

Abstract
Although diet has long been known to contribute to the pathogenesis of cardiovascular disease (CVD), research over the past decade has revealed an unexpected interplay between nutrient intake, gut microbial metabolism and the host to modify the risk of developing CVD. Microbial-associated molecular patterns are sensed by host pattern recognition receptors and have been suggested to drive CVD pathogenesis. In addition, the host microbiota produces various metabolites, such as trimethylamine-N-oxide, short chain fatty acids and secondary bile acids, that affect CVD pathogenesis. These recent advances support the notion that targeting the interactions between the host and microorganisms may hold promise for the prevention or treatment of CVD. In this Review, we summarize our current knowledge of the gut microbial mechanisms that drive CVD, with special emphasis on therapeutic interventions, and we highlight the need to establish causal links between microbial pathways and CVD pathogenesis
Introduction
Although highly effective lipid-lowering drugs are available, cardiovascular disease (CVD) remains the number one cause of death in developed countries1, and there remains a clear and unmet therapeutic need for the identification of new therapeutic targets for CVD. Genetic contributions have a strong role in disease pathogenesis; yet large scale genetic studies revealed that genetic variation is only a minor contributor (< 20%) to the risk of developing CVD 2,3. Thus, environmental factors have a predominant role in CVD pathogenesis, and it has long been known that diet is a major contributor to the risk of developing CVD4. Resident microbial communities in the intestinal tract represent a key ‘metabolic filter’ of our diet, as they can convert common nutrients into metabolites, some of which have been linked to CVD5,6. Indeed, both epidemiological and animal model studies have provided strong support for the idea that the interplay between microorganisms and the host have a contributory role to atherosclerotic CVD6–8. Chronic infection with pathogenic bacteria, as well as intestinal dysbiosis, has been associated with the progression of CVD11–19. Bacterial molecular patterns can directly engage host pattern recognition receptors in the intestine, but also in the vasculature5, promoting chronic inflammatory processes in the host. In addition to the direct stimulation of host immunity, the host microbiota can produce a wide variety of small-molecule metabolites that are sensed by host receptor systems to affect CVD pathogenesis in a manner that does not require direct microorganism-host interactions..
In fact, recent evidence suggests that both the susceptibility to atherosclerosis7 and thrombosis8 can be transmitted via gut microbial transplantation in mice. Research over the past decade has uncovered several key microbial metabolites such as trimethylamine N-oxide (TMAO), short chain fatty acids (SCFAs), and secondary bile acids that uniquely affect the progression of CVD. Although large antibiotic intervention trials have not shown clear benefit in CVD20–22, drug discovery is entering a new and exciting phase where targeting gut microbial metabolites represents a new tractable therapeutic strategy.
In this Review, we discuss the relevant transmissible microbial communities and taxa that are associated with, and microorganism-derived metabolites directly involved with CVD pathogenesis. With the major technological advances in microbial profiling methods and other ‘omic’ analytic platforms, we are now uniquely positioned to discover candidate microorganisms and their derived metabolites of relevance to CVD-related phenotypes, and pursue studies aimed at understanding mechanisms by which our microbial inhabitants impact human disease. We review microorganism-driven pathways that were reported to have an impact on CVD pathogenesis, and discuss how gut microorganism-targeted therapeutics hold untapped promise as new therapies for the prevention and/or treatment of CVD.
Microorganism-associated molecular patterns
It is now well appreciated that diverse microbial communities reside within the intestinal tract, on the skin surface, and on nearly every exposed surface of the human body5. Although still controversial, there is also a growing body of literature suggesting that increased permeability of oral and intestinal epithelial barriers may enable a small number of bacteria to enter into the systemic circulation, where they can ultimately enter into host tissues to promote disease9–11. Whether residing in or on exposed surfaces, or when they access the systemic circulation, bacteria can directly engage the innate immune system to not only elicit appropriate bacteriocidal responses, but to also regulate host metabolism and inflammatory pathways relevant to CVD. This type of microorganism-host cross talk is driven by direct interaction of microorganism-associated molecular patterns (MAMPs) and host pattern recognition receptors (PRRs) (FIG.1). There are now a number of examples linking MAMP – PRR driven signaling pathways to heart disease, and in fact chronic infections. In addition, evidence of prior exposure to certain bacterial pathogens has been associated with increased CVD risk11–19.
Figure 1. Direct engagement of pattern recognition receptors by microbial-associated molecular patterns driving CVD.
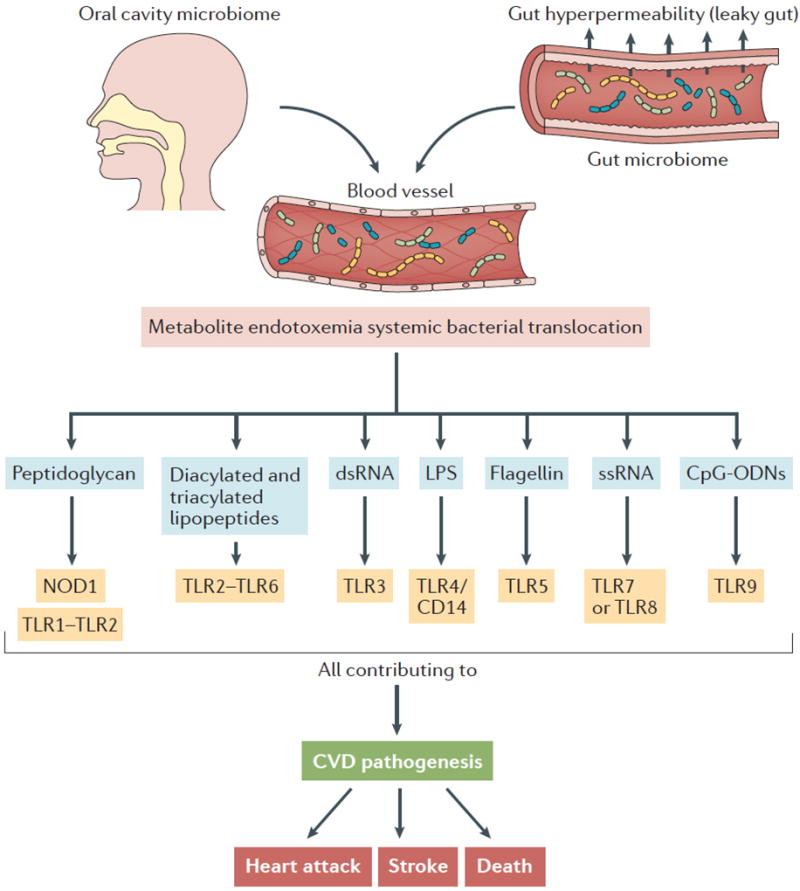
Microbial-associated molecular patterns (MAMPs) can promote CVD via the direct engagement of host pattern recognition receptors (PRRs), promoting chronic inflammatory processes in the host. In the context of cardiovascular disease, dysbiosis in both the oral and gut microbiome can elicit local MAMP-PRR signaling within those microenvironments (not shown). In addition, systemic bacterial translocation can promote CVD by MAMP-PRR signaling in distant sites, including the liver and artery wall. Abbreviations: CD14, cluster of differentiation 14; CpG ODNs, CpG oligodeoxynucleotides; LPS, lipopolysaccharide; MI, myocardial infarction; NOD1, nucleotide oligomerization domain-containing 1; TLR, toll-like receptor.
Several recent studies have analyzed microbiota communities in human oral, gut and atherosclerotic plaques from individuals with established atherosclerosis, and have found a reproducible correlation between CVD and bacterial pathogens, including Chlamydia pneumoniae, Porphyromonas gingivalis, Helicobacter pylori and Aggregibacter actinomycetemcomitans12–15. Importantly, some of these bacteria have been found in the intestinal tract as expected, but also within atherosclerotic plaques13–15, which indicates the possibility of engagement of PRRs in highly distinct microenvironments. In a metagenome-wide association study (MWAS) for atherosclerosis it was recently reported that the genus Collinsella spp. is enriched in subjects with atherosclerosis, while Eubacterium spp. and Roseburia spp. are more abundant among healthy controls16. Whether these associations are causally linked to disease pathogenesis, or alternatively, are driven by the presence of disease or disease-associated comorbidities and/or therapeutics remains to be determined.
Initially described in the Helsinki Heart Study, the common microbial pathogen, C. pneumoniae, has repeatedly been associated with CVD17–22. In addition, C. pneumoniae has been shown in vitro to elicit cell autonomous proinflammatory effects in isolated macrophages, endothelial cells and smooth muscle cells, which would be consistent with a link to atherosclerotic CVD18,19. However, primary infection with C. pneumonia has inconsistently altered atherosclerosis phenotypes in mouse models, and antibiotic treatments effective at treating C. pneumonia have proven ineffective in clinical studies, leaving a causal role for the bacteria in disease pathogenesis still a matter of debate20–23. More evidence for a bacteria-CVD link was provided from studies investigating the pathogen Porphyromonas gingivalis, which has been investigated because of associations between periodontal disease and CVD risk24–29. Either oral or systemic infection with P. gingivalis accelerates atherosclerosis in animal models, in part by promoting macrophage foam cell formation and inducing platelet aggregration24–29. Also, there may be a link between Helicobacter pylori infection and CVD risk30–32. However, careful primary infection studies in mouse models of atherosclerosis have yielded conflicting results30–32. Collectively, there are a number of reports suggesting that pathogenic bacteria may be causally linked to atherosclerotic CVD. However, whether CVD can be viewed as an infectious disease is still matter of intense debate because, as noted above, a number of large randomized prospective human antibiotic and anti-viral trials have failed to show any benefit of treatment or prophylaxis therapy in CVD or mortality20–23.
