast genomes and developed abundant genetic resources including cp hotspots, cpSSRs and polymorphic gSSRs. The cp genomes had a typical quadripartite structure with a conserved genome arrangement, and the evolutionary pattern of cp genomes in Saxifragaceae was also examined utilizing four representative genera. In addition, the intergeneric gSSRs we randomly selected have shown that Oresitrophe and Mukdenia exhibited a significant genetic structure. The genomic patterns and genetic resources presented in this study will contribute to further studies on population genetic, phylogeny and conservation biology in Saxifragaceae.
Additional files
Table S3. Locality and voucher information for populations of Oresitrophe rupifraga and Mukdenia rossii used in this study. Voucher specimens are deposited at the herbarium of Zhejiang University (HZU), Hangzhou, Zhejiang, China. (DOCX 17 kb)
Figure S1. The distribution and presence of simple sequence repeats (SSRs) in the cp genome of Oresitrophe rupifraga-HNYD (A) and Mukdenia rossii (B). (PDF 392 kb)
Table S1. The detail information of polymorphic gSSRs identified within Oresitrophe and between Oresitrophe and Mukdenia. (XLSX 103 kb)
Table S2. Primer pairs designed for each detected polymorphic gSSRs within Oresitrophe and between Oresitrophe and Mukdenia. (XLSX 157 kb)
Figure S2. Summary of STRUCTURE analyses based on the gSSR data. (A) Mean ln posterior probabilities of each K, LnP(D). (B) The corresponding ΔK statistics calculated according to Evanno et al. (2005). (PDF 354 kb)
Table S4. cpSSRs identified from comparative analysis of chloroplast genome for Oresitrophe and Mukdenia. (DOCX 22 kb)
Acknowledgments
We sincerely thank Zhechen Qi, Ruisen Lu, Yu Feng for their help with the plant materials.
Funding
This research was supported by the National Natural Science Foundation of China (Grant No. 31500184), the first-grade Postdoctoral Fund of Henan 2017, the NSFC-NSF Dimensions of Biodiversity program (Grant No. 31461123001), and the Fundamental Research Funds for the Central Universities (Grant No. 2018QNA6003).
Availability of data and materials
The raw data for assembling the chloroplast genomes of Oresitrophe rupifraga and Mukdenia rossii and the contigs of them for genomic resources are available from the corresponding author upon reasonable request.
Authors’ contributions
PL, CXF and DES designed the study. PL, PZH and JL conducted the field sampling. LXL, YWW and PZH produced and analyzed the data. LXL and YWW wrote the manuscript. PL, DES, CXF and JL revised the manuscript. All authors approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4633-x) contains supplementary material, which is available to authorized users.
Contributor Information
Luxian Liu, Email: liushuangcx2007@126.com.
Yuewen Wang, Email: 1589677715@qq.com.
Peizi He, Email: 1723755463@qq.com.
Pan Li, Email: panli_zju@126.com.
Joongku Lee, Email: jklee6@daum.net.
Douglas E. Soltis, Email: dsoltis@ufl.edu
Chengxin Fu, Email: cxfu@zju.edu.cn.
