

Abstract
Liver fibrosis is a pathological scarring response to chronic hepatocellular injury and hepatic stellate cells (HSCs) are key players in this process. PNPLA3 I148M is a common variant robustly associated with liver fibrosis but the mechanisms underlying this association are unknown. We aimed to examine a) the effect of fibrogenic and proliferative stimuli on PNPLA3 levels in HSCs and b) the role of wild type and mutant PNPLA3 overexpression on markers of HSC activation and fibrosis.
Here, we show that PNPLA3 is upregulated by the fibrogenic cytokine transforming growth factor-beta (TGF-β), but not by platelet-derived growth factor (PDGF), and is involved in the TGF-β-induced reduction in lipid droplets in primary human HSCs. Furthermore, we show that retinol release from human HSCs ex vivo is lower in cells with the loss-of-function PNPLA3 148M compared with 148I wild type protein. Stable overexpression of PNPLA3 148I wild type, but not 148M mutant, in human HSCs (LX-2 cells) induces a reduction in the secretion of matrix metallopeptidase 2 (MMP2), tissue inhibitor of metalloproteinase 1 and 2 (TIMP1 and TIMP2), which is mediated by retinoid metabolism. In conclusion, we show a role for PNPLA3 in HSC activation in response to fibrogenic stimuli. Moreover, we provide evidence to indicate that PNPLA3-mediated retinol release may protect against liver fibrosis by inducing a specific signature of proteins involved in extracellular matrix remodelling.
Introduction
Liver fibrosis is a pathological scarring response to chronic hepatocellular injury in individuals with chronic liver disease. Liver fibrosis results from an imbalance between the production and degradation of extracellular matrix and is potentially reversible if the liver injury is eliminated (1). Conversely, if the liver injury persists, liver fibrosis can progress to cirrhosis, which is associated with nodule formation and the development of sinusoidal portal hypertension (2). However, there is a large inter-individual heterogeneity in the susceptibility to develop clinically significant fibrosis.
Hepatic stellate cells (HSCs) play a key role in the development of liver fibrosis (3). In physiological conditions, HSCs are the main reservoir of the body’s retinoids (vitamin A), which are stored in their lipid droplets (4). After liver damage, HSCs lose their retinol content and transdifferentiate into activated myofibroblasts. Active HSCs produce the collagen fibres, the scaffold of the extracellular matrix, and inflammatory and fibrogenic factors involved in matrix remodelling (5). Several proteins modulate hepatic fibrogenesis by acting on HSCs (5,6). Transforming growth factor-beta (TGF-β) is the major fibrogenic cytokine in liver disease and it is increased in animal models of liver fibrosis and in patients with chronic liver disease (7,8). PDGF is the most potent mitogen for HSCs and is upregulated in the fibrotic liver (9).
Patatin-like phospholipase domain-containing 3 (PNPLA3, also called adiponutrin, ADPN) is a membrane-bound protein with predominant lipase activity (10,11). We have shown that PNPLA3 also has retinyl-esterase activity and is highly expressed in human HSCs, where it is involved in retinol metabolism (12). In particular, PNPLA3 expression in HSCs is regulated by retinol availability and it plays an important role in lipid droplet remodelling, promoting the extracellular release of retinol (12). A naturally occurring sequence variation (rs738409) resulting in an isoleucine (I) to methionine (M) substitution at position 148 (I148M) (13) of the protein is the most robustly replicated genetic variant associated with liver disease (14), hepatic fat accumulation (15) and progression to fibrosis and chronic liver disease (16–19). Epidemiological evidence suggests that the impact of the I148M mutation on liver fibrosis progression may be independent of the predisposition to hepatic fat accumulation (16,20). However, the mechanism underlying the direct effect on the progression to liver fibrosis still remains obscure.
In this study, we examined the effect of the PNPLA3 148I wild type and the 148M mutant protein on HSC activation and the acquisition of fibrogenic phenotype. We found that PNPLA3 is upregulated by TGF-β and that overexpression of the mutant protein results in a specific signature of proteins involved in extracellular matrix remodelling.
Results
TGF-β reduces intracellular retinyl ester content by PNPLA3 upregulation in pHSCs
To examine if TGF-β regulates PNPLA3 synthesis, we incubated primary HSCs with TGF-β (10 ng/ml) and examined PNPLA3 protein levels at different time points. We found that PNPLA3 was upregulated by this cytokine (Fig. 1A). Next we examined whether PDGF, a potent inducer of proliferation, influences PNPLA3 synthesis. We did not find any differences in PNPLA3 synthesis after incubation with 10 ng/ml PDGF (Fig. 1B).
Figure 1.