Even with the disappointing news with large antibiotic trials, there is ever expanding evidence that host PRRs are important regulators of CVD pathogenesis. For instance, single nucleotide polymorphisms (SNPs) in the host peptidoglycan receptor NOD1 are linked to early onset coronary heart disease33, and Nod1−/− mice are protected against atherosclerosis on an apolipoprotein E null (apoE−/−) background, which is a commonly used mouse model of atherosclerosis driven by severe accumulation of cholesterol in circulating remnant lipoproteins34. The heterodimeric complex of toll like receptors (TLR) 2 and TLR6 (TLR2-TLR6) is central to sensing bacterial di- and tri-acylated peptides35,36. A recently described SNP in the Tlr6 gene has been linked to left ventricular wall thickening and markers of inflammation37,38, and Tlr2−/− mice have strikingly reduced atherosclerosis, especially when challenged with synthetic TLR2 agonists36. Furthermore, transplantation of bone marrow from mice lacking the double-strand DNA receptor TLR3 into LDLr−/−, which is a commonly used mouse model of atherosclerosis driven by elevated low density lipoprotein cholesterol levels mice results in reduced atherosclerosis burden39. Among all of the CVD-associated MAMP-PRR pathways, the lipopolysaccharide (LPS) receptor, TLR4, has been most thoroughly investigated40–44. There have been a large number of reported TLR4 SNPs, with some having no association, some with increased, and some with decreased association to CVD risks40–44. However, genetic deficiency of TLR4 in either LDLr−/− or apoE−/− mice is reported to reduce atherosclerotic burden41–43. Importantly, many other non-bacterial ‘endogenous’ TLR4 agonists such as oxidized low density lipoproteins42, saturated fatty acids43, and high mobility group box 1 (HMGB1)44 have been shown to promote atherosclerosis progression. Additionally, a SNP in TLR7, a receptor for single-strand RNA, has been linked to ischemic stroke45, yet intriguingly, TLR7 deficiency in an apoE−/− background worsens atherosclerosis53. In a similar fashion, mice lacking the unmethylated CpG DNA receptor TLR9 in an apoE−/− background have exacerbated atherosclerosis46. Collectively, there is little doubt that activation of a number of host MAMP-PRR pathways can affect atherosclerosis progression. However, it remains doubtful that either activating or inhibiting PRRs is a viable option because of the central role of these receptors in innate immunity.
Box 1: Therapeutic strategies.
The majority of microbiome drug discovery efforts tend to focus on prebiotic or probiotic approaches, as the causative mediators and microbial species involved in disease pathogenesis remain to be determined for most host phenotypes under investigation. Given the mounting evidence linking the microorganism-dependent metabolite TMAO to CVD pathogenesis, there is strong likelihood that the TMAO pathway will be one of the first microbial pathways selectively targeted with non-lethal small molecules for the amelioration of human CVD. In strong support of this, a recent study showed the first proof of concept studies using a small-molecule inhibitor of gut microbial TMA lyase enzymes that led to a reduction in TMA and TMAO levels, and protection of mice against diet-induced atherosclerosis58. Regardless of which therapeutic approach is taken, because of the numerous mechanistic links between TMAO and the pathogenesis of disease, and strong corroborative associations from multiple human clinical studies, the gut microbial TMAO pathway holds tremendous potential as a therapeutic target for CVD treatment and prevention.
Microbial metabolites in CVD
In addition to surface antigens (MAMPs) on commensal and pathogenic bacteria, gut microorganisms enzymatically produce various metabolites that can both act locally in the gut, as well as travel systemically to affect host physiology in healthy and disease states. In fact, our gut microbiome represents a crucial filter of our diet, and effectively chemically diversifies each meal we consume. This simple idea, that microorganisms themselves, or products secreted from microorganisms, can cause disease or improve health (that is, probiotics), has been bolstered tremendously over the past decade. Recent advances in the fields of untargeted metabolomics in well-characterized clinical cohorts, coupled with mechanistic animal model studies have been used to discover several human disease-associated gut microbial metabolites. In fact, we now know that there are a large number of circulating metabolites that are derived solely or in part from bacterial metabolism of either dietary nutrients or diet-derived xenobiotics47–50. The rapidly expanding microorganism-dependent metabolome includes many methylamines, polyamines, polysaccharides, short chain fatty acids (SCFA), secondary bile acids, B vitamins, uremic toxins like p-cresol sulfate and indoxyl-sulfate, 4-ethylphenylsulfate, dihydrodigoxin, and a long list of xenobiotic-derived metabolites47–50. At this point, several microbial metabolites are recognized, the levels of which are associated with CVD-related phenotypes, but as association does not equal causation, the potential mechanistic participation of these metabolites in CVD remains unclear. A major limitation in progress on mechanistic studies, beyond their laborious nature, is that many of the circulating candidate analytes are yet chemically undefined. Moreover, even for those that are structurally resolved and available for study, we have almost no information about how microorganism-generated metabolites are sensed, whether it is through dedicated host receptors systems or whether they have non-receptor mediated mechanisms to affect CVD pathogenesis (for example, as allosteric modifiers).
Currently, there are three main classes of gut microorganism-dependent metabolites that have been linked to CVD risk either in humans or mice models - trimethylamines, short chain fatty acids and secondary bile acids. We focus on current mechanistic understandings through which these gut microorganism-dependent metabolites signal to the host and affect CVD pathogenesis.
The gut microbe-derived metabolite TMAO
Since its discovery and first reported link to CVD pathogenesis51, the gut microbial co-metabolite trimethylamine-N-oxide (TMAO) has quickly gained traction as both a biomarker for human CVD risk, and as a promoter of atherothrombotic diseases5–8,51–65. In fact, numerous human and mouse studies have support the notion that the TMAO pathway is one of the first bona fide gut microbiome centered CVD drug targets5,6,51–77. The TMAO pathway is a meta-organismal metabolic pathway whereby nutrients that are present in high fat foods (phosphatidylcholine, choline, L-carnitine and likely other trimethylamine-containing nutrients) can be metabolized by several distinct gut microbial enzyme complexes (CutC/D, CntA/B, YeaW/X)to generate the primary gut microbial metabolite TMA. TMA can be synthesized by either the choline utilization TMA lyase system (CutC/D)76, the carnitine Rieske-type oxgenase/reductase system (CntA/B)77, or the YeaW/X system which can utilize multiple substrate sources55. TMA then enters the portal circulation and is further metabolized by host enzymes in the liver, called flavin-containing monooxygenases (predominantly the FMO3 isoform), to produce TMAO (FIG. 2). Several recent reviews have highlighted the clinical relevance and therapeutic potential of the TMAO pathway in CVD5,6,50, so this will not be discussed in detail here. Briefly, a wealth of human and animal model data supports the conclusion that the gut microorganism-derived metabolite TMAO has both strong clinical prognostic value, and dietary provision of TMAO can promote atherosclerosis and thrombotic vascular disease in mouse models5–8,51–65. In addition to roles in atherosclerosis and thrombosis, the TMAO pathway has also been associated with other closely linked cardiometabolic diseases in humans including cardiac hypertrophy, cardiac fibrosis, chronic kidney disease, type 2 diabetes and obesity66–75.
Figure 2. The metaorganismal TMAO pathway as a driver of CVD.
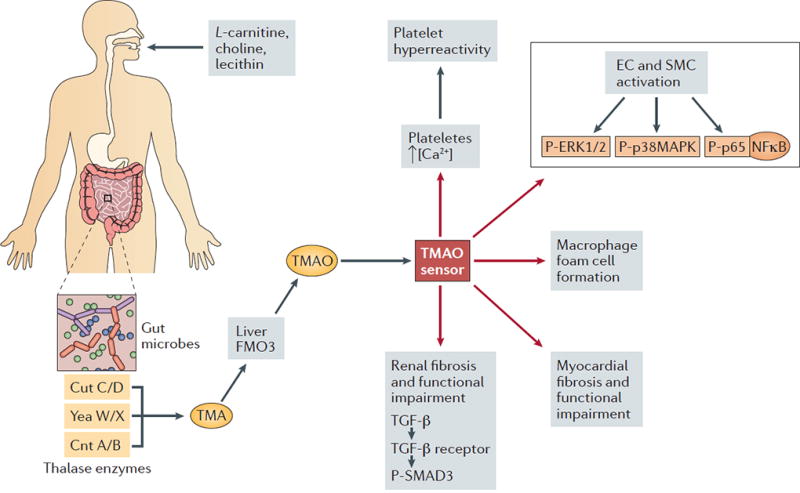
Postprandial delivery of choline, phosphatidylcholine (PC), carnitine, γ-butyrobetaine, and likely other methylamine-containing source nutrient gut microbes provides substrate for the gut-microbial-mediated production of trimethylamine (TMA). Microbial TMA lyase enzymes (CutC/D, CntA/B, and YeaW/X) can then generate TMA, which enters the portal circulation and is ultimately delivered to the host liver. The host flavin-containing monooxygenase (FMO) family of enzymes, especially FMO3, can then convert TMA to TMAO. TMAO can then promote atherosclerosis, thrombosis, heart failure, insulin resistance, and kidney disease via tissue or cell type-specific reprogramming. Through an unknown receptor-mediated sensing mechanism (indicated as TMAO sensor), TMAO drives cell-specific signaling events that promote CVD pathogenesis. In platelets, TMAO rapidly enhances stimulus-induced calcium (Ca2+) release, which signals to drive pro-thrombotic programmes. In endothelial and smooth muscle cells TMAO rapidly activates mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-kB) to promote the expression of adhesion molecules such as ICAM and E-selectin. In addition, TMAO can signal through currently unidentified pathways to regulate increased macrophage foam cell formation. TMAO can also initiate profibrotic programmes in the heart and kidney via a transforming growth factor β (TGFβ) – phospho-SMAD3 signaling axis. Collectively, these cellular events converge to promote atherosclerosis, thrombotic vascular disease, and associated renal impairment.
The gut microbiome-driven metabolite TMAO represents a promising gut microbiome-centered drug target 50 (Box 1). In fact, with the recent seminal discoveries of the microbial enzymes catalyzing the production of TMA76,77, we are now poised to therapeutically intervene this pathway at the level of gut microbial enzymology. However, at this point molecular mechanisms whereby TMAO promotes atherosclerosis, thrombosis, heart failure, renal dysfunction, insulin resistance and obesity are still being elucidated. This is, in large part, due to the lack of understanding of the molecular sensor for TMAO (that is, the TMAO receptor). Although there is evidence that TMAO can function as an important osmolyte in biological systems78,79, more recent evidence suggest that TMAO can rapidly signal to cells within minutes8,80. In isolated platelets, brief exposures to physiological levels of TMAO can potentiate thrombin-induced calcium release8. Likewise, treatment with TMAO in endothelial cells or smooth muscle cells, or direct injection in vivo, was shown to rapidly induce the activation of mitogen-activated protein kinases and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), or the upregulation of downstream adhesion proteins on aortic endothelial cells80. The kinetics of these TMAO-induced signaling responses strongly suggest that host cells may possess a dedicated receptor or sensor system to transduce the TMAO signal to an appropriate cellular response (FIG 2). Likewise, elevated TMAO levels are associated with elevated phosphorylation of the protein Smad3, which is a key effector of transforming growth factor β (TGFβ) signaling70 Identification of a host TMAO receptor is a strong possibility, given that structurally similar TMA has been shown to be a high affinity agonist for a cell surface G protein coupled receptor known as trace amine-associated receptor 5 (Taar5)81,82. In fact, there are now many examples of dedicated host receptor systems for metabolites originating from gut microbes47–50. Identification of a dedicated TMAO receptor would have broad implications, given the clear links of TMAO in human disease. It is tempting to speculate that TMAO receptor antagonists could be an effective therapeutic strategy to prevent or even treat established CVD.
