

 AIMD simulations using the fragment-based coupled cluster theory accurately reveal the structural and dynamical properties of liquid water.
AIMD simulations using the fragment-based coupled cluster theory accurately reveal the structural and dynamical properties of liquid water.
Abstract
An accurate and efficient ab initio molecular dynamics (AIMD) simulation of liquid water was made possible using the fragment-based approach (J. F. Liu, X. He and J. Z. H. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 11931–11936). In this study, we advance the AIMD simulations using the fragment-based coupled cluster (CC) theory, more accurately revealing the structural and dynamical properties of liquid water under ambient conditions. The results show that the double-donor hydrogen-bond configurations in liquid water are nearly in balance with the single-donor configurations, with a slight bias towards the former. Our observation is in contrast to the traditional tetrahedral water structure. The hydrogen-bond switching dynamics in liquid water are very fast, with a hydrogen-bond life time of around 0.78 picoseconds, determined using AIMD simulation at the CCD/aug-cc-pVDZ level. This time scale is remarkably shorter than the ∼3.0 picoseconds that is commonly obtained from traditional nonpolarized force fields and density functional theory (DFT) based first-principles simulations. Additionally, the obtained radial distribution functions, triplet oxygen angular distribution, diffusion coefficient, and the dipole moment of the water molecule are uniformly in good agreement with the experimental observations. The current high-level AIMD simulation sheds light on the understanding of the structural and dynamical properties of liquid water.
1. Introduction
As the most abundant liquid on Earth, water is essential for life and is involved in nearly all biological, geological, and chemical processes.1 Despite being extensively studied for several decades, there is still ongoing debate on the dynamical picture of the liquid water structure under ambient conditions.1–11 For a microscopic understanding of liquid water, one of the most essential questions to address is the structure and dynamics of the hydrogen-bonding network in water which determine the unique water properties. This collective network fluctuation involving hydrogen-bond breaking and forming is of great importance for elucidating the dynamical picture of liquid water.
Understanding the nature of the dynamical hydrogen-bonding network in liquid water under ambient conditions is a challenge for both experimental and theoretical researchers. According to a series of spectral experiments and theoretical simulations,12–17 liquid water is usually regarded as a tetrahedral structure involving minimally distorted hydrogen-bonding configurations. However, based on X-ray absorption and Raman scattering experiments, Wernet et al. concluded that the first coordination shell around a water molecule in liquid water has two hydrogen-bonding partners on average with one donor and one acceptor.18,19 It favors a “ring-and-chain”-like structure in water, which is in contrast to the conventionally accepted tetrahedral structure of liquid water. It remains a topic of intense debate as to whether water has a tetrahedral or “ring-and-chain”-like structure.20
Another important issue is the hydrogen-bond switching dynamics in water. The dynamics of this network occur over a wide range of time scales, from femtosecond fluctuations that involve a few molecules to picosecond diffusive motions.21,22 Specifically, Bakker et al. observed that orientational relaxation of the HDO molecules dissolved in D2O occurred on either a very slow or a very fast time scale, with corresponding time constants of 13.0 and 0.7 picoseconds, respectively.23–26 Fecko et al. showed that the hydrogen-bond vibrational correlations decay with a period of 0.17 and 1.2 picoseconds due to the underdamped oscillation of hydrogen-bond and collective structural reorganizations, respectively.22 There are many other related experimental and theoretical investigations10,27–29 demonstrating large variations in the time constants of the hydrogen-bond relaxation.
With experimental methods, it is normally difficult to yield detailed information on the hydrogen-bond dynamics in water.30 Understanding the specific molecular structure and dynamics of liquid water at the atomistic level depends on molecular dynamics (MD) simulations. MD with empirical force fields have already provided fundamental insights into the microstructural and dynamical properties of water, with continuous improvement of the traditional force fields.31–38 Ab initio molecular dynamics (AIMD) simulations, with the molecular potentials described by the first-principles electronic structure theory, are a significant improvement over the empirical force fields.39–45 Density functional theory (DFT) has been the most widely used electronic structure method in AIMD simulations with reasonable accuracy and moderate computational cost.46 However, the performance of DFT-based AIMD simulations is dramatically affected by the choice of density functionals.46,47 High-level wavefunction theories, such as the second order Møller–Plesset perturbation (MP2) method, have also been utilized to obtain an accurate description of the structural and dynamical properties of liquid water.43–45,48 In our previous work, a MP2-based AIMD simulation of liquid water was carried out on a large body of water molecules (∼140 water molecules) through the fragmentation quantum mechanical (QM) method,49–53 and the simulated water structural and dynamical properties were uniformly in good agreement with experimental observations.53
It is of great importance and necessity to apply high-level wavefunction theories for an accurate simulation of the microstructural and dynamical properties of liquid water. To the best of our knowledge, the coupled cluster (CC) theory has not been utilized to treat a large body of water molecules for an AIMD simulation of liquid water, owing to the high scaling of the CC method with tremendous computational cost. Here we report substantial progress toward an accurate liquid water dynamics simulation using the fragment-based CC method. The nuclear quantum effect (NQE) is not included in this study. The NQE of a hydrogen atom in liquid water is mainly associated with O–H vibration-related properties, and most of the statistical properties of water such as diffusion or the O–O radial distribution function (RDF) are not much affected by the NQE as demonstrated in recent work by Marsalek and Markland.54 The good agreement between the experimental observations and the calculated properties of water without explicit inclusion of the NQE in previous studies46,53 implied that the influence of the NQE on hydrogen atoms has subtle effects on most of the statistical properties of liquid water. This study sheds light on a detailed understanding of the water structure and hydrogen-bonding dynamics in liquid water from AIMD simulations using the fragment-based CC method.
