

Abstract
Background and aims
Little is known about specific genetic determinants of carotid-intima-media thickness (CIMT) and carotid plaque in subjects with rheumatoid arthritis (RA). We have used the Metabochip array to fine map and replicate loci that influence variation in these phenotypes in Mexican Americans (MAs) and European Americans (EAs).
Methods
CIMT and plaque were measured using ultrasound from 700 MA and 415 EA patients with RA and we conducted association analyses with the Metabochip single nucleotide polymorphism (SNP) data using PLINK.
Results
In MAs, 12 SNPs from 11 chromosomes and 6 SNPs from 6 chromosomes show suggestive associations (p <1 × 10-4) with CIMT and plaque, respectively. The strongest association was observed between CIMT and rs17526722 (SLC17A2 gene) (β ± SE = −0.84 ± 0.18, p = 3.80 × 10−6). In EAs, 9 SNPs from 7 chromosomes and 7 SNPs from 7 chromosomes showed suggestive associations with CIMT and plaque, respectively. The top association for CIMT was observed with rs1867148 (PPCDC gene, β ± SE = −0.28 ± 0.06, p = 5.11 × 10−6). We also observed strong association between plaque and two novel loci: rs496916 from COL4A1 gene (OR = 0.51, p = 3.15 × 10−6) in MAs and rs515291 from SLCA13 gene (OR = 0.50, p = 3.09 × 10−5) in EAs.
Conclusions
We identified novel associations between CIMT and variants in genes SLC17A2 and PPCDC, and between plaque and variants from COL4A1 and SLCA13 that may pinpoint new candidate risk loci for subclinical atherosclerosis associated with RA.
Keywords: carotid plaque, subclinical atherosclerosis, genetic association, genes, single nucleotide polymorphisms
Introduction
Atherosclerosis is the precursor to coronary heart disease (CHD) and stroke, which is characterized by an accumulation of cholesterol-rich material in the arterial intimal-medial layers [1]. CIMT of the common carotid artery, as measured by high-resolution B-mode ultrasonography, is a useful noninvasive anatomic structural measure of cardiovascular disease [1, 2]. CIMT is not only an excellent surrogate marker of macrovascular atherosclerotic disease, but also widely used to study the early structural changes in the arterial wall including endothelium, connective tissue, and smooth muscle [1, 3]. Several studies have indicated that increased common CIMT is a better indicator of generalized atherosclerosis and coronary artery disease than earlier methods [3–6]. Another diagnostic feature of cardiovascular disease, carotid plaque, is defined as a distinct protrusion, greater than 1.5 mm, into the vessel lumen [7]. Studies have also indicated that the presence of plaque is associated with CHD independent of CIMT [5, 8, 9] even though CIMT includes the site of lipid deposition and plaque formation [1, 2, 10, 11]. Carotid plaque has been shown to be more closely related to coronary artery disease (CAD) and a better measure to predict coronary events than CIMT [5, 12, 13]. Nonetheless, CIMT measurement and carotid plaque assessment are both widely used for identifying subclinical atherosclerotic disease.
Rheumatoid arthritis (RA) is a disease characterized by chronic high-grade systemic inflammation, and a predisposition to accelerated atherosclerosis and cardiovascular events [14–17], with several studies showing increased CIMT in RA patients relative to non-RA controls [18, 19]. We have previously examined the association of carotid atherosclerosis with CV events in the RA cohort and found significant association [20]. In addition to traditional atherosclerosis risk factors, longer duration of RA disease may help predict the development of a severe morphological expression of atherosclerosis [21]. It is now well established that genetic and environmental factors influence CIMT and plaque. While several family studies have reported, moderate to high heritabilities (35%–67% for CIMT [2, 22–24] and low to moderate heritabilities (28% – 47%) for plaque [2, 25, 26], specific causal variants have not been identified, particularly in RA subjects, which would contribute significantly to understanding the mechanisms involved in the RA-mediated development of subclinical atherosclerosis and cardiovascular disease. In genome-wide association studies (GWAS), there have been few attempts to localize genetic variants that influence variation in CIMT and plaque in RA. Despite the identification of several loci for CHD, including ones associated with CIMT and plaque, most have been detected in populations of European ancestry and very few were observed in Mexican Americans, particularly in subjects with RA. Furthermore, most of the GWAS-identified common genetic variants have modest effect sizes, and the functional relevance of most of these variants has not been established [2, 27, 28]. Therefore, identification of causal variants would contribute significantly to our understanding of the mechanisms involved in the RA-mediated development of subclinical atherosclerosis and cardiovascular disease.
In this study, we used the Metabochip, a custom genotyping array for genetic studies of cardiometabolic diseases, to localize genetic variants influencing CIMT and plaque in RA patients from a bi-ethnic United States (US) population representing European Americans (EAs) and Mexican Americans (MAs), which is a major subgroup of Hispanics and the largest minority population in the US. Metabochip array is a valuable tool for replicating and fine-mapping the known cardiometabolic disease-related susceptibility loci, and additionally for localizing susceptibility loci for use as prognostic biomarkers of subclinical atherosclerotic phenotypes such as CIMT and plaque in RA [29, 30].
Patients and Methods
Subjects
We used existing samples/data from the ORALE (Outcome of Rheumatoid Arthritis Longitudinal Evaluation) study, involving 700 unrelated MAs and unrelated 415 EAs. From 1996 to 2009, we recruited consecutive patients who met the 1987 criteria for RA [31] from 11 private and public rheumatology outpatient clinics in San Antonio, Texas, at the time of a scheduled visit with a rheumatologist. All patients participated in a comprehensive baseline evaluation of their clinical characteristics as described in previous publications [32]. Serum IgM rheumatoid factor (RF) and IgG anti–CCP antibodies were measured on a stored serum specimen obtained at the time of the baseline evaluation, using solid phase enzyme immunoassays (TheraTest, Inc., Chicago, IL). For the RF and the anti-CCP, we quantified the antibodies titers to dilution. All research procedures were approved by the Institutional Review Board of the University of Texas Health San Antonio (UTHSA) and a written informed consent was obtained from the patient participants prior to the initiation of evaluations.
Phenotyping
Carotid Ultrasound
A single technician performed a duplex scan of the carotid arteries in all patients, following a standardized vascular protocol developed for the Multi-Ethnic Study of Atherosclerosis. We used an ATL HDI-3000 High Resolution Imaging machine with a L7-4 Transducer (Philips Medical Systems North America Company, Bothell, WA). The technician acquired 4 standardized B-mode images and a Doppler flow measurement from both sides of the neck. The first image was of the distal common carotid artery, and the three others were centered on the site of maximum near and far wall thickening in the proximal internal carotid artery or carotid bulb. Results were recorded on Super VHS tapes and mailed to a central facility (Ultrasound Reading Center, New England Medical Center, Boston, MA), for grading of the carotid artery intima-media thickness and carotid plaque. At the reading center, the images were digitized at 30 frames per second, and arterial diameter fluctuations with the cardiac cycle observed. Images were selected and read by a single, certified reader masked to subject characteristics. The CIMT levels were subjected to inverse normal transformation for the association analyses.
