

Abstract
Purpose
We investigate the function of the V83I polymorphism (m.6150G>A, rs879053914) in the mitochondrial cytochrome c oxidase subunit 1 (MT-CO1) gene and its role in African American (AA) primary open-angle glaucoma (POAG).
Methods
This study used Sanger sequencing (1339 cases, 850 controls), phenotypic characterization of Primary Open-Angle African American Glaucoma Genetics study (POAAGG) cases, a masked chart review of CO1 missense cases (V83I plus M117T, n = 29) versus wild type cases (n = 29), a yeast 2-hybrid (Y2H) cDNA library screen, and quantification of protein–protein interactions by Y2H and ELISA.
Results
The association of V83I with POAG in AA was highly significant for men (odds ratio [OR] 6.5; 95% confidence interval [CI] 2.0–21.3, P = 0.0001), but not for women (OR 1.1; 95% CI, 0.62–2.00, P = 0.78). POAG cases having CO1 double missense mutation (V83I + M117T, L1c2 haplogroup) had a higher cup-to-disc ratio (0.77 vs. 0.71, P = 0.04) and significantly worse visual function (average pattern standard deviation, 6.5 vs. 4.3, P = 0.009; average mean deviation −10.4 vs. −4.5, P = 0.006) when compared to matched wild type cases (L1b haplogroup). Interaction of the V83I region of CO1 with amyloid beta peptide (Aβ) was confirmed by ELISA assay, and this interaction was abrogated by V83I. A Y2H screen of an adult human brain cDNA library with the V83 region of CO1 as bait retrieved the UBQLN1 gene.
Conclusions
The V83I polymorphism was associated strongly with POAG in AA men and disrupts Aβ-binding to CO1. This region also interacts with a neuroprotective protein, UBQLN1.
Keywords: glaucoma, mitochondrial DNA, genetic diseases, haplogroups, open-angle glacuoma
Primary open-angle glaucoma (POAG) is characterized by chronic and progressive optic nerve degeneration and retinal ganglion cell loss, accompanied by corresponding visual field defects. Older age, positive family history, and African ancestry are well-known risk factors. POAG is highly prevalent in African Americans (AA), with a recent survey estimating that 9.2% of Philadelphian AA over age 50 have POAG.1 AA also present with more severe disease characterized by higher IOP and cup-to-disc ratio (CDR).2
The MT-CO1 gene, located in mitochondrial DNA (mtDNA), encodes the cytochrome c oxidase subunit 1 (CO1) protein. This protein is localized to the mitochondrial inner membrane, and is an essential component of Complex IV. Reductions in CO1 expression caused by mutations in CO1's translational activator, TACO1, result in late-onset Leigh disease, with a variable phenotype that includes optic neuropathy and visual impairments in humans3 and mice.4 Germline and somatic missense variants in MT-CO1 also have been implicated in prostate cancer.5–8
The Primary Open-Angle African American Glaucoma Genetics (POAAGG) study9 previously reported disease-associated missense mutations in the N-terminal region of MT-CO1, and found three variants (V83I, M117T, V193I) to be associated with POAG.10,11 The V83I missense mutation (m.6150G>A, p.V83I, rs879053914) is common in the POAAGG study population, with a minor allele frequency of approximately 5%, and was significantly enriched in AA POAG cases versus AA controls (odds ratio [OR] = 1.8, P = 0.01).10
The V83I mutation is of particular interest because it lies within a region of CO1 known to interact with amyloid beta (Aβ),12 a product of the amyloid precursor protein (APP) gene, that figures prominently in the pathology of Alzheimer's disease (AD). CO1 also has been reported to interact with α-synuclein,13 which is found with ubiqulins in Lewy bodies, and is associated with Parkinson's disease (PD) and Lewy body dementia. Substantial evidence indicates that Aβ may be involved in POAG pathology, and that AD and POAG may share etiologies.14–16
We examined whether the previously observed associations of MT-CO1 missense mutations with POAG in AA differed by sex, and then characterized the phenotypes of POAG patients possessing the V83I mutation relative to V83 (wild type) POAG patients. We also tested whether the reported CO1/Aβ in vitro interaction is affected by the V83I amino acid replacement, and sought to identify other CO1 interactors.
Methods
Study Subject Recruitment
The baseline demographics, and inclusion and exclusion criteria of the POAAGG study have been described previously.9 Subjects were identified from within all comprehensive and subspecialty ophthalmology clinics at the University of Pennsylvania (Scheie Eye Institute, Perelman Center for Advanced Medicine, Mercy Fitzgerald Hospital), Lewis Katz School of Medicine at Temple University, and a private practice (Windell Murphy, MD). Subjects were age 35 years or older, and self-identified as Black, AA, or as having African ancestry. All eligible patients underwent an onsite ophthalmic exam and interview. The full onsite exam included: (1) verification of name, age, date of birth, street address, sex, and informed consent with signature; (2) completion of a questionnaire in-clinic; (3) evaluation of height and weight; (4) explanation of procedure for blood or saliva collection for DNA analysis; (5) visual acuity (VA) measured using the Snellen chart at 20 feet; (6) automated refraction with a Reichert Phoropter RS Automatic Refractor (Reichert Technologies, Depew, NY, USA) if the presented VA was not 20/20 in either eye, followed by manual refraction; (7) IOP measured with a Goldmann applanation tonometer; (8) anterior and posterior segment examinations by slit-lamp with a 90-diopter (D) lens for optic nerve examination and indirect ophthalmoscopy; (9) gonioscopy confirming the presence of an open-angle; (10) central corneal thickness and axial length measurements assessed with an ultrasonic A-scan/pachymeter DGH 4000B SBH IOL Computation module (DGH Tech, Inc., Exton, PA, USA); (11) visual field test using the Humphrey Automated Field Analyzer (Standard 24-2 Swedish interactive thresholding algorithm); (12) stereo disc photos and fundus photography using the Topcon TRC 50EX Retinal Camera (Topcon Corp. of America, Paramus, NJ, USA); and (13) optical coherence topography (OCT) using either Cirrus or Stratus OCT (Carl Zeiss Meditec, Dublin, CA, USA). The outcomes of the procedures and all diagnoses were discussed with the patient at the conclusion of the examination. All enrolled subjects provided a signed informed consent and genomic DNA, which was extracted from peripheral blood or saliva.
Glaucoma specialists classified subjects as cases, controls, or suspects. POAG cases were defined as having an open iridocorneal angle and: (1) characteristic glaucomatous optic nerve findings in one or both eyes consisting of at least one of the following: notching, neuroretinal rim thinning, excavation, or a nerve fiber layer defect; (2) characteristic visual field defects on two consecutive reliable visual field tests in at least one eye, which were consistent with the observed optic nerve defects in that eye, as determined by fellowship-trained glaucoma specialists; and (3) all secondary causes of glaucoma excluded. Normal controls were defined as subjects older than 35, without: (1) high myopia (greater than −8.00 D); (2) high presbyopia (+8.00 D); (3) family history of POAG; (4) abnormal visual field; (5) IOP greater than 21 mm Hg; (6) neuroretinal rim thinning, excavation, notching or nerve fiber layer defects; (7) optic nerves asymmetry; or (8) a cup-to-disc ratio difference between eyes greater than 0.2. Subjects classified as glaucoma suspects were excluded from all analyses. Research was approved by an institutional review board at the University of Pennsylvania, and was conducted in accordance with the Declaration of Helsinki.17
PCR, DNA Sequencing and Mutation Analysis
Amplicons corresponding to the MT-CO1 and MT-RNR2 genes had been PCR amplified and Sanger sequenced, with methods and results reported previously.10 The original Sanger sequencing cohort contained POAG cases, controls, and some glaucoma suspects (excluded from analyses). Because some subjects' disease had progressed since the cohort was sequenced, subject disease status was updated to be current as of August 2016, before reanalysis for association with POAG by sex, and data from more recently enrolled subjects were included (total n = 2189; 1339 cases, 850 controls). Version 5.2 of Sequencher software was used to score sequencing chromatograms for missense variants. Missense variants were annotated using version 1.0 of the MitImpact resource (available in the public domain at http://mitimpact.css-mendel.it/) and the MITOMAP compendium to identify potentially deleterious mutations.