2. Results and discussion
2.1. Water properties
2.1.1. RDF
In order to characterize the structure of water, we first calculated its intermolecular oxygen–oxygen (gOO), oxygen–hydrogen (gOH), and hydrogen–hydrogen (gHH) radial distribution functions (RDFs). The obtained data were compared to the experimental observations,55,56 as well as the results obtained from the authors’ previous study performed at the MP2/aug-cc-pVDZ level.53 As shown in Fig. 1, the simulated gOO curve obtained using the CCD method is in excellent agreement with the experimental results for both the positions and intensities of the first two peaks. The gOO curve obtained using the MP2 method is also in good agreement with the experimental results, except that the trough between the first two peaks is slightly lower compared to the CCD and experimental results. Moreover, the MP2 RDF curve is not as smooth as that given by CCD owing to the relatively shorter simulation time at the MP2 level in the authors’ previous study. For the gOH and gHH curves, both of the intensities of the first peaks obtained from the MP2 simulation are overestimated in comparison with the experimental results, which is mainly due to the lack of the NQE in the simulation. In comparison, the CCD results significantly reduce the intensities of the first peaks for the gOH and gHH curves, although they are still overestimated in reference to the experimental results. For both the MP2 and CCD results, the second peaks of gOH and gHH slightly shift to higher radial positions in comparison with the experimental values, while the positions of the third peaks for gOH and gHH are both in good agreement with the experimental results. Overall, our AIMD simulated results using the fragment-based CCD/aug-cc-pVDZ method in this study are in good accordance with the experimental observations.
Fig. 1. (a) Oxygen–oxygen, (b) oxygen–hydrogen and (c) hydrogen–hydrogen radial distribution functions (RDF) of liquid water under ambient conditions obtained using the fragment-based AIMD simulations at the MP2/aug-cc-pVDZ53 and CCD/aug-cc-pVDZ levels, respectively.

Recently, Hirata and co-workers suggested that with such a small basis set as aug-cc-pVDZ, RDFs cannot generally be reproduced accurately.45 Furthermore, their subsequent study revealed a precise mechanism by which the adjustments of density, temperature, and/or pressure would lead to correct RDFs in MP2 and DFT water simulations.57 According to their study, the MP2 simulated water tends to be denser (ρ > 1.00 g cm–3) under ambient conditions due to the overestimated dispersion interaction, and thus requires a lower temperature and negative pressure to generate the experimentally observed RDF of liquid water. In this study, the initial density of the system was equilibrated to 1.002 g cm–3. During the AIMD simulation, the average water density of the QM region was 1.008 g cm–3, which was slightly larger than the experimental value under ambient conditions. There are some approximations made in this study, and the simulated water properties may result from a variety of factors, such as the utilized canonical NVT ensemble for the whole system, Langevin thermostat for adjusting the system temperature and the mechanical embedding scheme for treating the QM/MM coupling. The AIMD simulation details are summarized in the theory and computation section and in the ESI.† The agreement of the results with the experimental results is partially due to the fortuitously accurate intermolecular interaction potential described at the MP2/aug-cc-pVDZ or CCD/aug-cc-pVDZ level, with reference to the potential calculated at the CCSD(T)/aug-cc-pVQZ level (see Fig. S1 of the ESI†).
2.1.2. Oxygen–oxygen–oxygen triplet angles
To further analyze the local arrangement of the water molecules in the liquid phase, we calculated the distribution of oxygen–oxygen–oxygen triplet angles within the first coordination shell and tetrahedral order parameter q for the QM water molecules in simulation. The CCD result obtained in this study and the MP2 simulated result from the authors’ previous study53 are compared in Fig. 2 along with the experimental curve. Three oxygen atoms were considered as a given triplet if two of the oxygen atoms are within a predefined distance cutoff from the third. The computational details are given in the ESI.† The experimental angular distribution shows a shoulder at around 60° and a broad and strong peak at around 100°.56 The CCD simulated angular distribution curve is in good agreement with the experimental curve, with one distinct shoulder at around 70° and a strong peak at around 100°, and the intensity of the strong peak exactly matches the experimental observation. In contrast, for the MP2 simulated results, the intensity of the strong peak is underestimated and the shoulder is overestimated. The tetrahedral order parameter q, an index to describe the similarity of the simulated water structure with the perfect tetrahedral structure, would yield a value of 1 for the perfect tetrahedral structure and 0 for the ideal gas. The tetrahedral order parameter q is 0.515 based on the fragment-based CCD/aug-cc-pVDZ simulation, close to our previous MP2 simulated value of 0.520 and the experimental value of 0.570. The current result is more accurate than 0.670, which was obtained from a DFT-based Car–Parrinello molecular dynamics (CPMD) simulation using PBE0 + TS-vdW(SC).47
Fig. 2. The oxygen–oxygen–oxygen triplet angular distribution and tetrahedral order parameter of liquid water obtained from the fragment-based AIMD simulation at the MP2/aug-cc-pVDZ53 and CCD/aug-cc-pVDZ levels, respectively. The water molecules whose oxygen atoms are less than or equal to 8 Å away from the center of the water box were used for this analysis.