Carotid plaque was identified as a discrete projection of 50% or more from the adjacent wall into the vessel lumen. For CIMT, we measured the end diastole at each of the near and far walls of the right and left common carotid arteries, and the anterior oblique, lateral and posterior oblique views of the internal carotid artery, for a total of 16 CIMT measurements per person. The maximal CIMT of the common and internal carotid arteries were obtained by averaging the maximal measurement from the near and far walls at each projection, from the right and left sides. Then the composite maximal CIMT was calculated by averaging the common and internal carotid maximal CIMT values. The result is one CIMT value per person, expressed in millimeters. Our study involved a single ultrasonographer and a single reader. Nevertheless, to assess the technique’s reliability, our reader re-read 50 images, and a different reader re-read a separate set of 50 images. The intra-reader intraclass correlation coefficient for CIMT was 0.99, and the inter-reader coefficient was 0.94. For plaque, the intra-reader kappa statistic was 1.0, while the inter-reader kappa was 0.94.
Genotyping
The Metabochip (Illumina) is a custom BeadChip targeting 196,725 genetic variants. Common and less common genetic variants were chosen from among the first iteration of the 1000 Genomes Project and represent index GWAS-identified variants regardless of disease or phenotype as of 2009 [30]. As previously described [30], it was primarily designed for fine mapping of metabolic and cardiovascular disease-related loci, and replication of susceptibility loci for specific GWAS-identified regions associated with cardio-metabolic disease and related phenotypes. We performed the genotyping according to the Illumina protocol and initial data handling and analysis was performed using Genome Studio v1.7.4 (Illumina).
Sample and SNP Quality Control Measures
Several quality control measures were applied to the genotypic data of each ethnic group, and only the autosomal SNPs that passed QC were considered for this study. Subjects with low call rates (< 0.95) were removed (MA = 13 and EA = 0). To identify and exclude highly related individuals or duplicate samples, we performed the relationship inference analytical procedure as implemented in the computer program KING [Kinship-based Inference for Genome-wide association studies, [33]] and identified related individuals. Subsequently, using the program PLINK [34] and the identity-by-descent (IBD) analysis, closely related individuals up to 3rd degree relatives (IBD > 0.185) were removed (MA = 17 and EA = 3). To detect ethnic outliers, we used EIGENSTRAT c3.0 software package [35] to employ principal components analysis to a subset of autosomal SNPs in our data that were in low LD (r2 < 0.2) and the HapMap samples as reference for the ethnic groups. Plots were generated using the first two principal components (PCs) for visual inspection. Using our data by ethnic group, samples were identified as population outliers, defined by 4SD from the mean of each of the 2 PCs that explained the majority of variation in the data, and were subsequently removed (MA = 2 and EA = 0). SNPs with a genotyping call rate less than 95% were removed using PLINK [34]. In addition, SNPs with Hardy-Weinberg Equilibrium (HWE) values of p < 10−4 [(MA = 236 (CIMT) and 120 (plaque); and EA = 114 (CIMT), and 38 (plaque)] and with minor allele frequency (MAF) < 0.01 (MA = 57, 323 and EA = 60, 168) were removed from the analysis. After filtering and genotyping pruning, 122,549 SNPs from 668 MAs and 120,827 from 415 EAs were remained in the association analyses.
Statistical Genetic Analyses
We performed association analyses between the transformed CIMT as a quantitative trait and SNP genotypes in both MA and EA samples after QCs, using PLINK software version 1.07[34]. Principal Components (PCs) were derived using EIGENSTRAT principal component analysis [35] to adjust for potential population stratification influences. A linear regression additive genetic model (SNPs coded as 0,1, or 2 based on the minor allele dosage) adjusted for the effects of covariates age, sex, RA duration, medication status (statin use, and hypertension, [htn, medication]), and the first two PC1 and PC2, was used for association testing of CIMT, a quantitative trait. Association statistics for plaque, a discrete trait, were calculated using logistic regression assuming an additive model. Manhattan plots were constructed using the computer programs PLINK and Haploview and the regional association plots were generated using the program LocusZoom [36] and the Q-Q plots were done using Efficient and Parallelizable Association Container Toolbox software [EPACTS, http://genome.sph.umich.edu/wiki/EPACTS]. Association p values were adjusted for multiple testing using the conservative Bonferroni correction: 4.08 × 10−7 for MA and 4.14 × 10−7 for EA. Using Haploview, combined p values were obtained using Fisher’s method. Linkage disequilibrium (LD) between SNP pairs was estimated using r2 values.
Polygenic Risk Score (PRS)
To further investigate the genetic architecture of CIMT and plaque, PRS analyses were conducted, with scores representing summations of CIMT- and plaque-associated alleles from the Metabochip array. Scoring routines were determined from the association test results for the MA cohort, with risk alleles identified based on varying p-value thresholds (1,000 different p-value (Pts) thresholds, representing increments of p = 0.001), each weighted by their estimated effect sizes on CIMT or plaque. Scores were then computed in the independent EA cohort, and evaluated as predictors of CIMT or plaque via linear regression models (covariates age, sex, PCs 1 and 2, RA duration, statin use, and htn). SNPs were initially pruned using PLINK's clumping methodology based on linkage disequilibrium (LD; r2 = 0.1), distance (250 Kb), and association P-values for CIMT or plaque in the MA cohort (http://pngu.mgh.harvard.edu/~purcell/plink/clump.shtml), reducing the number of SNPs utilized in the scoring routines from 122,549 to 36,630 for the CIMT data, and 122,649 to 36,748 for the plaque data. This work was performed using PRSice v.1.23, a polygenic risk score software [37].
Results
We analyzed Metabochip array data for 668 Mexican American (MA) and 415 European American (EA) individuals with RA from the ORALE study using association analyses by ethnic group to identify genetic variants that contribute to variation in CIMT (inverse normalized) and plaque, which are the best surrogates for atherosclerosis and predictors of risk of stroke and myocardial infarction. The characteristics of the study participants are shown in Table 1. The mean age of MAs was 59.3 years (females 79.0%), whereas for EAs, it was 64.4 years (females 65.1%). The mean CIMT value was lower in MAs (0.98) than in EAs (1.15), and the average RA duration was slightly longer in EAs (16.7 years) compared to that in MAs (15.1 years). In contrast, 53.2% MAs had plaque while EAs showed slightly higher occurrence of plaque (64.2%). Using EPACTS, we generated the quantile-quantile (Q-Q) plots of the transformed CIMT in both MAs and EAs (Supplementary Figure 1), where the genomic inflation factor (λ) was calculated to be 1.009 (CIMT) and 1.022 (plaque) for MA data and 1.008 (CIMT) and 1.0 (plaque) for EA data. Thus Q-Q plots exhibit a roughly straight line through the origin with a unit slope indicating almost no inflation.
