

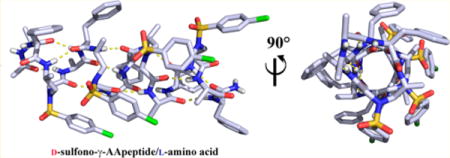
Abstract
New types of foldamer scaffolds are formidably challenging to design and synthesize, yet highly desirable as structural mimics of peptides/proteins with a wide repertoire of functions. In particular, the development of peptidomimetic helical foldamers holds promise for new biomaterials, catalysts, and drug molecules. Unnatural L-sulfono-γ-AApeptides were recently developed and shown to have potential applications in both biomedical and material sciences. However, D-sulfono-γ-AApeptides, the enantiomers of L-sulfono-γ-AApeptides, have never been studied due to the lack of high-resolution three-dimensional structures to guide structure-based design. Herein, we report the first synthesis and X-ray crystal structures of a series of 2:1 L-amino acid/D-sulfono-γ-AApeptide hybrid foldamers, and elucidate their folded conformation at the atomic level. Single-crystal X-ray crystallography indicates that this class of oligomers folds into well-defined right-handed helices with unique helical parameters. The helical structures were consistent with data obtained from solution 2D NMR, CD studies, and molecular dynamics simulations. Our findings are expected to inspire the structure-based design of this type of unique folding biopolymers for biomaterials and biomedical applications.
Graphical abstract
INTRODUCTION
Synthetic unnatural oligomers, so-called foldamers,1 are capable of mimicking the three-dimensional structure and function of natural biopolymers, and have advantages of enhanced resistance to proteolytic degradation and sequence diversity, as well as promise in medical and material application. Moreover, folding into conformationally stable structures, they could interact with various targets such as proteins,2 membranes,3 and so on. Up to now, oligomeric architectures including β-peptides,4 peptoids,5 β-peptoids,6 oligoureas,7 aza-peptides,8 α-aminoisobutyric acid foldamers,9 oligoproline,10 aromatic amide foldamers,11 and others have been well documented on the basis of either homogeneous or heterogeneous backbones, leading to various applications in molecular self-assembly and recognition.12 Certain peptides consisting of both D-amino acids and L-amino acids were studied for their helicity;13 however, it is very challenging to predict the folding propensity of the mixed peptides due to the intrinsic opposite folding propensity of D- and L-amino acids.4b,14 For instance, Gramicidin A containing alternating L- and D-amino acid is capable of forming a right-handed helix,13d,15 while another oligo-L-Val-D-Val peptide was found to form a left-handed helix.13c It is therefore attractive to explore new helical foldameric scaffolds of this type to better understand the helical propensity of unnatural oligomers bearing D-form synthetic residues, so as to design new molecular entities that could modulate specific biological process, in the long-term journey of biopolymer-mimicking foldamer research.
Recently, we have reported the development of a class of peptidomimetics-AApeptides (N-acetylated-N-aminoethyl amino acids-based oligomers), which are stemming from the chiral PNA backbone16 and taking advantage of chemical diversity with arbitrary side chains as well as resistance to proteolytic degradation (Figure 1a).17 AApeptides have started to show promise in biomedical and material applications. Hitherto, all of the AApeptides were derived from natural L-amino acids, and we thus ended up with L-AApeptides. The folding propensity of oligomers bearing a L-sulfono-γ-AApeptide backbone has been investigated in solution by 2D-NMR studies,18 which suggests homogeneous L-sulfono-γ-AApeptides and 1:1 α/L-sulfono-γ-AA hybrid peptides adopt helical conformations. However, it remains yet unknown the impact of the corresponding D-enantiomers of L-γ-AApeptide on the formation of helical secondary structures.
Figure 1.
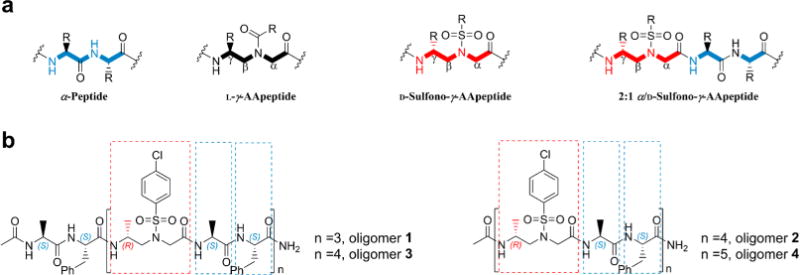
(a) General structures of α-peptides, L-γ-AApeptides, D-sulfono-γ-AApeptides, and 2:1 α/D-sulfono-γ-AA peptides. (b) 2:1 α/D-sulfono-γ-AA peptidic oligomers prepared for structural and spectroscopic evaluation in this study.