As TMAO pathway drug discovery efforts advance, it will be imperative to understand the microbial taxa responsible for regulating flux through the TMA - TMAO pathway. For example, very little is known regarding which species or broader communities are the main producers of TMA in mouse and human ecosystems. In mouse studies in which dietary source nutrients for TMAO were specifically altered (for example, dietary provision of choline, L-carnitine or γ-butyrobetaine), a refined list of taxa has been associated with circulating TMAO levels, including the broad order of RF39 (r=0,49; false discovery rate, FDR=0.04), families of Erysipelotrichaceae (r=0.40; FDR=0.01), Lachnospiracea (r=0.779; FDR=0.084), and at the genus levels Prevotella (r=0.44; FDR=0.001), Anaeroplasma (r=0.677; FDR=0.016), Porphomonadaceae (r=-0.581; FDR=0.052), and Akkermansia muciniphila (r=0.634; FDR=0.009)51,52,55. Follow-up studies in humans comparing vegetarians and omnivores showed that several taxa of fecal microorganisms are associated with circulating TMAO levels, including Prevotella, Clostridiaceae, Incertae_Sedis_XII, Peptostreptococcaceae, Clostridium, Fusibacter, Lachospira, and Sporobacter52. Such associations between bacterial taxa and circulating TMAO levels are only useful in hypothesis generation, but do not establish a direct link between the taxa and TMAO production. A more fruitful method of identifying bona fide TMA-producing bacterial species is to screen reference genomes to identify the genetic potential to generate TMA (for example, presence of TMA lyase enzyme operons that encode CutC/D, CntA/B and YeaW/X), followed by functional studies. An elegant example of such reference genome mining was recently described83, supporting the power of genetics in predicting relevant metabolite-producing bacterial taxa. However, when it comes to metabolite production it is ultimately key to prove quantifiable biochemical potential. Coupling brute force microbial culture with biochemistry, a recent study screened 79 culturable human commensal bacteria for the potential to produce TMA from choline in vitro84. This important study identified eight distinct bacterial strains with the biochemical capacity to produce TMA from choline including Anarococcus hydrogenalis, Clostridium asparagiforme, Clostridium hathewayi, Clostridium sporogenes, Edwardsiella tarda, Escherichia fergusonii, Proteus penneri and Providencia rettgeri84. More importantly, low level colonization with a TMA producer (C. sporogenes) in the defined background of a core community that lacked TMA lyase activity (Collinsella aerofaciens, Bacteroides caccae, Bacteroides ovatus, Bacteroides thetaiotaomicron, and Eubacterium rectale) resulted in both TMA and TMAO generation in vivo in recolonized gnotobiotic mice, as well as reductions in choline levels in the host84. Another recent study found that the choline utilization cluster (Cut) is widely distributed across many gut microbial phyla in human stool metagenomes85. In addition to human stool metagenomes, CutC can be found with similar frequency in oral mucosa and supragingival plaque86, making it tempting to speculate that oral TMA production may be associated with the link between periodontal disease and CVD risk. Finally, a recent study has identified the human gut microbial taxa possessing the choline TMA lyase (CutC) and the carnitine oxygenase (cntA) using a multi-level screening approach87. Therefore, there has been rapid growth in our understanding surrounding which human commensals have the capacity to produce TMA from major source nutrients.
In addition to TMA producing bacteria, there is also mounting evidence that certain microbial species can metabolize TMAO and TMA for maintenance of trophic chains in both colonic and marine bacterial ecosystems88–91, but the relevant microorganisms and operons involved are still being elucidated. One major pathway facilitating catabolism of TMAO is driven by the bacterial torCAD operon, which encodes a TMAO reductase (TorA), a c-type cytochrome (TorC), and a TorA-specific chaperone (TorD)88. Additionally, bacteria present in marine ecosystems can also degrade TMAO through a TMAO demethylase enzyme (Tdm)89. Certain marine bacteria from the Roseobacter clade can use both TMA and TMAO as energy sources, which is facilitated by importing these metabolites via a ATP-binding cassette transporter TmoXWV for subsequent catabolism890,91. Certain methanogenic bacteria present in the human colonic ecosystem such as Methanomassiliicoccus luminyensis can reduce TMA as a substrate for methanogenesis92. It remains possible that these bacterial TMA-TMAO catabolic pathways are another potential avenue of therapeutic intervention, and may be an ideal probiotic strategy. There is little doubt that identification of both TMA producing and TMA/TMAO catabolizing human gut commensals will be particularly informative in the area of prebiotics and probiotics.
Although the vast majority of studies support a mechanistic link between the TMAO pathway and CVD, It is important to note that not all studies have show clear association between dietary intake of TMAO source nutrients (choline or carnitine) intake and CVD risk93–99. One recent study showed that dietary L-carnitine supplementation, which modestly raised circulating TMAO levels, was associated with a ~15% decrease in aortic atherosclerotic lesion area without altering aortic cholesterol content in apoE−/− mice transgenically expressing human cholesteryl ester transfer protein93. Furthermore, a large (n=29,079) population study in Japan demonstrated that dietary choline and betaine intak, estimated by a semi-quantitative food questionnaire, was not associated with CVD mortality94. Likewise, in the large (n=14,430) American-based Atherosclerosis Risk in Communities (ARIC) study it was found that dietary choline intake was not significantly associated with CVD95. In agreement, a large (n=16,165) Dutch study showed that dietary intake of choline or betaine was not associated with CVD risk96. Unfortunately, none of these large population studies examining the relationship between dietary choline, betaine, or carnitine intake at CVD outcomes measured circulating TMAO levels. Therefore, additional studies are needed to determine whether TMAO itself correlates with CVD risk in these same large population studies. This is likely to be the case given that a recent large (n=19,256) meta-analysis of hard CVD endpoint studies showed that circulating TMAO levels was a much stronger predictor of major adverse cardiovascular events (MACE) than its source nutrients (choline, betaine L-carnitine)97. Although the association between circulating TMAO levels and MACE are highly reproducible, some studies have suggested that this association in driven in part by impaired kidney function98. Also, a recent study showed that TMAO levels may not be equally associated with all types of vascular disease, especially in the case of large artery ischemic stroke and transient ischemic attack99. Collectively, although most studies suggest a strong association between TMAO levels and atherosclerotic coronary heart disease, these recent findings suggest that additional work is needed to understand which patient populations would benefit from TMAO lowering therapeutic strategies.
SCFAs and other fermentation products in CVD pathogenesis
The products of gut microbial fermentation are by far the most well-studied gut microbial metabolites, having key roles in both the maintenance of gut microbial ecology, and in fine-tuning host immunity and metabolic disease100–104. The major products of microbial fermentation of dietary fibers are SCFAs, with the most abundant metabolites being acetate, butyrate and proprionate. Although additional studies are needed to identify the most abundant human commensal producers, some information is available regarding which taxa are responsible for the majority of SCFA production. Acetate is produced by a large number of enteric bacteria, with major contributors such as Ruminococcus spp., Prevotella spp., Bifidobacterium spp., Bacteroides spp., Akkermansia muciniphila, Blautia hydrogenotrophica, Clostridium spp., and Streptococcus spp.105,106. Proprionate can be generated via three distinct biochemical pathways from Bacteroides spp., Phascolarctobacterium succinatutens, Dialister spp., Veillonella spp., Megasphaera elsdenii, Coprococcus catus, Salmonella spp., Roseburia inulinivorans, and Ruminococcus obeum105,107. Butyrate can be generated from locally-generated acetate substrate by Coprococcus comes, Coprococcus catus, Coprococcus eutactus, Anaerostipes spp., Eubacterium rectale, Eubacterium hallii, Faecaibacterium prausnitzii and Roseburia spp.105,107. Butyrate can also be generated via carbohydrate fermentation by several members of the Lachnospiraceae, Ruminococcaceae, Acidaminococcaceae families108. Undoubtedly, the major drivers of SCFA production in both human and rodent gut microbiomes remain completely understood, and reference genomes predict that many additional SCFA producers are yet to be discovered. SCFAs can function as a macronutrient energy source and hormone-like signaling molecules that enter the portal circulation to ultimately signal through dedicated host receptor systems to regulate innate immunity and host metabolism. Some of the currently identified host SCFA receptors include G protein receptor 41 (GPR41)109, G protein receptor 43 (GPR43)109, G protein receptor 109A (GPR109A)110, and olfactory receptor 78 (OLF78)111 (see below). Although SCFAs are the most well-studied gut microorganism-derived metabolites, it is still unclear whether SCFAs are causally linked to the development of human diseases, especially CVD, and their manipulation to modulate disease processes and susceptibility remains under investigation.
The vast majority of literature linking the gut microbiota to human disease implicate SCFAs as potential disease preventing or mitigating factors in obesity, diabetes, intestinal immunity, hypertension, kidney disease, cancer, and both alcoholic and non-alcoholic liver disease (NAFLD)100–104. Although there is a substantial body of literature linking microorganism-derived SCFAs to CVD risk factors, there is a paucity of studies showing clear association with circulating SCFAs and CVD risk or mortality in humans. In fact, most of the links between SCFAs and CVD risk factors such as obesity, diabetes, hypertension, renal dysfunction, and liver disease have been established in animals models. Therefore, unlike TMAO, the potential for SCFAs to be a therapeutic target for CVD is not strongly supported by independently replicated clinical studies. Gut microorganism-derived SCFA and their links to metabolic disease in animal models have been recently covered in several excellent reviews100–104. Below, we therefore only highlight several SCFA intervention human trials, and we discuss recent results from studies using animal models.