Table 1.
Characteristics of ORALE Study Subjects with Rheumatoid Arthritis (RA)
| Variables | Mean ± SD or n, % | |
|---|---|---|
| Population | Mexican Americans (MAs) | European Americans (EAs) |
| No. of subjects phenotyped and genotypeda | 700 | 414 |
| Age (years) | 59.3 ± 11.3 | 64.4 ± 11.3 |
| Females | 79.0 % | 65.1 % |
| RA durationb (years) | 15.1 ± 10.3 | 16.7 ± 11.2 |
| IgM rheumatoid factor (RF) (IU/ml) | 765.5 ± 1643.7 | 410.6 ± 946.4 |
| IgM RF ≥ 25 (IU/ml)c | 637 (88.4%) | 333 (78.5%) |
| Anti-CCP (IU/ml)d | 390.2 ± 1468.3 | 252.7 ± 358.3 |
| Anti-CCP > 6.9 (IU/ml) | 597 (82.9%) | 319 (75.2%) |
| CIMTe | 0.98 ± 0.44 | 1.15 ± 0.59 |
| CIMTf | − 0.12 ± 0.92 | 0.17 ± 1.01 |
| Plaque | 53.2% | 64.2% |
After sample and SNP QC analyses, the phenotypic and genotypic (i.e., SNPs with MAF < 0.01 were excluded) data available for association analysis by ethnic group;
Rheumatoid Arthritis (RA) duration;
IgM RF = IgM rheumatoid factor (RF);
Anti-CCP = Anti-cyclic citrullinated peptide;
CIMT original values;
CIMT values were inverse-normalized for association analyses.
Association results of the transformed CIMT after adjustment for covariate effects in MAs are shown in Figure 2. As reported in Table 2a, we identified 12 SNPs from 11 chromosomes that exhibited association with CIMT in MAs at p < 1 × 10−4, and the p values ranged from 9.95 × 10−5 to 3.80 × 10−6. The best association in our MA data was observed with the marker rs17526722 (β ± SE = −0.84 ± 0.18, p = 3.80 × 10−6), an intronic variant within the SLC17A2 (Solute Carrier Family 17, member 2) gene on chromosome 6 (6p22.2, MIM: 611049). The mean CIMT by genotype categories were, −0.80/AG, and 0.01/GG, respectively. Although the minor allele homozygotes were absent, the carriers of the minor allele A were found to have reduced CIMT, compared to the major allele (G), indicating that the minor allele is associated with reduced CIMT and therefore the variants appear to have a protective effect. The regional association plot containing the lead SNP (rs17526722) associated with CIMT in MAs is depicted in Figure 1.C (a). Loci associated with plaque in MAs are shown in Figure 1B and Table 2b. We identified 6 SNPs from chromosomes 1, 6, 9, 13, 15, and 22, showing associations at p <1 × 10−4, and the p-values ranged from 9.76 × 10−5 to 3.15 × 10−6. The top SNP (rs496916; OR (95% CI) = 0.514 (0.39 – 0.68), p = 3.15 × 10−6) is an intronic variant within COL4A1 (collagen type IV alpha 1 chain) gene on chromosome 13 (13q34, MIM: 120130). The regional association plot for rs496916 is depicted in Figure 1. C (b).
Figure 2.
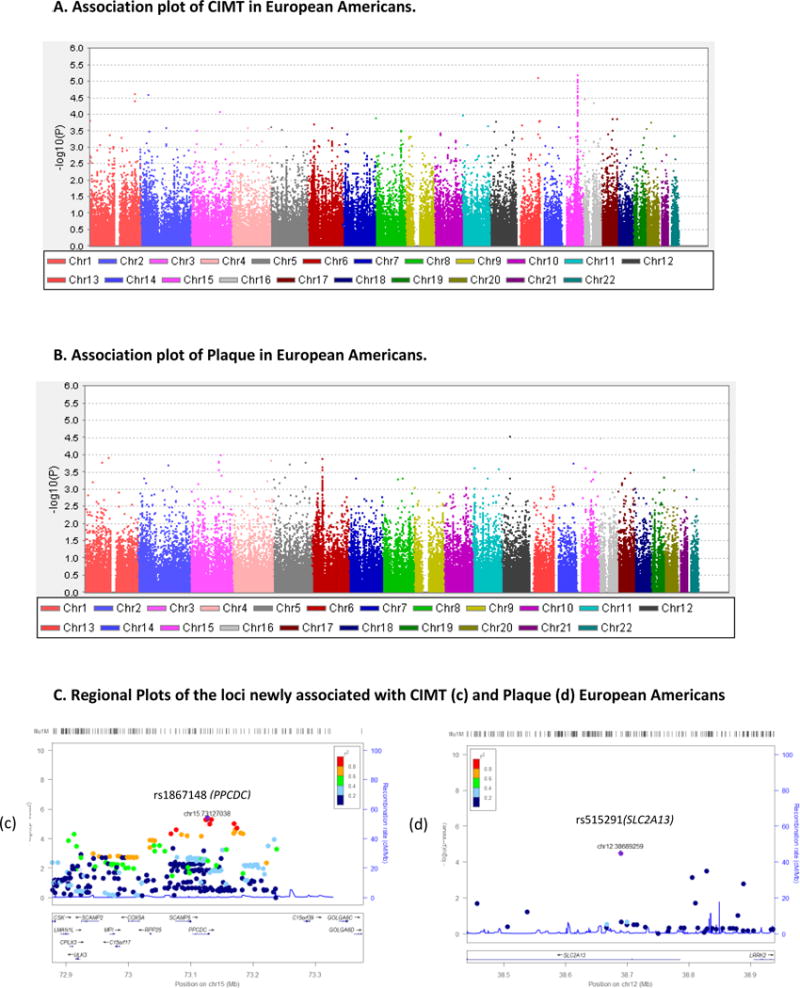
CIMT and Plaque Association and Regional Plots in European Americans
Table 2.