2.1.3. Diffusion coefficient
The dynamical properties of liquid water can be measured from the diffusion coefficient. The convergence of the diffusion coefficient as a function of the simulation time and the QM water cluster size are shown in Fig. S2 and S3 of the ESI.† The calculated diffusion coefficient at 300 K converged to 0.20 Å2 ps–1 in the last 7.0 ps AIMD simulation, as compared to the experimental value of 0.23 Å2 ps–1.58 This result is consistent with our previous study.53 The empirical force fields usually predict a rather larger diffusion coefficient (>0.26 Å2 ps–1),59 while the DFT-based AIMD simulations generally give a relatively smaller value (<0.20 Å2 ps–1).46,47
2.1.4. Dipole moment
The dipole moment of a water molecule in the liquid phase has been under debate for a long time, partially because of the uncertainty on its value (2.6–3.0 Debye) measured from the X-ray scattering form factors.60–62 The current AIMD simulation also reveals the distribution of the molecular dipole of water as shown in Fig. S4 of the ESI.† The molecular dipole is broadly distributed from 1.8 to 3.2 Debye with an average value of 2.53 Debye, reflecting the diversity of the electrostatic fields of the environment and the local geometry fluctuation of the water molecules. The predicted dipole moment from the DFT-based CPMD simulation is relatively larger (around 2.90 Debye),47 and it also heavily depends on the choice of density functionals.
Hence, for these basic properties of liquid water, the fragment-based AIMD simulation at the CCD/aug-cc-pVDZ level gives reasonable results that are uniformly in good agreement with the experimental observations. Based on these results, we further investigated the detailed microstructure and hydrogen-bond dynamics of liquid water from the AIMD simulated trajectory.
2.2. Water microstructure
Controversy regarding whether liquid water has a tetrahedral or “ring-and-chain”-like microstructure still persists.19,20 According to Wernet et al.,18 water molecules with single hydrogen-bond donors (SD) are dominant in liquid water. However, nearly all of the theoretical studies based on force fields or DFT overwhelmingly favor the tetrahedral structure, in which water molecules with the double hydrogen-bond donor (DD) configurations are dominant in the liquid phase.47 From our previous fragment-based AIMD study at the MP2/aug-cc-pVDZ level,53 we found that both of these two configurations (SD and DD) were almost equally distributed in the liquid phase, with a slight bias towards the tetrahedral structure.
In this study, we made quantitative statistics of the number and types of hydrogen-bond configurations in liquid water by utilizing the popular hydrogen-bond definition proposed by Luzar and Chandler.63 The defining parameters for hydrogen bonds include both distance and angular criteria, namely, rOO < 3.5 Å and θ∠OA···OD–HD < 30° (Fig. 3a). Using this definition, we first analyzed the difference among the hydrogen-bonds in CCD simulated liquid water using the 2D weighted histogram analysis method (WHAM-2D)64 (see Fig. 3b). We also compared it with the MP2 result from the authors’ previous study53 (Fig. 3c). The free energy landscape clearly shows that the most probable region corresponding to the ideal hydrogen-bond in CCD simulated liquid water is with the distance rOO in the range of 2.8–3.0 Å and the angle θ around 8–15° (not 0°), respectively. The other regions under the hydrogen-bond definition represent the non-ideal or bent hydrogen-bond configurations. In comparison with the CCD result, the hydrogen-bonds formed in the MP2 simulated liquid water show a very similar pattern except that the MP2 simulated ideal hydrogen-bond has a relatively shorter rOO distance (in the range of 2.7–2.9 Å), which is caused by the relatively stronger intermolecular interaction calculated using MP2/aug-cc-pVDZ (see Fig. S1†). The ideal hydrogen-bonds are usually more stable than those non-ideal or bent hydrogen-bonds. The deviation of the distance rOO and angle θ from their most probable region would decrease the stability of the hydrogen bonds in liquid water. Hence, these strong and weak hydrogen-bonds in liquid water would lead to different hydrogen-bond dynamics.
Fig. 3. (a) The definition of a hydrogen-bond between two water molecules and the free energy landscape of the hydrogen-bonds in the (b) CCD/aug-cc-pVDZ and (c) MP2/aug-cc-pVDZ simulated liquid water. The definition for a hydrogen-bond is that the distance between two oxygen atoms roo < 3.5 Å and θ∠OA···OD–HD < 30°.

The fractions of double-donor (DD), single-donor (SD), and non-donor (ND) configurations from the last 7 ps AIMD simulation are given in Table 1. According to Wernet et al.,18 around 80% of water molecules in the liquid phase donate only one hydrogen to form a hydrogen bond with their neighbors, whereas DD and ND configurations only account for 15% and 5%, respectively. However, the DFT-based CPMD simulation47 predicted the opposite trend, i.e., 79% of water molecules are in the DD configuration and only 20% of them are in the SD configuration. In addition, the result obtained from the empirical force field (SPCFW) also shows a similar pattern, with dominant 70% DD configurations and only 27% SD configurations. The present fragment-based AIMD simulation at the CCD/aug-cc-pVDZ level predicts a more balanced picture of the hydrogen-bond structure in liquid water. Specifically, our result shows that the DD configuration accounts for just 50% of the overall population, which is closely followed by 42% SD configurations (Table 1). This result is also in accordance with the predicted tetrahedral order parameter of 0.515, which means that the structure of water presents a mixture of two configurations with a slight bias toward the DD configuration (tetrahedral structure) over the SD configuration (“ring-and-chain”-like structure). Our previous fragment-based AIMD simulation at the MP2/aug-cc-pVDZ level53 also reached a similar conclusion.
Table 1. The percentage (%) of hydrogen-bond configurations: double-donor (DD), single-donor (SD), and non-donor (ND) configurations in liquid water from different methods.
| Type | Exp a | Method |
|||
| CCD b | MP2 c | SPCFW | CPMD d | ||
| DD | 15 ± 25 | 50 | 53 | 70 | 79 |
| SD | 80 ± 20 | 42 | 40 | 27 | 20 |
| ND | 5 ± 5 | 8 | 7 | 3 | 1 |
aThe experimentally fitted percentage of hydrogen-bond configurations in liquid water from ref. 18.
bThe EE-GMF approach at the CCD/aug-cc-pVDZ level from this study.
cThe EE-GMF approach at the MP2/aug-cc-pVDZ level from ref. 53.
dCPMD simulation results (using PBE0+TS-vdW(SC)) from ref. 47, which utilized the same definition of hydrogen bonds as this study.