References
- 1.Petit R, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, et al. Glacial refugia: hotspots but not melting pots of genetic diversity. Science. 2003;300(5625):1563–1565. doi: 10.1126/science.1083264. [DOI] [PubMed] [Google Scholar]
- 2.Hewitt GM. Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc Lond Ser B Biol Sci. 2004;359(1442):183–195. doi: 10.1098/rstb.2003.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hewitt G. The genetic legacy of the quaternary ice ages. Nature. 2000;405:907. doi: 10.1038/35016000. [DOI] [PubMed] [Google Scholar]
- 4.Lascoux M, Palme AE, Cheddadi R, Latta RG. Impact of ice ages on the genetic structure of trees and shrubs. Philos Trans R Soc Lond Ser B Biol Sci. 2004;359(1442):197–207. doi: 10.1098/rstb.2003.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bao L, Kudureti A, Bai W, Chen R, Wang T, Wang H, Ge J. Contributions of multiple refugia during the last glacial period to current mainland populations of Korean pine (Pinus koraiensis) Sci Rep. 2015;5:18608. doi: 10.1038/srep18608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen JM, Liu F, Wang QF, Motley TJ. Phylogeography of a marsh herb Sagittaria trifolia (Alismataceae) in China inferred from cpDNA atpB-rbcL intergenic spacers. Mol Phylogenet Evol. 2008;48(1):168–175. doi: 10.1016/j.ympev.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Wang HW, Ge S. Phylogeography of the endangered Cathaya argyrophylla (Pinaceae) inferred from sequence variation of mitochondrial and nuclear DNA. Mol Ecol. 2006;15(13):4109–4122. doi: 10.1111/j.1365-294X.2006.03086.x. [DOI] [PubMed] [Google Scholar]
- 8.Harrison SP, Yu G, Takahara H, Prentice IC. Diversity of temperate plants in East Asia. Nature. 2001;413:129. doi: 10.1038/35093166. [DOI] [PubMed] [Google Scholar]
- 9.Qiu YX, Fu CX, Comes HP. Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of quaternary climate and environmental change in the World's most diverse temperate flora. Mol Phylogenet Evol. 2011;59(1):225–244. doi: 10.1016/j.ympev.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Cao X, Herzschuh U, Ni J, Zhao Y, Böhmer T. Spatial and temporal distributions of major tree taxa in eastern continental Asia during the last 22,000 years. The Holocene. 2014;25(1):79–91. [Google Scholar]
- 11.Bai WN, Liao WJ, Zhang DY. Nuclear and chloroplast DNA phylogeography reveal two refuge areas with asymmetrical gene flow in a temperate walnut tree from East Asia. The New Phytologist. 2010;188(3):892–901. doi: 10.1111/j.1469-8137.2010.03407.x. [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Tsuda Y, Shen H, Hu L, Saito Y, Ide Y. Genetic structure and hierarchical population divergence history of Acer mono var. mono in south and Northeast China. PLoS One. 2014;9(1):e87187. doi: 10.1371/journal.pone.0087187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng YF, Wang WT, Liao WJ, Wang HF, Zhang DY. Multiple glacial refugia for cool-temperate deciduous trees in northern East Asia: the mongolian oak as a case study. Mol Ecol. 2015;24(22):5676. doi: 10.1111/mec.13408. [DOI] [PubMed] [Google Scholar]
- 14.Soltis DE, Kuzoff RK, Mort ME, Zanis M, Fishbein M, Hufford L, Koontz J, Arroyo MK. Elucidating deep-level phylogenetic relationships in Saxifragaceae using sequences for six chloroplastic and nuclear DNA regions. Ann Mo Bot Gard. 2001;88(4):669–693. [Google Scholar]
- 15.Deng JB, Drew BT, Mavrodiev EV, Gitzendanner MA, Soltis PS, Soltis DE. Phylogeny, divergence times, and historical biogeography of the angiosperm family Saxifragaceae. Mol Phylogenet Evol. 2015;83:86–98. doi: 10.1016/j.ympev.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 16.Wu Z, Raven P. Flora of China, Vol. 8: Brassicaceae through Saxifragaceae. Beijing: science press and St. Louis: Missouri Botanical Garden Press; 2001. p. 506. [Google Scholar]
- 17.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 18.Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, McCown B, Harbut R, Simon P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot. 2012;99(2):193–208. doi: 10.3732/ajb.1100394. [DOI] [PubMed] [Google Scholar]