| a. Genetic Variants Associated with Carotid IMT in Mexican Americans (p < 1.0E-4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chr. | SNP | Position, bpa | Gene/Nearest Gene | Loc. | A1b | MAFc | BETA | SE | Pf |
| 6 | rs17526722 | 26026834 | SLC17A2 | I | A | 0.10 | −0.8377 | 0.1797 | 3.80E-06 |
| 2 | rs4894108 | 180112792 | ZNF385B | I | G | 0.19 | −0.2501 | 0.05751 | 1.59E-05 |
| 17 | rs2672901 | 76411261 | KIAA1303 | I | A | 0.32 | − 0.1929 | 0.04489 | 2.00E-05 |
| 13 | rs12873154 | 109718853 | COL4A1 | I | G | 0.20 | −0.3918 | 0.09165 | 2.21E-05 |
| 10 | rs61850526 | 63190012 | C10orf107 | I | T | 0.02 | −0.4624 | 0.1126 | 4.56E-05 |
| 11 | rs76599700 | 61401283 | FADS3 | I | T | 0.26 | −0.7317 | 0.1814 | 6.17E-05 |
| 16 | rs11860529 | 82330023 | CDH13 | I | T | 0.08 | 0.4112 | 0.1028 | 7.06E-05 |
| 1 | rs17436982 | 219622889 | HLX/DUSP10 | IG | T | 0.22 | 0.2827 | 0.07173 | 8.98E-05 |
| 5 | rs250216 | 50317115 | PARP8/LOC642366 | IG | C | 0.07 | 0.2322 | 0.05899 | 9.20E-05 |
| 7 | rs11761467 | 27828130 | TAX1BP1 | I | T | 0.10 | −0.3244 | 0.08256 | 9.44E-05 |
| 6 | rs11966018 | 12317214 | HIVEP1/EDN1 | IG | C | 0.03 | 0.691 | 0.1763 | 9.82E-05 |
| 15 | rs7177074 | 97357475 | LOC145814 | I | A | 0.05 | −0.5138 | 0.1312 | 9.95E-05 |
| b. Genetic Variants Associated with Plaque in Mexican Americans (p < 1.0E-4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chr. | SNP | Position, bpa | Gene/Nearest Gene | Loc. | A1b | MAFc | ORd | 95% CIe | Pf |
| 13 | rs496916 | 109649015 | COL4A1 | I | C | 0.41 | 0.514 | 0.39–0.68 | 3.15E-06 |
| 15 | rs9806753 | 46953709 | SHC4/EID1 | IG | A | 0.26 | 1.739 | 1.27–2.21 | 6.71E-06 |
| 6 | rs9463110 | 12890588 | PHACTR1 | I | G | 0.44 | 1.699 | 1.3–2.16 | 1.45E-05 |
| 22 | rs2092179 | 38364539 | CACNA1I | I | C | 0.25 | 0.588 | 0.46 – 0.77 | 3.85E-05 |
| 9 | rs7869506 | 98127033 | SLC35D2 | I | T | 0.26 | 1.688 | 1.31–2.18 | 5.41E-05 |
| 1 | rs6667860 | 36730800 | CSF3R/GRIK3 | IG | C | 0.48 | 1.658 | 1.29–2.14 | 9.76E-05 |
Chr. = chromosome; SNP = single nucleotide polymorphism;
Based on National Center for Biotechnology Information (NCBI) Build36/130 (hg18); Loc = Location; I = Intron; IG = Intergenic;
A1 = minor allele;
MAF = minor allele frequency;
OR = Odds Ratio;
CI = Confidence Interval;
p values ranked from low to high.
Figure 1.
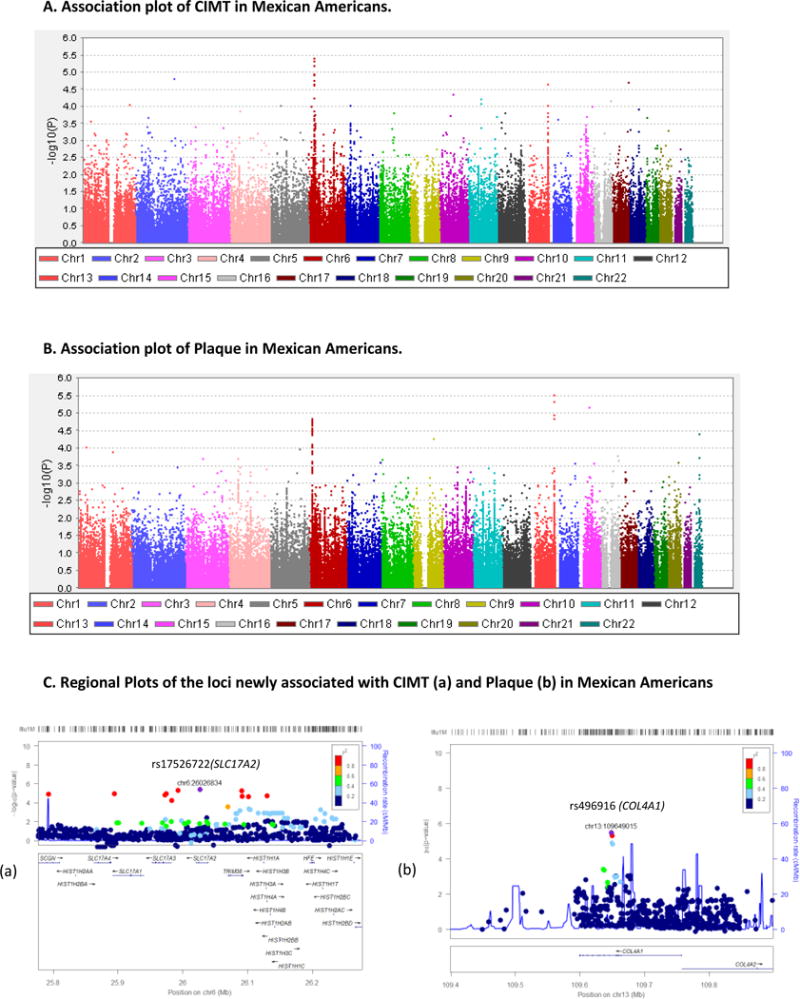
CIMT and Plaque Association and Regional Plots in Mexican Americans
The association findings related to EAs are depicted in Figure 2 and Table 3. As can be seen from Table 3a Figure 2A (a), after adjusting for covariate effects, 9 SNPs from chromosomes 1, 2, 6, 11, 13, 15, and 16, showed association with CIMT at <1 × 10−4, and the p values ranged from 9.45 × 10−5 to 5.11 × 10−6. The top associated SNP was rs1867148 (β ± SE = − 0.280 ± 0.06, p = 5.11 × 10−6), an intronic variant located in the PPCDC (Phosphopantothenoylcysteine Decarboxylase) gene on chromosome 15 (15q24.2, MIM: 609854). Of the 9 SNPs, 3 have low minor allele frequencies (MAFs), ranging from 0.01–0.05. For example, in the best associated marker rs1867148, as revealed by the mean CIMT by genotype classes (i.e., −0.19/CC, 0.20/CT, and 0.43/TT), the carriers of the low frequency allele C were found to have lower CIMT. Figure 2C (c) shows the regional association plot containing rs1867148 associated with CIMT in EAs. Top two SNPs (rs1867148 and rs7163636) associated with CIMT in EAs are in complete LD (r2 = 1). As shown in Table 3b and Figure 2A (b), 7 loci associated with plaque in EAs, showed moderate associations (p<1 × 10−4). The top SNP (rs515291; OR = 0.4987, 95% CI= 0.36–0.69, p 3.09 × 10−5) is an intronic variant within the SLC2A13 (solute carrier family 2 member 13) gene on chromosome 12q12 (MIM: 611036). As shown in Figure 2C (d), the regional association plot shows the SNPs that are associated with plaque in EAs include the lead SNP rs515291 (SLC2A13) followed by an intergenic variant (rs10501399) located between MYEOV-CCND1 genes on chromosome 11.