PNPLA3 is upregulated by TGF-β and not by PDGF in primary hepatic stellate cells (pHSCs). Western blotting analysis of PNPLA3 expression in primary HSCs incubated with (A) TGF-β (10 ng/ml) or (B) PDGF (10 ng/ml) for 0, 4, 8 and 24 h. The intensity of the western blotting bands was measured by Image Lab Software (Bio-Rad) and expressed as relative density. The value obtained at 0 h was assigned as 1.
Next we examined whether incubation with TGF-β induces a reduction in intracellular neutral lipid content. Primary HSCs were incubated in medium containing retinol (10 µM) and palmitic acid (100 µM) for 24 h; after this time point, the medium was changed and cells were incubated with and without TGF-β (10 ng/ml) in the absence of retinol and palmitic acid for up to 24 h (Fig. 2A). We observed a faster reduction in the intracellular lipid content in cells exposed to TGF-β compared to no exposure to TGF-β (Fig. 2B). In contrast, we did not observe any changes in the intracellular lipid droplet content in response to PDGF 10 ng/ml (Supplementary Material, Fig. S1).
Figure 2.
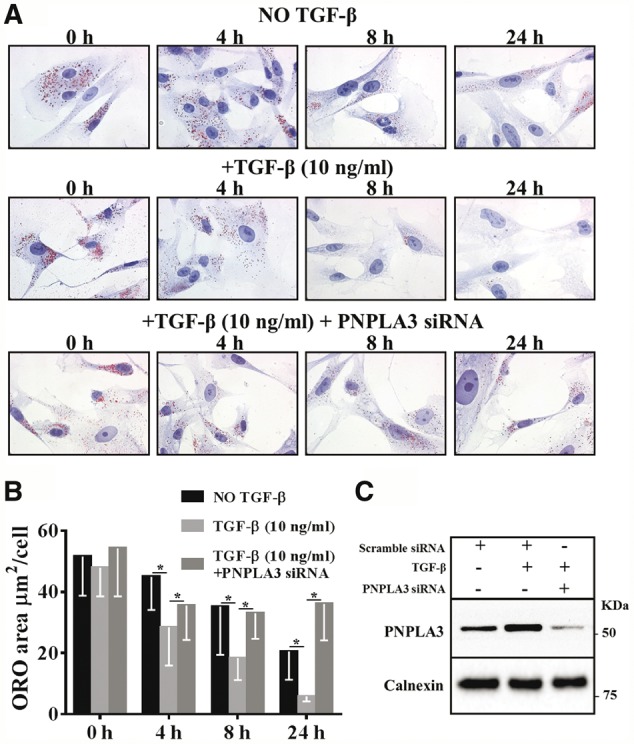
PNPLA3 mediates the TGF-β-induced reduction in lipid droplet content in human pHSCs. (A) Intracellular neutral lipid content visualized by ORO staining in pHSCs incubated with retinol and palmitic acid for 24 h and then in medium without retinol-palmitic acid for the indicated times without TGF-β (upper panel), with TGF-β 10 ng/ml (middle panel) and with TGF-β 10 ng/ml plus PNPLA3 siRNA (lower panel). (B) Intracellular ORO-stained area of pHSCs without TGF-β, with TGF-β and with TGF-β plus PNPLA3 siRNA quantified by BioPix. (C) Western blotting analysis of PNPLA3 expression in pHSCs without TGF-β, with TGF-β and with TGF-β plus PNPLA3 siRNA after 24 h.
To test whether PNPLA3 was responsible for the reduction in intracellular lipid content after TGF-β incubation, we tested whether the reduction in intracellular lipid content induced by TGF-β was influenced by PNPLA3 knockdown by siRNA (Fig. 2A–C). RNA interference downregulated PNPLA3 expression by 3-fold (Fig. 2C). Importantly, PNPLA3 downregulation prevented the TGF-β-induced reduction in intracellular lipid content (Fig. 2A and B).
Generation and characterization of a stable cell line overexpressing PNPLA3 148I wild type and 148M mutant
To examine the role of PNPLA3 148I wild type and its 148M mutant in the secretion of proteins involved in fibrogenesis, we generated and characterized a LX-2 cell line stably overexpressing the human PNPLA3 wild type and mutant protein (Fig. 3A–C). In agreement with previous results of acute overexpression of PNPLA3 (12), we observed a reduction in the intracellular lipid content of LX-2 cells overexpressing the wild type but not the mutant protein after retinol and palmitic acid incubation for 48 h (Fig. 3A and B), suggesting that only the wild type protein was able to reduce the intracellular lipid content. To confirm the robustness of our model, we examined the intracellular lipid content in cells after: a) retinol and palmitic acid incubation for 48 h followed by retinol depletion in retinol free medium for 24 h (Supplementary Material, Fig. S2); b) retinol and palmitic acid incubation for 24 h (Supplementary Material, Fig. S3A); c) retinol and palmitic acid incubation for 24 h followed by retinol depletion in the retinol-free medium for 12 h (Supplementary Material, Fig. S3B); and d) retinol and palmitic acid incubation for 24 h followed by retinol depletion in the retinol-free medium for 24 h (Supplementary Material, Fig. S3C). Consistently, we observed lower intracellular lipid droplet content in cells overexpressing the wild type but not the mutant protein under all the tested conditions.