- 19.Montes I, Conklin D, Albaina A, Creer S, Carvalho GR, Santos M, Estonba A. SNP discovery in European anchovy (Engraulis encrasicolus, L) by high-throughput transcriptome and genome sequencing. PLoS One. 2013;8(8):e70051. doi: 10.1371/journal.pone.0070051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gitzendanner MA, Soltis PS, Wong KS, Ruhfel BR, Soltis DE. Plastid phylogenomic analysis of green plants: a billion years of evolutionary history. Am J Bot. 2017; in press [DOI] [PubMed]
- 21.Palmer JD. Plastid chromosomes: structure and evolution. The Molecular Biology of Plastids. 1991;7:5–53. [Google Scholar]
- 22.Raubeson L, Jansen R. In: Chloroplast genomes of plants. Plant diversity and evolution: genotypic and phenotypic variation in higher plants. Henry RJ, editor. London: CABI; 2005. pp. 45–68. [Google Scholar]
- 23.Drouin G, Daoud H, Xia J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol Phylogenet Evol. 2008;49(3):827–831. doi: 10.1016/j.ympev.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Dong W, Xu C, Cheng T, Lin K, Zhou S. Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol Evol. 2013;5(5):989–997. doi: 10.1093/gbe/evt063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu LX, Li R, Worth JRP, Li X, Li P, Cameron KM, Fu CX. The complete chloroplast genome of Chinese bayberry (Morella rubra, Myricaceae): implications for understanding the evolution of Fagales. Front Plant Sci. 2017;8:968. doi: 10.3389/fpls.2017.00968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomson RC, Wang IJ, Johnson JR. Genome-enabled development of DNA markers for ecology, evolution and conservation. Mol Ecol. 2010;19(11):2184–2195. doi: 10.1111/j.1365-294X.2010.04650.x. [DOI] [PubMed] [Google Scholar]
- 27.Greiner S, Sobanski J, Bock R. Why are most organelle genomes transmitted maternally? BioEssays. 2015;37(1):80–94. doi: 10.1002/bies.201400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed I, Biggs PJ, Matthews PJ, Collins LJ, Hendy MD, Lockhart PJ. Mutational dynamics of aroid chloroplast genomes. Genome Biol Evol. 2012;4(12):1316–1323. doi: 10.1093/gbe/evs110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doorduin L, Gravendeel B, Lammers Y, Ariyurek Y, Chin AWT, Vrieling K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011;18(2):93–105. doi: 10.1093/dnares/dsr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Yang Y, Henry RJ, Rossetto M, Wang Y, Chen S, Plant DNA. Barcoding: from gene to genome. Biol Rev Camb Philos Soc. 2015;90(1):157–166. doi: 10.1111/brv.12104. [DOI] [PubMed] [Google Scholar]
- 31.Alkan C, Sajjadian S, Eichler EE. Limitations of next-generation genome sequence assembly. Nat Methods. 2011;8(1):61–65. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li P, Lu R-S, Xu W-Q, Ohi-Toma T, Cai M-Q, Qiu Y-X, Cameron KM, Fu C-X. Comparative genomics and phylogenomics of east Asian tulips (Amana, Liliaceae) Front Plant Sci. 2017;8:451. doi: 10.3389/fpls.2017.00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang LK, Yan HD, Zhao XX, Zhang XQ, Wang J, Frazier T, Yin G, Huang X, Yan DF, Zang WJ, et al. Identifying differentially expressed genes under heat stress and developing molecular markers in orchardgrass (Dactylis glomerata L.) through transcriptome analysis. Mol Ecol Resour. 2015;15(6):1497–1509. doi: 10.1111/1755-0998.12418. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Cheng Y-F, Deng C, Ma Y, Wang Z-W, Chen X-H, Xue L-B. Comparative transcriptome analysis of eggplant (Solanum melongena L.) and Turkey berry (Solanum torvum Sw.): phylogenomics and disease resistance analysis. BMC Genomics. 2014;15(1):412. doi: 10.1186/1471-2164-15-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li YC, Korol AB, Fahima T, Beiles A, Nevo E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol. 2003, 11:2453–65. [DOI] [PubMed]
- 36.Zhang L, Yuan D, Yu S, Li Z, Cao Y, Miao Z, Qian H, Tang K. Preference of simple sequence repeats in coding and non-coding regions of Arabidopsis thaliana. Bioinformatics. 2004;20(7):1081–1086. doi: 10.1093/bioinformatics/bth043. [DOI] [PubMed] [Google Scholar]
- 37.Kashi Y, King D, Soller M. Simple sequence repeats as a source of quantitative genetic variation. Trends Genet. 1997;13(2):74. doi: 10.1016/s0168-9525(97)01008-1. [DOI] [PubMed] [Google Scholar]
