

Abstract
Brown fat dissipates energy as heat and protects against obesity. Here, we identified nuclear factor I-A (NFIA) as a transcriptional regulator of brown fat by a genome-wide open chromatin analysis of murine brown and white fat followed by motif analysis of brown-fat-specific open chromatin regions. NFIA and the master transcriptional regulator of adipogenesis, PPARγ, co-localize at the brown-fat-specific enhancers. Moreover, the binding of NFIA precedes and facilitates the binding of PPARγ, leading to increased chromatin accessibility and active transcription. Introduction of NFIA into myoblasts results in brown adipocyte differentiation. Conversely, the brown fat of NFIA knockout mice displays impaired expression of the brown-fat-specific genes and reciprocal elevation of muscle genes. Finally, expression of NFIA and the brown-fat-specific genes is positively correlated in human brown fat. These results indicate that NFIA activates the cell-type-specific enhancers and facilitates the binding of PPARγ for controlling the brown fat gene program.
Introduction
Obesity and its complications including diabetes amount to a world-wide epidemic. While white adipose tissue (WAT) stores energy as lipids and expands in obesity, brown adipose tissue (BAT) is specialized to dissipate energy through the uncoupling protein-1 (UCP1) on the mitochondrial inner membrane. When activated, UCP1 dissipates the electrochemical gradient that is normally used for adenosine triphosphate (ATP) synthesis, resulting in energy expenditure in the form of heat1. Although the existence of human BAT was controversial until recently, since the re-discovery of BAT in human adults2–6, it has been considered a potential target in the treatment of obesity. Human BAT activity is inversely correlated with body mass index3, and studies have shown that chronic cold exposure7–8 and β3 adrenergic receptor agonist administration9 successfully recruit human BAT and increase systemic energy expenditure.
Lineage tracing has demonstrated that brown fat and skeletal muscle share a common progenitor, but brown fat and white fat do not10. Both brown fat and skeletal muscle derive from a Myf5-positive precursor, and a transcriptional cofactor PRD1-BF1-RIZ1 homologous domain containing 16 (PRDM16) works as a cell-fate switch10–12. The master transcriptional regulator of adipogenesis—peroxisome proliferator-activated receptor γ (PPARγ) —and its agonist was also shown to activate the brown fat gene program13,14. Motif analysis of PPARγ binding sites in BAT identified early B cell factor 2 (EBF2) as a transcriptional regulator of brown fat15. However, much remains elusive in the genome-wide landscape of brown fat development.
To gain insight into the underlying mechanism of brown fat development in a global and unbiased manner, we performed formaldehyde-assisted isolation of regulatory elements (FAIRE) coupled with high-throughput sequencing16 on murine BAT and WAT to profile the tissue-specific accessible chromatin regions. Through motif analysis of BAT-specific open chromatin regions, we identified NFIA as a transcriptional regulator of brown fat. NFIA exerts its effects by co-localizing with PPARγ at cell-type-specific enhancers.
Results
The NFI motif within BAT open chromatin
BAT and WAT share a common transcriptional program regulated by PPARγ and CCAAT/enhancer binding proteins (C/EBPs). However, these tissues also have depot-selective gene programs that are responsible for their specific functions11,15,17. Regulatory elements controlling gene expression are characterized by open chromatin structures accessible to transcription factors and cofactors. We performed FAIRE-seq analyses of murine interscapular BAT, inguinal WAT (iWAT) and epididymal WAT (eWAT) to map open chromatin regions genome-wide, and we identified 24,322 FAIRE peaks for BAT, 10,012 for iWAT, and 12,656 for eWAT (Fig. 1a, b). Genes near BAT-specific FAIRE peaks were associated with gene ontology (GO) terms such as brown fat cell differentiation (Supplementary Fig. 1a, b), suggesting that the FAIRE-seq experiments unbiasedly identified functionally active, depot-specific accessible chromatin regions. Through motif analysis, in addition to known regulators such as C/EBPβ, EBF2, and PPARγ13,15,18, we found that the binding motif for the NFI transcription factor was the most highly enriched within BAT-specific open chromatin regions (Fig. 1c).
Figure 1. The NFI binding motif is highly enriched in brown-fat-specific open chromatin regions.
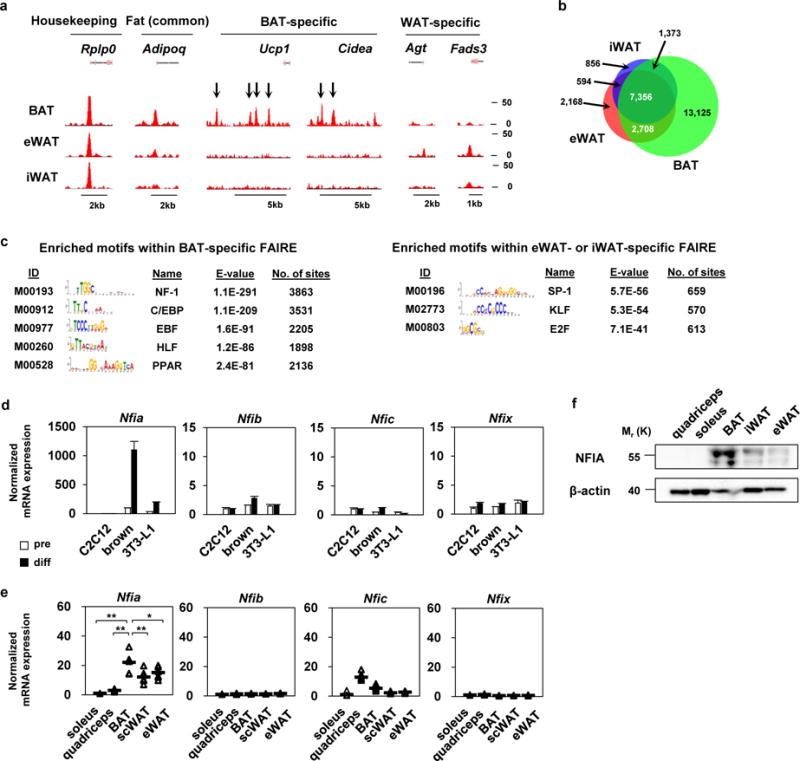
a, Representative FAIRE-seq tracks of murine BAT, iWAT and eWAT. b, Venn diagram showing overlap of BAT, iWAT and eWAT FAIRE-peaks. c, Motifs enriched in BAT-specific FAIRE peaks and eWAT or iWAT-specific FAIRE-peaks. The motif analysis was performed once based on the FAIRE-seq dataset. d, mRNA expression levels of the NFI family in C2C12 myoblasts, immortalized brown adipocytes and 3T3-L1 white adipocytes before and after differentiation (mean +/− S.E.M.; N = 3 independent samples). e, mRNA expression levels of the NFI family in soleus muscle, quadriceps muscle, BAT, iWAT and eWAT (mean +/− S.E.M.; N = 7 independent samples; * p <0.05, ** p <0.01). f, Western blot analysis of NFIA in representative samples from soleus muscle, quadriceps muscle, BAT, iWAT and eWAT. β-actin was used as a loading control. Representative images of two independent experiments are shown.
Of the four isoforms of the NFI family19, we found that Nfia was highly expressed in brown adipocytes compared with its expression in 3T3-L1 white adipocytes or C2C12 myoblasts. Moreover, the gene expression level of Nfia was robustly induced during brown adipocyte differentiation (Fig. 1d). NFIA was also highly expressed in BAT compared with WAT or skeletal muscle at both the RNA and protein levels (Fig. 1e, f). Furthermore, the expression level of Nfia was induced when mice are challenged by exposure to cold or β3-agonist CL316,243 (Supplementary Fig. 1c–e). These findings indicate that NFIA is a candidate transcriptional regulator that defines brown adipocyte identity.
NFIA induces brown adipogenesis
To examine whether NFIA can induce adipocyte differentiation from myoblasts, we introduced NFIA into C2C12 myoblast cell lines using retroviral vectors (Fig. 2a). The cells were grown to confluence and treated with an adipogenic cocktail. Strikingly, NFIA-expressing cells differentiated into lipid-filled adipocytes (Fig. 2b). Consistent with cell morphology, NFIA induced expression of the master regulator Pparg and the general adipocyte marker Fabp4 (Fig. 2d). In contrast, myogenic genes such as Myod1 and Myog were suppressed by NFIA (Fig. 2c). NFIA also induced brown-fat-specific genes including Cidea and Ppargc1a as well as the thermogenic gene Ucp1 in response to elevated cAMP through forskolin treatment (Fig. 2e). Functionally, NFIA-expressing cells showed induced uncoupled respiration (Fig. 2f).
Figure 2. NFIA is capable of—and required for—driving brown adipocyte differentiation.