Here, we describe the first structures of heterogeneous oligomers consisting of a 2:1 pattern of L-α/D-sulfono-γ-AA amino acids. High-resolution X-ray crystal structures of these 2:1 α/D-AApeptide hybrid foldamers ambiguously delineate their sequence–structure relationships, revealing the well-folded, three-dimensional right-handed helical structures of the entire set of oligomers. These results provide a basis for designing de novo foldameric structures of this type as ordered biopolymers and potential therapeutic agents in the future.
RESULTS AND DISCUSSION
Sequence Design
We incorporated D-sulfono-γ-AA building blocks into the hybrid peptidic oligomers and designed the peptidomimetic sequences with alternative L-amino acid/D-sulfono-γ-AA residues in the 2:1 pattern. To simplify the sequences and exclude the potential impact of side chains on the folding propensity, we initially chose to design oligomers of variable lengths that have the same repeating units, L-Phe, L-Ala, and 4-chlorobenzenesulfonyl-containing D-sulfono-γ-AA residues (Figure 1b). Altogether four sequences were obtained initially, each of which being acetylated on the N-terminus.
Solid-phase Fmoc chemistry was employed to synthesize all heterogeneous peptides according to our previous protocol.18b Gratifyingly, the reactions took place very efficiently without acetic anhydride capping after every coupling cycle, and ultimately the desired L-amino acid/D-sulfono-γ-AApeptide hybrids were obtained with decent yield after HPLC purification.
High-Resolution Crystallographic Studies of Oligomers 1, 3, and 4
Oligomers 1, 3, and 4 were obtained as crystalline solid from a similar solvent system by slow evaporation, and their structures were successfully solved by single-crystal X-ray crystallography with resolutions of 0.83, 0.85, and 1.12 Å, respectively (Figure 2). The longest sequence oligomer 4 formed needle-shaped crystals first from acetonitrile (ACN) in the presence of 0.5% trifluoroacetic acid (TFA), consistent with our expectation that the longer oligomer is more prone to form a stable helical structure. The crystals of 1 and 3 were obtained subsequently from ACN/TFA and ACN/THF/H2O, respectively, and their structures were also obtained by single-crystal X-ray crystallography. Gratifyingly, oligomer 1, which is comparable to a 14-mer peptide in length, could adopt a well-defined helical structure as well. The persistent and unified intramolecular H-bond network and organized packing of side chain unambiguously indicated that this class of oligomers provides a particularly strong stabilization of this novel secondary structure motif.
Figure 2.
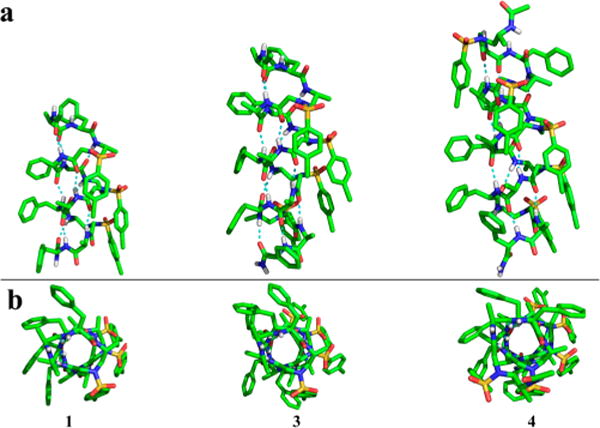
Single-crystal structures. (a) Side views of crystals 1, 3, and 4. Hydrogen bonding is shown in cyan. (b) Top views of crystals 1, 3, and 4 along helix axis.
All of the crystals reveal a right-handed helical structure with virtually unanimous helical radius of 2.6 Å and helical pitch of 5.1 Å, with 4.5 residues per turn (Figure 3a). This is somehow surprising, as D-peptides are known to adopt a left-handed helical conformation. In addition, all crystals show the same 16–16–14-hydrogen-bonding pattern (Figure 3), formed between the N–H group of the α-amino acid residue and the C=O group of the α-amino acid four residues earlier (type A 16-hydrogen bonding), or between the N–H group of the α-amino acid residue and the C=O group of the D-sulfono-γ-AA four residues earlier (type B 16-hydrogen bonding), or between the N–H group of the D-sulfono-γ-AA residue and the C=O group of the D-sulfono-γ-AA four residues later (14-hydrogen bonding), i + 4 → i hydrogen bonding with a distance of 2.1 Å (H⋯O distance). Hence, the name 4.516–14 helix is designated, indicating the number of residues per helical turn, and 16 or 14 atoms being involved in the ring formed by the intramolecular hydrogen bonds. This 4.516–14 helix is less tightly packed as compared to the 310 helix and α-helix, but slightly more tightly packed than the π-helix. A comparison of this helix to other types of natural helical peptides (Table 1) reveals a close resemblance to the π-helix, where the same number of residues is included in each hydrogen-bond loop together with similar helical pitch and diameter. As the π-helix has recently been found to play an important role in biological functions,19 our results may provide a novel approach of π-helix mimicry in the future. Additionally, side chains of this class of helix are projecting away from the helical axis. However, it is interesting that the side chains of the α-amino acid residues point toward the N-terminus and splay out slightly, while the side chains on the D-sulfono-γ-AA residues point toward the C-terminus due to the (R)-configuration of the chiral side chains in the sulfono-γ-AA residues in the scaffold.