- 38.Jones E, Dupal M, Dumsday J, Hughes L, Forster J. An SSR-based genetic linkage map for perennial ryegrass (Lolium perenne L.) Theor & Appl Genet. 2002;105(4):577–584. doi: 10.1007/s00122-002-0907-3. [DOI] [PubMed] [Google Scholar]
- 39.Yuan N, Sun Y, Comes HP, Fu CX, Qiu YX. Understanding population structure and historical demography in a conservation context: population genetics of the endangered Kirengeshoma palmata (Hydrangeaceae) Am J Bot. 2014;101(3):521–529. doi: 10.3732/ajb.1400043. [DOI] [PubMed] [Google Scholar]
- 40.Testolin R, Marrazzo T, Cipriani G, Quarta R, Verde I, Dettori MT, Pancaldi M, Sansavini S. Microsatellite DNA in peach (Prunus persica L. Batsch) and its use in fingerprinting and testing the genetic origin of cultivars. Genome. 2000;43(3):512–520. [PubMed] [Google Scholar]
- 41.Delplancke M, Alvarez N, Benoit L, Espindola A, IJ H, Neuenschwander S, Arrigo N. Evolutionary history of almond tree domestication in the Mediterranean basin. Mol Ecol. 2013;22(4):1092–1104. doi: 10.1111/mec.12129. [DOI] [PubMed] [Google Scholar]
- 42.Xia EH, Yao QY, Zhang HB, Jiang JJ, Zhang LP, Gao LZ. CandiSSR: an efficient pipeline used for identifying candidate polymorphic SSRs based on multiple assembled sequences. Front Plant Sci. 2015;6:1171. doi: 10.3389/fpls.2015.01171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blair MW, Giraldo MC, Buendía HF, Tovar E, Duque MC, Beebe SE. Microsatellite marker diversity in common bean (Phaseolus vulgaris L.) Theor Appl Genet. 2006;113(1):100. doi: 10.1007/s00122-006-0276-4. [DOI] [PubMed] [Google Scholar]
- 44.Bae KM, Sim SC, Hong JH, Choi KJ, Kim DH, Kwon YS. Development of genomic SSR markers and genetic diversity analysis in cultivated radish (Raphanus sativus L.). Oliver and Boyd. 2015:216–24.
- 45.Wang X, Wang L. GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Front Plant Sci. 2016;7:1350. doi: 10.3389/fpls.2016.01350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- 47.Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33(suppl 2):W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Research. 2013;41(W1):W575–81. [DOI] [PMC free article] [PubMed]
- 49.Soltis DE, Mort ME, Latvis M, Mavrodiev EV, O'Meara BC, Soltis PS, Burleigh JG, Rubio de Casas R. Phylogenetic relationships and character evolution analysis of Saxifragales using a supermatrix approach. Am J Bot. 2013;100(5):916–929. doi: 10.3732/ajb.1300044. [DOI] [PubMed] [Google Scholar]
- 50.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32(suppl 2):W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurtz S, Schleiermacher C. REPuter-fast computation of maximal repeats in complete genomes. Bioinformatics. 1999;15(5):426–427. doi: 10.1093/bioinformatics/15.5.426. [DOI] [PubMed] [Google Scholar]
- 53.Faircloth BC. Msatcommander: detection of microsatellite repeat arrays and automated, locus-specific primer design. Mol Ecol Resour. 2008;8(1):92–94. doi: 10.1111/j.1471-8286.2007.01884.x. [DOI] [PubMed] [Google Scholar]
- 54.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol & Evol. 2013;30(4):772. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 56.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23(10):1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 57.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalinowski ST, Taper ML, Marshall TC. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol Ecol. 2007;16(5):1099–1106. doi: 10.1111/j.1365-294X.2007.03089.x. [DOI] [PubMed] [Google Scholar]
- 59.Rousset F. Genepop'007: a complete re-implementation of the genepop software for windows and Linux. Mol Ecol Resour. 2008;8(1):103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 60.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes. 2007;7(4):574–578. doi: 10.1111/j.1471-8286.2007.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gilbert KJ, Andrew RL, Dan GB, Franklin MT, Kane NC, Moore JS, Moyers BT, Renaut S, Rennison DJ, Veen T. Recommendations for utilizing and reporting population genetic analyses: the reproducibility of genetic clustering using the program structure. Mol Ecol. 2012;21(20):4925–4930. doi: 10.1111/j.1365-294X.2012.05754.x. [DOI] [PubMed] [Google Scholar]