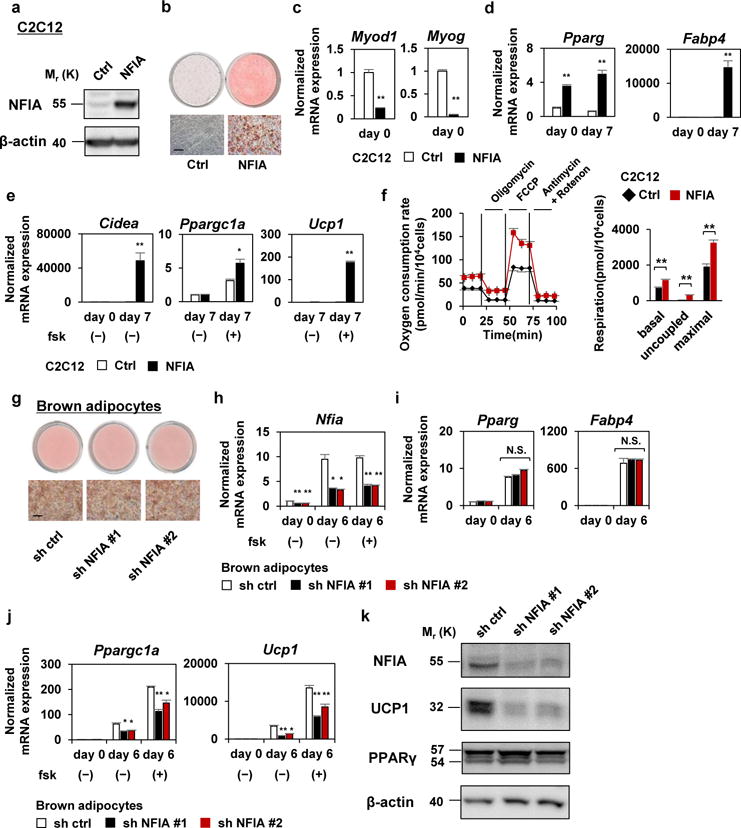
a, Western blot analysis of NFIA in control and NFIA-expressing cells. β-actin was used as a loading control. b, Control and NFIA-expressing C2C12 myoblasts were stained with Oil Red O seven days after inducing adipocyte differentiation. Scale bar, 50 μm. c–e, Myogenic genes (c), common adipocyte genes (d) and brown-fat-specific genes (e) were quantified by RT-qPCR at the indicated time course (mean +/− S.E.M.; N = 3 independent samples; * p <0.05, ** p <0.01). f, Oxygen consumption of control and NFIA-expressing C2C12 myoblasts (mean +/− S.E.M.; N = 10 independent samples; * p<0.05, ** p<0.01). g, Control sh RNA or sh RNA for NFIA was introduced into immortalized brown adipocytes and the cells were stained with Oil Red O six days after inducing adipocyte differentiation. Scale bar, 50 μm. h–j, Nfia (h), Pparg and Fabp4 (i), Ppargc1a and Ucp1 (j) were quantified by RT-qPCR at the indicated time course (mean +/− S.E.M.; N = 3 independent samples; * p <0.05, ** p <0.01). k, Western blot analysis of NFIA, UCP1, PPARγ in indicated cells. β-actin was used as a loading control. Representative images of two independent experiments are shown.
Hierarchal clustering of genes quantified by RNA-seq showed the global changes in gene expression caused by the introduction of NFIA (Supplementary Fig 2a). When we defined genes selective for BAT and skeletal muscle (SKM) by fold changes of expression levels between these tissues, BAT-selective genes were enriched in the cluster of genes up-regulated by NFIA (p = 9.9 × 10−28, chi-square test), while SKM-selective genes were enriched in the cluster of genes down-regulated by NFIA (p = 2.3 × 10−32). And GO analysis independently supported this observation (Supplementary Fig 2b). Taken together, these data indicate that introduction of NFIA into myoblasts drives brown adipocyte differentiation while inhibiting myogenic differentiation.
We also tried introduction of NFIA into 3T3-F442A white preadipocyte cells (Supplementary Fig. 2d). The effect of NFIA on lipid accumulation and on common adipocyte gene expression after differentiation was modest (Supplementary Fig. 2c, e). However, NFIA very strongly increased the expression levels of the brown-fat-specific genes (Supplementary Fig. 2f), suggesting that NFIA can drive the brown fat gene program also in white preadipocytes.
To test the endogenous role of NFIA, we next performed loss-of-function experiments. We introduced a short hairpin (sh) RNA for NFIA into brown adipocytes and achieved significant knockdown throughout the differentiation (Fig. 2h, k). While the effect of NFIA knockdown on cell morphology and on common adipocyte gene expression were not significant (Fig. 2g, i), expression levels of the brown-fat-specific genes such as Ppargc1a and Ucp1 were significantly reduced (Fig. 2j). Expression of UCP1 protein was also highly reduced (Fig. 2k). Similarly, when we introduced a small interfering (si) RNA for NFIA by electroporation into fully-differentiated brown adipocytes, expression levels of the brown-fat-specific genes were significantly reduced (Supplementary Fig. 2g–j). These results suggest that NFIA is required for both activation and maintenance of the brown-fat-specific gene expression. Altogether, our gain- and loss-of-function experiments show that NFIA is capable of and required for controlling the brown fat gene program.
NFIA and PRDM16 work in parallel with each other
The effect of NFIA on the brown fat gene program prompted us to examine the relationship with and requirement for PRDM16, which perform a similar function in this context. Although mass-spectrometric analysis suggested that NFIA is included in PRDM16 protein complex18, our co-immunoprecipitation experiments showed that NFIA does not bind physically to PRDM16 (Fig. 3a). Introduction of NFIA into myoblasts did not induce Prdm16 expression. However, introduction of PRDM16 did induce Nfia expression while introduction of PPARγ did not (Fig. 3b, c). Importantly, PRDM16 was dispensable for the effect of NFIA, because NFIA was capable of stimulating adipocyte differentiation and stimulating the brown-fat-specific gene expression even when PRDM16 was knocked down (Fig. 3d–g). Interestingly, the opposite was also true (Fig. 3h–k). Overall, these results suggest that NFIA and PRDM16 work in parallel with each other.
Figure 3. PRDM16 is dispensable for the effect of NFIA.
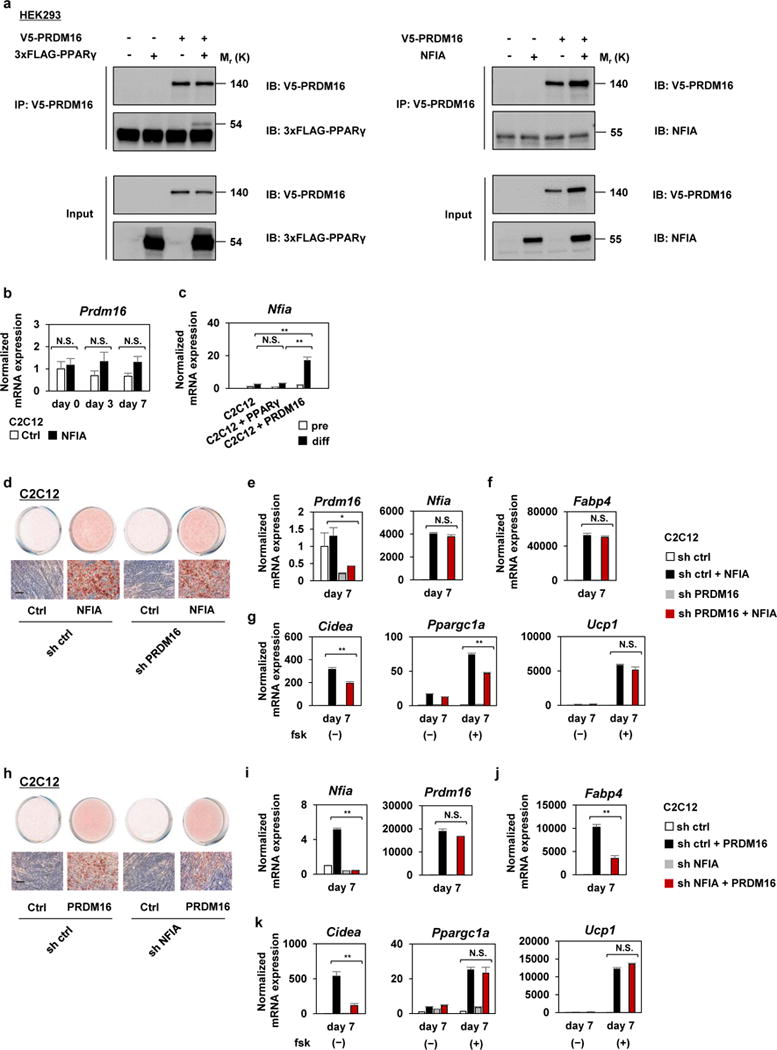
a, To the left, immunoprecipitation of V5-tagged PRDM16 from HEK293 cells expressing V5-tagged PRDM16 and/or 3xFLAG-tagged PPARγ followed by western blot analysis to detect 3xFLAG-tagged PPARγ, as a positive control10. To the right, immunoprecipitation of V5-tagged PRDM16 from HEK293 cells expressing V5-tagged PRDM16 and/or NFIA followed by western blot analysis to detect NFIA. Representative images of two independent experiments are shown. b, RT-qPCR analysis of Prdm16 in control or NFIA-expressing C2C12 cells during the induction of adipocyte differentiation (mean +/− S.E.M.; N = 3 independent samples; * p <0.05, ** p <0.01). c, RT-qPCR analysis of Nfia in control, PPARγ - or PRDM16-expressing C2C12 cells (mean +/− S.E.M.; N = 3 independent samples ; * p <0.05, ** p <0.01). d, Control sh RNA or sh RNA for PRDM16 was introduced into control or NFIA-expressing C2C12 myoblasts, and stained with Oil Red O seven days after inducing adipocyte differentiation. Scale bar, 50 μm. e–g, Prdm16 and Nfia (e), general adipocyte marker Fabp4 (f) and the brown-fat-specific genes (g) were quantified by RT-qPCR at the indicated time course (mean +/− S.E.M.; N = 3 independent samples ; * p <0.05, ** p <0.01). h, Control sh RNA or sh RNA for NFIA were introduced into control or PRDM16-expressing C2C12 myoblasts, and stained with Oil Red O seven days after inducing adipocyte differentiation. Scale bar, 50 μm. i–k, Nfia and Prdm16 (i), general adipocyte marker Fabp4 (j) and the brown-fat-specific genes (k) were quantified by RT-qPCR at the indicated time course (mean +/− S.E.M.; N = 3 independent samples ; * p <0.05, ** p <0.01).