Figure 3.
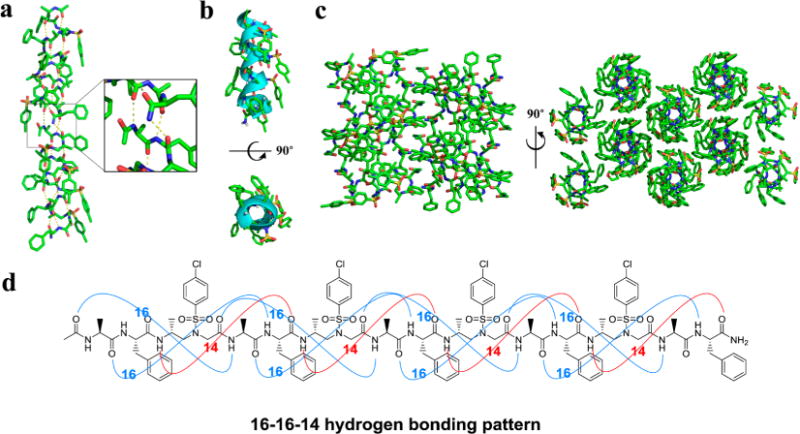
Helical scaffold of oligomer 3. (a) Structure of crystals 3 packing along peptide axis; intermolecular hydrogen-bonding pattern was shown in inset for clarity. (b) Cartoon representation of 3 shown in oval to further clarify the helix. (c) Crystal packing of oligomer 1 viewed perpendicular and then down to the helical axis. (d) 16–16–14-hydrogen-bonding pattern detected in the crystal structure of 3.
Table 1.
Parameters of Helical Structures Found in Proteins and 4.516–14 Helix
| secondary structure | handedness | helical pitch p (Å) | radius of helix r (Å) |
|---|---|---|---|
| α-helix | right-handed | 5.4 | 2.3 |
| 310 helix | right-handed | 6.0 | 1.9 |
| π-helix | right-handed | 5.0 | 2.8 |
| 4.516–14 helix | right-handed | 5.1 | 2.6 |
In crystal packing, the individual helical segments are arranged in a hydrogen-bonding-driven head-to-tail manner to give regularly elongated helical treads (Figure 3a). Helical cartoon representation clearly presents the robust 4.516–14 helix structures (Figure 3b). Altogether combined with highly ordered, tight packing of the helical and side chains (Figure 3c) suggests that the 4.516–14 helix could provide an unprecedented opportunity to construct new helical structures considering the unique residue parameters and orientations.
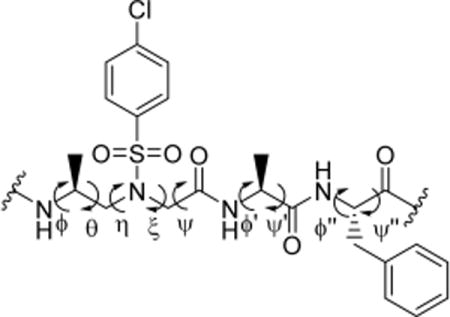
Moreover, the average backbone torsion angles ϕ, θ, η, ξ, ψ, ϕ′, ψ′, ϕ″, and ψ″ are quite unanimous in all of the helical structures (Table 2). The torsion angles of α-Ala units in these oligomers with −62 ± 3° and −39 ± 7° for ϕ′ and ψ′, respectively, are similar to those of canonical α-helices (−64 ± 7°, −41 ± 7°); however, the backbone torsion angles ϕ″ and ψ″ of α-Phe units here are −108 ± 4° and 125 ± 9°, which are somewhat close to those of β-strands (ϕ, ψ″ −130 ± 10°, 125 ± 10°). The torsion angles of D-sulfono-γ-AA residues reasonably differ from those of α-helices, β-sheets, and the previously reported natural or synthetic peptides.5b,6,14e,20 These unambiguous torsion angle data, along with the clear arrangement of side chain and hydrogen pattern, could shed light on the creation of either finite helical bundles in materials or the rational design of helical structure targeting membrane receptors or protein–protein interactions in the future.