Phylogeny and Localization of Amino Acid Replacements
Build 17 of PhyloTree (available in the public domain at www.phylotree.org) and the MITOMAP and MITOMASTER databases (available in the public domain at www.mitomap.org) were used to associate the observed mtDNA variants with mitochondrial haplogroups. The subcellular localizations of MT-CO1 missense mutations were predicted by sequence analysis using the UniProt resource (available in the public domain at www.uniprot.org).
Phenotypic Characterization of V83I POAG Cases
Phenotypic data for 1070 AA POAG cases were extracted from the POAAGG study's Research Electronic Data Capture (REDCap) database,18 and cases were grouped for analysis by MT-CO1 genotypes: m.6150G (V83, wild type), m.6150G>A (V83I mutation), m.6548C (V83 wild type), and m.6548C>T (V83 wild type, with silent substitution, L215L, indicative of African haplogroup L1b).
The masked comparison included 29 cases with V83I and M117T (“double missense”) mutations, associated with mtDNA haplogroup L1c2. “Triple missense” cases, possessing a third variant, V193I, associated with an L1c2 sublineage, L1c2b1b, were excluded. The control group included 29 cases with the L1b-associated silent substitution (m.6548C>, L215L) and lacking all three missense variants. Each L1c2 case was paired with an L1b control case having the same sex and reported family history of glaucoma (yes or no), and similar age at enrollment into the POAAGG study. Haplogroup was masked during chart review.
The following data were collected from the medical records: demographic characteristics, including age, sex, family history of glaucoma; glaucoma phenotypes at visit closest to enrollment, including ICD-9 codes for glaucoma severity, maximum IOP, visual acuity, central corneal thickness (CCT), retinal nerve fiber layer thickness (RNFLT) on OCT, CDR, mean deviation (MD), and pattern standard deviation (PSD) on Humphrey visual field test. A 20% cutoff for false-positive and false-negative response rates and fixation losses was used for visual fields, and the OCT with the best signal strength (minimum 7/10) was chosen for each patient. We used both eyes for this analysis, adjusting for the correlation between eyes.
Yeast Two-Hybrid (Y2H) Studies
All Y2H studies were performed by Hybrigenics Services (available in the public domain at www.hybrigenics-services.com, Paris, France). The coding sequence of the human MT-CO1 fragment (amino acids 41–101, GenBank accession number gi: 251831109) was PCR-amplified and cloned into vectors pB27 and pB66 as C-terminal fusions to LexA (LexA-CO1) and the Gal4 DNA-binding domain (Gal4-CO1), respectively. Codon use of the CO1 insert was optimized for yeast expression, and to prevent spurious amino acid changes caused by differences in the human mitochondrial and yeast nuclear genetic codes. The constructs were checked by sequencing, and used as a bait to screen a random-primed Human Adult Brain cDNA library constructed19 into pP6, using RNA from a 27-year-old male (#D6030-15, lot A308079; Invitrogen, Carlsbad, CA, USA). Vector pB27 derives from the original pBTM116 vector,20 pB66 derives from the original pAS2 ΔΔ vector21 and pP6 is based on the pGADGH plasmid.22
The N-Gal4-CO1-C bait construct was used for the cDNA library screen, and 47 million clones, 4-fold the complexity of the library, were screened using a mating approach20 with YHGX13 (Y187 ade2-101::loxP-kanMX-loxP, matα) and CG1945 (mata) yeast strains. A total of 323 His+ colonies were selected on a medium lacking tryptophan, leucine, and histidine. The prey fragments of the positive clones were amplified by PCR and sequenced at their 5′ and 3′ junctions. The resulting sequences were used to identify the corresponding interacting proteins in the GenBank database (NCBI), using a fully automated procedure. A confidence score (predicted biological score [PBS]) was attributed to each interaction as described previously.19 The prey fragment for the human APP gene corresponded to amino acids 672 to 713, GenBank accession number gi: 228008403, corresponding to Aβ (1-42). The prey fragment was cloned in frame with the Gal4 Activation Domain into plasmid pP7. The pP7 prey plasmid is derived from the pP6 plasmid. The coding sequence of the mutant (V83I) CO1 fragments (amino acids 41–101) was synthesized and cloned in frame with the Gal4 DNA binding domain into plasmid pB66 (N-Gal4- CO1mut-C). The constructs were checked by sequencing the entire insert.
The prey fragment for UBQLN1 (amino acids108–388, GenBank accession number gi:194328681) was obtained from the ULTImate Y2H screening of wild type CO1 (amino acids 41–101) against the Human Adult Brain cDNA library. The bait constructs, LexA-CO1, Gal4-CO1, and Gal4-CO1mut, were transformed into the yeast haploid strains L40 ΔGal4 (mata) and CG1945 (mata) respectively. The preys, APP and UBQLN1, were transformed into YHGX13 (Y187 ade2-101::loxP-kanMX-loxP, matα) strain. The diploid yeast cells were obtained using a mating protocol with both yeast strains. These assays are based on the HIS3 reporter gene, for growth assay without histidine.
Interaction pairs were tested in triplicate, using a growth assay on ± histidine and ± 3-Amino-1,2,4-triazole (3-AT) plates, and streaks from three independent clones from each diploid were picked for the growth assay. DO-2 selective medium lacking tryptophan and leucine was used as a growth control and to verify the presence of the bait and prey plasmids. The DO-3 selective medium without tryptophan, leucine, and histidine selects for the interaction between bait and prey. Interaction pairs were tested in quadruplicate with four independent clones from each diploid picked for the growth assay. For each interaction, several dilutions (10−1, 10−2, 10−3, 10−4) of the diploid yeast cells (culture normalized at 5 × 104 cells) and expressing bait and prey constructs were spotted on several selective media. The different dilutions also were spotted on a selective medium without tryptophan, leucine, and histidine (DO-3). Four different concentrations of 3-AT were added to the DO-3 plates to increase stringency. The following 3-AT concentrations were tested: 1, 5, 10, and 50 mM.