NFIA binds to the brown fat enhancers
To dissect the genome-wide binding landscape of NFIA in brown adipocyte differentiation, we performed ChIP-seq analysis using the NFI antibody, which reacts primarily with NFIA, but also with NFIC and NFIX. We also performed ChIP-seq for PPARγ, C/EBPα, C/EBPβ, EBF2 and H3K27 acetylation (H3K27Ac). We additionally performed assay for transposase-accessible chromatin coupled with high throughput sequencing (ATAC-seq) for accessible chromatin regions20. We further performed ChIP-seq for NFIA with the FLAG M2 antibody in C2C12 myoblasts that expressed FLAG-tagged NFIA. We identified 12,486 and 12,748 NFI binding sites, respectively, on day 0 and day 6 of differentiation and the majority of NFI binding sites were located distal to genes, as is the case with PPARγ (Fig. 4a and Supplementary Fig. 3a). Motif analysis showed that NFI binding sites in brown adipocytes and NFIA binding sites in NFIA-expressing C2C12 myoblasts were strongly enriched with NFI motif (Fig. 4b and Supplementary Fig. 3b, c), in agreement with direct DNA binding. We observed that NFI binds to the enhancers of the master regulator Pparg and the brown-fat-specific genes such as Cidea and Ucp1 (Fig. 4c). Importantly, most of the NFI binding sites in brown adipocytes and NFIA binding sites in NFIA-expressing C2C12 myoblasts overlapped each other at BAT FAIRE peaks (Supplementary Fig. 3e). Furthermore, when we defined BAT- and WAT-selective genes by fold changes of expression levels between these tissues, BAT-selective genes were closer to NFI binding sites than were WAT-selective genes (Fig. 4d), and BAT-selective genes harbored more NFI binding sites than did WAT-selective genes or all genes within +/− 50 kb of the TSS. (Fig. 4e). And the NFI binding signal was highly enriched near BAT-selective genes compared with that signal near WAT-selective genes or all genes (Fig. 4f). These results indicate that NFI binding is enriched at brown-fat-specific enhancers.
Figure 4. NFI binding is enriched at brown-fat-specific enhancers.
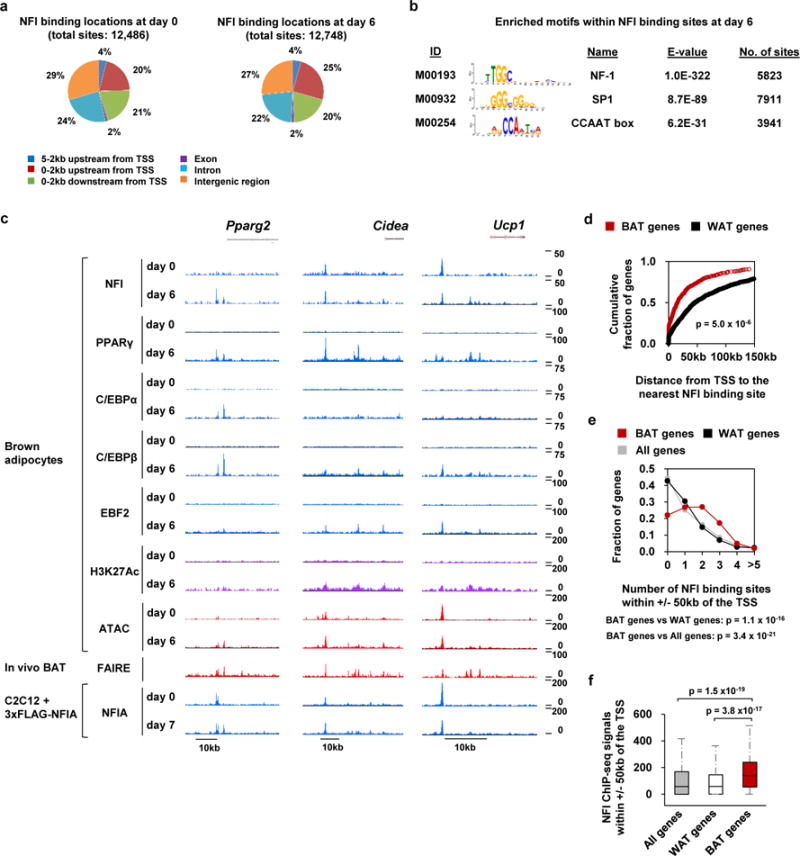
a, Genomic location of NFI binding sites in brown adipocytes at day 0 and day 6 of differentiation. b, Enriched known motifs within NFI binding sites in brown adipocytes at day 6 of differentiation. c, Representative tracks of ChIP-seq for NFI, PPARγ, C/EBPα, C/EBPβ, EBF2 and H3K27Ac in brown adipocytes, ATAC-seq of brown adipocytes, FAIRE-seq of in vivo BAT, and ChIP-seq for NFIA in NFIA-expressing C2C12 myoblasts at Pparg, Cidea and Ucp1 loci. d, The distance from TSS to the nearest NFI binding site is shown for BAT- and WAT- selective genes. The definition of BAT- and WAT- selective genes (N = 549 and N = 849, respectively) are shown in the methods. e, Number of NFI binding sites within +/− 50 kb of the TSS are shown for BAT- and WAT- selective genes (N = 549 and N = 849, respectively). f, Box plot showing the strength of the NFI binding signal (MACS score) near all genes (N=21,258), BAT- and WAT-selective genes (N = 549 and N = 849, respectively). The box shows median, first- and third- quartile. The whisker shows the value still within one-and-a-half times the interquartile range. The genome-wide analyses were performed once based on the ChIP-seq dataset.
Co-localization of NFIA and PPARγ
We also found that the binding sites of NFI and PPARγ often overlapped each other. Since co-localization of transcription factors at the chromatin is crucial for both cell-fate decision21 and cell-type-specific signaling22, we investigated co-localization of NFI and PPARγ genome-wide. We found that the binding sites of NFI overlapped those of PPARγ at 63% of all binding sites in differentiated brown adipocytes (8,001 of 12,748 sites, Fig. 5a). Most strikingly, the majority of the co-localizing peaks were pre-occupied by NFI but not occupied by PPARγ, C/EBPα, C/EBPβ nor EBF2 before differentiation, and these sites exhibited a high level of H3K27 acetylation and chromatin accessibility, markers of active enhancers even before differentiation (Fig. 5b, c). And the binding sites of NFI were substantially concordant between day 0 and day 6 of differentiation, unlike other transcription factors examined (Supplementary Fig. 3f–j).
Figure 5. Co-localization of NFI and PPARγ at the brown-fat-specific enhancers.