In one of the first direct administration studies in humans, physiologically relevant doses of acetate or proprionate were given to examine acute effects on metabolism112. This study found no effect on the levels of blood glucose or insulin, but both SCFAs reduced the levels of circulating free fatty acids112. Moreover, independent studies showed that although acute (3 hours) gastric infusion of SCFAs had no effect on glycemia , the levels of free fatty acid ere reduced113. Follow-up studies demonstrated that SCFA administration increased serum glucagon levels, which may have indirectly altered the levels of circulating free fatty acid114. In a subsequent long-term study in which proprionate was supplemented for seven weeks, there was a modest decrease in fasting glucose and alterations in glucose-stimulated insulin secretion during an oral glucose tolerance test115. Furthermore, provision of dietary supplementation of non-digestible polysaccharides yielded contradicting results116--122. However, it is important to note that although these studies often claim the observed metabolic effects are driven by alterations in the levels of SCFA, fiber-rich diets have pleotropic effects that extend beyond SCFA biology. Recently, one of the first randomized double-blind placebo-controlled antibiotic trials was conducted in which the levels of SCFA and a number of metabolic parameters were carefully monitored123. As expected, the antibiotic (vancomycin) treatment group showed drug selective reorganization of gut microbial diversity and composition, which was associated with statistically significant reductions in the levels of SCFA123. Surprisingly, even with large effects on the levels of SCFA no apparent alterations in energy or glucose homeostasis, which indicates that the phenotypic effects of SCFA seen in mice are not so easily translated into humans123. Collectively, these studies highlight the critical need for additional clinical investigation into whether modulation of SCFAs would hold any benefit in patients with CVD.
Even with these seemingly negative results in humans, there are some emerging discoveries with SCFAs and other microbial fermentation products that deserve additional investigation. For instance, a recent report described a pathway by which SCFAs can engage the olfactory receptor OLF78 and GPR41 in the kidney to regulate renin secretion and blood pressure97. In addition, another study demonstrated that obstructive sleep apnea-induced hypertension is transmissible by gut microbial transplantation111. In addition to SCFAs, the gut microbial fermentation product lactic acid has been reported to activate the host G protein-coupled receptor GPR81 to suppress adipose tissue lipolysis125. Interestingly, inhibition of the lactate receptor GRP81 was shown to protect against ischemic brain injury in mice126. As noted above, another emerging biologically active non-SCFA bacterial fermentation product is succinate127–129. Succinate is produced in large quantities in mammalian cells during the citric acid cycle, but it can also be generated by colonic microorganisms, including Prevotella129. Prevotella-generated succinate is presumed responsible for the observed beneficial effect on mice intestinal glucose production following provision of Prevotella copri129. Succinate can activate the host G protein-coupled receptor GPR91127–1295, which indicates a potential new gut microbial metabolite-host receptor interaction that is relevant to CVD pathogenesis. With the exception of the butyrate receptor GPR109A130, there have been no studies showing that SCFA, lactate or succinate receptor knockout mice exhibit alterations in atherosclerosis susceptibility, and therefore additional preclinical studies are needed.
The gut microbial bile acid metabolome in CVD
Gut microbial metabolism of bile acids represents one of the more intriguing stories of metaorganismal symbiosis, whereby an intimate bi-directional circuit produces essential detergents for intestinal fat absorption in the host, and provides a host-derived signal to maintain microbiome community structure131–133. Bile acids are now recognized to function as diverse signaling molecules that regulate host macronutient metabolism and energy expenditure (FIG. 3) (reviewed in REFs 131–140). Once delivered to the colonic microenvironment, bile salts can elicit cytotoxic effects to certain members of the gut microbiome ecosystem131–140. Although not completely understood, primary bile salts and locally-produced secondary bile acids are thought to regulate microbial community structure via membrane detergent effects, and also by inducing DNA damage in certain members of microbial communities 141–144. Interestingly, certain bacteria have evolved bile salt resistance mechanisms via alterations in efflux pumps141,142, and alterations in membrane lipid and protein composition143–147. For example, B. longum BBMN68 possesses a hemolysin-like protein that enables for selective resistance to taurine-conjugated bile salts145. Some bacteria such as Camphylobacter spp. and Salmonella typhimurium can be readily detected in gallbladder bile, where bile acid concentrations are extremely high148. The ability of bile salts and bile acids to dictate gut microbiome community structure likely has a major role in downstream effects on host metabolism. The microorganisms that are able to persist in the bile salt-rich colonic microenvironment can then chemically diversify bile salts into a number of biologically active species that can then be sensed by the host. One of the best-studied microorganism-driven biotransformations is the enzymatic hydrolysis of the C-24 N-acyl bond of glycine- or taurine-conjugated bile salts into free bile acids by bile salt hydrolase (BSH)135–138. BSH activity is widespread through both Gram-positive and Gram-negative species, including Clostridium, Bifidobacterium, Enterococcus, Lactobacillus, Bacteroides, Methanobrevibacter smithii, Methanosphera stadmanae, and likely many others135–138. In addition to deconjugation, gut microorganisms are the sole source of 7α and 7β dehydoxylase activity, which generates ‘secondary’ bile acids such as deoxycholic acid (DCA), lithocholic acid (LCA), hyodeoxycholic acid (HDCA) and ursodeoxycholic acid (UDCA)134–136. Unfortunately, there is currently limited information in regards to the bacteria genera that possess 7α and 7β dehydoxylase activity, and additional studies are needed in this area. In fact, the vast majority of genetic and biochemical studies have focused primarily on substrains Clostridium scindens, in which the bile acid inducible (bai) operon has been well characterized149. In addition to deconjugation and 7α and 7β dehydoxylation reactions, gut microbes can oxidize or epimerize bile acids via several distinct enzymes of the hydroxysteroid dehydrogenase (HSDH) family150,151. HSDH activity has been confirmed in a diverse variety of bacteria, including Bacteroides, Clostridium, Escherichia, Egghertella, Eubacterium, Peptostreptococcus and Ruminococcus. Formation of ethyl esters and long-chain fatty acid esters from bile acids can be driven by gut bacteria such as Bacteroides, Eubacterium and Lactobacillus152,153. However, the bacterial enzymes responsible for these esterification reactions are unknown. Finally, gut microorganisms can remove sulfate from either 3α or 3β-sulfated bile acids, and this sulfatase activity has been seen in Clostridium, Peptococcus, Fusobacterium, and Pseudomonas154. Collectively, through deconjugation, oxidation, epimerization, 7α/7β dehydoxylation, esterification and desulfation gut microorganisms chemically diversify the bile acid pool, and then the secondary bile acids can enter the portal circulation to functions as endocrine-like signaling molecules with potent effects on host physiology and disease.
Figure 3. Microbial production of secondary bile acids in CVD.
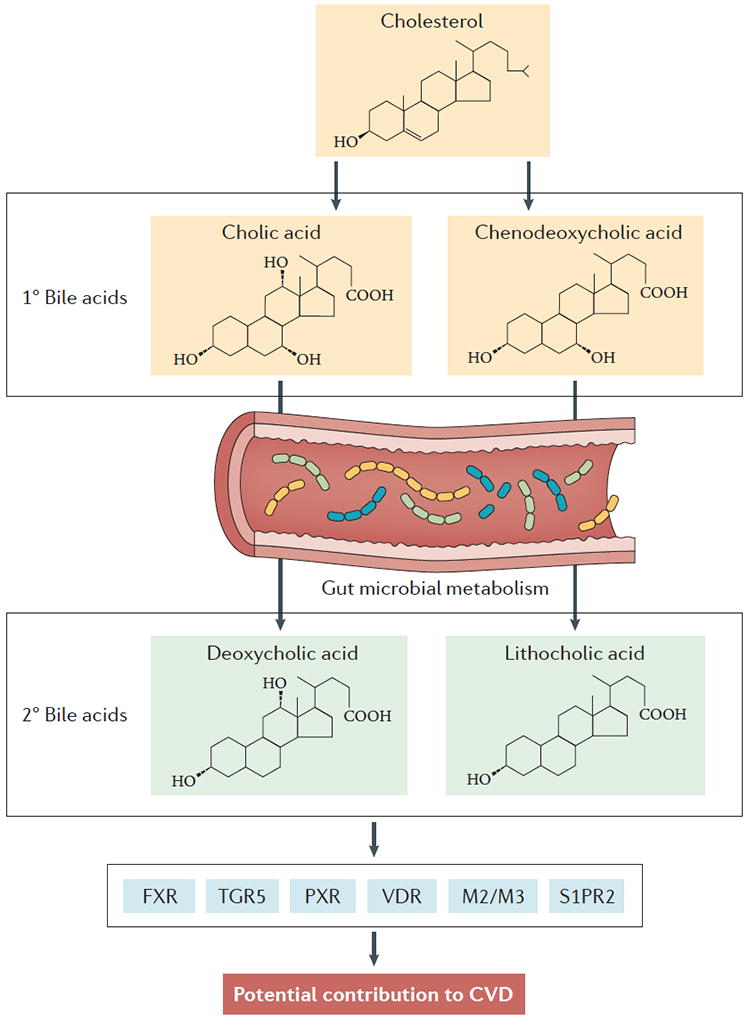
Initially, primary bile acids are synthesized in the host liver from cholesterol. De novo synthesized primary bile acids such as cholic acid, chenodeoxycholic acid (CDCA) and muricholic acid (MCA; only produced in rodents) are then conjugated with either glycine (humans) or taurine (humans and mice) at the C-24 carboxyl position. Following conjugation, resulting bile salts are secreted into bile along with cholesterol and phospholipids to form mixed micelles, which are transiently stored in the gall bladder. When a meal is ingested, the gall bladder contracts to release mixed micelles into the proximal intestine where they function as essential emulsifiers to enable proper absorption of hydrophobic molecules such as fatty acids and fat-soluble vitamins (such as vitamin A, vitamin D, vitamin E and vitamin K) (not shown). Importantly, bile salts are left behind in the intestinal lumen where they ultimately traverse to the colon. Once in the colonic microenvironment, they participate in a bi-directional interplay regulating microbial community structure, and they are subsequent microorganism-driven metabolism of primary bile salts into secondary bile acids (deoxycholic acid and lithocholic acid), which can have an impact on host physiology and disease susceptibility. Importantly, after aiding in intestinal lipid absorption both primary bile salts and secondary bacterial metabolites are almost quantitatively re-absorbed (>95% recovered) in the ileum via dedicated host transporters in ileal enterocytes (not shown). This reabsorptive process provides newly diversified bile acid species, which can then signal to the host through dedicated receptor systems, including farnesoid X receptor (FXR), protein-coupled bile acid receptor 1 (TGR5), pregnane X receptor (PXR), vitamin D receptor (VDR), muscarinic receptors 2 and 3 (M2/M3), and sphingosine-1-phosphate receptor 2 (S1PR2).