Table 3.
| a. Genetic Variants Associated with Carotid IMT in European Americans (p < 1.0E-4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chr. | SNP | Position, bpa | Gene/Nearest Gene | Loc. | A1b | MAFc | BETA | SE | Pf |
| 15 | rs1867148 | 73127038 | PPCDC | I | C | 0.44 | −0.2804 | 0.06064 | 5.11E-06 |
| 13 | rs323453 | 104232379 | LOC728183/DAOA | IG | G | 0.34 | −0.2724 | 0.06346 | 2.23E-05 |
| 1 | rs4846566 | 217797888 | LOC728510/ZC3H11B | IG | T | 0.01 | −1.186 | 0.2777 | 2.46E-05 |
| 2 | rs12987042 | 38518674 | ARL6IP2/RPLPO-like | IG | A | 0.36 | −0.2608 | 0.0619 | 3.13E-05 |
| 15 | rs6495122 | 72912698 | CPLX3/ULK3 | IG | A | 0.42 | −0.2525 | 0.06149 | 4.90E-05 |
| 1 | rs2645091 | 2214505 | SKI | I | T | 0.15 | −0.3511 | 0.08741 | 7.07E-05 |
| 16 | rs17821532 | 52504199 | FTO | I | A | 0.05 | −0.7006 | 0.1751 | 7.58E-05 |
| 6 | rs7742814 | 119185974 | C6orf204/ASF1A | IG | G | 0.36 | 0.2555 | 0.06462 | 9.09E-05 |
| 11 | rs4387380 | 5824023 | OR52E6/OR52E8 | IG | C | 0.03 | −0.8226 | 0.2085 | 9.45E-05 |
| b. Genetic Variants Associated with Plaque in European Americans (p < 1.0E-4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chr. | SNP | Position, bpa | Gene/Nearest Gene | Loc | A1b | MAFc | ORd | 95% CIe | Pf |
| 12 | rs515291 | 38689259 | SLC2A13 | I | G | 0.32 | 0.4987 | 0.36–0.69 | 3.09E-05 |
| 11 | rs10501399 | 68979039 | MYEOV/CCND1 | IG | T | 0.09 | 0.2938 | 0.16–0.52 | 3.34E-05 |
| 15 | rs692390 | 39274282 | EXDL1 | I | A | 0.19 | 0.4138 | 0.27–0.63 | 4.30E-05 |
| 5 | rs6887230 | 148706339 | GRPEL2 | I | G | 0.30 | 2.02 | 1.4–2.8 | 5.61E-05 |
| 17 | rs2070776 | 59361230 | CD79B | C (ns) | T | 0.33 | 0.5033 | 0.36–0.71 | 6.68E-05 |
| 16 | rs3785233 | 7607511 | A2BP1 | I | C | 0.16 | 0.419 | 0.27–0.65 | 9.49E-05 |
| 6 | rs10948573 | 50800310 | TFAP2D | I | G | 0.37 | 2.044 | 1.4–2.9 | 9.68E-05 |
Chr. = chromosome; SNP = single nucleotide polymorphism;
Based on NCBI Build36/130 (hg18); Loc = Location; I = Intron; IG = Intergenic;
A1 = minor allele;
MAF = minor allele frequency;
OR = Odds Ratio;
CI = Confidence Interval;
p values ranked from low to high.
Although a majority of the genetic variants contributing to variation in CIMT and plaque in MAs and EAs appear to be population-specific, some of the associated loci were commonly found in both populations. As shown in Table 4, six SNPs were found with combined p values < 1 × 10−5, where the same SNPs were found to be associated with both CIMT and plaque at p of < 0.05 in both MAs and EAs. The best associated marker was chr15:73071294, which was more strongly associated in EAs compared with MAs and the minor allele appears to have a protective effect. Of the top SNPs shown to be associated with CIMT in MAs (Table 2), none of the SNPs showed association with CIMT in EAs (Table 3) and vice versa. Furthermore, in our study, none of the SNPs survived the conservative Bonferroni’s correction, which required a significance threshold of 4.1 × 10−7 by ethnicity. On the other hand, of the 23 moderately associated SNPs (combined p < 1.0 × 10−4) with CIMT in MAs (not reported), only 4 SNPs were found to be jointly associated (p < 0.05) in both MAs and EAs. Whereas for plaque, of the 37 strongly associated SNPs (combined p < 1.0 × 10−4) in MAs (not reported), only 8 markers were found to be jointly associated with plaque (p < 0.05) in both MAs and EAs.
Table 4.
Common Association Signals with CIMT and Plaque in both Mexican American and European Americans
| Chr. | SNP | Location, bpa | Gene/Nearest Gene | Type | Mexican Americans | European Americans | Combined p valuec | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1b | MAF | β-SNP (SE) | p-value | A1b | MA F | β-SNP (SE) | p-value | ||||||
| 15 | chr15:73071294 | 73071294 | RPP25/SCAMP5 | IG | A | 0.02 | −0.5279 (0.1834) |
0.004133 | A | 0.02 | −0.7115 (0.2012) |
4.54×10−4 | 2.66×10−5 |
| 11 | rs582037 | 125723080 | DCPS/ST3GAL4 | IG | A | 0.40 | 0.1308 (0.0386) |
0.005611 | A | 0.33 | 0.234 (0.0648) |
3.48×10−4 | 2.76 × 10−5 |
| 3 | rs4839634 | 144619509 | SLC9A9 | I | G | 0.15 | 0.1592 (0.0633) |
0.01215 | G | 0.14 | 0.3274 (0.0897) |
2.96×10−4 | 4.87 × 10−5 |
| Plaque | OR (SE) | OR (SE) | |||||||||||
| 1 | rs6694848 | 63276194 | LOC199897/FOXD3 | IG | A | 0.33 | 1.516 (0.1309) |
0.001473 | A | 0.46 | 1.782 (0.1718) |
7.69×10−4 | 1.66×10−5 |
| 3 | rs9809344 | 135825448 | KY | I | T | 0.23 | 0.7201 (0.1428) |
0.02147 | T | 0.39 | 0.5426 (0.1676) |
2.64×10−4 | 7.41×10−5 |
| 13 | rs7327080 | 22689882 | SGCG | I | G | 0.33 | 1.404 (0.1259) |
0.007004 | G | 0.32 | 0.5607 (0.1774) |
0.001111 | 9.93×10−5 |
Chr. = chromosome; SNP = single nucleotide polymorphism; MAF = minor allele frequency;
Based on National Center for Biotechnology Information (NCBI) Build36 (hg18);
A1 = minor allele;
combined p values ranked from low to high;
As shown in Supplementary Figure S2, the high-resolution plots (S2.a and S2.b) for cumulative 0.001 p-value thresholds (PTs) reveal major peaks at pt < 0.054 (one-sided p = 0.0177; β = 104.81 (SE = 49.66); R2 = 0.0073; n = 3,159 risk alleles) and pt < 0.002 (one-sided p = 0.020; β = 17.43 (SE = 8.30); R2 = 0.0131; n = 123 risk alleles) for CIMT and plaque, respectively. Although accounting for a small portion of CIMT and plaque variation in EAs, the PRSs based on MA data suggest underlying genetic architecture that is, at least in part, common to both MAs and EAs. This is reflected in the nominally significant SNP associations (p < 0.05) observed in the two populations, as noted above.