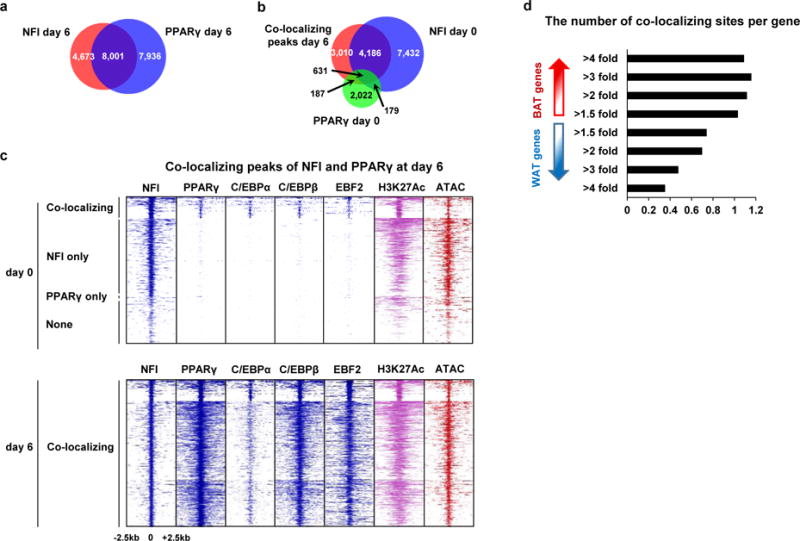
a, Venn diagram showing the overlap of NFI and PPARγ ChIP-seq peaks in brown adipocytes at day 6 of differentiation. b, Venn diagram showing the overlap of the co-localizing peaks at day 6 of differentiation, NFI ChIP-seq peaks at day 0 and PPARγ ChIP-seq peaks at day 0. c, Heat map showing ChIP-seq for NFI, PPARγ, C/EBPα, C/EBPβ, H3K27Ac and ATAC-seq tag densities at the co-localizing peaks of NFI and PPARγ at day 6. d, Bar graph showing the number of co-localizing sites per gene within +/− 50 kb of BAT- and WAT-selective genes stratified by the fold changes of gene expression. The genome-wide analyses were performed once based on the ChIP-seq dataset.
To test whether the co-localization of NFI and PPARγ is associated with gene expression, we counted the number of the co-localizing sites per gene within +/−50 kb regions around the BAT- and WAT-selective genes stratified by the fold change of expression. The results showed that the more the genes were expressed in brown fat compared with expression in white fat, the higher the number of co-localizing sites per gene (Fig. 5d). We observed that the co-localization was enriched near BAT-selective genes also in white adipocytes (Supplementary Fig. 3k, l). NFI binding sites near BAT-selective genes were closer to DR-1 motifs (the consensus motif for PPARγ) compared with those near WAT-selective genes (Supplementary Fig. 3m), suggesting that the co-localization near BAT-selective genes is, at least in part, determined by the DNA sequence itself.
To reveal the functional consequences of NFIA and PPARγ co-localization, we utilized a model system in which we introduced into C2C12 myoblasts either PPARγ alone or both PPARγ and NFIA. As previously reported, introduction of PPARγ alone is sufficient to promote adipocyte differentiation10. Co-expression of NFIA with PPARγ did not alter the degree of differentiation—as judged by both Oil Red O staining and general adipocyte marker Fabp4 expression (Fig. 6a, b). We confirmed that NFIA binds to the brown-fat-specific enhancers in this model system (Fig. 6d). Note especially that the binding of PPARγ to those enhancers near Cidea, Ppara, Ppargc1a and Ucp1 was significantly facilitated when NFIA co-localized with PPARγ (Fig. 6e), even though PPARγ protein levels were similar in both cells (Fig. 6c). We observed that the binding of PPARγ to some of those enhancers was already facilitated before differentiation (Supplementary Fig. 4a, b). Conversely, the binding of NFIA to those enhancers was independent of the co-localization of PPARγ, since NFIA in cells without PPARγ was able to bind to these loci as strongly as NFIA in cells with PPARγ (Supplementary Fig. 4c, d and Fig. 6d). Moreover, chromatin accessibility of those enhancers was dramatically increased when NFIA and PPARγ were co-localized (Fig. 6f). Finally, the co-localization robustly activated transcription of those genes (Fig. 6g). Together, these results demonstrate that co-localization of NFIA facilitates PPARγ binding to the brown-fat-specific enhancers for controlling the brown-fat-specific gene expression.
Figure 6. Co-localization of NFIA facilitates PPARγ binding to the brown-fat-specific enhancers and drives active transcription.
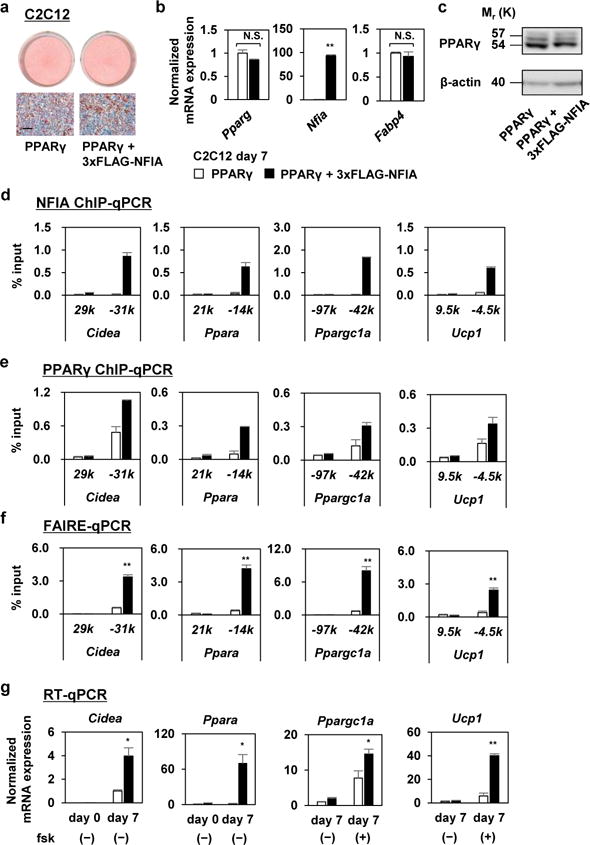
a, C2C12 myoblasts expressing only PPARγ—and those expressing both PPARγ and 3xFLAG-NFIA—were stained with Oil Red O seven days after inducing adipocyte differentiation. Scale bar, 50 μm. b, Indicated genes were quantified by RT-qPCR at day 7 after adipocyte differentiation (mean +/− S.E.M.; N = 3 independent samples; * p <0.05, ** p <0.01). c, Western blot analysis of PPARγ protein in PPARγ- or PPARγ + 3xFLAG-NFIA expressing C2C12 myoblasts. β-actin was used as a loading control. d, ChIP-qPCR analysis of NFIA. Cidea 29k, Ppara 21k, Ppargc1a -97k, and Ucp1 9.5k are background sites. The representative result of three independent experiments is shown (N = 2 independent samples; mean +/− S.E.M.). e, ChIP-qPCR analysis of PPARγ. Cidea 29k, Ppara 21k, Ppargc1a -97k, and Ucp1 9.5k are background sites. The representative result of three independent experiments is shown (N = 2 independent samples; mean +/− S.E.M.). Source data for Figure 6d and Figure 6e are provided in Supplementary Table 2. f, FAIRE-qPCR analysis. Cidea 29k, Ppara 21k, Ppargc1a -97k, and Ucp1 9.5k are background sites (mean +/− S.E.M.; N = 3 independent samples; * p <0.05, ** p<0.01). g, RT-qPCR analysis of indicated genes (mean +/− S.E.M.; N = 3 independent samples; * p <0.05, ** p <0.01).
Role of NFIA in BAT in vivo
To evaluate the physiological relevance of NFIA in BAT in vivo, we analyzed BAT in NFIA knockout mice. NFIA knockout mice are born in Mendelian ratios but die within a week of birth due to neurological deficits including agenesis of corpus callosum and hydrocephalus23. We therefore analyzed the BAT of neonates soon after birth. BAT masses and morphology were comparable among three genotypes (Fig. 7a). However, we observed significantly decreased expression of Ucp1 mRNA (Fig. 7b) and UCP1 protein (Fig. 7c) in NFIA-KO tissues. By ChIP-qPCR analysis of those tissues, we also observed severely impaired PPARγ binding to the Ucp1 -4.5kb enhancer (Fig. 7d). Notably, we observed co-localization of NFIA and PPARγ at the Ucp1 -4.5kb enhancer in our model system (Fig. 6d, e) and expression levels of Pparg were comparable between wild-type and NFIA-KO tissues (Supplementary Fig. 5a). Consistently, the binding of PPARγ to the Ucp1 -4.5kb and Ucp1 -11.7kb enhancer was highly decreased when NFIA was knocked down in brown adipocytes (Supplementary Fig. 5b). These results suggest that co-localization of NFIA and PPARγ is necessary for optimal expression of the Ucp1 gene in vivo.