ELISA Studies
Sandwich ELISA assays were performed with biotinylated synthetic peptides to confirm the previously reported affinity of Aβ1–42 peptide with the N-terminal region of CO1, and to quantify the effect of the V83I amino acid replacement on this interaction. The synthetic Aβ1–42 (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA) and scrambled Aβ (AIAEGDSHVLKEGAYMEIFDVQGHVFGGKIFRVVDLGSHNVA) peptides (AnaSpec, Fremont, CA, USA) were incubated with wild type CO1 (Biotin-Ahx-LGQPGNLLGNDHIYNVIVTAHAFVIIFFIVIPIIIGGFGNWLVPLIIGAPDMAFPRINNIS) or the mutant CO1 with V83I amino acid replacement (Biotin-Ahx- LGQPGNLLGNDHIYNVIVTAHAFVIIFFIVIPIIIGGFGNWLIPLIIGAPDMAFPRINNIS) (NeoBioLab, Cambridge, MA, USA). Purified ApoE protein (OriGene Technologies, Rockville, MD, USA) and a 25 amino acid NADH dehydrogenase subunit 3 (MT-ND3) peptide encoded by mtDNA (Biotin-TTNLPLMVMSSLLLIIILALSLAYE), was used as a positive control for interaction. The scrambled Aβ peptide and protein (OriGene Technologies) were used as negative controls for evaluating interaction with Aβ. Sandwich ELISA was performed by binding Aβ1–42 or scrambled Aβ1–42 to the plate overnight at 4°C, followed by blocking with 5% nonfat dry milk and 1% BSA for 90 minutes at room temperature (RT). After blocking, the wells were incubated with biotinylated-wild type-Cox1 peptide, Biotinylated-Mut (V83I)-Cox1 peptide, ApoE protein (positive control) or TBX3 (negative control) for two hours at RT. Each experimental well was washed thrice with 1X TBST for 5 minutes at RT. The interacting complex was detected by incubation with Avidin-peroxidase conjugate followed by ABTS (50 μl/well) for color development. Approximately 30 μl/well of 1% SDS in PBS with Tween 20 (PBST) was added to stop the color development and the ELISA plate was read at 410 nm in an ELISA microplate reader (Molecular Devices, Sunnyvale, CA, USA). All ELISA experiments were done in triplicate.
Statistical Analysis
All P values were calculated using Fisher's exact test, except for phenotypic characteristics, which were calculated using logistic regression, with generalized estimating equations (GEE)23 to account for correlation between eyes for ocular characteristics. All analyses were performed in SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
Characteristics of the Study Population
Phenotypic data from 1070 AA POAG cases were analyzed, and the majority (59%) of these subjects were female. The mean age of wild type (V83) cases was 71.1 years, vs. 70.8 years for V83I cases (P = 0.81). Sanger sequencing of the m.6150 region of MT-CO1 was obtained for 2189 AA individuals (1339 POAG cases, 850 controls), and 4.5% possessed the V83I mutation. The 850 non-POAG controls also were mostly female (65%), but significantly younger (61 ± 11.8 years, P < 0.001) than the POAG group. The total number of cases sequenced was larger than 1070 because an additional 269 cases were sequenced subsequent to phenotypic analysis.
Sequencing Identified Additional Missense Variants in MT-CO1
Sanger sequencing of the N-terminal region of MT-CO1 identified 21 missense variants, in addition to the three variants detected previously in the POAAGG cohort (m.6150G>A [V83I], m.6253T>C [M117T], m.6480G>A [V193I]; Supplemental Table S1).10,11 None of the missense variants differed significantly between cases and controls; however, most were rare, and the sample size was insufficient to detect differences between cases and controls for rare variants.
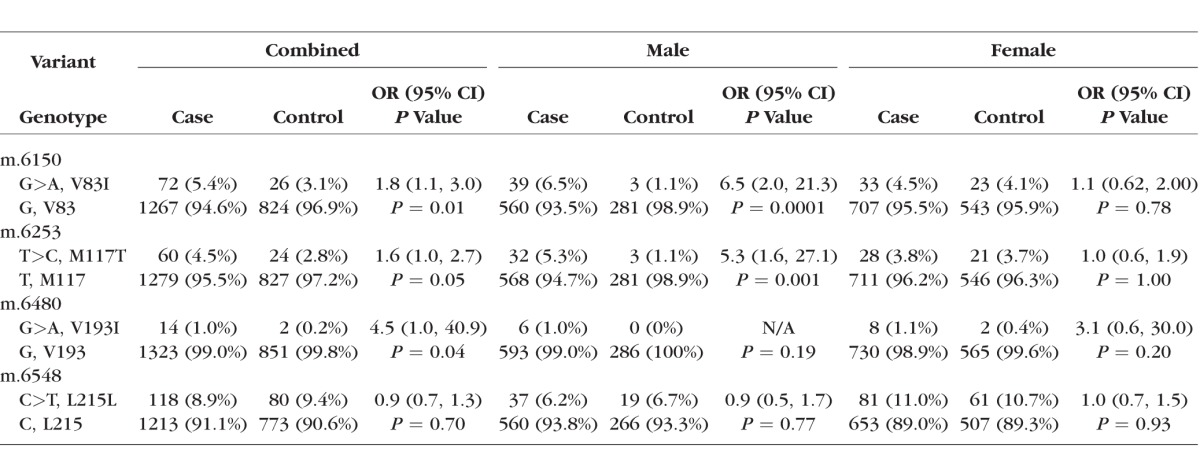
Association of Four MT-CO1 Variants With POAG in AA Males and Females
Common mtDNA variants are associated with one or more mtDNA lineages (haplogroups), with African haplogroups represented by the letter “L”, with L0, L1, L2, L4, and so forth representing the deepest branches. Supplementary Figure S1 depicts the mtDNA family tree in schematic form, and indicates the relative divergence times of selected MT-CO1 variants, including V83I (mostly L1c2 haplogroup). V83I also is found in a subset of POAAGG subjects belonging to an L2 haplogroup (not shown); these subjects possessed an L2-associated variant, m.2416T>C in the MT-RNR2 gene, but lacked the L1c2-associated variant M117T. The synonymous variant, m.6548C>T (L215L), is uniquely associated with the L1b African haplogroup, which lacks all three missense variants.
Sanger sequencing data for four variants in MT-CO1 were analyzed after stratifying cases and controls by sex (Table 1). The associations of the three missense variants with POAG were reported previously to be significant when males and females were analyzed together.10 When analyzed separately by sex, V83I was not observed more frequently in female cases than female controls (OR 1.1; 95% confidence interval [CI], 0.62–2.00; P = 0.8). However, when male cases were compared to male controls, the difference was highly significant (OR, 6.5; 95% CI, 2.0–213, P = 0.0001, Table 1). Association of M117T with POAG was significant for males only (OR, 5.3; 95% CI, 1.6–27.1, P = 0.001). Association of V193I with POAG was significant in the combined group, but not for males or females when analyzed separately. Association of the fourth variant, the L1b haplogroup-linked synonymous substitution m.6548C>T (L215L), with POAG was not significant in the combined group, or for males or females (Table 1).
Table 1.
Association of MT-CO1 Variants With POAG in AA Males and Females
Characteristics of V83I POAG Patients
We compared glaucoma-related traits, for example, IOP, CCT, visual fields, and family history of POAG, among cases (n = 1070) after grouping by genotype (V83 wild type versus V83I). For a parallel comparison, we also grouped cases by the synonymous variant m.6548C>T (L215L), which is associated with a related L1 haplogroup, L1b, lacking the V83I mutation (Supplementary Figure S1). The V83I patients demonstrated worse visual field defects, differing significantly in mean PSD (P = 0.008) and average MD (P = 0.02; Table 2). When analyzed separately by sex, mean PD was higher in V83I males and V83I females, but the difference was significant in males (P = 0.047), but not females. Despite worse disease, the V83I patients had significantly lower IOP (P = 0.03). Mean CCT was higher in the V83I group, but this difference was not significant. However the distribution of CCT across three bins was nominally significant (P = 0.049), with the largest fraction of V83I patients having CCT greater than 540 μm. Of the V83I cases, 73% reported a positive family history of glaucoma, versus 57% of V83 (wild type) cases, and this difference was statistically significant (P = 0.03). Maternal family history of glaucoma also was reported more often in V83I cases (33% vs. 20% for V83 wild type, P = 0.03).