Discussion
We conducted a population-based CIMT and plaque association analysis in a bi-ethnic population of MAs and EAs with RA. We sought to assess the extent to which fine-mapped regions of multiple cardiometabolic disease related association signals, containing common and low frequency/rare variants, have relevance to susceptibility to CV risk factors and CV events. Our study by ethnic group revealed several loci suggestively associated (i.e., p < 1 × 10−4) with CIMT and plaque after adjusting for the influences of age, sex, RA duration and medical status, and population stratification, which were found to be mostly ethnic-specific. We found novel associations with loci on chromosomes 6 and 13 that were suggestively associated with CIMT and plaque in MAs and on chromosomes 15 and 12 that were suggestively associated with CIMT and plaque in EAs, respectively. In MAs, CIMT showed the best association with marker rs17526722, an intronic SNP in the SLC17A2 gene, and the carriers of the minor allele were found to have lower CIMT. This SNP is in strong LD with the marker rs36014129 in the gene SLC17A3, which has been shown to be suggestively associated with CIMT in a meta-analysis of GWASs from the CHARGE consortium [2]. This gene belongs to the sodium/anion cotransporter family and involved in sodium dependent phosphate transport. It has been associated with uric acid levels in recent GWASs [38, 39], which have in turn been shown to be associated with cardiovascular disease although their role in subclinical atherosclerosis is yet to be established [40]. Interestingly, this SNP was one of the 10 SNPs on chromosome 6 suggestively associated with CIMT that were located in an extended LD region containing the SLC17A2, SLC17A3-SLC17A2, TRIM38, SLC17A1, SLC17A3, SCGN, HIST1H3A-HIST1H4A, and TRIM38-HIST1H1A genes (Supplementary Table S1). SLC17A2 is associated with diseases such as nephrolithiasis/osteoporosis, hypophosphatemic, 1 and fanconi renotubular syndrome 2 (http://www.genecards.org). Some of the variants from genes (SLC17A1, SLC17A2, SCGN, TRIM38) of this region have been shown to be associated (p < 5 × 10−5) with serum iron phenotypes in premenopausal women of European descent [41].
Of the 6 loci suggestively associated with plaque in MAs, evidence for strong association with the marker rs496916 in the gene COL4A1 suggests a potential role in subclinical atherosclerosis in MAs. This gene encodes a type IV collagen alpha protein, which is an integral component of basement membrane and is involved in angiogenesis and blood vessel morphogenesis [42]. Notably, its functions include conferring elasticity to extracellular matrix and binding platelet derived growth factor. Mutations in this gene have been associated with cerebrovascular disease, and renal and muscular defects (http://www.genecards.org). It is interesting to note that two other SNPs from this gene have been previously shown to be associated with incident coronary heart disease in African Americans [29]. In addition, certain genetic variants in the COL4A1-COL4A2 gene region represented on the Metabochip were shown to be associated with CHD in the PAGE African American individuals [29] as well as in individuals with European ancestry. Another gene (PHACTR1) region on chromosome 6p24.1 (MIM 608723) showing suggestive evidence of association with plaque is also a replication of earlier association with CHD in African American individuals [29]. Although these replications of reported associations with CHD are interesting, their specific role in atherosclerotic vascular disease is not clear.
In EAs, a strong association of CIMT was observed with marker rs1867148, which is in the PPCDC gene, and carriers of the minor allele (C) were found to have decreased CIMT. This gene is necessary for the biosynthesis of coenzyme A from pantothenic acid (Vitamin B5) and may be indirectly involved in zinc metabolism [43]. Notably, lowered zinc levels are associated with higher IMT scores [44]. Thus, the implicated gene region appears to be involved in the subclinical atherosclerosis in EAs, though the exact function is not known. However, a SNP (rs6495122) from the CPLX3 gene (MIM 609585) region was suggestively associated with CIMT in EAs and has been reported to be associated with blood pressure and hypertension in individuals with European ancestry [45].
For plaque in EAs, we found several loci with suggestive associations with the lead SNP from SLC2A13 gene, which is a H(+)/myo-inositol cotransporter that plays an important role in various cellular processes such as transporting glucose and sugars, bile salts and organic acids, metal ions and amine compounds, involved in glucose metabolism, and exhibits insulin-mimetic properties [46]. Abnormalities of inositol metabolism are associated with insulin resistance and microvascular complications of diabetes [46]. A second region of interest showing suggestive evidence of association with plaque in EAs is on chromosome 11 marked by an intergenic SNP (rs10501399) located between MYEOV and CCND1 genes [47]. These genes are involved in the proliferation of human coronary artery smooth muscle cells (HCASMCs) such as cardiac fibroblasts and deficient proliferation of other cell types (e.g. vascular endothelial cells) [48].
However, despite the correlation between CIMT and plaque, different trait-specific genetic determinants can be expected due to their differing pathobiology and associated phenotypic severity of plaque as shown by our earlier study as well as other studies [49, 50]. In addition, genetic differences are expected between the populations of European background and admixed populations such as the Mexican Americans that have both European and Native American ancestries. As revealed by our study, top SNPs that are associated with CIMT and plaque are different in significance levels but exhibit associations with either phenotype at nominal significance levels (p < 0.05) as shown in the revised tables S2–S5 in the supplementary material for review purpose. As noted above, it is well known that some variants exhibit unique associations with a given phenotype (CIMT or plaque) while other variants exhibit common associations with both phenotypes (CIMT and plaque). Furthermore, results from previous studies also support our findings [51, 52]. Thus, common variants in several genes exhibited significant associations with CIMT and plaque in both MAs and EAs. These findings may help understand the genetic architecture of subclinical atherosclerosis in these populations.
Finally, the significant results from the polygenic risk scoring for CIMT and plaque indicate the following: 1) the SNP-based association results for CIMT and plaque for the MA cohort were found to be predictive of CIMT levels in the independent EA dataset in an aggregate manner; 2) parts of the allelic architecture underlying CIMT and carotid plaque are common to both study populations; and 3) the best-fit PRS models for EA CIMT measures and plaque diagnoses occur at low association PTs in the MA dataset, especially for plaque, suggesting that potential causal alleles with smaller, interethnic effect sizes collectively play a more limited role than they do in other complex, highly polygenic phenotypes, such as psychiatric disorders, where the best-fit PRS models occur at substantially higher thresholds [53, 54]; and 4) since direct replication of findings is lacking, PRS association findings with CIMT in both MAs and EAs may be indicative of evidence for the presence of common loci associated with CIMT in both populations.