Figure 7. Deficiency of NFIA causes an impaired brown fat gene signature and reciprocal elevation of skeletal muscle gene expression in vivo.
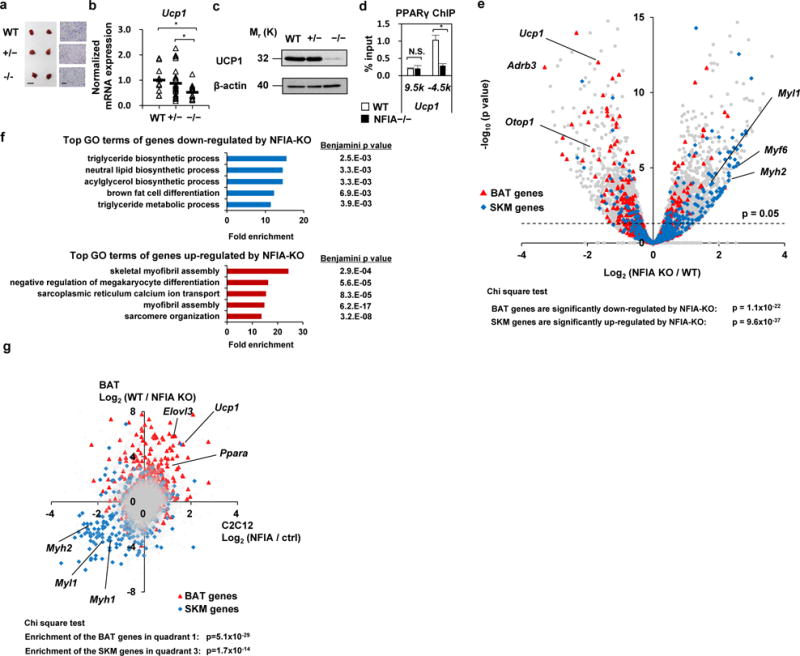
a, Macroscopic pictures (scale bar, 2.5 mm) and HE staining (scale bar, 100 μm) of BAT sections from neonates. b, RT-qPCR analysis of Ucp1 gene (mean +/− S.E.M.; N = 11 mice for WT, 24 mice for NFIA +/−, and 15 mice for NFIA −/−, respectively; * p <0.05, ** p <0.01). c, Western blot analysis of UCP1 protein. β-actin was used as a loading control. Representative images of two independent experiments are shown. d, ChIP-qPCR analysis of in vivo BAT. Ucp1 9.5k is a background site (mean +/− S.E.M.; N = 3 independent samples; * p <0.05, ** p <0.01). Representative results of two independent experiments are shown. e, Volcano plot of RNA-seq analysis. BAT- and SKM- selective genes are depicted in red and blue, respectively. The definition of BAT- and SKM- selective genes (N = 254 and N = 312, respectively) are shown in the methods. f, Top GO terms of genes down- or up-regulated by NFIA-KO. g, Scatter plot showing fold changes of gene expression by NFIA introduction into C2C12 myoblasts and NFIA knockout in BAT. BAT- and SKM- selective genes are depicted in red and blue, respectively. The genome-wide analyses were performed once based on the RNA-seq dataset.
To evaluate genome-wide changes in gene expression by NFIA-KO, we performed RNA-seq analysis (Fig. 7e). The expression levels of a battery of brown-fat-specific genes and mitochondrial genes were significantly decreased, while common fat genes were relatively maintained. In contrast, muscle-specific genes were reciprocally elevated by NFIA-KO (Supplementary Fig. 5c–f). Genes down-regulated by NFIA-KO were associated with GO terms such as triglyceride biosynthetic process and brown fat differentiation, and genes up-regulated by NFIA-KO were associated with GO terms such as skeletal myofibril assembly (Fig. 7f). A scatter plot of gene expression changes that were due to introduction of NFIA into myoblasts and NFIA-KO in BAT (Fig. 7g) demonstrated that BAT-selective genes (p = 5.1 × 10−29, chi-square test) and SKM-selective genes (p = 1.7 × 10−14) are reciprocally regulated by both gain- and loss-of-function studies. Finally, in db/db mice—a mouse model of obesity and diabetes—we found that expression levels of both Nfia and Ucp1 in BAT were suppressed compared with levels in C57BL6/J mice (Supplementary Fig. 5g), suggesting that downregulation of NFIA may play a pathophysiological role in the development of obesity and diabetes. Together, these results demonstrate that NFIA is required for the optimal activation of the brown fat gene program and repression of the muscle gene program in vivo.
NFIA in human brown fat
To explore the possible role of NFIA in human BAT, we analyzed the perirenal BAT of human patients with pheochromocytoma and with non-functional adrenal tumors. Human perirenal BAT is considered relatively close to murine classical BAT in terms of developmental origin and gene signature24. Perirenal BAT is activated in patients with pheochromocytoma, and the gene expression pattern is similar to that of classical BAT in mice25. We observed that NFIA expression was higher in patients with pheochromocytoma compared with those who had non-functional adrenal tumors (Fig. 8a). Furthermore, expression levels of the brown-fat-specific genes including UCP1 and PPARGC1A were positively and significantly correlated with NFIA expression (Fig. 8b). We also analyzed human brown and white adipocytes, obtained from supraclavicular and subcutaneous region, respectively26. Expression levels of NFIA were higher in human brown adipocytes compared with white adipocytes throughout the differentiation (Fig. 8c). Finally, we measured the expression levels of NFIA in BAT and WAT of human necks27. The expression of NFIA was numerically higher in BAT than expression in WAT, although the difference did not reach statistical significance, possibly due to limited sample size (Fig. 8d). These results indicate that NFIA may also control the brown fat gene program in humans.
Figure 8. Expression of NFIA and the brown-fat-specific genes is positively correlated in perirenal brown fat of human patients with pheochromocytoma.
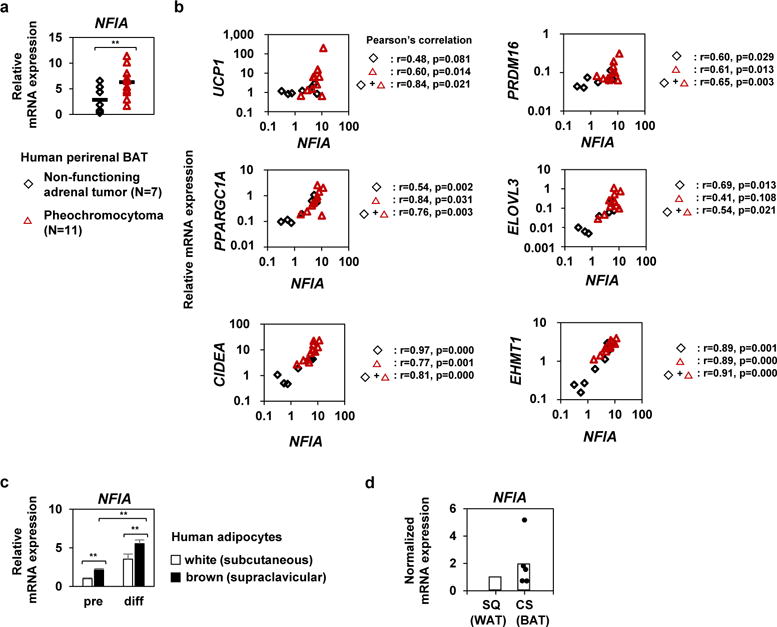
a, Expression levels of NFIA in perirenal brown fat of human patients with pheochromocytoma or non-functional adrenal tumors (mean +/− S.E.M.; N = 7 independent samples for non-functional adrenal tumors and N = 11 independent samples for pheochromocytoma ; * p <0.05, ** p <0.01). b, Correlation of expression of the brown-fat-specific genes and NFIA expression. c, Expression levels of NFIA in human brown (supraclavicular) and white (abdominal subcutaneous) adipocytes before and after differentiation. (mean +/− S.E.M.; N = 5 independent samples for white adipocytes and N = 6 independent samples for brown adipocytes; * p <0.05, ** p <0.01). d, RT-qPCR analysis of NFIA in human neck WAT (subcutaneous) and BAT (carotid sheath). (N = 5 independent samples). The analyses were performed once because human samples were highly limited.
Discussion
PPARγ is the master transcriptional regulator of adipocyte differentiation28. Here, we show that NFIA co-localizes with PPARγ at the brown-fat-specific enhancers for controlling the brown fat gene program. The binding of NFIA precedes and facilitates the binding of PPARγ. NFIA may recruit chromatin remodeling complexes such as Swi/Snf, as reported in case of human adrenal cells29. Alternatively, NFIA may work as a pioneer factor30 by facilitating structural changes of chromatin that increase accessibility and by recruiting PPARγ. Consistently, knockdown of PPARγ results in the almost complete loss of the effect of NFIA (Supplementary Fig. 6a–d). Supportively, genetic variations which alter NF-1 motifs were reported to affect not only the binding of NFI, but also the binding of PPARγ31, chromatin accessibility32, and the enhancer activity33. Proximally co-occupied transcription factors often compete with a nucleosome to access DNA34, and co-localization of NFIA and PPARγ more likely to result in nucleosome displacement than binding of NFIA or PPARγ alone, probably leading to increased chromatin accessibility, enhancer activity and gene expression. Nevertheless, further studies are needed to clarify the whole picture of chromatin remodeling during brown fat development caused by NFIA and other factors.