Table 2.
Typical Torsion Angles in Helical Structures 1, 3, and 4 Based on Single Crystals
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| angle | ϕ | θ | η | ξ | ψ | ϕ′ | ψ′ | ϕ″ | ψ″ |
| 1 | 122.8° | −80.7° | 78.2° | 60.7° | −167.8° | −62.2° | −36.0° | −111.5° | 127.5° |
| 3 | 107.9° | −74.6° | 79.5° | 63.6° | −168.6° | −58.9° | −46.2° | −105.0° | 116.3° |
| 4 | 121.1° | −77.1° | 80.4° | 56.2° | −170.7° | −64.9° | −33.8° | −108.4° | 131.4° |
NMR Studies of Oligomer 5
To further investigate the solution conformation of this type of foldamers, we synthesized sequence 5 with equal length of 3 but with different side groups to facilitate good proton resonance dispersion in 2D NMR spectra, and to improve the solubility in solution, as well as to represent the generality of the sequence of this class. A series of 2D NMR were conducted at a concentration of 5 mM in CD3OH at 10 °C. gDQFCOSY and zTOCSY spectra were recorded to assign the NMR peaks with the combination of NOESY, among which the assignments were achieved straightforwardly because there are regular α amino acid residues over the sequence.
Three types of long-range of NOEs between protons on the scaffold were detected (Figure 4), including (a) i, i+1 NOEs, correlations from methylene/side CH3/amide protons of both α-amino acid and D-sulfono-γ-AApeptide, and amide protons on adjacent residue; (b) i, i+2 NOEs, correlations between amide protons of α-amino acid and amide protons/side chain protons two residues later of the α-amino acid, or correlations between methenyl protons of D-sulfono-γ-AApeptide and the amide protons of α-amino acid two residues earlier or later; and (c) chimeric i, i+3 NOEs, correlations between α protons of the D-sulfono-γ-AApeptide and amide protons of the α-amino acid three residues later, or correlations between side chain protons of the α-amino acid and amide protons of the α-amino acid three residues later, with two α-amino acid and one D-sulfono-γ-AApeptide unit in between. All together, these NOEs are consistent with the crystal structures and suggest that this L-amino acid/D-sulfono-γ-AApeptide foldamer is helical in methanol.
Figure 4.
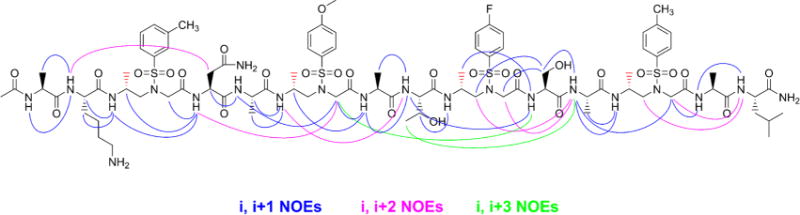
Summary of detected NOESY crosspeaks of 5 mM oligomer 5 between protons on nonadjacent residues in CD3OH. Three types of NOEs are displayed in different colors. Each D-sulfono-γ-AApeptide unit is considered as one residue.
CD Studies
Circular dichroism (CD) spectroscopy of sequences was conducted to evaluate the helical propensity in solution. All four oligomers displayed similar CD signatures in trifluoroethanol (80 μM). Our compounds revealed a pronounced maximum at 198–202 nm and a minimum at 212 nm (Figure 5a). A similar pattern was found in the α-helix, but a blue shift takes place with the incorporation of D-sulfono-γ-AA residues. Although the maximum at 200 nm for oligomer 1 is smaller than others, the maximums at 200 nm for oligomers 2, 3, and 4 do not differ significantly, suggesting they have comparable folding propensity. Similarly, the minimum at 212 nm with Cotton effect does not change dramatically with increasing length of the oligomers, further indicating that the helicity is stable regardless of the oligomer length.
Figure 5.
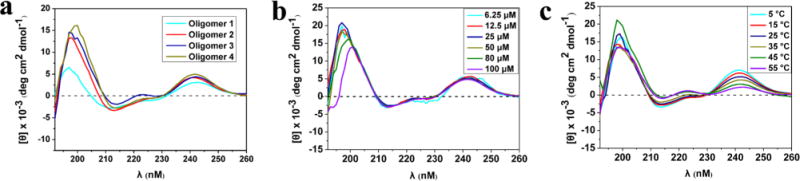
(a) CD spectra of oligomers 1–4 (80 μM) measured at room temperature in TFE. (b) CD spectra of oligomer 4 in TFE at various concentration (6.25–100 μM) at room temperature. (c) CD spectra of oligomer 4 (80 μM) in TFE at various temperatures.