Our study also suggests that the genetic variants contributing to variation in CIMT and plaque are largely population-specific. However, PRS showed a significant, although small, shared polygenic component for CIMT. Importantly, we were able to replicate association signals for chromosomal regions on 6, 12 and 13 influencing CIMT from a large GWAS meta-analysis despite the use of relatively small sample sizes. Most of these associations are with either intergenic or intronic variants, suggesting that they may be tagging nearby rare variants that were not detected by population based GWAS[4]. Limitations include the availability of relatively modest samples of RA patients, particularly EAs. Our ability to search for preclinical atherosclerotic risk variants was confined to the pre-selected genomic regions on the Metabochip.
In summary, this genetic association study of CIMT and plaque implicated some known genetic risk loci for subclinical atherosclerosis, and highlighted a subset of the loci previously associated with cardiovascular disease as potential molecular drivers of accelerated atherosclerosis features, and discovered new genetic variants associated with premature cardiovascular disease components. These novel findings provide additional insights into the pathophysiology of subclinical atherosclerosis in RA that will require validation and functional investigation. Strengths of our study include the use of both CIMT and plaque as surrogates for subclinical atherosclerosis from the RA patient sample of the well-characterized ORALE study’s bi-ethnic population in the United States (US), involving Mexican Americans and European Americans. Subclinical atherosclerotic phenotype such as CIMT can offer greater power for gene localization/identification than a dichotomous disease status. To our knowledge, this is the first study to localize risk loci contributing to variation in CIMT and plaque in a US bi-ethnic RA population.
Supplementary Material
Highlights.
-
-
Identified new loci associated with CIMT and plaque in RA patients
-
-
Known genetic risk loci implicated in subclinical atherosclerosis
-
-
Evidence for loci as potential molecular drivers of accelerated atherosclerosis
-
-
Replicated previous associations with CIMT and plaque
-
-
Polygenic Risk Score analysis found common loci for CIMT and Plaque in MAs and EAs.
Acknowledgments
We sincerely thank the participants of the ORALE study.
Financial Support: This work was supported by grants from the US National Institutes of Health (R01-HL-085742, R01-HD-037151, and UL1-RR-25767).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributors: All authors reviewed and approved the final version of the manuscript. RA, AE, and IdR have full access to both genotypic and phenotypic data of this study and take full responsibility for the accuracy of the data. AE, IdR, conceived, and obtained funding for the study. EA, IdR, JFR, and DFB were involved in patient recruitment, acquisition of phenotypic data, and monitored data collection for the whole study. RA, RD, AE, IdR VSF were involved in study conception and design. VSF, SK, RA, RD, MdA, JC, DW, MF performed Metabochip genotyping, developed quality control measures, and performed molecular genetic analyses. RA, VSF, AE, RD, MdA, IdR, CPJ, SM, JB, MZK were involved in statistical genetic analyses and interpretation of results, and RA drafted the manuscript.
Ethics approval: All procedures performed in this study involving human participants were in accordance with the ethical standards of the Institutional Review Board, UT Health San Antonio.
Conflict of Interest:
None of the authors had a conflict of interest
References
- 1.Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006;47(8 Suppl):C7–12. doi: 10.1016/j.jacc.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 2.Bis JC, et al. Meta-analysis of genome-wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nat Genet. 2011;43(10):940–947. doi: 10.1038/ng.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lorenz MW, et al. Prediction of clinical cardiovascular events with carotid intima-media thickness: a systematic review and meta-analysis. Circulation. 2007;115(4):459–467. doi: 10.1161/CIRCULATIONAHA.106.628875. [DOI] [PubMed] [Google Scholar]
- 4.Melton PE, et al. Genetic architecture of carotid artery intima-media thickness in Mexican Americans. Circ Cardiovasc Genet. 2013;6(2):211–221. doi: 10.1161/CIRCGENETICS.113.000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nambi V, et al. Carotid intima-media thickness and presence or absence of plaque improves prediction of coronary heart disease risk: the ARIC (Atherosclerosis Risk In Communities) study. J Am Coll Cardiol. 2010;55(15):1600–1607. doi: 10.1016/j.jacc.2009.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naqvi TZ, Lee MS. Carotid intima-media thickness and plaque in cardiovascular risk assessment. JACC Cardiovasc Imaging. 2014;7(10):1025–38. doi: 10.1016/j.jcmg.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Juanatey C, et al. Increased prevalence of severe subclinical atherosclerotic findings in long-term treated rheumatoid arthritis patients without clinically evident atherosclerotic disease. Medicine (Baltimore) 2003;82(6):407–413. doi: 10.1097/01.md.0000101572.76273.60. [DOI] [PubMed] [Google Scholar]
- 8.Beecham GW, et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014;10(9):e1004606. doi: 10.1371/journal.pgen.1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathiesen EB, et al. Carotid plaque area and intima-media thickness in prediction of first-ever ischemic stroke: a 10-year follow-up of 6584 men and women: the Tromso Study. Stroke. 2011;42(4):972–978. doi: 10.1161/STROKEAHA.110.589754. [DOI] [PubMed] [Google Scholar]
- 10.Insull W., Jr The pathology of atherosclerosis: plaque development and plaque responses to medical treatment. Am J Med. 2009;122(1 Suppl):S3–S14. doi: 10.1016/j.amjmed.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Isaacs A, et al. Risk scores of common genetic variants for lipid levels influence atherosclerosis and incident coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33(9):2233–2239. doi: 10.1161/ATVBAHA.113.301236. [DOI] [PubMed] [Google Scholar]
- 12.Brook RD, et al. A negative carotid plaque area test is superior to other noninvasive atherosclerosis studies for reducing the likelihood of having underlying significant coronary artery disease. Arterioscler Thromb Vasc Biol. 2006;26(3):656–662. doi: 10.1161/01.ATV.0000200079.18690.60. [DOI] [PubMed] [Google Scholar]
- 13.Spence JD. Measurement of intima-media thickness vs. carotid plaque: uses in patient care, genetic research and evaluation of new therapies. Int J Stroke. 2006;1(4):216–221. doi: 10.1111/j.1747-4949.2006.00068.x. [DOI] [PubMed] [Google Scholar]
- 14.del Rincon ID, et al. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44(12):2737–2745. doi: 10.1002/1529-0131(200112)44:12<2737::AID-ART460>3.0.CO;2-%23. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Gay MA, Gonzalez-Juanatey C, Martin J. Rheumatoid arthritis: a disease associated with accelerated atherogenesis. Semin Arthritis Rheum. 2005;35(1):8–17. doi: 10.1016/j.semarthrit.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez-Juanatey C, et al. Carotid intima-media thickness predicts the development of cardiovascular events in patients with rheumatoid arthritis. Semin Arthritis Rheum. 2009;38(5):366–371. doi: 10.1016/j.semarthrit.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Maradit-Kremers H, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum. 2005;52(2):402–411. doi: 10.1002/art.20853. [DOI] [PubMed] [Google Scholar]