Once bacterially modified bile acids enter the portal blood, typically in the postprandial state, they can engage an ever-growing number of recognized host bile acid receptors (FIG. 2). The best known host bile acid receptor is the nuclear hormone receptor farnesoid X receptor (FXR), which regulates the transcription of key genes involved in primary bile acid biosynthesis155. Importantly, polymorphisms in FXR have been linked with hyperglycemia and circulating free fatty acid levels156, and genetic deletion of FXR in mice alters the progression of atherosclerosis in mice157. Recent studies have also shown that FXR is required for the metabolic reprogramming initiated by human microbial transplantation158,159. Interestingly, transplantation of human microbiota into conventional mice can alter mouse bile acid composition and induce FXR activation by reducing the levels of the FXR antagonist tauro-beta-muricholic acid159 Another host bile acid receptor gaining attention in recent years is the G protein-coupled receptor TGR5160,161. TGR5 activation has been linked to increased energy expenditure146, and TGR5 knockout mice are protected against atherosclerosis development160. Secondary bile acids such as lithocholic acid can activate the nuclear hormone receptor pregnane X receptor (PXR), which is a well known transcriptional regulator of xenobiotic and lipid metabolism162. Genetic deletion of the lithocholic acid receptor PXR in mice reduces the progression of atherosclerosis163. Interestingly, certain bacterially-modified bile acids (3-oxo-lithocholic acid and lithocholic acid) can also activate the vitamin D receptor (VDR)164, and genetic studies in both humans and mice have shown a role for VDR activation in CVD-related disease164–166. Taurine-conjugated bile acids have been shown to activate specific muscarinic receptors167, and polymorphisms in the muscarinic acetylcholine receptor M2 isoform have recently been associated with reduced heart rate and death by myocardial infarction168. Finally, all conjugated bile acids can activate signaling through the sphingosine-1-phosphate receptor 2 (S1PR2)169, and S1PR2 knockout mice have been shown to have reduced atherosclerosis burden in the apolipoprotein E deficient background170. Collectively, microorganism-derived bile acid metabolites represent diverse endocrine signals that hold tremendous therapeutic potential. In fact, in May 2016, a semi-synthetic bile acid analogue, obeticholic acid (trade name Ocaliva) was approved by the United States Food and Drug Administration for the treatment of liver disease171. This success story is likely one of many to come at the microbial metabolite-host receptor interface.
Conclusions
Although drug discovery has historically focused on targeting human enzymes, we are entering a new era of microbial pharmacology as biomedical research is aiming to target the microorganisms that live within us to improve human health). In particular, CVD is uniquely positioned for success with several gut microbial metabolites clearly associated with CVD risk. Successes in drug discovery at the microbial metabolite-host receptor interface have already been realized, and preclinical proof of concept is now established for non-lethal small-molecule inhibitors of TMAO production58. In parallel, there are substantial efforts to develop prebiotic, probiotic and fecal transplantation strategies to interrupt microorganism-host pathways that are involved in CVD pathogenesis50. As we forge ahead to determine microbial contributions to the origin and progression of human diseases, it is imperative that we move away from the traditional microbiome profiling approaches that simply ask who is there. Instead, we need to ask what disease-causing microbial products can be identified. Once we identify relevant microorganism-derived molecules, we must adopt classic principals of endocrinology to identify the host receptor systems involved in disease pathogenesis. The diversity of chemicals and metabolites derived from our gut microbial symbionts is quite astounding, and provides an essentially untapped source of new drug targets for cardiometabolic disease.
Acknowledgments
The authors are supported by grants from the National Heart Lung and Blood Institute (NHLBI), the Office of Dietary Supplements (ODS), and the National Institute on Alcohol Abuse and Alcoholism (NIAAA): R01HL122283 (J.M.B.), P50AA024333 (J.M.B.), R01HL103866 (S.L.H.), P01HL076491 (S.L.H.), R01HL126827 (S.L.H.) and R01DK106000 (S.L.H.) and the Cleveland Clinic Liver Tumor Center of Excellence.
Glossary
- pattern recognition receptor
host sensors which detect molecules typical for pathogens
- atherosclerosis
a disease process in with the inside of an artery narrows due to the build up of plaque
- thrombosis
the formation of a clot inside of a vessel
- hyperlipidemic
a condition where blood lipids are elevated
- ischemic stroke
a stroke that occurs when a blood vessel to the brain is blocked by a blood clot
- metabolome
the complete set of small molecule chemicals found within a biological sample
- transient ischemic attack
also called a “mini-stroke”a transient ischemic attack is a brief episode of neurological dysfunction cause by lack of blood flow to the brain
- glycemia
the level of glucose in one’s blood
- detergents
a surfactant or mix of surfactants that has cleaning or membrane disturbing properties
- taurine
a major sulfur-containing amino acid
- postprandial state
the state immediately following a meal
Footnotes
Author contributions
J.M.B. and S.L.H substantially contributed to the discussion of content, and review and editing of the manuscript before submission.
Competing interests statement: The authors declare competing interests.
Competing interests statement.
S.L.H. is named as inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics. He is also a paid consultant for Esperion and P&G, and has received research funds from Astra Zeneca, P&G, Pfizer Inc., Roche Diagnostics, and Takeda. S.L.H. has also received royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland HeartLab, Esperion and Siemens.
J. M. B declares no competing interests.
References
- 1.Mozaffarian D, et al. Executive summary: heart disease and stroke statistics - 2016 update: a report from the American Heart Assocation. Circulation. 2016;133:447–454. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 2.Ardissino D, et al. Influence of 9p21.3 genetic variants on clinical and angiographic outcomes in early-onset myocardial infarction. J. Am. Coll. Cardiol. 2011;58:426–34. doi: 10.1016/j.jacc.2010.11.075. [DOI] [PubMed] [Google Scholar]
- 3.Ripatti S, et al. A multilocus genetic risk score for coronary heart diease: case-control and prospective cohort analyses. Lancet. 2010;376:1393–400. doi: 10.1016/S0140-6736(10)61267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu E, et al. Diet, lifestyle, biomarkers, genetic factors, and risk of cardiovascular disease in the Nurses’ health study. Am. J. Public Health. 2016;106:1616–1623. doi: 10.2105/AJPH.2016.303316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JM, Hazen SL. The gut microbial endocrine organ: bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015;66:343–359. doi: 10.1146/annurev-med-060513-093205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J. Clin Invest. 2014;124:4204–4211. doi: 10.1172/JCI72331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregory JC, et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J. Biol. Chem. 2015;290:5647–5660. doi: 10.1074/jbc.M114.618249. This study demonstrates for the first time that atherosclerosis susceptibility can be transmitted by gut microbial transplantation, fulfilling a key Koch’s postulate for microbial contribution to disease causation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu W, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165:111–124. doi: 10.1016/j.cell.2016.02.011. This study shows the first cell autonomous effects of the gut microbe-derived metabolite TMAO on platelet activation and thrombosis potential in vivo, that microbial transplantation transmits TMAO levels and thrombosis potential in vivo, and that thrombotic event risk in subjects tracks with TMAO levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leung C, et al. The role of gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016;13:412–425. doi: 10.1038/nrgastro.2016.85. [DOI] [PubMed] [Google Scholar]
- 10.Munford RS. Endotoxemia-menace, marker, or mistake? J. Leukoc. Biol. 2016;100:687–698. doi: 10.1189/jlb.3RU0316-151R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kholy KE, Genco RJ, Van Dyke TE. Oral infections and cardiovascular disease. Trends Endocrinol. Metab. 2015;26:315–321. doi: 10.1016/j.tem.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Filardo S, et al. Chlamydia pneumoniae-mediated inflammation in atherosclerosis: A meta-analysis. Mediators Inflamm. 2015;2015:378658. doi: 10.1155/2015/378658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koren O, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA. 2011;108(Suppl. 1):4592–4598. doi: 10.1073/pnas.1011383107. This study provides evidence that human atherosclerotic plaques contain bacteria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitra S, et al. In silico analyses of metagenomes from human atherosclerotic plaque samples. Microbiome. 2015;3:38. doi: 10.1186/s40168-015-0100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caladrini CA, et al. Microbial composition of atherosclerotic plaques. Oral Dis. 2014;20:e128–e134. doi: 10.1111/odi.12205. [DOI] [PubMed] [Google Scholar]
- 16.Karlsson FH, et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012;3:1245. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saikku P, et al. Chronic Chlamydia pneumoniae infection as a risk factor for coronary heart disease in the Helsinki Heart Study. Ann. Intern. Med. 1992;15:273–278. doi: 10.7326/0003-4819-116-4-273. [DOI] [PubMed] [Google Scholar]
- 18.Kol A, et al. Chlamydia and human heat shock protein 60s activate human vascular endothelium, smooth muscle cells, and macrophages. J. Clin. Invest. 1999;103:571–577. doi: 10.1172/JCI5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen S, et al. Chlamydia pneumoniae-induced foam cell formation requires MyD88-dependent and -independent signaling and is reciprocally modulated by liver X receptor activation. J. Immunol. 2008;181:7186–7193. doi: 10.4049/jimmunol.181.10.7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grayston JT, et al. Azithromycin for the secondary prevention of coronary events. N. Engl. J. Med. 2005;352:1637–1645. doi: 10.1056/NEJMoa043526. [DOI] [PubMed] [Google Scholar]
- 21.Cannon CP, et al. Antibiotic treatment of Chlamydia pneumoniae after acute coronary syndrome. N. Engl. J. Med. 2005;352:1646–1654. doi: 10.1056/NEJMoa043528. [DOI] [PubMed] [Google Scholar]