Table 2.
Phenotypic Characteristics of AA POAG Cases With and Without the V83I Mutation
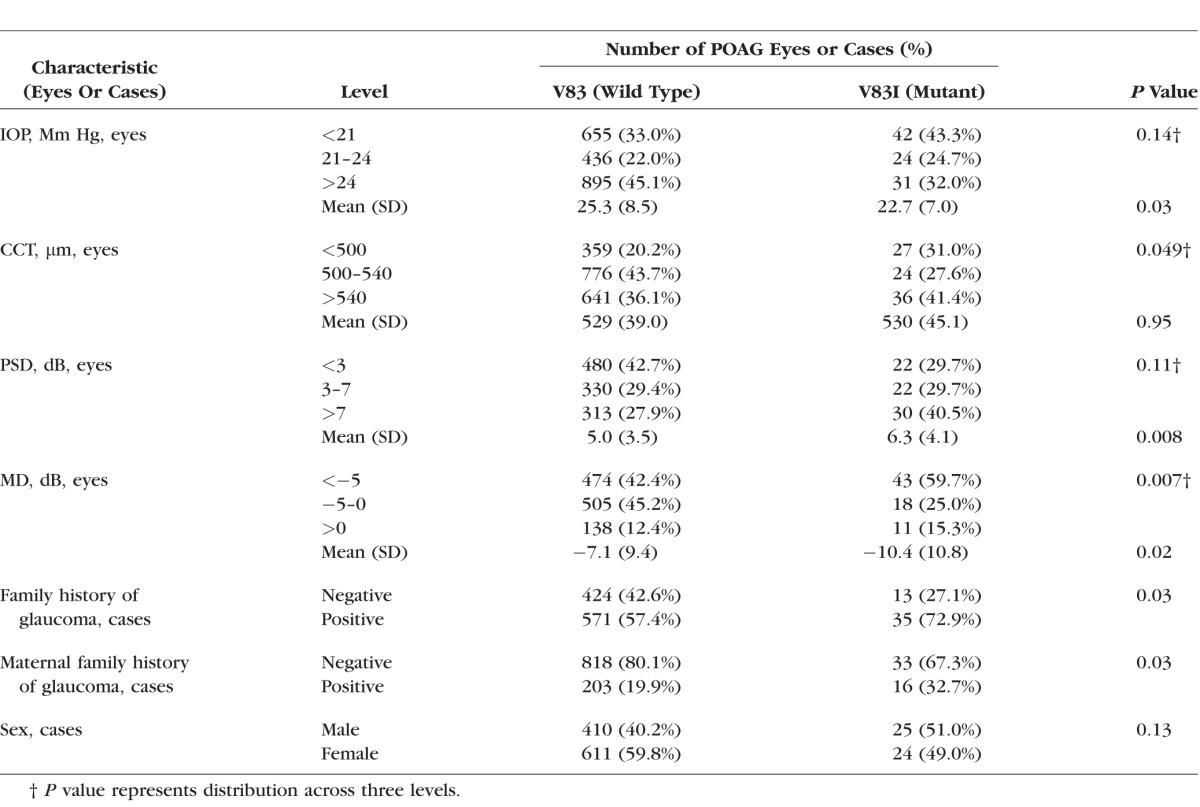
Female cases outnumbered male cases in the cohort as a whole, by approximately 1.5:1, whereas the V83I group contained nearly equal numbers of males and females. In other words, the V83I case group was disproportionately male. The mean age at enrollment of V83I cases was 68.8 years for males and 72.8 for females, versus 70.1 and 71.8, respectively, in the corresponding wildtype (V83) groups. Mean age did not differ significantly between the V83I and wildtype groups (P = 0.58 and P = 0.63 for males and females, respectively). Prostate cancer was noted more frequently in the V83I group, 10.2% vs. 5.6%, but this difference was not significant, and likely explained by the higher proportion of males in the V83I group.
The L215L (m.6548C>T) reference group, representing haplogroup L1b and lacking all three missense variants, differed significantly only in that it was disproportionately female (P = 0.049), and had a significantly lower incidence of prostate cancer (P = 0.04). This is presumably due to a larger proportion of females in the L215L group (data not shown).
Masked Chart Reviews of Haplogroup L1c2 (V83I + M177T) Versus L1b Patients
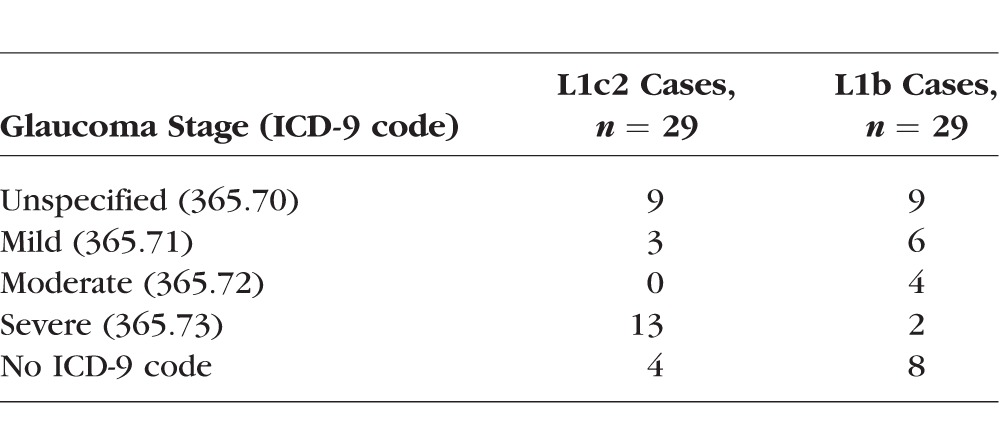
Because the V83I patients had significantly worse visual function than V83 (wild type) patients, despite lower IOP, we performed a masked chart review, focused on L1c2 patients (n = 29). In order to control for non-L1 mtDNA ancestry, patients from the L1b haplogroup (wild type for V83 and M117) were used as the reference group (n = 29). The two groups also were matched for age, sex, and family history of POAG (yes or no) to control for these potential confounders. The chart review was conducted with a fellowship-trained glaucoma specialist (AL). In parallel with the chart review, we determined that the L1c2 group had significantly worse disease, as assessed by ICD-9 codes for glaucoma severity previously entered in the electronic medical records. Of the L1c2 cases having disease stage codes, 81% had “severe” disease (ICD-9 365.73) as opposed to 17% of L1b cases (P = 0.002, Table 3).
Table 3.