- 62.Lu R, Li P, Qiu Y. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front Plant Sci. 2016;7:2054. doi: 10.3389/fpls.2016.02054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 65.Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc Natl Acad Sci. 2010;107(10):4623–4628. doi: 10.1073/pnas.0907801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mcneal JR, Kuehl JV, Boore JL, de Pamphilis CW. Complete plastid genome sequences suggest strong selection for retention of photosynthetic genes in the parasitic plant genus Cuscuta. BMC Plant Biol. 2007;7(1):57. doi: 10.1186/1471-2229-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haberle RC, Fourcade HM, Boore JL, Jansen RK. Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J Mol Evol. 2008;66(4):350–361. doi: 10.1007/s00239-008-9086-4. [DOI] [PubMed] [Google Scholar]
- 68.Zhong B, Yonezawa T, Zhong Y, Hasegawa M. Episodic evolution and adaptation of chloroplast genomes in ancestral grasses. PLoS One. 2009;4(4):e5297. doi: 10.1371/journal.pone.0005297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guisinger MM, Chumley TW, Kuehl JV, Boore JL, Jansen RK. Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in Poaceae. J Mol Evol. 2010;70(2):149–166. doi: 10.1007/s00239-009-9317-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Millen RS, Olmstead RG, Adams KL, Palmer JD, Lao NT, Heggie L, Kavanagh TA, Hibberd JM, Gray JC, Morden CW. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell. 2001;13(3):645–658. doi: 10.1105/tpc.13.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Downie SR, Olmstead RG, Zurawski G, Soltis DE, Soltis PS, Watson JC, Palmer JD. Six independent losses of the chloroplast DNA rpl2 intron in dicotyledons: molecular and phylogenetic implications. Evolution. 1991;45(5):1245. doi: 10.1111/j.1558-5646.1991.tb04390.x. [DOI] [PubMed] [Google Scholar]
- 72.Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, Jansen RK. The complete chloroplast genome sequence of Pelargonium x hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol & Evol. 2006;23(11):2175–2190. doi: 10.1093/molbev/msl089. [DOI] [PubMed] [Google Scholar]
- 73.Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchishinozaki K. The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 1986;5(9):2043. doi: 10.1002/j.1460-2075.1986.tb04464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wicke S, Schneeweiss GM, Depamphilis CW, Kai FM, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3–5):273. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li Z, Long H, Zhang L, Liu Z, Cao H, Shi M, Tan X. The complete chloroplast genome sequence of tung tree (Vernicia fordii): organization and phylogenetic relationships with other angiosperms. Sci Rep. 2017;7(1):1869. doi: 10.1038/s41598-017-02076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94(3):275–288. doi: 10.3732/ajb.94.3.275. [DOI] [PubMed] [Google Scholar]
- 77.Dong W, Liu J, Yu J, Wang L, Zhou S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One. 2012;7(4):e35071. doi: 10.1371/journal.pone.0035071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mariotti R, Cultrera NG, Díez CM, Baldoni L, Rubini A. Identification of new polymorphic regions and differentiation of cultivated olives (Olea europaea L.) through plastome sequence comparison. BMC Plant Biol. 2010;10(1):211. doi: 10.1186/1471-2229-10-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bodin SS, Kim JS, Kim JH. Complete chloroplast genome of Chionographis japonica (Willd.) maxim. (Melanthiaceae): comparative genomics and evaluation of universal primers for Liliales. Plant Mol Biol Report. 2013;31(6):1407–1421. [Google Scholar]
- 80.Mucciarelli M, Fay MF, Plastid DNA. Fingerprinting of the rare Fritillaria moggridgei (Liliaceae) reveals population differentiation and genetic isolation within the Fritillaria tubiformis complex. Phytotaxa. 2013;91(1):1–23. [Google Scholar]
- 81.Leonard OR: Using comparative plastomics to identify potentially informative non-coding regions for basal angiosperms, with a focus on Illicium (Schisandraceae). Dissertations & Theses - Gradworks 2015.