- 18.Del R I, et al. Association between carotid atherosclerosis and markers of inflammation in rheumatoid arthritis patients and healthy subjects. Arthritis Rheum. 2003;48(7):1833–1840. doi: 10.1002/art.11078. [DOI] [PubMed] [Google Scholar]
- 19.Tyrrell PN, et al. Rheumatic disease and carotid intima-media thickness: a systematic review and meta-analysis. Arterioscler Thromb Vasc Biol. 2010;30(5):1014–1026. doi: 10.1161/ATVBAHA.109.198424. [DOI] [PubMed] [Google Scholar]
- 20.Evans MR, et al. Carotid atherosclerosis predicts incident acute coronary syndromes in rheumatoid arthritis. Arthritis Rheum. 2011;63(5):1211–20. doi: 10.1002/art.30265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abu-Shakra M, et al. Duplex study of the carotid and femoral arteries of patients with rheumatoid arthritis: a controlled study. Semin Arthritis Rheum. 2005;35(1):18–23. doi: 10.1016/j.semarthrit.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Duggirala R, et al. Genetic basis of variation in carotid artery wall thickness. Stroke. 1996;27(5):833–837. doi: 10.1161/01.str.27.5.833. [DOI] [PubMed] [Google Scholar]
- 23.Fox CS, et al. Genetic and environmental contributions to atherosclerosis phenotypes in men and women: heritability of carotid intima-media thickness in the Framingham Heart Study. Stroke. 2003;34(2):397–401. doi: 10.1161/01.str.0000048214.56981.6f. [DOI] [PubMed] [Google Scholar]
- 24.Wang D, et al. A genome-wide scan for carotid artery intima-media thickness: the Mexican-American Coronary Artery Disease family study. Stroke. 2005;36(3):540–545. doi: 10.1161/01.STR.0000155746.65185.4e. [DOI] [PubMed] [Google Scholar]
- 25.Hunt KJ, et al. Genetic basis of variation in carotid artery plaque in the San Antonio Family Heart Study. Stroke. 2002;33(12):2775–2780. doi: 10.1161/01.str.0000043827.03966.ef. [DOI] [PubMed] [Google Scholar]
- 26.Moskau S, et al. Heritability of carotid artery atherosclerotic lesions: an ultrasound study in 154 families. Stroke. 2005;36(1):5–8. doi: 10.1161/01.STR.0000149936.33498.83. [DOI] [PubMed] [Google Scholar]
- 27.den H H, et al. GWAS-identified loci for coronary heart disease are associated with intima-media thickness and plaque presence at the carotid artery bulb. Atherosclerosis. 2015;239(2):304–310. doi: 10.1016/j.atherosclerosis.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts R, Stewart AF. Genes and coronary artery disease: where are we? J Am Coll Cardiol. 2012;60(18):1715–1721. doi: 10.1016/j.jacc.2011.12.062. [DOI] [PubMed] [Google Scholar]
- 29.Franceschini N, et al. Prospective associations of coronary heart disease loci in African Americans using the MetaboChip: the PAGE study. PLoS One. 2014;9(12):e113203. doi: 10.1371/journal.pone.0113203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voight BF, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8(8):e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnett FC, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 32.Del R I, et al. Glucocorticoid dose thresholds associated with all-cause and cardiovascular mortality in rheumatoid arthritis. Arthritis Rheumatol. 2014;66(2):264–272. doi: 10.1002/art.38210. [DOI] [PubMed] [Google Scholar]
- 33.Manichaikul A, et al. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26(22):2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 36.Pruim RJ, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Euesden J, Lewis CM, O’Reilly PF. PRSice: Polygenic Risk Score software. Bioinformatics. 2015;31(9):1466–1468. doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dehghan A, et al. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet. 2008;372(9654):1953–1961. doi: 10.1016/S0140-6736(08)61343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolz M, et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5(6):e1000504. doi: 10.1371/journal.pgen.1000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stark K, et al. Common polymorphisms influencing serum uric acid levels contribute to susceptibility to gout, but not to coronary artery disease. PLoS One. 2009;4(11):e7729. doi: 10.1371/journal.pone.0007729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koller DL, et al. Genome-wide association study of serum iron phenotypes in premenopausal women of European descent. Blood Cells Mol Dis. 2016;57:50–3. doi: 10.1016/j.bcmd.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuo DS, Labelle-Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet. 2012;21(R1):R97–110. doi: 10.1093/hmg/dds346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evans DM, et al. Genome-wide association study identifies loci affecting blood copper, selenium and zinc. Hum Mol Genet. 2013;22(19):3998–4006. doi: 10.1093/hmg/ddt239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masley SC, et al. Emerging risk factors as markers for carotid intima media thickness scores. J Am Coll Nutr. 2015;34(2):100–107. doi: 10.1080/07315724.2014.916238. [DOI] [PubMed] [Google Scholar]
- 45.Meyre D, et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009;41(2):157–159. doi: 10.1038/ng.301. [DOI] [PubMed] [Google Scholar]
- 46.Croze ML, Soulage CO. Potential role and therapeutic interests of myo-inositol in metabolic diseases. Biochimie. 2013;95(10):1811–1827. doi: 10.1016/j.biochi.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 47.Janssen JW, et al. MYEOV, a gene at 11q13, is coamplified with CCND1, but epigenetically inactivated in a subset of esophageal squamous cell carcinomas. J Hum Genet. 2002;47(9):460–464. doi: 10.1007/s100380200065. [DOI] [PubMed] [Google Scholar]
- 48.Dubey RK, et al. Adenosine Attenuates Human Coronary Artery Smooth Muscle Cell Proliferation by Inhibiting Multiple Signaling Pathways That Converge on Cyclin D. Hypertension. 2015;66(6):1207–1219. doi: 10.1161/HYPERTENSIONAHA.115.05912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bjorkegren JL, et al. Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J Am Coll Cardiol. 2015;65(8):830–45. doi: 10.1016/j.jacc.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunt KJ, et al. Genetic basis of variation in carotid artery plaque in the San Antonio Family Heart Study. Stroke. 2002;33(12):2775–80. doi: 10.1161/01.str.0000043827.03966.ef. [DOI] [PubMed] [Google Scholar]
- 51.Bis JC, et al. Sequencing of 2 subclinical atherosclerosis candidate regions in 3669 individuals: Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium Targeted Sequencing Study. Circ Cardiovasc Genet. 2014;7(3):359–64. doi: 10.1161/CIRCGENETICS.113.000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang L, et al. Lack of associations of ten candidate coronary heart disease risk genetic variants and subclinical atherosclerosis in four US populations: the Population Architecture using Genomics and Epidemiology (PAGE) study. Atherosclerosis. 2013;228(2):390–9. doi: 10.1016/j.atherosclerosis.2013.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kos MZ, et al. Common biological networks underlie genetic risk for alcoholism in African- and European-American populations. Genes Brain Behav. 2013;12(5):532–542. doi: 10.1111/gbb.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.