Glaucoma Severity Codes (ICD-9) of Matched L1c2 vs. L1b POAG Cases
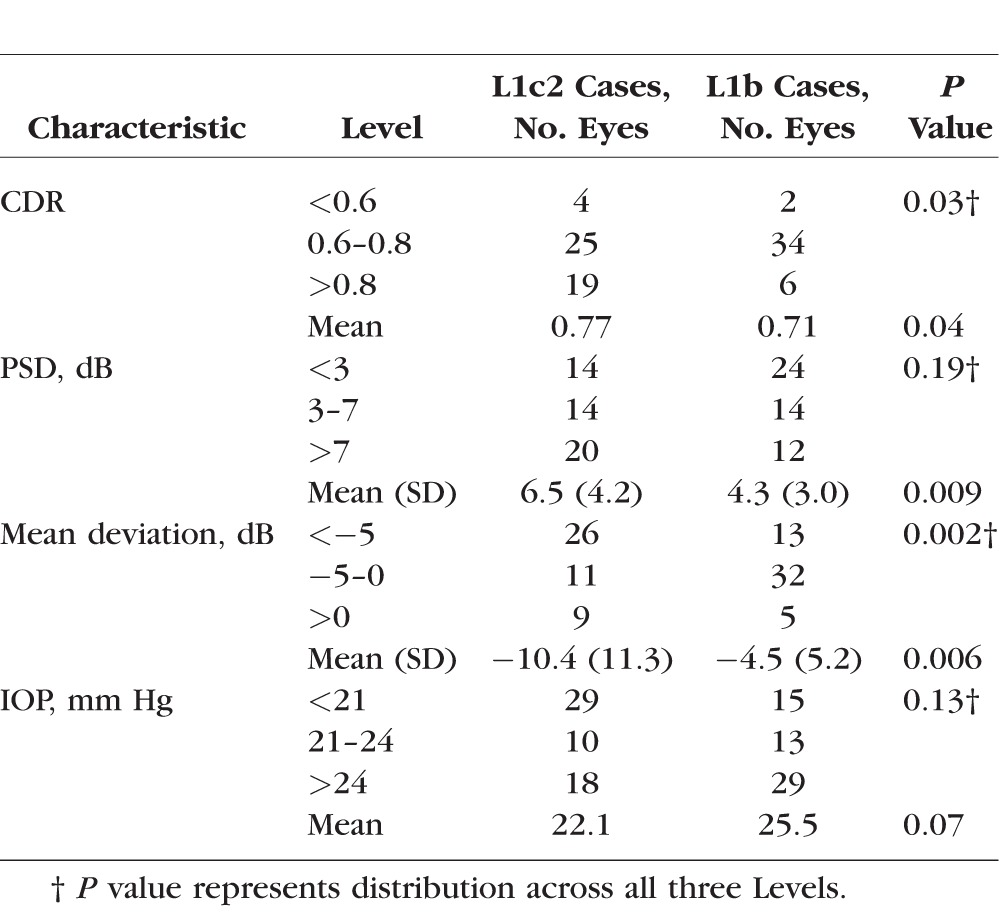
The L1c2 group had significantly higher mean CDR (P = 0.04) and worse visual function as assessed by average PSD (P = 0.009) and MD (P = 0.006) at the visit closest to diagnosis (Table 4). IOP was lower in the L1c2 group despite their worse disease, although the difference did not reach statistical significance (P = 0.07). CCT (not shown) did not differ significantly between the L1c2 and L1b groups.
Table 4.
Phenotypic Traits of Matched L1c2 Versus L1b POAG Patients
Subcellular Localization of V83, M117 and V193 Amino Acid Residues
Sequence analysis by UniProt predicted the V83I replacement affects a residue located on the inner side of the inner mitochondrial membrane, which is exposed to the mitochondrial matrix, whereas M117T and V193I were predicted to be located inside the mitochondrial inner membrane. The domain organization of the CO1 protein and locations of these residues are depicted in Supplementary Figure S2. The reported Aβ-interacting region of the CO1 protein, which includes V83, spans an intermembrane, transmembrane, and mitochondrial matrix region.
Y2H test of CO1/Aβ Interaction, and cDNA Library Screen For CO1 Interactors
We could not demonstrate the protein–protein interaction of CO1 and Aβ in the Y2H system. Accordingly, we were unable to study the effect of the V83I amino acid replacement on the CO1/Aβ interaction by Y2H, and used an ELISA assay instead, described below. The screen of the human adult brain cDNA library, using an N-terminal CO1 wild type (V83) fragment as bait, retrieved seven unique clones containing fragments of ubiquilin1, the product of the UBQLN1 gene. The Predicted Biological Scores19 for these Y2H interactions were classified as “very high confidence,” and UBQLN1 is a known interactor and chaperone of mitochondrial membrane proteins.
Quantification of Wild Type and Mutant (V83I) CO1 Fragment With UBQLN1 Fragment in the Y2H System
The CO1 (WT)/UBQLN1 interaction was confirmed to be very strong; however, the mutant CO1 (V83I)/UBQLN1 interaction was equally strong, so V83I did not affect this interaction in the Y2H system. Both interactions resisted up to 10 mM 3-AT (a competitive inhibitor of the HIS3 gene product), which was the highest concentration tested (Supplementary Figure S3).
Interaction of CO1 and Aβ Peptides
The reported affinity of wild type CO1 (V83) peptide for Aβ1–4212 was confirmed by ELISA, whereas mutant CO1 (V83I) was found to interact very weakly with Aβ (Fig.). At the highest peptide concentrations tested, V83I reduced the interaction by 92%, to a level comparable to the negative control. However, wild type and mutant (V83I) CO1 peptides had similar lack of affinity for the scrambled Aβ negative control peptides, suggesting the interaction of CO1 and Aβ is specific, and is disrupted by V83I. We were unable to study the interaction of CO1 peptide with the fragment of UBQLN1 retrieved in the cDNA library screen by ELISA, on account of an inability to synthesize the hydrophobic UBQLN1 peptide.
Figure.

ELISA assay for interaction of mutant and wild type CO1 peptides with Aβ1–42 or scrambled Aβ1–42. APOE protein was the positive control, and TBX3 protein and scrambled Aβ peptide (ScrAβ1–42) were negative controls.