CD studies of oligomer 4 at variable concentrations as well as the solvent effect on helical stability were also carried out. It is notable that the oligomer adopts a helical conformation well even at 6.25 μM, showing helical propensity almost consistently from 6.25 to 100 μM (Figure 5b). The helical stability of sequence was also evaluated by temperature-dependent CD studies (Figure 5c). There is no change in CD shape of oligomer 4 over the temperature range 5–55 °C, and only a slight decrease of signal intensity at 212 nm, indicating high thermal stability of helical sequence of this type in solution.
It should also be noted that the sequence retains a good degree of helicity in the presence of water, although the population is somewhat less than that in trifluoroethanol. Nevertheless, the peak at 240 nm changed dramatically in the presence of methanol or acetonitrile, and we hypothesized that the curve at 240 nm is not indicative of secondary structure, which instead could be attributed to the presence of aromatic residue in the structure. To test our hypothesis, oligomer 6 that does not bear any aromatic residue was synthesized and investigated by CD study. There is no signal at 240 nm but a supremely dominant blue shift takes place, which is similar to the CD curve of oligomer 5 in which the percentage of aromatic groups is also low (Figure 6b and Figure S7). Gratifyingly, oligomer 6 was able to crystallize to reveal the same helical folding pattern as others (Figure 6c). This is an exciting result considering that sometimes a side chain could play a crucial role in the crystallization. The results here suggest that the folding propensity of this type of L-amino acid/D-sulfono-γ-AApeptide hybrid foldamer is not limited by side-chain diversity in crystalline state. In addition, our results indicate that CD results of this class of foldamer need to be cautiously interpreted in the future depending on the nature of their side chains.
Figure 6.
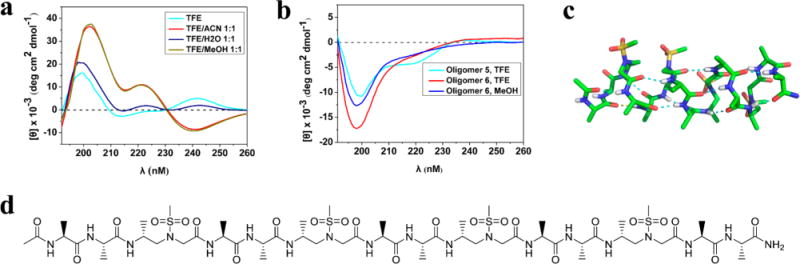
(a) CD spectra of oligomer 4 (80 μM) in various solvents at room temperature. (b) CD spectra of oligomers 5 and 6 (80 μM) in TFE or MeOH. (c) Crystal structure of oligomer 6. (d) Sequence structure of oligomer 6.
Molecular Dynamics Simulations
The stability of the helical structure of oligomer 3 in solution was probed by molecular dynamics simulations at various temperatures. The most common structures were identified by a clustering analysis based on the heavy atom root-mean-square deviation (RMSD) with the crystal structure. The clusters and the RMSD as a function of simulation time are shown in Figure 7a; the occurrence of these clusters during the simulations is indicated by the colors of the RMSD curves. A total of seven clusters were identified. The simulations at 250 and 300 K were dominated by clusters 1–4, which were folded, helical, and similar to the crystal structure. An overlay of these clusters with the crystal structure (Figure 7b) shows that clusters 1–4 contain a structurally highly preserved helical core, consisting of hydrogen bonds 3–9 (defined in Figure 8a). This helical core was highly stable in all 250 and 300 K simulations. The two termini (defined by hydrogen bonds 1, 2, 10, 11, and 12, Figure 8a) were more flexible. This flexibility frequently led to a loss of contacts between the termini and the helical core, as seen in clusters 2–4. Hydrogen-bond stability for the 300 K simulations is shown in Figure 8b. Hydrogen bonds in the helical core were present for more than 80% of the time, while hydrogen bonds of the termini formed less than 40% of the time. At simulated temperatures of 350 and 400 K, the central hydrogen bonds broke, and the helices unfolded, resulting in RMSDs > 6 Å (Figure 7a). Clusters 5–7, unfolded and nonhelical structures, were prevalent in these high temperature simulations. Movies of the 300 K simulations are presented in the Supporting Information.
Figure 7.