- 22.O’Connor CM, et al. Azithromycin for the secondary prevention of coronary heart disease events - The Wizard Study: A randomized controlled trial. JAMA. 2003;290:1459–1466. doi: 10.1001/jama.290.11.1459. [DOI] [PubMed] [Google Scholar]
- 23.Caligiuri G, et al. Chlamydia pneumoniae infection does not induce or modify atherosclerosis in mice. Circulation. 2001;103:2834–2838. doi: 10.1161/01.cir.103.23.2834. [DOI] [PubMed] [Google Scholar]
- 24.Hussain M, Stover CM, Dupont AP. gingivalis in periodontal disease and atherosclerosis - scenes of action for antimicrobial peptides and complement. Front. Immunol. 2015;6:45. doi: 10.3389/fimmu.2015.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giacona MB, et al. Porphyromonas gingivalis induces its uptake by human macrophages and promotes foam cell formation in vitro. FEMS Microbiol. Lett. 2004;241:95–101. doi: 10.1016/j.femsle.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 26.Naito M, et al. Porphyromonas gingivalis-induced platelet aggregation in plasma depends on Hgp44 adhesion but not Rgp proteinase. Mol. Microbiol. 2006;59:152–167. doi: 10.1111/j.1365-2958.2005.04942.x. [DOI] [PubMed] [Google Scholar]
- 27.Li L, et al. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation. 2002;105:861–867. doi: 10.1161/hc0702.104178. [DOI] [PubMed] [Google Scholar]
- 28.Brodala N, et al. Porphyromonas gingivalis bacteremia induces coronary and aortic atherosclerosis in normocholesterolemic and hypercholesterolemic pigs. Arterioscler. Thromb. Vasc. Biol. 2005;25:1446–1451. doi: 10.1161/01.ATV.0000167525.69400.9c. [DOI] [PubMed] [Google Scholar]
- 29.Lalla E, et al. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2003;23:1405–1411. doi: 10.1161/01.ATV.0000082462.26258.FE. [DOI] [PubMed] [Google Scholar]
- 30.Mach F, et al. Influence of Helicobacter pylori infection during atherogenesis in vivo in mice. Circ. Res. 2002;90:E1–E4. doi: 10.1161/hh0102.102270. [DOI] [PubMed] [Google Scholar]
- 31.Chen XH, et al. Helicobacter pylori infection enhances atherosclerosis in high-cholesterol diet fed C57BL/6 mice. Zhonghua Xin Xue Guan Bing Za Shi. 2010;38:259–263. [PubMed] [Google Scholar]
- 32.Liuba P, et al. Co-infection with Clamydia pneumoniae and Helicobacter pylori results in vascular endothelial dysfunction and enhanced VCAM-1 expression in apoE-knockout mice. J. Vasc. Res. 2003;40:115–122. doi: 10.1159/000070708. [DOI] [PubMed] [Google Scholar]
- 33.El Moktari NE, Ott SJ, Nebel A, Simon R, Schreiber S. A functional variant in the CARD4 gene and risk of premature coronary heart disease. Int. J. Immunogenet. 2006;33:307–311. doi: 10.1111/j.1744-313X.2006.00618.x. [DOI] [PubMed] [Google Scholar]
- 34.Kanno S, et al. Activation of an innate immune receptor, Nod1, accelerates atherogenesis in Apoe−/− mice. J. Immunol. 2015;194:773–780. doi: 10.4049/jimmunol.1302841. [DOI] [PubMed] [Google Scholar]
- 35.Schenk M, Belisle JT, Modlin RL. TLR2 looks at lipoproteins. Immunity. 2009;31:847–849. doi: 10.1016/j.immuni.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 36.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sale ML, et al. Toll-like receptor 6 Ser249Pro polymorphism is associated with lower left ventricular wall thickness and inflammatory response in hypertensive women. Am. J. Hypertens. 2010;23:649–654. doi: 10.1038/ajh.2010.24. [DOI] [PubMed] [Google Scholar]
- 38.Hamann L, et al. Association of a common TLR-6 polymorphism with coronary artery disease - implications for healthy ageing? Immun. Ageing. 2013;10:43. doi: 10.1186/1742-4933-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lundberg AM, et al. Toll-like receptor 3 and 4 signaling through TRIF and TRAM adaptors in haematopoetic cells promotes atherosclerosis. Cardiovasc. Res. 2013;99:364–373. doi: 10.1093/cvr/cvt033. [DOI] [PubMed] [Google Scholar]
- 40.Ballistreri CR, et al. TLR4 polymorphisms and ageing: implications for the pathophysiology of age-related diseases. J. Clin. Immunol. 2009;29:406–415. doi: 10.1007/s10875-009-9297-5. [DOI] [PubMed] [Google Scholar]
- 41.Michelsen KS, et al. Lack of toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart CR, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding Y, et al. Toll-like receptor 4 deficiency decreases atherosclerosis but does not protect against inflammation in obese low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012;32:1596–1604. doi: 10.1161/ATVBAHA.112.249847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanellakis P, et al. High-mobility group box protein 1 neutralization reduces development of diet-induced atherosclerosis in apolipoprotein e-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011;31:313–319. doi: 10.1161/ATVBAHA.110.218669. [DOI] [PubMed] [Google Scholar]
- 45.Salagianni M, et al. Toll-like receptor 7 protects from atherosclerosis by constraining “inflammatory” macrophage activation. Circulation. 2012;126:952–962. doi: 10.1161/CIRCULATIONAHA.111.067678. [DOI] [PubMed] [Google Scholar]
- 46.Koulis C, et al. Protective role for Toll-like receptor-9 in the development of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2014;34:516–525. doi: 10.1161/ATVBAHA.113.302407. [DOI] [PubMed] [Google Scholar]
- 47.Lee WJ, Hase K. Gut microbiota-generated metabolites in animal health and disease. Nat. Chem. Biol. 2014;10:416–424. doi: 10.1038/nchembio.1535. [DOI] [PubMed] [Google Scholar]
- 48.Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016;16:341–352. doi: 10.1038/nri.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spanogiannopoulos P, et al. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 2016;14:273–287. doi: 10.1038/nrmicro.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown JM, Hazen SL. Targeting of microbe-derived metabolites to improve human health: The next frontier for drug discovery. J. Biol. Chem. 2017;292:8560–8568. doi: 10.1074/jbc.R116.765388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. This paper was the first to directly demonstrate a pathogenic role for a gut microbe-dependent metabolite, TMAO, in cardiovascular disease pathogenesis using both mouse models of disease, and large scale clinical studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koeth RA, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013;19:576–85. doi: 10.1038/nm.3145. This study demonstrates a nutrient (carnitine) gut microbial - host metaorganismal pathway (the TMAO pathway) providing a mechanistic link between red meat consumption and cardiovascular disease pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang WH, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013;368:1575–1578. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z, et al. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur. Heart J. 2014;35:904–910. doi: 10.1093/eurheartj/ehu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koeth RA, et al. gamma-butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 2014;20:799–812. doi: 10.1016/j.cmet.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Senthong V, et al. Plasma trimethylamine N-oxide, a gut microbe-generated phosphatidylcholine metabolite, is associated with atherosclerotic burden. J. Am. Coll. Cardiol. 2016;67:2620–2628. doi: 10.1016/j.jacc.2016.03.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Senthong V, et al. Intestinal microbiota-generated metabolite trimethylamine N-oxide and 5-year mortality risk in stable coronary artery disease: the contributory role of intestinal microbiota in a COURAGE-like patient cohort. J. Am. Heart Assoc. 2016;5:e002816. doi: 10.1161/JAHA.115.002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Z, et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell. 2015;163:1585–1595. doi: 10.1016/j.cell.2015.11.055. This paper is the first to show that a non-lethal small molecule inhibitor of a gut microbial enzyme (TMA lyases) can protect mice against diet enhanced atherosclerosis development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng Y, et al. Dietary phosphatidylcholine and risk of all-cause and cardiovascular-specific mortality among US women and men. Am. J. Clin. Nutr. 2016;104:173–180. doi: 10.3945/ajcn.116.131771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Randrianarisoa E, et al. Relationship of serum trimethylamine N-oxide (TMAO) levels with early atherosclerosis in humans. Sci. Rep. 2016;6:26745. doi: 10.1038/srep26745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim RB, et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 2016;89:1144–1152. doi: 10.1016/j.kint.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 62.Skagen K, et al. The carnitine-butyrobetaine-trimethylamine-oxide pathway and its association with cardiovascular mortality in patients with carotid atherosclerosis. Atherosclerosis. 2016;247:64–69. doi: 10.1016/j.atherosclerosis.2016.01.033. [DOI] [PubMed] [Google Scholar]
- 63.Missailidis C, et al. Serum trimethylamine-N-oxide is strongly related to renal function and predicts outcome in chronic kidney disease. PLoS One. 2016;11:e0141738. doi: 10.1371/journal.pone.0141738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mafune A, et al. Associations among serum trimethylamine-N-oxide (TMAO) levels, kidney function and infarcted coronary artery number in patients undergoing cardiovascular surgery: a cross-sectional study. Clin. Exp. Nephrol. 2105 doi: 10.1007/s10157-015-1207-y. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu W, Wang Z, Tang WH, Hazen SL. Gut microbe-generated TMAO from dietary choline is prothrombotic in subjects. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.116.025338. (In Press – embargo date 04/24/17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Troseid M, et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J. Intern. Med. 2015;277:717–726. doi: 10.1111/joim.12328. [DOI] [PubMed] [Google Scholar]
- 67.Organ CL, et al. Choline diet and its gut microbe-derive metabolite, Trimethylamine N-oxide, exacerbate pressure overload-induced heart failure. CircHeart Fail. 2016;9:e002314. doi: 10.1161/CIRCHEARTFAILURE.115.002314. This paper demonstrates a role for the gut microbial metabolite, TMAO, in pressure overload-induced heart failure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rhee EP, et al. A combined epidemiologic and metabolomics approach improves CKD prediction. J. Am. Soc. Nephrol. 2013;24:1330–1338. doi: 10.1681/ASN.2012101006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bell JD, et al. Nuclear magnetic resonance studies of blood plasma and urine from subjects with chronic renal failure: identification of trimethylamine-N-oxide. Biochim. Biophys. Acta. 1991;1096:101–107. doi: 10.1016/0925-4439(91)90046-c. [DOI] [PubMed] [Google Scholar]