Although NFIA and PRDM16 perform a similar function in brown adipocyte differentiation, we show that these two factors work in parallel with each other. And both NFIA and PRDM16 are indispensable for the full activation of the brown fat gene program. Indeed, expression levels of Prdm16 were relatively unaffected in NFIA-KO mice (Supplementary Fig. 5c), and the opposite was also the case35 (fold change of Nfia expression in KO/WT was 0.99). Additionally, PGC1α is also dispensable for the effect of NFIA, because the effect of NFIA is totally maintained even when PGC1α is knocked down (Supplementary Fig. 6e–h). Although expression levels of Adrb3 were severely reduced by NFIA-KO, Adrb3 was also dispensable for the effect of NFIA (Supplementary Fig. 6i–l).
The BAT of NFIA-KO mice showed impaired expression of the brown fat gene program. However, the appearance and mass of the BAT was relatively preserved, suggesting that co-localization of NFIA and PPARγ alone may not be sufficient for determining brown fat cell fate. Cell-fate determination may be achieved through the contribution of multiple factors, and deficiency of NFIA alone could be somewhat compensated for. The investigation at the prenatal stage36 and lineage tracing experiments10 will help characterize the role of NFIA more definitively. Additionally, tissue-specific deletion of NFIA in mice will be required for investigating the role of NFIA in adult BAT in systemic metabolism.
Since brown fat and skeletal muscle share a common progenitor, repressing the muscle gene program is required in addition to inducing the brown fat gene program to direct the cell-fate determination toward brown fat. However, many of the myogenic genes do not have NFIA binding sites at their enhancers, and the repressive effect of NFIA on muscle genes may be indirect. Note that PPARγ is reported to suppress MyoD—a master regulator of myogenesis—through enhanced ubiquitination and degradation of the MyoD protein37. NFIA may suppress myogenesis by inducing PPARγ; uncharacterized, PPARγ-independent mechanisms may also be involved.
Originally, NFI was found to bind to an enhancer of the Fabp4 gene38, and we previously reported that the NF-1 motif is enriched within open chromatin regions of differentiated white adipocytes compared with undifferentiated cells39. In this study, we found that the NF-1 motif is enriched in BAT-specific—not WAT-specific—open chromatin regions in vivo. NFIA is important for general adipogenesis—probably by inducing PPARγ—and NFIA is more important for the BAT-selective gene program than the WAT-selective gene program. Indeed, the overlap of NFI binding sites with BAT FAIRE peaks is higher than that of NFI binding sites with eWAT FAIRE peaks (Supplementary Fig. 7a, b). Supportively, a transcriptome analysis independently suggested that NFIA would be a positive regulator of brown adipocyte differentiation40. Interestingly, it is reported that NFIC negatively regulates adipocyte differentiation41. Future studies will be needed to understand the mechanisms underlying differential regulation of brown adipocyte differentiation by different isoforms of NFI family.
Developing a therapy for obesity through activating BAT is highly anticipated. Recently, an epigenome-wide association study showed that DNA methylation of a CpG site at the intron of NFIA in human adipose tissue shows a positive and significant association (p = 4.0 × 10−20) with obesity42, suggesting that down-regulation of NFIA may contribute to pathophysiology of obesity in humans. Identifying an upstream regulator of NFIA may open a door for BAT-targeted anti-obesity therapy.
Methods
Retroviral expression system
For gain-of-function experiments, we used pMXs retroviral expression system as previously described39. The sequences used for shRNA-mediated loss-of-function experiments were: shNFIA #1, 5′-CCUUCUCAACUCUGUAACA-3′; shNFIA #2, 5′-GUCAGCAGUUACAUACAUA-3′; shPPARγ, 5′-CAAGAGAUCACAGAGUAUG-3′; shPRDM16, 5′-GAAGAGCGUGAGUACAAAU-3′. The corresponding double-stranded DNA sequences were subcloned into pLMP retroviral vector (Open Biosystems) using XhoI and EcoRI restriction enzyme sites. For retrovirus production, Platinum E packaging cells (Cell Biolabs) were transfected with the vector using lipofectamine 2000 (Invitrogen). Two days afterwards, conditioned medium was centrifuged at 2,000 rpm for 5 minutes, and the supernatant was supplemented with 10 mg/ml polybrene and used for overnight infection. Subsequently, infected cells were selected using appropriate antibiotics.
Cell culture
C2C12 myoblasts and 3T3-L1 adipocytes were purchased from American Type Culture Cell Collection (ATCC). 3T3-F442A adipocytes were gifts from Shingo Kajimura11. Immortalized brown adipocytes were gifts from Kohjiro Ueki43 and Shingo Kajimura44. For adipocyte differentiation of C2C12 transfected with NFIA and/or PPARγ expression vector, cells were treated for 48 hours in medium containing 10% FBS, 0.5 mM isobuylmethylxanthine, 125 nM indomethacin, 1 mM dexamethosone, 850 nM insulin, 1 nM T3 and 1 mM rosiglitazone. After 48 hours, cells were switched to medium containing 10% FBS, 850 nM insulin, 1 nM T3 and 1 mM rosiglitazone. To stimulate thermogenic gene expression, cells were incubated with 10 μM forskoline (fsk) for 4 hours.
siRNA-mediated gene knockdown
For NFIA knockdown experiments by electroporation, a control siRNA and a siRNA for NFIA was purchased from Sigma (Mission_SIC-001 and Mm_Nfia_9630). Differentiated brown adipocytes at day 6 of differentiation were washed, trypsinized, centrifuged and transfected by the Neon transfection system (Invitrogen). The cells were harvested 2 days after the transfection. For PGC1α or Adrb3 knockdown experiments by lipofection, a control siRNA and a siRNA for PGC1α or Adrb3 was purchased from Santa Cruz Biotechnology (sc-37007, sc-38885 and sc-39869). The siRNA were transfected using lipofectamine RNAiMAX (Invitrogen) 2 days before confluence, according to the manufacture’s instruction.
ChIP
Samples were treated by nuclear extraction buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1% IGEPAL CA-630) for 10 minutes and immediately cross-linked with 1 % formaldehyde for 7.5 minutes at room temperature. Cross-linking was quenched using 125 mM glycine for 5 minutes. The chromatin was sheared by a probe sonicator (Branson) and was spun at 15,000 rpm for 5 minutes. Antibodies were added for overnight incubation at 4 °C. Mixes of Protein A and Protein G Sepharose (GE) were added to samples for 4 hours at 4 °C. Subsequent procedures were performed as described previously39. The antibodies used were FLAG M2 (1 μg per IP, Sigma, F3165), NFI (12 μg per IP, Santa Cruz Biotechnology, sc-30198), PPARγ (4 μg per IP, mix of Santa Cruz Biotechnology, sc-7273, and Perseus Proteomics, A3409A), C/EBPα (4 μg per IP, Santa Cruz Biotechnology, sc-61), C/EBPβ (4 μg per IP, Santa Cruz Biotechnology, sc-150), EBF2 (6 μg per IP, R&D Systems, AF7006) and H3K27Ac (4 μg per IP, Abcam, ab4729). ChIP-seq libraries were prepared using KAPA hyper prep kit (KAPA Biosystems) according to the manufacturer’s instructions.
FAIRE
FAIRE was performed as described previously39, with optimization for experiment using in vivo tissues. Briefly, freshly collected adipose tissues obtained from 8-week-old male C57BL/6J mice were minced with scissors and cross-linked with 1 % formaldehyde for 7 minutes at room temperature, followed by quenching with 125 mM glycine for 5 minutes. A Pasteur pipette was used to carefully wash the floating minced samples with cold PBS. The chromatin was sheered by using a homogenizer and then a probe sonicator (Branson). Subsequent procedures were performed as described previously39. FAIRE-seq Libraries were prepared using ChIP-Seq Sample Prep Kit (Illumina) according to the manufacturer’s instructions.
ATAC
50,000 nuclei of brown adipocytes before and after differentiation were transposed using Tn5 transposase as previously described20. Briefly, cells were lysed using ice-cold lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1 % IGEPAL CA-630) and were spun at 500 g for 10 minutes. The pellet was resuspended in the transposase reaction mix and incubated at 37 °C for 30 minutes. The sample was column-purified and amplified by 15 cycles of PCR before high-throughput sequencing.