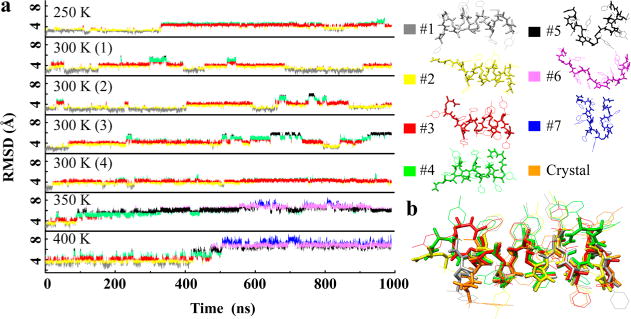
Molecular dynamics simulations of oligomer 3 in solution. (a) RMSD of simulation versus time, with the color of the RMSD curve representing the configurational clusters. (b) Overlay of folded clusters 1–4 with the crystal structure. Side-chain hydrogens are hidden for clarity.
Figure 8.
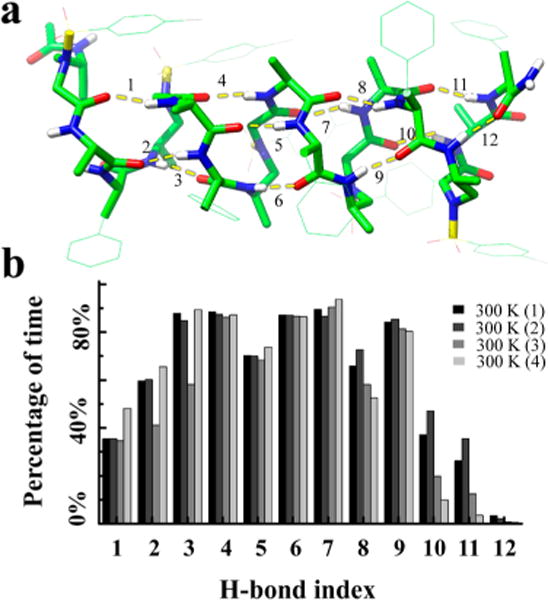
Backbone H-bond stability of oligomer 3 in the four 300 K simulations. (a) Backbone H-bond index and their location on the backbone. Side-chain hydrogens are hidden for clarity. (b) Percentage of time the H-bonds were formed in the 1 ms simulations.
CONCLUSIONS
We reported the first examples of a high-resolution structure of de novo D-sulfono-γ-AA/L-amino acid hybrid foldamers, an unprecedented secondary structure based on the AApeptides scaffold. It is particularly intriguing that D-sulfono-γ-AApeptides have never been made previously and studied for their folding propensity, and thus our results may inspire further exploration of this class of peptidomimetics. A series of crystals adopt well-defined right-handed helical conformations with the pattern of 4.516–14 helix in the solid state, which were successfully fulfilled by synthesis of heterogeneous oligomers containing incrementally increasing lengths of readily accessible D-sulfono-γ-AA units. Both X-ray crystal structures and NMR solution studies revealed a highly conserved secondary structure with close resemblance to π-helix. The foldameric structure was further supported by CD spectroscopic data in various solvents. Moreover, long molecular dynamic simulations conduced in acetonitrile at room temperature indicated a highly stable central helical core, with limited motion of the two termini. The unique foldamer reported here could shed light on the design of new folding biopolymers and materials of this type in the future. Investigations of L-amino acid/L-sulfono-γ-AApeptide hybrids are also underway in our lab to clarify the secondary structure of this type of counterpart foldamers.
Supplementary Material
Acknowledgments
We thank M. Kumar for support with HRMS sample analysis and the USF Interdisciplinary NMR Facility for NMR sample analysis. This work was generously supported by NSF CAREER 1351265 and NIH 1R01GM112652-01A1. Computer time was provided by USF Research Computing, sponsored in part by NSF MRI CHE-1531590. The X-ray diffraction data for 6 were measured using synchrotron radiation at ChemMat-CARS Sector 15/APS/Argonne National Laboratory. Chem-MatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1346572. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under contract no. DE-AC02-06CH11357.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b03007.
Experimental procedures and characterization of all sequences; detailed 2D NMR, protons assignment, and summary of NOEs; crystal data and structure refinement of four samples in table; and computational modeling methods (PDF)
X-ray crystallographic data for oligomer 1 (CIF)
X-ray crystallographic data for oligomer 3 (CIF)
X-ray crystallographic data for oligomer 4 (CIF)
X-ray crystallographic data for oligomer 6 (CIF)
Movies of the 300 K molecular dynamics simulation of oligomer 3 in acetonitrile (MPG)
ORCID
Arjan van der Vaart: 0000-0002-8950-1850
Hai Xu: 0000-0003-1610-9556
Jianfeng Cai: 0000-0003-3106-3306
Notes
The authors declare no competing financial interest.