- 70.Tang WH, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015;116:448–455. doi: 10.1161/CIRCRESAHA.116.305360. This paper demonstrates that the gut microbial metabolite TMAO can induce renal fibrosis, functional impairment, and contribute to adverse CVD outcomes in subjects with chronic kidney disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dambrova M, et al. Diabetes is associated with higher trimethylamine N-oxide plasma levels. Exp. Clin. Endocrinol. Diabetes. 2016;124:251–256. doi: 10.1055/s-0035-1569330. [DOI] [PubMed] [Google Scholar]
- 72.Lever M, et al. Betaine and trimethylamine-N-oxide as predictors of cardiovascular outcomes show different patterns in diabetes mellitus: an observational study. PLoS One. 2014;9:e114969. doi: 10.1371/journal.pone.0114969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tang WH, Wang Z, Li XS, Fan Y, Li DS, Wu Y, Hazen SL. Increased trimethylamine N-oxide portends high mortality risk independent of glycemic control in patients with type 2 diabetes mellitus. Clin. Chem. 2017;63:297–306. doi: 10.1373/clinchem.2016.263640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miao J, et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 2015;6:6498. doi: 10.1038/ncomms7498. Through unbiased metabolomics screening, this paper demonstrates for the first time a role for the TMAO-producing enzyme flaving-containing monooxygenase 3 in insulin action. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schugar RC, et al. The TMAO-producing enzyme flavin-containing monooxygenase 3 regulates obesity and the beiging of white adipose tissue. Cell Rep. 2017;19:2451–2461. doi: 10.1016/j.celrep.2017.05.077. This paper is the first to show that pharmacologic inhibition or genetic deletion of flavin-containing monooxygenase 3 protects mice from high fat diet-induced obesity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. USA. 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu Y, et al. Carnitine metabolism to trimethylamine by an usual Rieske-type oxygenase from human microbiota. Proc. Natl. Acad. Sci. USA. 2014;111:4268–4273. doi: 10.1073/pnas.1316569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yancey PH, et al. Living with water stress: evolution of osmolyte systems. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 79.Lin TY, Timasheff SN. Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine-N-oxide interactions with protein. Biochemistry. 1994;33:12695–12701. doi: 10.1021/bi00208a021. [DOI] [PubMed] [Google Scholar]
- 80.Seldin MM, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor kB. J. Am. Heart Assoc. 2016;5:e002767. doi: 10.1161/JAHA.115.002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Q, et al. Synchronous evolution of an odor biosynthesis pathway and behavioral response. Curr. Biol. 2013;23:11–20. doi: 10.1016/j.cub.2012.10.047. This paper demonstrates that the gut microbial metabolite trimethylamine (TMA) can activate a G protein-coupled receptor to regulate behavioral responses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wallrabenstein I, et al. Human trace amine-associated receptor TAAR5 can be activated by trimethylamine. PLoS One. 2013;8:e54950. doi: 10.1371/journal.pone.0054950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Falony G, Vleira-Silva S, Raes J. Microbiology meets big data: The case of gut microbe-derived trimethylamine. Annu. Rev. Microbiol. 2015;69:305–321. doi: 10.1146/annurev-micro-091014-104422. This innovative paper describes the power of mining reference genomes for prediction of bacterial enzymatic activity. [DOI] [PubMed] [Google Scholar]
- 84.Romano KA, Vivas EI, Amador-Noguez D, Rey FE. Instestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio. 2015;6:e02481. doi: 10.1128/mBio.02481-14. This paper identified human commensal bacteria that possess TMA lyase activity, and demonstrates that transplantation of these strain can alter host choline homeostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martinez-del Campo A, et al. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. MBio. 2015;6:e00042–15. doi: 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Levin BJ, et al. A prominent glycyl radical enzyme in human gut microbiomes metabolizes trans-4-hydroxy-l-proline. Science. 2017;355:eaai8386. doi: 10.1126/science.aai8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rath S, et al. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome. 2017;5:54. doi: 10.1186/s40168-017-0271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mejean V, et al. TMAO anaerobic respiration in Escherichia coli: involvement of the tor operon. Mol. Microbiol. 1994;11:1169–1179. doi: 10.1111/j.1365-2958.1994.tb00393.x. [DOI] [PubMed] [Google Scholar]
- 89.Lidbury I, Murrell JC, Chen Y. Trimethylamine N-oxide metabolism by abundant marine heterotrophic bacteria. Proc. Natl. Acad. Sci. USA. 2014;111:2710–2715. doi: 10.1073/pnas.1317834111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lidbury ID, Murrell JC, Chen Y. Trimethylamine and trimethylamine N-oxide are supplementary energy sources for marine carbon and nitrogen cycling. ISME J. 2015;9:760–769. doi: 10.1038/ismej.2014.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li CY, et al. Mechanistic insight into trimethylamine N-oxide recognition by the marine bacterium Ruegeria pomeroyi DSS-3. J. Bacteriol. 2015;197:3378–3387. doi: 10.1128/JB.00542-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brugere JF, et al. Archaebiotics: proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes. 2014;5:5–10. doi: 10.4161/gmic.26749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Collins HL, et al. L-carnitine intake and high trimethylamine N-oxide levels correlate with low aortic lesions in ApoE(−/−) transgenic mice expressing CETP. Atherosclerosis. 2016;244:29–37. doi: 10.1016/j.atherosclerosis.2015.10.108. [DOI] [PubMed] [Google Scholar]
- 94.Nagata C, et al. Choline and betaine intakes are not associated with cardiovascular disease mortality risk in Japanese men and women. J. Nutr. 2015;145:1787–1792. doi: 10.3945/jn.114.209296. [DOI] [PubMed] [Google Scholar]
- 95.Bidulescu A, et al. Usual choline and betaine dietary intake and incident coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) study. BMC Cardiovasc. Disord. 2007;7:20. doi: 10.1186/1471-2261-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dalmeijer GW, et al. Prospective study on dietary intakes of folate, betaine, and choline and cardiovascular disease risk in women. Eur. J. Clin. Nutr. 2008;62:386. doi: 10.1038/sj.ejcn.1602725. [DOI] [PubMed] [Google Scholar]
- 97.Heianza Y, et al. Gut microbiota metabolites and risk of major adverse cardiovascular events and death: a systematic review and meta-analysis of prospective studies. J. Am. Heart Assoc. 6:e004947. doi: 10.1161/JAHA.116.004947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mueller DM, et al. Plasma levels of trimethylamine-N-oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis. 2015;243:638–644. doi: 10.1016/j.atherosclerosis.2015.10.091. [DOI] [PubMed] [Google Scholar]
- 99.Yin J, et al. Dysbiosis of gut microbiota with reduced trimethylamine-N-oxide level in patients with large-artery atherosclerotic stroke or transient ischemic attack. J. Am. Heart Assoc. 4:e002699. doi: 10.1161/JAHA.115.002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165:1332–1345. doi: 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 101.Canfora EE, Jocken JW, Blaak EE. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015;11:577–591. doi: 10.1038/nrendo.2015.128. [DOI] [PubMed] [Google Scholar]
- 102.Ramezani A, et al. Role of the gut microbiome in uremia: a potential therapeutic target. Am. J. Kidney Dis. 2016;67:483–498. doi: 10.1053/j.ajkd.2015.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Miyamoto J, et al. The role of short-chain fatty acid on blood pressure regulation. Curr. Opin. Nephrol. Hypertens. 2016;25:379–383. doi: 10.1097/MNH.0000000000000246. [DOI] [PubMed] [Google Scholar]
- 104.Leung C, Rivera L, Furness JB, Angus PW. The role of gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016;13:412–425. doi: 10.1038/nrgastro.2016.85. [DOI] [PubMed] [Google Scholar]
- 105.Rey FE, et al. Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 2010;285:22082–22090. doi: 10.1074/jbc.M110.117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Scott KP, Martin JC, Campbell G, Mayer C-D, Flint HJ. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium “Roseburia inulinivorans. J. Bacteriol. 2006;188:4340–4349. doi: 10.1128/JB.00137-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ. Acetate utilization and butyryl coenzyme A (CoA):acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl. Environ. Microbiol. 2002;68:5186–5190. doi: 10.1128/AEM.68.10.5186-5190.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Louis P, Flint HJ. Formation of proprionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017;19:29–41. doi: 10.1111/1462-2920.13589. [DOI] [PubMed] [Google Scholar]
- 109.Brown AJ, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by proprionate and other short chain carboxylic acids. J. Biol. Chem. 2003;278:11312–11319. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- 110.Thangaraju M, et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009;69:2826–2832. doi: 10.1158/0008-5472.CAN-08-4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pluznick JL, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA. 2013;110:4410–4415. doi: 10.1073/pnas.1215927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wolever T, Spadafora P, Eshuis H. Interaction between colonic acetate and proprionate in humans. Am. J. Clin. Nutr. 1991;53:681–687. doi: 10.1093/ajcn/53.3.681. [DOI] [PubMed] [Google Scholar]
- 113.Laurent C, et al. Effect of acetate and proprionate on fasting hepatic glucose production in humans. Eur. J. Clin. Nutr. 1995;49:484–491. [PubMed] [Google Scholar]
- 114.Fernandes J, Vogt J, Wolever TM. Intravenous acetate elicits a greater free fatty acid rebound in normal than hyperinsulinemic humans. Eur. J. Clin. Nutr. 2012;66:1029–1034. doi: 10.1038/ejcn.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Venter CS, Vorster HH, Cummings JH. Effects of dietary proprionate on carbohydrate and lipid metabolism in healthy volunteers. Am. J. Gastroenterol. 1990;85:549–553. [PubMed] [Google Scholar]
- 116.Robertson MD, Bickerton AS, Dennis AL, Vidal H, Frayn KN. Insulin-sensitizing effects of dietary resistant starch and effects on skeletal muscle and adipose tissue metabolism. Am. J. Clin. Nutr. 2005;82:559–567. doi: 10.1093/ajcn.82.3.559. [DOI] [PubMed] [Google Scholar]
- 117.Cani PD, et al. Gut microbiota fermentation of prebiotics increases satieogenic and incretin gut peptide production with consequences for appetite sensation and glucose response after a meal. Am. J. Clin. Nutr. 2009;90:1236–1243. doi: 10.3945/ajcn.2009.28095. [DOI] [PubMed] [Google Scholar]