To provide more insight into the microstructure of liquid water, some representative hydrogen-bond network structures were extracted from our AIMD simulated trajectory as shown in Fig. 4 and S5.† Both the tetrahedral and “ring-and-chain”-like structures of water are present in this simulation. As shown in Fig. 4a, the traditional picture of a water molecule, accepting and donating two hydrogen bonds to form a tetrahedral structure, partly exists in our simulation. All three inner water molecules form tetrahedral structures in the snapshot shown in Fig. 4a. On the other hand, in Fig. 4b, there are ring-like structures which are formed by a group of DD and SD configurations of the water molecules. There are also unclosed ring-like (or chain-like) structures (see Fig. 4c), in which the central water molecule does not donate any hydrogen bond (in the ND configuration) at the end of the chain. The ND configuration may arise from local coordination of the hydrogen-bond network to form a temporarily meta-stable state. All of these hydrogen-bonding structures change dynamically throughout the entire AIMD simulation. The tetrahedral, ring-like and chain-like structures interchange with each other from time to time, resulting in a dynamical picture of a mixture of tetrahedral and “ring-and-chain”-like structures in liquid water as indicated from the current AIMD simulation.
Fig. 4. The representative hydrogen-bond structures in the simulated liquid water. The dashed line denotes the hydrogen-bond. The pink, yellow and grey circles label the double-donor (DD), single-donor (SD) and none-donor (ND) configurations of the hydrogen-bonded water molecules, respectively.

2.3. Hydrogen-bond dynamics
The hydrogen-bond dynamics in liquid water take place in a very fast manner.10 To quantitatively describe the dynamical properties of water, we first computed the residence time correlation function S(t) for the water molecules around the central water molecule and their first coordination shell in the QM region using the following equation,65
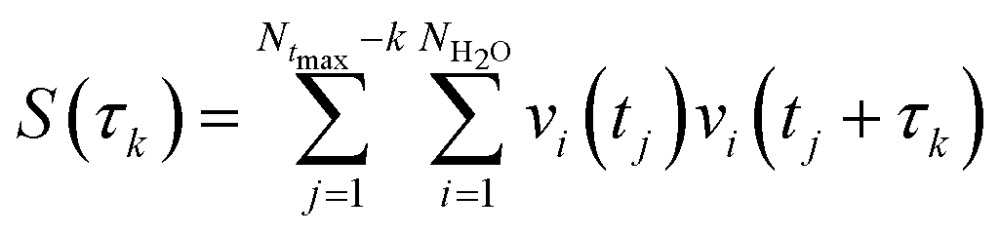 |
1 |
where τk is the kth time interval (τk = kΔτ; Δτ = 1 fs; k = 0, 1,…Ntmax – 1), vi(t) becomes 1 or 0 depending on the water molecule i within or outside the shell defined by a radius around a tagged water molecule at time t, and NH2O is the number of water molecules in the QM region. The S(t) measures the average number of water molecules which continuously stay around the tagged water molecule. In this simulation, S(t) decays in an exponential fashion, and the time constant associated with this decay gives a measure of the residence time of water around a specific site. Fig. 5a shows the calculated S(t) along with the exponential fit S(t) = exp(–t/τ) using a radius cutoff of 3.5 Å from the last 10 ps AIMD simulation. The residence time τ of a water molecule obtained from the exponential fit is 2.0 ps given by the fragment-based AIMD simulation at the CCD/aug-cc-pVDZ level, indicating that the diffusion of the water molecules in the liquid phase is very fast.
Fig. 5. (a) Residence time correlation function S(t) for the water molecules around the central water molecule and their first coordination shell in the QM region (normalized to S(tmin) = 1). (b) Hydrogen-bond correlation function C(t) for the water molecules in the reduced QM region. The red curve is the corresponding exponential fit.

Furthermore, we investigated the hydrogen-bond dynamics by introducing the hydrogen-bond correlation function66
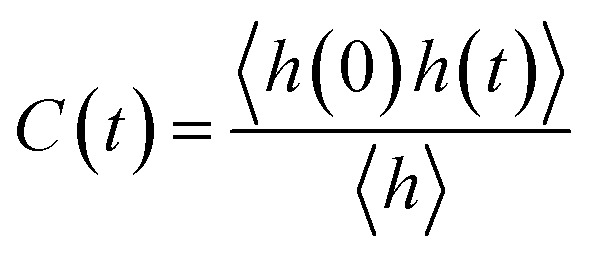 |
2 |
where h(t) is a hydrogen-bond population descriptor, which is unity when a tagged pair of molecules is hydrogen-bonded at time t and is zero otherwise. This correlation function is calculated for all QM water molecules defined to be hydrogen-bonded at both times 0 and t, which describes the probability of the tagged pair of molecules being hydrogen-bonded at time t given that the pair was hydrogen-bonded at time 0. The calculated hydrogen-bond correlation function is shown in Fig. 5b, which is best described by a series of two exponential decay functions. Two time constants obtained from the exponential fit are identified as the average hydrogen bond lifetime,2 and are 0.78 and 5.11 ps, respectively. This result indicates that the hydrogen-bond dynamics in liquid water occur on either a very fast or a slow time scale. This result is in agreement with Bakker and coworkers’ experiment,23 where they measured that the hydrogen-bonds in liquid water decay on either a very fast or a slow time scale (0.7 and 13.0 ps for HDO dissolved in D2O), corresponding to the weak and strong hydrogen bonds in liquid water, respectively. However, the hydrogen-bond life time obtained from the empirical force fields and previous DFT-based CPMD simulations are normally overestimated, with the fast and slow time scales around 3.0 ps and 18.0 ps, respectively.2 Therefore, we conclude that the fragment-based AIMD simulation at the CCD/aug-cc-pVDZ level can well describe the ultrafast hydrogen-bond dynamics in liquid water, which would help to better understand the dynamic properties of liquid water.