References
- 1.(a) Horne WS, Gellman SH. Acc Chem Res. 2008;41:1399–1408. doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Goodman CM, Choi S, Shandler S, DeGrado WF. Nat Chem Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Gademann K, Ernst M, Hoyer D, Seebach D. Angew Chem, Int Ed. 1999;38:1223–1226. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1223::AID-ANIE1223>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]; (b) Sadowsky JD, Schmitt MA, Lee HS, Umezawa N, Wang S, Tomita Y, Gellman SH. J Am Chem Soc. 2005;127:11966–11968. doi: 10.1021/ja053678t. [DOI] [PubMed] [Google Scholar]; (c) Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. J Am Chem Soc. 2004;126:9468–9469. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]
- 3.(a) Werder M, Hauser H, Abele S, Seebach D. Helv Chim Acta. 1999;82:1774–1783. [Google Scholar]; (b) Hamuro Y, Schneider JP, DeGrado WF. J Am Chem Soc. 1999;121:12200–12201. [Google Scholar]; (c) Porter EA, Wang X, Lee HS, Weisblum B, Gellman SH. Nature. 2000;404:565. doi: 10.1038/35007145. [DOI] [PubMed] [Google Scholar]; (d) Seurynck SL, Patch JA, Barron AE. Chem Biol. 2005;12:77–88. doi: 10.1016/j.chembiol.2004.10.014. [DOI] [PubMed] [Google Scholar]; (e) Patch JA, Barron AE. J Am Chem Soc. 2003;125:12092–12093. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]; (f) Choi S, Clements DJ, Pophristic V, Ivanov I, Vemparala S, Bennett JS, Klein ML, Winkler JD, DeGrado WF. Angew Chem. 2005;117:6843–6847. doi: 10.1002/anie.200501279. [DOI] [PubMed] [Google Scholar]; (g) De Poli M, Zawodny W, Quinonero O, Lorch M, Webb SJ, Clayden J. Science. 2016;352:575–580. doi: 10.1126/science.aad8352. [DOI] [PubMed] [Google Scholar]
- 4.(a) Cheng RP, Gellman SH, DeGrado WF. Chem Rev. 2001;101:3219–3232. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]; (b) Berlicki Ł, Pilsl L, Wébber E, Mándity IM, Cabrele C, Martinek TA, Fülöp F, Reiser O. Angew Chem, Int Ed. 2012;51:2208–2212. doi: 10.1002/anie.201107702. [DOI] [PubMed] [Google Scholar]
- 5.(a) Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK. Proc Natl Acad Sci U S A. 1992;89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stringer JR, Crapster JA, Guzei IA, Blackwell HE. J Am Chem Soc. 2011;133:15559–15567. doi: 10.1021/ja204755p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laursen JS, Harris P, Fristrup P, Olsen CA. Nat Commun. 2015;6:7013. doi: 10.1038/ncomms8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischer L, Claudon P, Pendem N, Miclet E, Didierjean C, Ennifar E, Guichard G. Angew Chem, Int Ed. 2010;49:1067–1070. doi: 10.1002/anie.200905592. [DOI] [PubMed] [Google Scholar]
- 8.(a) Malachowski WP, Tie C, Wang K, Broadrup RL. J Org Chem. 2002;67:8962–8969. doi: 10.1021/jo026280d. [DOI] [PubMed] [Google Scholar]; (b) Proulx C, Sabatino D, Hopewell R, Spiegel J, García Ramos Y, Lubell WD. Future Med Chem. 2011;3:1139–1164. doi: 10.4155/fmc.11.74. [DOI] [PubMed] [Google Scholar]
- 9.Jones JE, Diemer V, Adam C, Raftery J, Ruscoe RE, Sengel JT, Wallace MI, Bader A, Cockroft SL, Clayden J, Webb SJ. J Am Chem Soc. 2016;138:688–695. doi: 10.1021/jacs.5b12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilhelm P, Lewandowski B, Trapp N, Wennemers H. J Am Chem Soc. 2014;136:15829–15832. doi: 10.1021/ja507405j. [DOI] [PubMed] [Google Scholar]