Discussion
Male-Female Dimorphism in POAG
The association of sex with POAG is controversial with only some studies finding men at higher risk; however, a recent meta-analysis concluded men are at 1.3-fold higher risk of POAG than women.24 Leber's hereditary optic neuropathy (LHON) was the first disease shown to be caused by missense mutations in mtDNA, resulting in defects in mitochondrial energy production.25 LHON, like POAG, involves familial degeneration of the optic nerve. LHON affects males much more severely than females, with higher penetrance and an earlier age of onset in males. Most LHON cases are caused by three mtDNA pathogenic variants (m.3460G>A in MT-ND1, m.11778G>A in MT-ND4, or m.14484T>C in MT-ND6), all with incomplete penetrance and with a risk of symptoms developing ranging from 1.7- to 7.7-fold higher in men versus women (available in the public domain at https://www.ncbi.nlm.nih.gov/books/NBK1174/). The association of m.6150G>A (V83I) in MT-CO1 with POAG also exhibited an extreme male sex bias, with an OR 5.9-fold higher in men than in women (Table 1). Inheritance of mitochondria is matrilineal, so natural selection of mitochondria occurs only in females, and male-specific phenotypes may not have fitness consequences for mitochondria, resulting in male-female asymmetry in disease severity.26
A recent study of nuclear SNPs near 9p21 reported a significantly stronger association with normal tension glaucoma in females (OR 1.5) than males (OR 1.35).27 Collectively these findings suggest that the genetic architecture of POAG for males and females may differ substantially, with penetrance differing by sex. Male-female dimorphism in response to overexpression of mutant amyloid precursor protein has been reported in a mouse model of AD, with complex IV activity consistently higher in female control mice.14
The V83I POAG Phenotype
The V83I patients had worse visual function and degeneration of the optic nerve, despite lower IOP, even after controlling for age, sex, family history of POAG, AA ancestry, and L1 mtDNA haplogroup. This evidence is consistent with the proposal that the V83I missense mutation may contribute to POAG pathogenesis, and these patients may be more vulnerable to POAG in general, and at lower IOP than other AAs. Interestingly, a study of the association of mtDNA variations with dementia risk and Aβ in elderly AAs found African mitochondrial haplogroup L1 participants were at elevated risk for dementia (OR, 1.88; P = 0.004), lower plasma Aβ 1–42 levels (P = 0.03), and greater risk for intellectual decline, relative to the most common AA haplogroup, L3.28
Potential Functional Effects of MT-CO1 Missense Variants
Cytochrome c oxidase is a “bigenomic” protein machine with components encoded by the nuclear and mitochondrial genomes. The mtDNA-encoded proteins, for example, CO1, are functionally constrained by the requirement to interact with nuclear proteins. The three POAG-associated missense mutations may occur either alone, for example, V83I on an L2 mtDNA background, or in combination, for example, V83I and M117T on L1c2-related haplogroups, which also may carry V193I (Supplementary Figure S1). M117T has been predicted to be pathogenic based on 3D structural modeling suggesting it participates in bigenomic protein–protein interactions, and might influence the stability of Complex IV.29 The V193I variant originally was detected in a patient having cytochrome c oxidase deficiency, and was proposed to be pathogenic, based on its absence in 300 controls.30 However, V193I now is understood to be a common polymorphism associated with the L1c2b1b African haplogroup, which also carries V83I and M117T31 and with non-African haplogroups. V193I has been proposed as a “helper” variant, acting in synergy with the primary LHON mutation, m.11778G>A, in a Chinese family.32 V193I, like M117T, may affect interactions with Complex IV proteins encoded by nuclear genes.29
V83I is the stronger candidate for a functional variant in addition to a marker for POAG susceptibility. The association of V83I with POAG in AA men was stronger than for M117T or V193I (Table 1). This is because the association of V83I with POAG stemmed not only from L1c2 subjects also having M117T, but also from additional L2 haplogroup subjects who have V83I, but lack the other two missense mutations. V83I also is the only one of these mutations within the Aβ-binding region, and we showed this replacement greatly abrogated this interaction in the ELISA assay (Fig.). Another reported prostate cancer-linked mutation (M74T, m.6124T>C) is located nine residues from V83. This mutation was not observed in POAAGG subjects; however, it also is within the Aβ-binding region, and was shown to be functional, causing resistance to statin-induced apoptosis,8 increase reactive oxygen production, and enhanced cellular proliferation.7 M74T cybrid cells had a significantly faster doubling time than wild type cells. So it is reasonable to speculate that V83I or other nearby CO1 mutations also might influence cellular proliferation, albeit negatively, thereby promoting neurodegeneration. The retrieval of UBQLN1, a neuroprotective AD- and Aβ-associated gene, by a Y2H cDNA library screen suggests this region may function in the recognition of CO1 for sequestration by ubiquilins and degradation by the proteasome.
Potential for Interactions Among CO1, Aβ, and Ubiquilin-1
The POAG-associated V83I CO1 mutation diminished the interaction with Aβ1–42 in the ELISA assay (Fig.). CO1 and Aβ1–42 have been shown to coprecipitate from mitochondria of human neuroblastoma cells, so this interaction appears to occur in vivo.12 These observations suggest the interaction of soluble Aβ with CO1 is normal and potentially beneficial, whereas aggregation of Aβ may cause mitochondrial dysfunction, including oxidative damage by reactive oxygen species, induction of apoptosis and ion channel formation.15
The yeast two-hybrid library screen, using the Aβ-interacting region of CO1 as “bait” retrieved an AD-associated gene, ubiquilin1 (UBQLN1). UBQLN1 functions as a molecular chaperone of the amyloid precursor protein (APP), and modifies the intracellular trafficking of APP. Knockdown of UBQLN1 results in increased levels of Aβ.33,34 Sequence variation in UBQLN1 has been associated with AD,35 and overexpression of some UBQLN1 transcript variants exerts neuroprotective effects via the unfolded protein response (UPR).36 Ubiquilins, including UBQLN1, recently have been shown to chaperone and triage mitochondrial membrane proteins for degradation by the proteasome,37 suggesting the CO1/UBQLN1 interaction might occur in the cytosol, for example as a response to the release of CO1 from damaged mitochondria. The region of UBQLN1 retrieved by the two hybrid screen (amino acids 108–388) overlaps with the N-terminal part of its “M domain” (amino acids 180–470), which is required for binding transmembrane domains of mitochondrial membrane proteins.37 Genetic ablation of ubiquilins results in the accumulation of noninserted mitochondrial membrane protein precursors in the cytosol.37 UBQLN1 has not been implicated in POAG, but was among a small number of antiapoptotic genes found to be upregulated by platelet derived growth-factor CC, which protects retinal ganglion cells (RGC) from death in optic nerve crush-injured mouse retinae.38
Mechanisms for Mitochondrial Dysfunction in Glaucoma and AD
Glaucoma and AD have several features in common, including increasing incidence with age and loss of specific neuronal subpopulations. Glaucoma is more prevalent among Down syndrome patients, who have an extra copy of the APP gene, encoding Aβ. Down syndrome cases are prone to glaucoma development before the age of 40 and at normal pressure39 in addition to early-onset AD. This is consistent with overlapping molecular etiologies for AD and glaucoma involving Aβ.40 Intramitochondrial Aβ may directly cause neurotoxicity and mitochondrial dysfunction, including impairment of OXPHOS and interaction with mitochondrial proteins.41,42 Aβ is known to localize to the mitochondrial matrix and inner mitochondrial membrane, so there is a potential for Aβ to interact directly with Complex IV, which it is known to inhibit.14,43
Neuronal mitochondria offer potential targets for therapies that support mitochondrial function.44–47 Complex IV is a target for interventions that may protect RGCs from death as a result of glaucoma. For example, methylene blue has been shown to protect rat RGCs from toxic insults to mitochondria, such as rotenone, which can cause optic neuropathy.48 Administration of near-infrared light also is neuroprotective, possibly via the same mechanism as methylene blue: transfer of electrons directly to Complex IV.49 It was shown recently that Aβ-induced mitochondrial dysfunction involves the forkhead box O3a FOXO3A gene,50 which is present in neuronal mitochondria, binds mtDNA, and leads to decreased MT-CO1 expression, so there also may be indirect influences of Aβ on Complex IV.
Glaucomatous changes might arise from multiple mechanisms of mitochondrial damage, some of which might involve Aβ in addition to changes in mtDNA and nuclear genes.51 These factors might act alone, or in concert with elevated IOP, which may damage mitochondria directly.52 For example, elevated pressure causes release of cytochrome c and OPA1 release in RGC-5 cells, resulting in apoptotic cell death.53 Mitochondrial damage from IOP also was demonstrated in glaucomatous DBA/2J mice, where elevated IOP resulted in mitochondrial fission, matrix swelling, cytochrome c release, and a moderate reduction of expression of MT-CO1 mRNA.52 Oxidative stress caused by ozone exposure has been shown to increase Aβ1–42 production and accumulation of Aβ1–42 in mitochondria, with colocalization of OPA1, Aβ, and CO1.54 Increased expression of Aβ and abnormal APP metabolism is associated with RGC apoptosis in experimental glaucoma models, with colocalization of Aβ with apoptotic RGCs.14,16
Limitations of This Study
The association of V83I with POAG in AA men may be explained by unknown functional factors associated with the corresponding mtDNA ancestries (primarily L1c2 and L2 lineages) and/or by population stratification of the POAAGG study population. Additional work will be required to determine whether V83I or other mutations directly influence Complex IV activity or confer other functional phenotypes, and to assess the potential for involvement of UBQLN1 in POAG pathogenesis.