- 82.Ebert D, Peakall R. Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol Ecol Resour. 2009;9(3):673. doi: 10.1111/j.1755-0998.2008.02319.x. [DOI] [PubMed] [Google Scholar]
- 83.Provan J, Powell W, Hollingsworth PM. Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol Evol. 2001;16(3):142–147. doi: 10.1016/s0169-5347(00)02097-8. [DOI] [PubMed] [Google Scholar]
- 84.Huang J, Yang X, Zhang C, Yin X, Liu S, Li X. Development of chloroplast microsatellite markers and analysis of chloroplast diversity in Chinese jujube (Ziziphus jujuba mill.) and wild jujube (Ziziphus acidojujuba mill.) PLoS One. 2015;10(9):e0134519. doi: 10.1371/journal.pone.0134519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Diekmann K, Hodkinson TR, Barth S. New chloroplast microsatellite markers suitable for assessing genetic diversity of Lolium perenne and other related grass species. Ann Bot. 2012;110(6):1327. doi: 10.1093/aob/mcs044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pan L, Li Y, Guo R, Wu H, Hu Z, Chen C. Development of 12 chloroplast microsatellite markers in Vigna unguiculata (Fabaceae) and amplification in Phaseolus vulgaris. Appl in Plant Sciences. 2014;2(3) [DOI] [PMC free article] [PubMed]
- 87.Deng Q, Zhang H, He Y, Wang T, Su Y. Chloroplast microsatellite markers for Pseudotaxus chienii developed from the whole chloroplast genome of Taxus chinensis var. mairei (Taxaceae) Appl in Plant Sciences. 2017;5(3):1300075. doi: 10.3732/apps.1600153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Neale DB, Kremer A. Forest tree genomics: growing resources and applications. Nat Rev Genet. 2011;12(2):111–122. doi: 10.1038/nrg2931. [DOI] [PubMed] [Google Scholar]
- 89.Hodel RGJ, Gitzendanner MA, Germain-Aubrey CC, Liu X, Crowl AA, Sun M, Landis JB, Claudia SSM, Douglas NA, Chen S. A new resource for the development of SSR markers: millions of loci from a thousand plant transcriptomes. Appl in Plant Sciences. 2016;4(6):1600024. doi: 10.3732/apps.1600024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thiel T, Michalek W, Varshney R, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.) Theor & Appl Genet. 2003;106(3):411–422. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- 91.Robinson AJ, Love CG, Batley J, Barker G, Edwards D. Simple sequence repeat marker loci discovery using SSR primer. Bioinformatics. 2004;20(9):1475–1476. doi: 10.1093/bioinformatics/bth104. [DOI] [PubMed] [Google Scholar]
- 92.Da ML, Palmieri DA, de Souza VQ, Kopp MM, de Carvalho FI, Costa dOA: SSR locator: tool for simple sequence repeat discovery integrated with primer design and PCR simulation. Int J of Plant Genomics 2008, 2008(2008):412696. [DOI] [PMC free article] [PubMed]
- 93.de Casas RR, Mort ME, Soltis DE. The influence of habitat on the evolution of plants: a case study across Saxifragales. Ann Bot. 2016;118(7):1317. doi: 10.1093/aob/mcw160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qi ZC, Shen C, Han YW, Shen W, Yang M, Liu J, Liang ZS, Li P, Fu CX. Development of microsatellite loci in Mediterranean sarsaparilla (Smilax aspera; Smilacaceae) using transcriptome data. Appl in Plant Sciences. 2017;5(4):1700005. doi: 10.3732/apps.1700005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nybom H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol. 2004;13(5):1143–1155. doi: 10.1111/j.1365-294X.2004.02141.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Locality and voucher information for populations of Oresitrophe rupifraga and Mukdenia rossii used in this study. Voucher specimens are deposited at the herbarium of Zhejiang University (HZU), Hangzhou, Zhejiang, China. (DOCX 17 kb)
Figure S1. The distribution and presence of simple sequence repeats (SSRs) in the cp genome of Oresitrophe rupifraga-HNYD (A) and Mukdenia rossii (B). (PDF 392 kb)
Table S1. The detail information of polymorphic gSSRs identified within Oresitrophe and between Oresitrophe and Mukdenia. (XLSX 103 kb)
Table S2. Primer pairs designed for each detected polymorphic gSSRs within Oresitrophe and between Oresitrophe and Mukdenia. (XLSX 157 kb)
Figure S2. Summary of STRUCTURE analyses based on the gSSR data. (A) Mean ln posterior probabilities of each K, LnP(D). (B) The corresponding ΔK statistics calculated according to Evanno et al. (2005). (PDF 354 kb)
Table S4. cpSSRs identified from comparative analysis of chloroplast genome for Oresitrophe and Mukdenia. (DOCX 22 kb)
Data Availability Statement
The raw data for assembling the chloroplast genomes of Oresitrophe rupifraga and Mukdenia rossii and the contigs of them for genomic resources are available from the corresponding author upon reasonable request.