- 11.(a) Garric J, Leger JM, Huc I. Angew Chem, Int Ed. 2005;44:1954–1958. doi: 10.1002/anie.200462898. [DOI] [PubMed] [Google Scholar]; (b) Zhang DW, Zhao X, Hou JL, Li ZT. Chem Rev. 2012;112:5271–5316. doi: 10.1021/cr300116k. [DOI] [PubMed] [Google Scholar]
- 12.De Santis E, Ryadnov MG. Chem Soc Rev. 2015;44:8288–8300. doi: 10.1039/c5cs00470e. [DOI] [PubMed] [Google Scholar]
- 13.(a) Krause E, Bienert M, Schmieder P, Wenschuh H. J Am Chem Soc. 2000;122:4865–4870. [Google Scholar]; (b) Fairman R, Anthony-Cahill SJ, DeGrado WF. J Am Chem Soc. 1992;114:5458–5459. [Google Scholar]; (c) Benedetti E, Di Blasio B, Pedone C, Lorenzi GP, Tomasic L, Gramlich V. Nature. 1979;282:630. doi: 10.1038/282630a0. [DOI] [PubMed] [Google Scholar]; (d) Lomize AL, Orekhov V, Arsen’ev AS. Bioorg Khim. 1992;18:182–200. [PubMed] [Google Scholar]
- 14.(a) Seebach D, Gademann K, Schreiber JV, Matthews JL, Hintermann T, Jaun B, Oberer L, Hommel U, Widmer H. Helv Chim Acta. 1997;80:2033–2038. [Google Scholar]; (b) Sharma GVM, Reddy KR, Krishna PR, Sankar AR, Narsimulu K, Kumar SK, Jayaprakash P, Jagannadh B, Kunwar AC. J Am Chem Soc. 2003;125:13670–13671. doi: 10.1021/ja035752i. [DOI] [PubMed] [Google Scholar]; (c) Mándity IM, Wéber E, Martinek TA, Olajos G, Tóth GK, Vass E, Fülöp F. Angew Chem, Int Ed. 2009;48:2171–2175. doi: 10.1002/anie.200805095. [DOI] [PubMed] [Google Scholar]; (d) Martinek TA, Fulop F. Chem Soc Rev. 2012;41:687–702. doi: 10.1039/c1cs15097a. [DOI] [PubMed] [Google Scholar]; (e) Baldauf C, Günther R, Hofmann HJ. Angew Chem, Int Ed. 2004;43:1594–1597. doi: 10.1002/anie.200353249. [DOI] [PubMed] [Google Scholar]
- 15.Sham SS, Shobana S, Townsley LE, Jordan JB, Fernandez JQ, Andersen OS, Greathouse DV, Hinton JF. Biochemistry. 2003;42:1401–1409. doi: 10.1021/bi0204286. [DOI] [PubMed] [Google Scholar]
- 16.(a) Nielsen P, Egholm M, Berg R, Buchardt O. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]; (b) Wittung P, Nielsen PE, Buchardt O, Egholm M, Norden B. Nature. 1994;368:561–563. doi: 10.1038/368561a0. [DOI] [PubMed] [Google Scholar]
- 17.(a) Hu Y, Li X, Sebti SM, Chen J, Cai J. Bioorg Med Chem Lett. 2011;21:1469–1471. doi: 10.1016/j.bmcl.2011.01.005. [DOI] [PubMed] [Google Scholar]; (b) Shi Y, Teng P, Sang P, She F, Wei L, Cai J. Acc Chem Res. 2016;49:428–441. doi: 10.1021/acs.accounts.5b00492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Teng P, Shi Y, Sang P, Cai J. Chem – Eur J. 2016;22:5458–5466. doi: 10.1002/chem.201504936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Wu H, Qiao Q, Hu Y, Teng P, Gao W, Zuo X, Wojtas L, Larsen RW, Ma S, Cai J. Chem – Eur J. 2015;21:2501–2507. doi: 10.1002/chem.201406112. [DOI] [PubMed] [Google Scholar]; (b) Wu H, Qiao Q, Teng P, Hu Y, Antoniadis D, Zuo X, Cai J. Org Lett. 2015;17:3524–3527. doi: 10.1021/acs.orglett.5b01608. [DOI] [PubMed] [Google Scholar]
- 19.(a) Pauling L, Corey RB, Branson HR. Proc Natl Acad Sci U S A. 1951;37:205–211. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weaver TM. Protein Sci. 2000;9:201–206. doi: 10.1110/ps.9.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Seebach DL, Matthews J. Chem Commun. 1997:2015–2022. [Google Scholar]; (b) Shin YH, Mortenson DE, Satyshur KA, Forest KT, Gellman SH. J Am Chem Soc. 2013;135:8149–8152. doi: 10.1021/ja403319q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fremaux J, Fischer L, Arbogast T, Kauffmann B, Guichard G. Angew Chem, Int Ed. 2011;50:11382–11385. doi: 10.1002/anie.201105416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.