Conclusions
Because V83I is associated predominately with African mitochondrial ancestries (L1c2, L2, and others), this variant may contribute to the relatively high POAG risk in AAs. V83I disrupts a previously reported protein–protein interaction with Aβ, which could be significant in light of proposals that the etiology of POAG may overlap with other neurodegenerative conditions, particularly AD, with Aβ common to both. We propose that CO1 also interacts with UBQLN1, and that UBQLN1, which is a potential therapeutic target for AD,55 may also be relevant to POAG.
Interventions to treat optic neuropathies by supporting mitochondrial function are under development.56 Recently, metformin and the antioxidant mitotempo have been shown to protect human neural stem cells and cultured mouse neurons, respectively, against Aβ-induced mitochondrial dysfunction.57,58 Alternatively, emerging gene-based therapies now offer the ability to correct deleterious germline alterations in mtDNA.
Supplementary Material
Acknowledgments
The authors thank Jie He for assisance with sequencing mtDNA, and Gui-shuang Ying and Rebecca Salowe for helpful suggestions during preparation of the manuscript.
Supported by the National Eye Institute (1RO1EY023557-01), National Eye Institute Vision Core Grant (P30 EY01583-26), and National Institutes of Health (NIH; LM010098; Bethesda, MD, USA), as well as the F.M. Kirby Foundation, Research to Prevent Blindness, UPenn Hospital Board of Women Visitors, Paul Mackall and Evanina Bell MacKall Trust, and National Eye Institute, NIH, Department of Health and Human Services, under eyeGENE (contracts #HHSN260220700001C and HHSN263201200001C), and the Department of Ophthalmology at the University of Pennsylvania.
Disclosure: D.W. Collins, None; H.V. Gudiseva, None; V.R.M. Chavali, None; B. Trachtman, None; M. Ramakrishnan, None; W.T. Merritt III, None; M. Pistilli, None; R.A. Rossi, None; S. Blachon, None; P.S. Sankar, None; E. Miller-Ellis, None; A. Lehman, None; V. Addis, None; J.M. O'Brien, None
References
- 1. Waisbourd M, Pruzan NL, Johnson D,et al. . The Philadelphia glaucoma detection and treatment project: detection rates and initial management. Ophthalmology. 2016; 123: 1667– 1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedman DS, Jampel HD, Munoz B,et al. . The prevalence of open-angle glaucoma among blacks and whites 73 years and older: The Salisbury Eye Evaluation Glaucoma Study. Arch Ophthalmol. 2006; 124: 1625– 1630. [DOI] [PubMed] [Google Scholar]
- 3. Weraarpachai W, Antonicka H, Sasarman F,et al. . Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset leigh syndrome. Nat Genet. 2009; 41: 833– 837. [DOI] [PubMed] [Google Scholar]
- 4. Richman TR, Spahr H, Ermer JA,et al. . Loss of the RNA-binding protein TACO1 causes late-onset mitochondrial dysfunction in mice. Nat Commun. 2016; 7: 11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Petros JA, Baumann AK, Ruiz-Pesini E,et al. . mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A, 2005; 102: 719– 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ray AM, Zuhlke KA, Levin AM,et al. . Sequence variation in the mitochondrial gene cytochrome c oxidase subunit I and prostate cancer in African American men. Prostate. 2009; 69: 956– 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arnold RS, Sun Q, Sun CQ,et al. . An inherited heteroplasmic mutation in mitochondrial gene COI in a patient with prostate cancer alters reactive oxygen, reactive nitrogen and proliferation. Biomed Res Int. 2013; 20132: 39257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun Q, Arnold RS, Sun CQ,et al. . A mitochondrial DNA mutation influences the apoptotic effect of statins on prostate cancer. Prostate. 2015; 75: 1916– 1925. [DOI] [PubMed] [Google Scholar]
- 9. Charlson ES, Sankar PS, Miller-Ellis E,et al. . The primary open-angle African American glaucoma genetics study: baseline demographics. Ophthalmology. 2015; 122: 711– 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collins DW, Gudiseva HV, Trachtman B,et al. . Association of primary open-angle glaucoma with mitochondrial variants and haplogroups common in African Americans. Mol Vis. 2016; 22: 454– 471. [PMC free article] [PubMed] [Google Scholar]
- 11. Collins DW, Gudiseva HV, Trachtman BT,et al. . Mitochondrial sequence variation in African-American primary open-angle glaucoma patients. PLoS One. 2013; 8: e76627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hernandez-Zimbron LF, Luna-Munoz J, Mena R,et al. . Amyloid-beta peptide binds to cytochrome C oxidase subunit 1. PLoS One. 2012; 7: e42344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woods WS, Boettcher JM, Zhou DH,et al. . Conformation-specific binding of alpha-synuclein to novel protein partners detected by phage display and NMR spectroscopy. J Biol Chem. 2007; 282: 34555– 34567. [DOI] [PubMed] [Google Scholar]
- 14. Coskun P, Wyrembak J, Schriner SE,et al. . A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012; 1820: 553– 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghiso JA, Doudevski I, Ritch R,et al. . Alzheimer's disease and glaucoma: mechanistic similarities and differences. J Glaucoma. 2013; 22 suppl)5: S36– S38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gupta V, Gupta VB, Chitranshi N,et al. . One protein, multiple pathologies: multifaceted involvement of amyloid beta in neurodegenerative disorders of the brain and retina. Cell Mol Life Sci. 2016; 73: 4279– 4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. World Medical Association Declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. JAMA. 1997; 277: 925– 926. [PubMed] [Google Scholar]
- 18. Harris PA, Taylor R, Thielke R,et al. . Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009; 42: 377– 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Formstecher E, Aresta S, Collura V,et al. . Protein interaction mapping: a drosophila case study. Genome Res. 2005; 15: 376– 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vojtek AB, Hollenberg SM. . Ras-raf interaction: Two-hybrid analysis. Methods Enzymol. 1995; 255: 331– 342. [DOI] [PubMed] [Google Scholar]
- 21. Fromont-Racine M, Rain JC, Legrain P. . Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat Genet. 1997; 16: 277– 282. [DOI] [PubMed] [Google Scholar]
- 22. Bartel P, Chien CT, Sternglanz R,et al. . Elimination of false positives that arise in using the two-hybrid system. BioTechniques. 1993; 14: 920– 924. [PubMed] [Google Scholar]
- 23. Zeger SL, Liang KY, Albert PS. . Models for longitudinal data: a generalized estimating equation approach. Biometrics. 1988; 44: 1049– 1060. [PubMed] [Google Scholar]
- 24. Kapetanakis VV, Chan MP, Foster PJ,et al. . Global variations and time trends in the prevalence of primary open angle glaucoma (POAG): a systematic review and meta-analysis. Br J Ophthalmol. 2016; 100: 86– 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wallace DC, Singh G, Lott MT,et al. . Mitochondrial DNA mutation associated with leber's hereditary optic neuropathy. Science. 1988; 242: 1427– 30. [DOI] [PubMed] [Google Scholar]