RNA expression analysis
Total RNA from cultured cells or tissues was isolated using TRIzol reagent (Invitrogen) and RNeasy minicolumns (QIAGEN). Isolated RNA was reverse-transcribed using ReverTra Ace qPCR RT Master Mix kit (Takara). Real-time quantitative PCR (SYBR green) analysis was performed on a 7900HT Fast Real-Time PCR System or QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems). Rplp0 was used as an internal normalization control of murine samples. TBP (Figure 7a, b), RN18S (Figure 7c), and PPIA (Figure 7d) were used as an internal normalization control of human samples. For RNA-seq, libraries were prepared using TruSeq Stranded mRNA Library Prep Kit (Illumina) according to the manufacturer’s instructions.
High-Throughput Sequencing
High-throughput sequencing was performed by using the Illumina Genome Analyzer System or HiSeq 2500. Demultiplex and base calling were performed with the CASAVA 1.8.2 software (Illumina).
ChIP-seq, FAIRE-seq and ATAC-seq data processing
The sequence reads were mapped to UCSC build mm9 (NCBI Build 37) assembly of the mouse genome using the ELAND mapping program. Peak calling was performed using the MACS version 1.4 with default parameters45. Peaks of ChIP-seq, FAIRE-seq and ATAC-seq were visualized by a GenomeJack browser (version 3.1, Mitsubishi Space Software). Galaxy cistrome46 was used for genomic region handling. For Venn diagram, note that sum of the number of peaks in each component may not equal to the number of overall peaks, because a single peak in one sample could overlap with multiple peaks in another sample. Motif analysis was performed using CentriMo47 version 4.10.2 with default parameters. We used the licensed version of TRANSFAC database48. Gene ontology annotation analysis was performed using DAVID49. Biological process terms “GO_BP_FAT” were used and GO terms with FDR less than 0.05 were shown in descending order of the fold enrichment. A heat map representation was generated using in-house software.
RNA-seq data processing
The sequence reads were mapped to the mm9 mouse genome using TopHat. Fragments per kilobase of exon per million fragment mapped (FPKM) values were calculated for each gene using CuffLinks. Differentially expressed genes were analyzed using DeSeq250. Genes with FPKM value <1 in all the samples were excluded for the differential expression analysis. A heat map representation was generated using GenePattern online software51.
Definition of tissue-selective genes for genome-wide analysis
We defined BAT- and WAT- selective genes (N = 549 and N = 849, respectively) using the publically available microarray dataset GSE28440. BAT-selective genes were defined as genes expressed in BAT two-fold or more in BAT than in WAT with statistical significance (p < 0.05). We likewise defined WAT-selective genes. For BAT- and SKM- selective genes (N = 254 and N = 312, respectively), we used microarray dataset GSE70857 and defined the tissue-selective genes as above.
Western blotting
Tissues were lysed in radioimmunoprecipitation assay (RIPA) buffer containing 0.1 % SDS, 1 % NP-40, 0.5 % Na deoxycholate, 150 mM NaCl, 50 mM Tris-Cl (pH 8.0), 1 mM EDTA supplemented with protease inhibitor (Roche). Proteins were separated by SDS-PAGE, transferred to nitrocellulose membrane, and detected with the antibodies anti-NFIA (1:500 dilution, Santa Cruz Biotechnology, sc-133816), anti-UCP1 (1:2000 dilution, Abcam, ab10983), anti-PPARγ (1:500 dilution, Santa Cruz Biotechnology, sc-7196), anti-β actin (1:1000 dilution, Santa Cruz Biotechnology, sc-1616). FLAG M2 (1:2000 dilution, Sigma, F3165), and V5 (1:5000 dilution, Invitrogen, R960-25)
Co-immunoprecipitation
HEK293 cells were transfected with pcDNA 3.1-V5-PRDM16, pcDNA 3.1-FLAG-PPARγ, or pcDNA 3.1-NFIA expression vector as indicated in the figure and figure legends. pcDNA 3.1-V5-PRDM16 and -FLAG-PPARγ vectors were gifts from Claudio Villanueva17. Two days after transfection, cells were lysed using RIPA buffer supplemented with protease inhibitor. Dynabeads protein A (Thermo Fisher) were incubated with V5 antibody (2 μg per IP, Invitrogen) at 4 °C for 2 hours, and then the lysate and antibody-beads complex were incubated at 4 °C for overnight. The beads were washed with RIPA buffer 5 times. Eluted proteins were analyzed by Western blotting as described above.
Oxygen consumption assay
Control or NFIA-expressing C2C12 myoblasts were plated on gelatin-coated XF24 culture microplates (Seahorse Bioscience), grown into confluence and treated with an adipogenic cocktail. At day 7 of differentiation the medium was replaced with XF24 assay medium supplemented with 1 mM sodium pyruvate and 25 mM glucose. The oxygen consumption rate was measured using XF24 flux analyzer (Seahorse Bioscience). The cells were treated with 1 μM oligomycin, 0.5 μM FCCP and 1 μM antimycin/rotenone in succession.
Animal Studies
All animal work was conducted according to the institutional guidelines at The University of Tokyo. NFIA-KO mice (stock number: 010318-UNC)23 were purchased from MMRRC (Mutant Mouse Regional Resource Center). The founder of this stock was 129S6 and subsequently was backcrossed to C57BL6/J for more than 20 generations. All the experiments were performed using male mice. Age of the mice for each experiment was indicated in the main text or figure legends.
Human Studies
Perirenal BAT samples were obtained from eleven patients with pheochromocytoma and seven with non-functioning adrenal tumors, as previously described25. All procedures were approved by the Hiroshima University Ethics Committee. Supraclavicular brown adipocytes and abdominal subcutaneous white adipocytes were obtained by head or neck tumor surgery and gallbladder surgery, respectively, as previously described26. All procedures were approved by the Scientific-Ethics Committees of the Capital Region and of Copenhagen and Frederiksberg Municipalities Denmark, journal numbers H-A-2009-020, H-A-2008-081, and (KF) 01-141/04, respectively. BAT and WAT samples from necks were obtained during thyroidectomy or anterior cervical spine surgery, as previously described27. All procedures were approved by the Human Studies Institutional Review Boards of Beth Israel Deaconess Medical Center, Joslin Diabetes Center and Massachusetts General Hospital. The entire study was approved by the research ethics committee of the Graduate School of Medicine, the University of Tokyo. All the procedures described above were conducted according to the Declaration of Helsinki, and all the patients gave written informed consent before taking part in the study.
Human adipocyte culture and differentiation
At the confluence, preadipocytes were induced to undergo adipocyte differentiation and cultured for 12 days, as previously described26. Samples were collected when the cells were 45–65 % confluence and the cells were fully differentiated (12 days after inducing differentiation).
Statistics and reproducibility
At least five mice were used for all the animal studies. This group size was based on our previous studies. Randomization was not performed, and the investigators were not blinded to mouse genotype or type of the human samples. No samples were excluded for analysis. Two-tailed student’s t-test was performed to determine the statistical significance between two groups unless otherwise specified, with a p value of less than 0.05 considered significant. We checked that the data met the assumption of the statistic tests, and variances were similar between the groups being tested. Experiments independently repeated three or more times were Fig. 1d, e, Fig. 2a–e, Fig. 2f–j, Fig. 3b–k, Fig. 6a–g, Fig. 7a,b, Supplementary fig. 2c–j, Supplementary fig. 5a, b, and Supplementary fig. 6a–d. Experiments independently performed two times were Fig. 1f, Fig. 2f, Fig. 2k, Fig. 3a, Fig. 7c,d, Supplementary fig. 1c–e, Supplementary fig. 4a–d, Supplementary fig. 5g, and Supplementary fig. 6e–l. Experiments or analyses performed once were Fig. 1a, b, c (FAIRE-seq), Fig. 4a–f (ChIP-seq and ATAC-seq), Fig 5a–c (ChIP-seq and ATAC-seq), Fig. 7e–g (RNA-seq), Fig. 8a–d (experiments using human samples), Supplementary fig. 1a,b (RNA-seq), Supplementary fig. 2a, b (RNA-seq), Supplementary fig. 3a–m (ChIP-seq), Supplementary fig. 5c–f (RNA-seq), and Supplementary fig 7a, b (FAIRE-seq and ChIP-seq).
Code availability
The in-house software for a heat map representation of ChIP-seq is available from the corresponding authors on request.
Data availability
High-throughput sequencing data have been deposited at the Gene Expression Omnibus (GEO) under the accession number GSE83764. Uncropped pictures of the Western blot analysis are shown in Supplementary figure 8. Source data for Figure 6d and Figure 6e are provided in Supplementary Table 2. All other data supporting the findings of this work are available from the corresponding authors on reasonable request.