At present, there are still no consistent conclusions on the impact of the NQE on the structure and dynamics of water hydrogen-bonds. According to a series of studies,67–69 the competition between the intra- and intermolecular quantum effects in water further reduces the impact of neglecting the NQEs on the average statistical properties of liquid water. Li et al. carried out systematic examination of a wide range of hydrogen-bonded systems through the ab initio path integral molecular dynamics, and showed that NQEs weaken weak hydrogen bonds but strengthen relatively strong ones.70 Furthermore, from their study, the NQEs would accelerate the hydrogen dynamics for the weak hydrogen-bonds. On the other hand, for the strong hydrogen-bonds, the NQEs would slow down their hydrogen dynamics. More recently, Wilkins et al. combined classical and ring polymer molecular dynamics simulations to provide a molecular description of the NQEs on water reorientation and hydrogen-bond dynamics in liquid water. They showed that NQEs lead to a moderate acceleration (∼13%) of the hydrogen-bond dynamics as compared to a classical description.71
3. Conclusions
In this study, we investigated the structural and dynamical properties of liquid water under ambient conditions using fragment-based AIMD simulation at the CCD/aug-cc-pVDZ level. The water properties, such as the RDFs, diffusion coefficient, molecular dipole and oxygen–oxygen–oxygen triplet angular distribution, are uniformly in excellent agreement with the experimental observations, which demonstrates the robustness of the EE-GMF method for AIMD simulation with high-level wavefunction theories.
Furthermore, we investigated the hydrogen-bond structures and dynamics in liquid water and gave a clear dynamic picture of liquid water. We found that the tetrahedral and “ring-and-chain”-like structures were almost equally distributed in liquid water, with around 50% of water molecules donating two hydrogen-bond donors to their neighbors (DD configuration) and 42% donating just one hydrogen-bond donor (SD configuration). In addition, the hydrogen-bonds in liquid water decay on either a very fast or a slow time scale, which correspond to short and long hydrogen-bond life times of 0.78 and 5.11 ps, respectively.
This study suggests that, using the aug-cc-pVDZ basis set, the overall performance of CCD is just slightly better than MP2 in describing the structural and dynamical properties of liquid water. The larger basis sets (such as aug-cc-pVTZ and aug-cc-pVQZ) may be needed to systematically investigate the relative merits between MP2 and CCD. The EE-GMF approach could in principle implement a systematic series of diagrammatic many-body theories (even for the CCSD and CCSD(T) methods) in conjunction with also systematic basis sets, together converging towards the exact limits for liquid water simulation. It is general in the choice of electronic structure theory for monomers and dimers, and most importantly exhibits linear scaling in the computational cost with respect to the system size. However, for EE-GMF calculations using high-level ab initio theories, the larger basis sets would significantly increase the computational cost on each fragment QM calculation. In future studies, we will utilize more efficient techniques, such as the density fitting algorithm and other local methods in fragment QM calculations, to accelerate such high-level AIMD simulations.
The current approach also has limitations. First, the transition of potentials at the QM/MM boundary is not smooth when water molecules leave or enter the QM region. This may cause some artificial effects on the structural and dynamical properties of the simulated liquid water near the QM/MM boundary, although we used a reduced QM region for analyzing the liquid water properties to alleviate the boundary effect. Second, the NQE is not included in this work, which may have an impact on the hydrogen-bond dynamics simulation. Improvements on the fragment-based AIMD simulation along these lines are currently underway in our laboratory.
4. Theory and computation
In this study, we employed the electrostatically embedded generalized molecular fractionation (EE-GMF)51,52 to perform the ab initio calculations of a large body of water molecules. According to the EE-GMF scheme, the total energy of a water cluster can be expressed as follows,
 |
3 |
where N is the number of water molecules. The QM energy calculations of monomer i (one water molecule) and dimer ij (two water molecules) are performed in the embedded electrostatic fields of the rest of the water molecules, represented by the Coulomb field of atomic charges, to account for the electronic polarization effect from the surrounding environment. in eqn (3) denotes the sum of the self-energy of the fragment along with the interaction energy between the fragment and background charges of the remaining system. i and ij are the energies of the water monomer i and dimer ij, respectively. The first term of eqn (3) is the summation of one-body energies. The second term is the local two-body QM interaction when the distance Rij between any two oxygen atoms from monomers i and j is less than or equal to a predefined distance threshold λ.53 Otherwise, the interaction energy between two water molecules (Rij > λ) is described by the classical Coulomb interaction. Therefore, the monomer energy and local pairwise interactions are explicitly treated by QM, while the higher-ordered many-body electrostatic interactions are implicitly incorporated using the electrostatic embedding scheme. Because of the charge embedding scheme utilized in fragment QM calculations, the long range interactions are included in each fragment QM calculations. The doubly counted electrostatic interactions between distant water pairs (beyond the distance threshold λ) need to be deducted using the last term of eqn (3) through the classical Coulomb interactions, where qm(i) denotes the atomic charge of the mth atom in the ith water molecule. The atomic charges from the SPCFW72 water model were utilized as the embedding field in this study. The distance threshold λ was set to 5.0 Å for ensuring the convergence of the total EE-GMF energy of the water cluster, while achieving an efficient computation. The detailed description of the EE-GMF method can be found in a series of recent publications51–53 and the ESI.†
A truncated octahedron box with edges of 42.6 Å (containing a total of 1997 flexible SPCFW72 water molecules) under periodic boundary conditions was constructed for simulation in this study. Before the AIMD simulation, the density of the system was equilibrated to 1.002 g cm–3 using the classical MD simulation. To improve the computational efficiency, a QM/MM scheme is utilized (the same as our previous study53), in which water molecules with oxygen atoms that are less than or equal to 10 Å away from the center of the simulation box are treated by QM, while the rest of the water molecules are described by MM. The EE-GMF approach is employed to calculate the total energy (eqn (3)) and atomic forces (see the ESI†) of the QM region (approximately 140 water molecules). The coupling between the QM and MM regions is treated by mechanical embedding. For all water molecules, the intra-molecular bonds are fully flexible. The technical details of the AIMD simulation are given in the ESI.† To reduce the boundary effect, a buffer zone of about 2 Å from the QM/MM boundary was established. We used a reduced QM region (the radius of which is 8.0 Å from the center of the simulation box) for analyzing the physicochemical properties of liquid water.