- 26. Frank SA, Hurst LD. . Mitochondria and male disease. Nature. 1996; 383: 224. [DOI] [PubMed] [Google Scholar]
- 27. Ng SK, Burdon KP, Fitzgerald JT,et al. . Genetic association at the 9p21 glaucoma locus contributes to sex bias in normal-tension glaucoma. Invest Ophthalmol Vis Sci. 2016; 57: 3416– 3421. [DOI] [PubMed] [Google Scholar]
- 28. Tranah GJ, Yokoyama JS, Katzman SM,et al. . Mitochondrial DNA sequence associations with dementia and amyloid-beta in elderly African Americans. Neurobiol Aging. 2014; 35: 442.e1– 442.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lloyd RE, McGeehan JE. . Structural analysis of mitochondrial mutations reveals a role for bigenomic protein interactions in human disease. PLoS One. 2013; 8: e69003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jaksch M, Hofmann S, Kleinle S,et al. . A systematic mutation screen of 10 nuclear and 25 mitochondrial candidate genes in 21 patients with cytochrome c oxidase (COX) deficiency shows tRNA(ser)(UCN) mutations in a subgroup with syndromal encephalopathy. J Med Genet. 1998; 35: 895– 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Oven M, Kayser M. . Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009; 30: E386– E394. [DOI] [PubMed] [Google Scholar]
- 32. Cai W, Fu Q, Zhou X,et al. . Mitochondrial variants may influence the phenotypic manifestation of Leber's hereditary optic neuropathy-associated ND4 G11778A mutation. J Genet Genomics. 2008; 35: 649– 655. [DOI] [PubMed] [Google Scholar]
- 33. Stieren ES, El Ayadi A, Xiao Y,et al. . Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J Biol Chem. 2011; 286: 35: 689– 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hiltunen M, Lu A, Thomas AV,et al. . Ubiquilin 1 modulates amyloid precursor protein trafficking and abeta secretion. J Biol Chem. 2006; 281: 32: 240– 253. [DOI] [PubMed] [Google Scholar]
- 35. Bertram L, Hiltunen M, Parkinson M,et al. . Family-based association between Alzheimer's disease and variants in UBQLN1. N Engl J Med. 2005; 352: 884– 894. [DOI] [PubMed] [Google Scholar]
- 36. Lu A, Hiltunen M, Romano DM,et al. . Effects of ubiquilin 1 on the unfolded protein response. J Mol Neurosci. 2009; 38: 19– 30. [DOI] [PubMed] [Google Scholar]
- 37. Itakura E, Zavodszky E, Shao S,et al. . Ubiquilins chaperone and triage mitochondrial membrane proteins for degradation. Mol Cell. 2016; 63: 21– 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tang Z, Arjunan P, Lee C,et al. . Survival effect of PDGF-CC rescues neurons from apoptosis in both brain and retina by regulating GSK3beta phosphorylation. J Exp Med. 2010; 207: 867– 880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yokoyama T, Tamura H, Tsukamoto H,et al. . Prevalence of glaucoma in adults with Down's syndrome. Jpn J Ophthalmol. 2006; 50: 274– 276. [DOI] [PubMed] [Google Scholar]
- 40. Masuzzo A, Dinet V, Cavanagh C,et al. . Amyloidosis in retinal neurodegenerative diseases. Front Neurol. 2016; 7: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pagani L, Eckert A. . Amyloid-beta interaction with mitochondria. Int J Alzheimers Dis. 2011; 2011: 925050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bobba A, Amadoro G, Valenti D,et al. . Mitochondrial respiratory chain complexes I and IV are impaired by beta-amyloid via direct interaction and through complex I-dependent ROS production, respectively. Mitochondrion. 2013; 13: 298– 311. [DOI] [PubMed] [Google Scholar]
- 43. Kaminsky YG, Tikhonova LA, Kosenko EA. . Critical analysis of Alzheimer's amyloid-beta toxicity to mitochondria. Front Biosci (Landmark Ed). 2015; 20: 173– 197. [DOI] [PubMed] [Google Scholar]
- 44. Sivak JM. . The aging eye: common degenerative mechanisms between the Alzheimer's brain and retinal disease. Invest Ophthalmol Vis Sci. 2013; 54: 871– 880. [DOI] [PubMed] [Google Scholar]
- 45. Osborne NN, Alvarez CN, del Olmo Aguado S. . Targeting mitochondrial dysfunction as in aging and glaucoma. Drug Discov Today. 2014; 19: 1613– 1622. [DOI] [PubMed] [Google Scholar]
- 46. Lascaratos G, Chau KY, Zhu H,et al. . Resistance to the most common optic neuropathy is associated with systemic mitochondrial efficiency. Neurobiol Dis, 2015; 8: 278– 285. [DOI] [PubMed] [Google Scholar]
- 47. Osborne NN, Nunez-Alvarez C, Joglar B,et al. . Glaucoma: focus on mitochondria in relation to pathogenesis and neuroprotection. Eur J Pharmacol. 2016; 787: 127– 133. [DOI] [PubMed] [Google Scholar]
- 48. Daudt DR III, Mueller B, Park YH,et al. . Methylene blue protects primary rat retinal ganglion cells from cellular senescence. Invest Ophthalmol Vis Sci. 2012; 53: 4657– 4667. [DOI] [PubMed] [Google Scholar]
- 49. Gonzalez-Lima F, Auchter A. . Protection against neurodegeneration with low-dose methylene blue and near-infrared light. Front Cell Neurosci. 2015; 9: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shi C, Zhu J, Leng S,et al. . Mitochondrial FOXO3a is involved in amyloid beta peptide-induced mitochondrial dysfunction. J Bioenerg Biomembr. 2016; 48: 189– 196. [DOI] [PubMed] [Google Scholar]
- 51. Lascaratos G, Garway-Heath DF, Willoughby CE,et al. . Mitochondrial dysfunction in glaucoma: understanding genetic influences. Mitochondrion. 2012; 12: 202– 212. [DOI] [PubMed] [Google Scholar]
- 52. Ju WK, Kim KY, Lindsey JD,et al. . Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Invest Ophthalmol Vis Sci. 2008; 49: 4903– 4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ju WK, Kim KY, Lindsey JD,et al. . Elevated hydrostatic pressure triggers release of OPA1 and cytochrome C, and induces apoptotic cell death in differentiated RGC-5 cells. Mol Vis. 2009; 15: 120– 134. [PMC free article] [PubMed] [Google Scholar]
- 54. Hernandez-Zimbron LF, Rivas-Arancibia S. . Oxidative stress caused by ozone exposure induces beta-amyloid 1-42 overproduction and mitochondrial accumulation by activating the amyloidogenic pathway. Neuroscience. 2015; 304: 340– 348. [DOI] [PubMed] [Google Scholar]
- 55. Takalo M, Haapasalo A, Natunen T,et al. . Targeting ubiquilin-1 in Alzheimer's disease. Expert Opin Ther Targets. 2013; 17: 795– 810. [DOI] [PubMed] [Google Scholar]
- 56. Gueven N, Nadikudi M, Daniel A,et al. . Targeting mitochondrial function to treat optic neuropathy. Mitochondrion. 2017; 36: 7– 14. [DOI] [PubMed] [Google Scholar]
- 57. Chiang MC, Cheng YC, Chen SJ,et al. . Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against amyloid-beta-induced mitochondrial dysfunction. Exp Cell Res. 2016; 347: 322– 331. [DOI] [PubMed] [Google Scholar]
- 58. Hu H, Li M. . Mitochondria-targeted antioxidant mitotempo protects mitochondrial function against amyloid beta toxicity in primary cultured mouse neurons. Biochem Biophys Res Commun. 2016; 478: 174– 180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.