Supplementary Material
Acknowledgments
We are grateful to Susanne Mandrup for her suggestion about our study. We thank Kaori Shiina and Kenji Tatsuno for their help in library preparation for high-throughput sequencing. Shiro Fukuda and Toshihiro Umehara for their help in computational analysis. Kohjiro Ueki, Shingo Kajimura and Claudio Villanueva for providing cells and plasmids. Kinichi Nakashima for his suggestion regarding NFIA-KO mice. Takuya Sugiyama, Tetsuya Kubota and Naoto Kubota for their help in animal experiments. We also thank Takahito Wada for his technical assistance. This work is funded by an AMED-CREST research grant from the Japan Agency for Medical Research and Development (AMED) to T.Y.; by a grant-in-aid for scientific research (B) from the Japan Society for the Promotion of Science (JSPS), grant number 25293209 to H.W.; by a grant-in-aid for JSPS fellows from JSPS, grant number 15J02835 to Y.Hiraike.; by a junior scientist development grant from the Japan Diabetes Society to Y.Hiraike. Y.Hiraike has been supported by a research fellowship from JSPS. The Centre of Inflammation and Metabolism (CIM) and the Centre for Physical Activity Research (CFAS), Department of Infectious Diseases, Rigshospitalet is supported by a grant from TrygFonden. CIM/CFAS is a member of DD2 - the Danish Center for Strategic Research in Type 2 Diabetes (the Danish Council for Strategic Research, grant no. 09-067009 and 09-075724). T.J.L has been supported by a research grant from the Danish Diabetes Academy supported by the Novo Nordisk Foundation. The Novo Nordisk Foundation Center for Basic Metabolic Research is supported by an unconditional grant from the Novo Nordisk Foundation to University of Copenhagen.
Footnotes
Author contributions
Y. Hiraike and H.W. designed the study. Y. Hiraike., H.W., J.Y., K.M., H.K., W.S., Y. Hirota and T.A. performed experiments. M.N., R.N., K.S., S.Y., S.T., Y.Hiraike and H.W. performed computational analysis. G.N., H.O., K.O., and M.Y. performed analysis of human perirenal BAT. AP.W., YH.T. and AM.C. performed analysis of human neck BAT and WAT. TJ.L., NZ.J. and C.S. performed analysis of human brown and white adipocytes. Y. Hiraike and H.W. wrote the manuscript. H.A., T.Y. and T.K. supervised all aspects of this work.
We declare that none of the authors have a financial interest related to this work.
References
- 1.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 2.Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007;293:E444–52. doi: 10.1152/ajpendo.00691.2006. [DOI] [PubMed] [Google Scholar]
- 3.van Marken Lichtenbelt WD, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–8. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 4.Cypess AM, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–17. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Virtanen KA, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–25. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 6.Saito M, et al. High Incidence of Metabolically Active Brown Adipose Effects of Cold Exposure and Adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Lans AAJJ, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest. 2013;123:3395–403. doi: 10.1172/JCI68993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanssen MJW, et al. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med. 2015;21:6–10. doi: 10.1038/nm.3891. [DOI] [PubMed] [Google Scholar]
- 9.Cypess AM, et al. Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metab. 2015;21:33–8. doi: 10.1016/j.cmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seale P, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–7. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seale P, et al. Transcriptional Control of Brown Fat Determination by PRDM16. Cell Metab. 2007;6:38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harms MJ, et al. PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes Dev. 2015;29:298–307. doi: 10.1101/gad.252734.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohno H, Shinoda K, Spiegelman BM, Kajimura S. PPARγ agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab. 2012;15:395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siersbæk MS, et al. Genome-wide profiling of peroxisome proliferator-activated receptor γ in primary epididymal, inguinal, and brown adipocytes reveals depot-selective binding correlated with gene expression. Mol Cell Biol. 2012;32:3452–63. doi: 10.1128/MCB.00526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajakumari S, et al. EBF2 determines and maintains brown adipocyte identity. Cell Metab. 2013;17:562–574. doi: 10.1016/j.cmet.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17:877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villanueva CJ, et al. Adipose subtype-selective recruitment of TLE3 or Prdm16 by PPARγ specifies lipid storage versus thermogenic gene programs. Cell Metab. 2013;17:423–35. doi: 10.1016/j.cmet.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kajimura S, et al. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009;460:1154–8. doi: 10.1038/nature08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gronostajski RM. Roles of the NFI / CTF gene family in transcription and development. Development. 2000;249:31–45. doi: 10.1016/s0378-1119(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 20.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinz S, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullen AC, et al. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011;147:565–76. doi: 10.1016/j.cell.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.das Neves L, et al. Disruption of the murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. Proc Natl Acad Sci U S A. 1999;96:11946–51. doi: 10.1073/pnas.96.21.11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. 2013;19:1252–63. doi: 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- 25.Nagano G, et al. Activation of classical brown adipocytes in the adult human perirenal depot is highly correlated with PRDM16-EHMT1 complex expression. PLoS One. 2015;10:1–13. doi: 10.1371/journal.pone.0122584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jespersen NZ, et al. A Classical Brown Adipose Tissue mRNA Signature Partly Overlaps with Brite in the Supraclavicular Region of Adult Humans. Cell Metab. 2013;17:798–805. doi: 10.1016/j.cmet.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 27.Cypess AM, et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med. 2013;19:635–9. doi: 10.1038/nm.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–56. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 29.Liu R, et al. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell. 2001;106:309–318. doi: 10.1016/s0092-8674(01)00446-9. [DOI] [PubMed] [Google Scholar]
- 30.Zaret KS, Carroll JS. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soccio RE, et al. Genetic Variation Determines PPARγ Function and Anti-diabetic Drug Response in Vivo. Cell. 2015;162:33–44. doi: 10.1016/j.cell.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maurano MT, et al. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat Genet. 2015;47:1393–401. doi: 10.1038/ng.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grossman SR, et al. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc Natl Acad Sci U S A. 2017;114:E1291–E1300. doi: 10.1073/pnas.1621150114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deplancke B, Alpern D, Gardeux V. The Genetics of Transcription Factor DNA Binding Variation. Cell. 2016;166:538–54. doi: 10.1016/j.cell.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 35.Harms MJ, et al. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metab. 2014;19:593–604. doi: 10.1016/j.cmet.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, et al. Ebf2 is a selective marker of brown and beige adipogenic precursor cells. Proc Natl Acad Sci U S A. 2014;111:14466–71. doi: 10.1073/pnas.1412685111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sunadome K, Suzuki T, Usui M, Ashida Y, Nishida E. Antagonism between the Master Regulators of Differentiation Ensures the Discreteness and Robustness of Cell Fates. Mol Cell. 2014;54:526–535. doi: 10.1016/j.molcel.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 38.Graves RA, Tontonoz P, Ross SR, Spiegelman BM. Identification of a potent adipocyte-specific enhancer: involvement of an NF-1-like factor. Genes Dev. 1991;5:428–37. doi: 10.1101/gad.5.3.428. [DOI] [PubMed] [Google Scholar]
- 39.Waki H, et al. Global mapping of cell type-specific open chromatin by FAIRE-seq reveals the regulatory role of the NFI family in adipocyte differentiation. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pradhan RN, et al. Dissecting the brown adipogenic regulatory network using integrative genomics. Sci Rep. 2017;7:42130. doi: 10.1038/srep42130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou J, et al. Nuclear factor I-C reciprocally regulates adipocyte and osteoblast differentiation via control of canonical Wnt signaling. FASEB J. 2017;31:1939–1952. doi: 10.1096/fj.201600975RR. [DOI] [PubMed] [Google Scholar]
- 42.Wahl S, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–86. doi: 10.1038/nature20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tseng YH, Ueki K, Kriauciunas KM, Ronald Kahn C. Differential roles of insulin receptor substrates in the anti-apoptotic function of insulin-like growth factor-1 and insulin. J Biol Chem. 2002;277:31601–31611. doi: 10.1074/jbc.M202932200. [DOI] [PubMed] [Google Scholar]
- 44.Ohno H, Shinoda K, Ohyama K, Sharp LZ, Kajimura S. EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature. 2013;504:163–7. doi: 10.1038/nature12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, et al. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu T, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bailey TL, MacHanick P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 2012;40:1–10. doi: 10.1093/nar/gks433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wingender E, et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang DW, Lempicki RA, Sherman BT. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reich M, et al. GenePattern 2.0. Nat Genet. 2006;38:500–1. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data have been deposited at the Gene Expression Omnibus (GEO) under the accession number GSE83764. Uncropped pictures of the Western blot analysis are shown in Supplementary figure 8. Source data for Figure 6d and Figure 6e are provided in Supplementary Table 2. All other data supporting the findings of this work are available from the corresponding authors on reasonable request.