The CCD/aug-cc-pVDZ method was employed for the AIMD simulation of liquid water under ambient conditions. The wall clock time for each MD step was around 3.5 minutes on 30 computer nodes with the Intel Xeon E5-4640 2.4 GHz processor (28 cores per node). The interaction potential energy between two water molecules calculated at the CCD/aug-cc-pVDZ level is shown in Fig. S1 of the ESI.† Prior to the production run of AIMD, the initial water structure was pre-equilibrated by classical MD simulation using the SPCFW force field. Subsequently, a total of 15.0 ps AIMD simulation was carried out. This simulation time is found to be sufficient in converging the predicted water properties as shown in the ESI.† During the AIMD simulation, the transition of the water molecules between the QM and MM regions, which is quantified as the number of the water molecules in the QM region, is shown in Fig. S6 of the ESI.†
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant No. 2016YFA0501700), National Natural Science Foundation of China (No. 21703289, 91639115, 21673074 and 21433004), Youth Top-Notch Talent Support Program of Shanghai, and NYU-ECNU Center for Computational Chemistry at NYU Shanghai, Shanghai Putuo District (Grant 2014-A-02), and NYU Global Seed Grant for Collaborative Research. We thank the Supercomputer Center of East China Normal University for providing us with computational time. We also thank Dr Raphael N. Alolga for helpful comments on the manuscript.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04205a
References
- Pettersson L. G. M., Henchman R. H., Nilsson A. Chem. Rev. 2016;116:7459–7462. doi: 10.1021/acs.chemrev.6b00363. [DOI] [PubMed] [Google Scholar]
- Ding Y., Hassanali A. A., Parrinello M. Proc. Natl. Acad. Sci. U. S. A. 2014;111:3310–3315. doi: 10.1073/pnas.1400675111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary D. C. Science. 2016;351:1268–1269. doi: 10.1126/science.aaf3061. [DOI] [PubMed] [Google Scholar]
- Stirnemann G., Wernersson E., Jungwirth P., Laage D. J. Am. Chem. Soc. 2013;135:11824–11831. doi: 10.1021/ja405201s. [DOI] [PubMed] [Google Scholar]
- Hassanali A., Giberti F., Cuny J., Kuhne T. D., Parrinello M. Proc. Natl. Acad. Sci. U. S. A. 2013;110:13723–13728. doi: 10.1073/pnas.1306642110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus Y. Chem. Rev. 2009;109:1346–1370. doi: 10.1021/cr8003828. [DOI] [PubMed] [Google Scholar]
- Angell C. A. Science. 2008;319:582–587. doi: 10.1126/science.1131939. [DOI] [PubMed] [Google Scholar]
- Errington J. R., Debenedetti P. G. Nature. 2001;409:318–321. doi: 10.1038/35053024. [DOI] [PubMed] [Google Scholar]
- Bakker H. J., Skinner J. L. Chem. Rev. 2010;110:1498–1517. doi: 10.1021/cr9001879. [DOI] [PubMed] [Google Scholar]
- Laage D., Hynes J. T. Science. 2006;311:832–835. doi: 10.1126/science.1122154. [DOI] [PubMed] [Google Scholar]
- Tokmakoff A. Science. 2007;317:54–55. doi: 10.1126/science.1144515. [DOI] [PubMed] [Google Scholar]
- Smith J. D., Cappa C. D., Wilson K. R., Messer B. M., Cohen R. C., Saykally R. J. Science. 2004;306:851–853. doi: 10.1126/science.1102560. [DOI] [PubMed] [Google Scholar]
- Kuhne T. D., Krack M., Parrinello M. J. Chem. Theory Comput. 2009;5:235–241. doi: 10.1021/ct800417q. [DOI] [PubMed] [Google Scholar]
- Fernandez-Serra M. V., Artacho E. Phys. Rev. Lett. 2006;96:016404. doi: 10.1103/PhysRevLett.96.016404. [DOI] [PubMed] [Google Scholar]
- Prendergast D., Galli G. Phys. Rev. Lett. 2006;96:215502. doi: 10.1103/PhysRevLett.96.215502. [DOI] [PubMed] [Google Scholar]
- Perakis F., De Marco L., Shalit A., Tang F. J., Kann Z. R., Kuhne T. D., Torre R., Bonn M., Nagata Y. Chem. Rev. 2016;116:7590–7607. doi: 10.1021/acs.chemrev.5b00640. [DOI] [PubMed] [Google Scholar]
- Gasparotto P., Hassanali A. A., Ceriotti M. J. Chem. Theory Comput. 2016;12:1953–1964. doi: 10.1021/acs.jctc.5b01138. [DOI] [PubMed] [Google Scholar]