- 118.Parnell JA, Reimer RA. Weight loss during oligofructose supplementation is associated with decreased ghrelin and increased peptide YY in overweight and obese adults. Am. J. Clin. Nutr. 2009;89:1751–1759. doi: 10.3945/ajcn.2009.27465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dewulf E, et al. Insight into the prebiotic concept: lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut. 2013;62:1112–1121. doi: 10.1136/gutjnl-2012-303304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Daud NM, et al. The impact of oligofructose on stimulation of gut hormones, appetite regulation and adiposity. Obesity. 2014;22:1430–1438. doi: 10.1002/oby.20754. [DOI] [PubMed] [Google Scholar]
- 121.Vulvic J, Juric A, Tzortzis G, Gibson GR. A mixture of trans-galactooligosaccharides reduces markers of metabolic syndrome and modulates the fecal microbiota and immune function of overweight adults. J. Nutr. 2013;143:324–331. doi: 10.3945/jn.112.166132. [DOI] [PubMed] [Google Scholar]
- 122.Pedersen C, et al. Gut hormone release and appetite regulation in healthy non-obese participants following oligofructose intake. A dose escalation study. Appetite. 2013;66:44–53. doi: 10.1016/j.appet.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 123.Rejinders D, et al. Effects of gut microbiota manipulation by antibiotics on host metabolism in obese humans: a randomized double-blind placebo-controlled trial. Cell Metab. 2016;24:341. doi: 10.1016/j.cmet.2016.07.008. This study dissociates large alterations in short chain fatty acid levels to insulin sensitivity and energy metabolism in humans. [DOI] [PubMed] [Google Scholar]
- 124.Durgan DJ, et al. Role of the gut microbiome in obstructive sleep apnea-induced hypertension. Hypertension. 2016;67:469–474. doi: 10.1161/HYPERTENSIONAHA.115.06672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cai TQ, et al. Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biophys. Res. Commun. 2008;377:987–991. doi: 10.1016/j.bbrc.2008.10.088. [DOI] [PubMed] [Google Scholar]
- 126.Shen Z, et al. Inhibition of G protein-coupled receptor 81 (GPR81) protects against ischemic brain injury. CNS Neurosci. Ther. 2015;21:271–279. doi: 10.1111/cns.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.He W, et al. Citric acid cycle intermediates as ligands for ophan G-protein-coupled receptors. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 128.Rubic T, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008;9:1261–1269. doi: 10.1038/ni.1657. [DOI] [PubMed] [Google Scholar]
- 129.De Vadder F, et al. Microbiota-produced succinate improves glucose homeostasis via intestinal gluconeogenesis. Cell Metab. 2016;24:151–157. doi: 10.1016/j.cmet.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 130.Lukasova M, et al. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed in immune cells. J. Clin. Invest. 2011;121:1163. doi: 10.1172/JCI41651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Russell DW. Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 2009;(50 Suppl):S120–S125. doi: 10.1194/jlr.R800026-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hylemon PB, et al. Bile acids as regulatory molecules. J. Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kuipers F, Bloks VW, Groen AK. Beyond intestinal soap-bile acids in metabolic control. Nat. Rev. Endocrinol. 2014;10:488–498. doi: 10.1038/nrendo.2014.60. [DOI] [PubMed] [Google Scholar]
- 134.Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015;56:1085–1099. doi: 10.1194/jlr.R054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014;30:332–338. doi: 10.1097/MOG.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 137.Klaassen CD, Cui JY. Review: Mechanisms of how the intestinal microbiota alters the effects of drugs and bile acids. Drug Metab. Dispos. 2015;43:1505–1521. doi: 10.1124/dmd.115.065698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PE. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7:22–39. doi: 10.1080/19490976.2015.1127483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hofmann AF, Hagey LR, Krasowski MD. Bile salts of vertebrates: structural variation and possible evolutionary significance. J. Lipid Res. 2010;51:226–246. doi: 10.1194/jlr.R000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Joyce SA, Gahan CG. Disease-associated changes in bile acid profiles and links to altered gut microbiota. Dig. Dis. 2017;35:169–177. doi: 10.1159/000450907. [DOI] [PubMed] [Google Scholar]
- 141.Lin J, Sahin O, Michael LO, Zhang Q. Critical role of multidrug efflux pump CmeABC in bile resistance and in vivo colonization of Camphylobacter jejuni. Infect. Immunol. 2003;71:4250–4259. doi: 10.1128/IAI.71.8.4250-4259.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yokota A, Veenstra M, Kurdi P, van Veen HW, Konings WN. Cholate resistance in Lactococcus lactis is mediated by an ATP-dependent multispecific organic anion transporter. J. Bacteriol. 2000;182:5196–5201. doi: 10.1128/jb.182.18.5196-5201.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fernandez Murga ML, Bernick D, de Valdez GF, Disalvo AE. Permeability and stability properties of membranes formed by lipids extracted from Lactobacillus acidophilus grown at different temperatures. Arch. Biochem. Biophys. 1999;364:115–121. doi: 10.1006/abbi.1998.1093. [DOI] [PubMed] [Google Scholar]
- 144.Kimoto H, Ohmono S, Okamoto T. Enhancement of bile tolerance in Lactococci by Tween 80. J. Appl. Micro. 2002;92:41–46. doi: 10.1046/j.1365-2672.2002.01505.x. [DOI] [PubMed] [Google Scholar]
- 145.Liu Y, et al. Functional role of tlyC1 encoding a hemolysin-like protein from Bifidobacterium longum BBMN68 in bile tolerance. FEMS Microbiol. Lett. 2014;360:167–173. doi: 10.1111/1574-6968.12601. [DOI] [PubMed] [Google Scholar]
- 146.Ruiz L, et al. The cell-envelope proteome of Bifidobacterium longum in an in vivo bile environment. Microbiology. 2009;155:957–967. doi: 10.1099/mic.0.024273-0. [DOI] [PubMed] [Google Scholar]
- 147.Schubert R, Jaroni H, Schoelmerich J, Schmidt KH. Studies on the mechanism of bile salt-induced liposomal membrane damage. Digestion. 1983;28:181–190. doi: 10.1159/000198984. [DOI] [PubMed] [Google Scholar]
- 148.Prouty AM, Schwesinger WH, Gunn JS. Biofilm formation and interaction with surfaces of gallstones by Salmonella spp. Infect. Immunol. 2002;70:2640–2649. doi: 10.1128/IAI.70.5.2640-2649.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7:22–39. doi: 10.1080/19490976.2015.1127483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lepercq P, et al. Epimerization of chenodeoxycholic acid to ursodeoxycholic acid by Clostridium baratii isolated from human feces. FEMS Microbiol. Lett. 2004;235:65–72. doi: 10.1016/j.femsle.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 151.Lepercq P, et al. Isolates from normal human intestinal flora but not lactic acid bacteria exhibit 7alpha- and 7beta-hydroxysteroid dehydrogenase activities. Microb. Ecol. Health Dis. 2004;16:195–201. [Google Scholar]
- 152.Kelsey MI, Molina JE, Huang SK, Hwang KK. The identification of microbial metabolites of sulfolithocholic acid. J. Lipid Res. 1980;21:751–756. [PubMed] [Google Scholar]
- 153.Benson GM, et al. Polydeoxycholate in human and hamster feces: a major product of cholate metabolism. J. Lipid Res. 1993;34:2121–2134. [PubMed] [Google Scholar]
- 154.Tazuke Y, Matsuda K, Adachi K, Tsukada Y. Purification and properties of a novel sulfatase from Pseudomonas testosterone that hydrolyzed 3 beta-hydroxy-5-cholenoic acid 3-sulfate. Biosci. Biotech. Biochem. 1998;62:1739–1744. doi: 10.1271/bbb.62.1739. [DOI] [PubMed] [Google Scholar]
- 155.Makishima M, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. Landmark paper identifying the first ever nuclear receptor for bile acids. [DOI] [PubMed] [Google Scholar]
- 156.Heni M, et al. Genetic variation in NR1H4 encoding the bile acid receptor FXR determines fasting glucose and free fatty acid levels in humans. J. Clin. Endocrinol. Metab. 2013;98:E1224–E1229. doi: 10.1210/jc.2013-1177. [DOI] [PubMed] [Google Scholar]
- 157.Zhang Y, et al. FXR deficiency causes reduced atherosclerosis in Ldlr−/− mice. Arterioscler. Thromb. Vasc. Biol. 2006;26:2316–2321. doi: 10.1161/01.ATV.0000235697.35431.05. [DOI] [PubMed] [Google Scholar]
- 158.Wahlström A, Kovatcheva-Datchary P, Stahlman M, Bäckhed F, Marschall HU. Crosstalk between bile acids and gut microbiota and its impact on farnesoid X receptor signaling. Dig. Dis. 2017;35:246–250. doi: 10.1159/000450982. [DOI] [PubMed] [Google Scholar]
- 159.Wahlström A, Kovatcheva-Datchary P, Stahlman M, Khan MT, Bäckhed F, Marschall HU. Induction of farnesoid X receptor signaling in germ-free mice colonized with a human microbiota. J. Lipid Res. 2017;58:412–419. doi: 10.1194/jlr.M072819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Watanabe M, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. This paper was the first to describe the role for the cell surface bile acid receptor TGR5 in linking postprandial bile acid production to control of energy metabolism. [DOI] [PubMed] [Google Scholar]
- 161.Pols TW, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–757. doi: 10.1016/j.cmet.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Staudinger JL, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sui Y, Xu J, Rios-Pilier J, Zhou C. Deficiency of PXR decreases atherosclerosis in apoE-deficient mice. J. Lipid Res. 2011;52:1652–1659. doi: 10.1194/jlr.M017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Makishima M, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 165.Lu S, et al. The associations between the polymorphisms of vitamin D receptor and coronary artery disease: a systematic review and meta-analysis. Medicine (Baltimore) 2016;95:e3467. doi: 10.1097/MD.0000000000003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Szeto FL, et al. Vitamin D receptor signaling inhibits atherosclerosis in mice. Mol. Endocrinol. 2012;26:1091–1101. doi: 10.1210/me.2011-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Raufman JP, Cheng K, Zimniak P. Activation of muscarinic receptor signaling by bile acids: physiological and medical implications. Dig. Dis. Sci. 2003;48:1431–1444. doi: 10.1023/a:1024733500950. [DOI] [PubMed] [Google Scholar]
- 168.Hautala AJ, et al. Acetylcholine receptor M2 gene variants, heart rate recovery, and risk of cardiac death after an acute myocardial infarction. Ann. Med. 2009;41:197–207. doi: 10.1080/07853890802477866. [DOI] [PubMed] [Google Scholar]
- 169.Studer E, et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology. 2012;55:267–276. doi: 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Skoura A, et al. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011;31:81–85. doi: 10.1161/ATVBAHA.110.213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.De Magalhaes Filho CD, Downes M, Evans R. Bile acid analog intercepts liver fibrosis. Cell. 2016;166:789. doi: 10.1016/j.cell.2016.08.001. [DOI] [PubMed] [Google Scholar]