- Wernet P., Nordlund D., Bergmann U., Cavalleri M., Odelius M., Ogasawara H., Naslund L. A., Hirsch T. K., Ojamae L., Glatzel P., Pettersson L. G. M., Nilsson A. Science. 2004;304:995–999. doi: 10.1126/science.1096205. [DOI] [PubMed] [Google Scholar]
- Nilsson A., Pettersson L. G. M. Nat. Commun. 2015;6:8998. doi: 10.1038/ncomms9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head-Gordon T., Johnson M. E. Proc. Natl. Acad. Sci. U. S. A. 2006;103:7973–7977. doi: 10.1073/pnas.0510593103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C. P., Skinner J. L. J. Chem. Phys. 2003;118:264–272. [Google Scholar]
- Fecko C. J., Eaves J. D., Loparo J. J., Tokmakoff A., Geissler P. L. Science. 2003;301:1698–1702. doi: 10.1126/science.1087251. [DOI] [PubMed] [Google Scholar]
- Woutersen S., Emmerichs U., Bakker H. J. Science. 1997;278:658–660. [Google Scholar]
- Bakker H. J., Woutersen S., Nienhuys H. K. Chem. Phys. 2000;258:233–245. [Google Scholar]
- van der Post S. T., Bakker H. J. J. Phys. Chem. B. 2014;118:8179–8189. doi: 10.1021/jp501240e. [DOI] [PubMed] [Google Scholar]
- van der Post S. T., Hsieh C. S., Okuno M., Nagata Y., Bakker H. J., Bonn M., Hunger J. Nat. Commun. 2015;6:8384. doi: 10.1038/ncomms9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. S., Pieniazek P. A., Yang M., Skinner J. L. J. Chem. Phys. 2010;132:174505. doi: 10.1063/1.3409561. [DOI] [PubMed] [Google Scholar]
- Paesani F., Yoo S., Bakker H. J., Xantheas S. S. J. Phys. Chem. Lett. 2010;1:2316–2321. [Google Scholar]
- Nicodemus R. A., Corcelli S. A., Skinner J. L., Tokmakoff A. J. Phys. Chem. B. 2011;115:5604–5616. doi: 10.1021/jp111434u. [DOI] [PubMed] [Google Scholar]
- Voloshin V. P., Naberukhin Y. I. J. Struct. Chem. 2009;50:78–89. [Google Scholar]
- Kohagen M., Mason P. E., Jungwirth P. J. Phys. Chem. B. 2016;120:1454–1460. doi: 10.1021/acs.jpcb.5b05221. [DOI] [PubMed] [Google Scholar]
- Medders G. R., Babin V., Paesani F. J. Chem. Theory Comput. 2014;10:2906–2910. doi: 10.1021/ct5004115. [DOI] [PubMed] [Google Scholar]
- Laury M. L., Wang L. P., Pande V. S., Head-Gordon T., Ponder J. W. J. Phys. Chem. B. 2015;119:9423–9437. doi: 10.1021/jp510896n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. P., Head-Gordon T., Ponder J. W., Ren P., Chodera J. D., Eastman P. K., Martinez T. J., Pande V. S. J. Phys. Chem. B. 2013;117:9956–9972. doi: 10.1021/jp403802c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. C., Wang Y. M., Bowman J. M. J. Chem. Phys. 2015;142:194502. doi: 10.1063/1.4921045. [DOI] [PubMed] [Google Scholar]
- Cisneros G. A., Wikfeldt K. T., Ojamae L., Lu J. B., Xu Y., Torabifard H., Bartok A. P., Csanyi G., Molinero V., Paesani F. Chem. Rev. 2016;116:7501–7528. doi: 10.1021/acs.chemrev.5b00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paesani F. Acc. Chem. Res. 2016;49:1844–1851. doi: 10.1021/acs.accounts.6b00285. [DOI] [PubMed] [Google Scholar]
- Reddy S. K., Straight S. C., Bajaj P., Pham C. H., Riera M., Moberg D. R., Morales M. A., Knight C., Gotz A. W., Paesani F. J. Chem. Phys. 2016;145:194504. doi: 10.1063/1.4967719. [DOI] [PubMed] [Google Scholar]
- VandeVondele J., Mohamed F., Krack M., Hutter J., Sprik M., Parrinello M. J. Chem. Phys. 2005;122:014515. doi: 10.1063/1.1828433. [DOI] [PubMed] [Google Scholar]
- Laasonen K., Sprik M., Parrinello M., Car R. J. Chem. Phys. 1993;99:9080–9089. [Google Scholar]
- Yoo S., Xantheas S. S. J. Chem. Phys. 2011;134:121105. doi: 10.1063/1.3573375. [DOI] [PubMed] [Google Scholar]
- Baer M. D., Mundy C. J., McGrath M. J., Kuo I. F. W., Siepmann J. I., Tobias D. J. J. Chem. Phys. 2011;135:124712. doi: 10.1063/1.3633239. [DOI] [PubMed] [Google Scholar]
- Del Ben M., Schonherr M., Hutter J., VandeVondele J. J. Phys. Chem. Lett. 2013;4:3753–3759. doi: 10.1021/jz501672u. [DOI] [PubMed] [Google Scholar]
- Zen A., Luo Y., Mazzola G., Guidoni L., Sorella S. J. Chem. Phys. 2015;142:144111. doi: 10.1063/1.4917171. [DOI] [PubMed] [Google Scholar]
- Willow S. Y., Salim M. A., Kim K. S., Hirata S. Sci. Rep. 2015;5:14358. doi: 10.1038/srep14358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestana L. R., Mardirossian N., Head-Gordon M., Head-Gordon T. Chem. Sci. 2017;8:3554–3565. doi: 10.1039/c6sc04711d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiStasio R. A., Santra B., Li Z. F., Wu X. F., Car R. J. Chem. Phys. 2014;141:084502. doi: 10.1063/1.4893377. [DOI] [PubMed] [Google Scholar]
- Del Ben M., Hutter J., VandeVondele J. J. Chem. Phys. 2015;143:054506. doi: 10.1063/1.4927325. [DOI] [PubMed] [Google Scholar]
- He X., Zhu T., Wang X. W., Liu J. F., Zhang J. Z. H. Acc. Chem. Res. 2014;47:2748–2757. doi: 10.1021/ar500077t. [DOI] [PubMed] [Google Scholar]
- Wang X. W., Liu J. F., Zhang J. Z. H., He X. J. Phys. Chem. A. 2013;117:7149–7161. doi: 10.1021/jp400779t. [DOI] [PubMed] [Google Scholar]
- Liu J. F., He X. Phys. Chem. Chem. Phys. 2017;19:20657–20666. doi: 10.1039/c7cp03356g. [DOI] [PubMed] [Google Scholar]
- Liu J. F., Qi L. W., Zhang J. Z. H., He X. J. Chem. Theory Comput. 2017;13:2021–2034. doi: 10.1021/acs.jctc.7b00149. [DOI] [PubMed] [Google Scholar]
- Liu J. F., He X., Zhang J. Z. H. Phys. Chem. Chem. Phys. 2017;19:11931–11936. doi: 10.1039/c7cp00667e. [DOI] [PubMed] [Google Scholar]
- Marsalek O., Markland T. E. J. Chem. Phys. 2016;144:054112. doi: 10.1063/1.4941093. [DOI] [PubMed] [Google Scholar]
- Skinner L. B., Huang C. C., Schlesinger D., Pettersson L. G. M., Nilsson A., Benmore C. J. J. Chem. Phys. 2013;138:074506. doi: 10.1063/1.4790861. [DOI] [PubMed] [Google Scholar]
- Soper A. K., Benmore C. J. Phys. Rev. Lett. 2008;101:065502. doi: 10.1103/PhysRevLett.101.065502. [DOI] [PubMed] [Google Scholar]
- Willow S. Y., Zeng X. C., Xantheas S. S., Kim K. S., Hirata S. J. Phys. Chem. Lett. 2016;7:680–684. doi: 10.1021/acs.jpclett.5b02430. [DOI] [PubMed] [Google Scholar]
- Holz M., Heil S. R., Sacco A. Phys. Chem. Chem. Phys. 2000;2:4740–4742. [Google Scholar]
- Price D. J., Brooks C. L. J. Chem. Phys. 2004;121:10096–10103. doi: 10.1063/1.1808117. [DOI] [PubMed] [Google Scholar]
- Badyal Y. S., Saboungi M. L., Price D. L., Shastri S. D., Haeffner D. R., Soper A. K. J. Chem. Phys. 2000;112:9206–9208. [Google Scholar]
- Silvestrelli P. L., Parrinello M. Phys. Rev. Lett. 1999;82:3308–3311. [Google Scholar]
- Gregory J. K., Clary D. C., Liu K., Brown M. G., Saykally R. J. Science. 1997;275:814–817. doi: 10.1126/science.275.5301.814. [DOI] [PubMed] [Google Scholar]
- Luzar A., Chandler D. Phys. Rev. Lett. 1996;76:928–931. doi: 10.1103/PhysRevLett.76.928. [DOI] [PubMed] [Google Scholar]
- Kumar S., Rosenberg J. M., Bouzida D., Swendsen R. H., Kollman P. A. J. Comput. Chem. 1995;16:1339–1350. [Google Scholar]
- Makarov V. A., Andrews B. K., Smith P. E., Pettitt B. M. Biophys. J. 2000;79:2966–2974. doi: 10.1016/S0006-3495(00)76533-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. S., Tuckerman M. E. J. Chem. Phys. 2007;126:164501. doi: 10.1063/1.2718521. [DOI] [PubMed] [Google Scholar]
- Habershon S., Markland T. E., Manolopoulos D. E. J. Chem. Phys. 2009;131:024501. doi: 10.1063/1.3167790. [DOI] [PubMed] [Google Scholar]
- Markland T. E., Berne B. J. Proc. Natl. Acad. Sci. U. S. A. 2012;109:7988–7991. doi: 10.1073/pnas.1203365109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Andino R. S., Miller C. M., Chen X., Wilkins D. M., Ceriotti M., Manolopoulos D. E. J. Phys. Chem. C. 2013;117:2944–2951. [Google Scholar]
- Li X. Z., Walker B., Michaelides A. Proc. Natl. Acad. Sci. U. S. A. 2011;108:6369–6373. [Google Scholar]
- Wilkins D. M., Manolopoulos D. E., Pipolo S., Laage D., Hynes J. T. J. Phys. Chem. Lett. 2017;8:2602–2607. doi: 10.1021/acs.jpclett.7b00979. [DOI] [PubMed] [Google Scholar]
- Wu Y. J., Tepper H. L., Voth G. A. J. Chem. Phys. 2006;124:024503. doi: 10.1063/1.2136877. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.