Figure 3.

Stable overexpression of PNPLA3 wild type (148I) but not mutant (148M) reduces intracellular lipid content in immortalized human hepatic stellate cells (LX-2). (A) Lipid droplets visualized by ORO-staining in LX-2 cells stably expressing human V5-tagged 148I or 148M PNPLA3 and incubated with retinol (10 µM) and palmitic acid (100 µM) for 48 h. Empty vector (EV) was used as negative control. (B) ORO-stained area quantified by BioPix. (C) Immunoblot showing transfection efficiency. (D) Freshly isolated primary hHSCs, at third passage after isolation, were pre-treated with 300 µM palmitic acid and 10 µM retinol for 24 h and then incubated with quiescence medium (DMEM 0.5% FBS; wash-out) to evaluate their ability to release lipid droplets. Lipid droplets were visualized by ORO staining, and (E) ORO positive (+ve) area were quantified by ImageJ. Original magnification: 400X. n = 3 I148I PNPLA3 hHSCs and n = 2 M148M PNPLA3 hHSCs; * P < 0.05 compared to I148I genotype.
PNPLA3 148M mutant induces retention of the intracellular lipid content in primary human HSCs ex vivo
To further confirm the role of PNPLA3 in the retinol metabolism of HSCs, we isolated human HSCs from surgical samples of patients homozygous for the PNPLA3 wild type (n = 3) and the mutant allele (n = 2). We examined the effect of the mutation on intracellular neutral lipid content after incubation of these primary human HSCs with retinol and palmitic acid for 24 h followed by retinol depletion in retinol-free medium for 24 h. Consistently with the LX-2 cell model, we observed that wild type cells had lower intracellular neutral lipid content than the I148M mutant cells (Fig. 3D and E).
PNPLA3 148I wild type but not 148M mutant promotes reduced retinol-mediated secretion of MMP2, TIMP1 and TIMP2
To examine the effect of PNPLA3 wild type and mutant protein on factors involved in fibrosis, we examined the intracellular protein levels of several proteins belonging to different families. Retinoids (including retinol) regulate metabolic pathways through binding to nuclear receptors and are involved in fibrosis (21,22), and therefore we examined cells in the presence of retinol. Specifically, we incubated LX-2 cells with retinol and palmitic acid for 24 h followed by retinol depletion in the retinol-free medium for 24 h. Then, we tested for differences in intracellular markers of cell activation (alpha smooth muscle actin (α-SMA) and TGF-β receptor) and of extracellular matrix formation and remodelling (metalloproteinases (MMPs), tissue inhibitors of metalloproteinase (TIMPs) and collagen I) between cells expressing the wild type or the mutant protein. We did not observe any difference in intracellular protein levels between cells overexpressing the PNPLA3 148I wild type or the 148M mutant protein (Supplementary Material, Fig. S4A). Next, we examined the effect of the wild type and mutant protein overexpression on extracellular level of proteins involved in the matrix formation (collagen I) and remodelling (MMPs and TIMPs) after 24 h incubation with retinol and palmitic acid followed by retinol depletion in retinol free medium for 24 h. Concentrated medium (10X) from the cells overexpressing the PNPLA3 148I wild type but not the 148M mutant protein and incubated with retinol and palmitic acid had lower levels of MMP2, TIMP1 and TIMP2 (Fig. 4A).
Figure 4.

PNPLA3 wild type but not PNPLA3 148M mutant overexpression induces a reduction in the secretion of MMP2, TIMP1 and TIMP2. Concentrated (10X) medium fractions of LX-2 stable cell lines expressing empty vector (EV), PNPLA3 WT or PNPLA3 148 M incubated with (A) regular medium plus retinol 10 µM and palmitic acid 100 µM for 24 h and then in retinol-palmitic acid free medium without FBS for 24 h, and (B) regular medium without retinol and palmitic acid for 24 h and then in medium without FBS for 24 h. Calnexin was used as loading control. MMP14: matrix metalloproteinase14; MMP2: matrix metallopeptidase 2; MMP9: matrix metallopeptidase 9; TIMP1: tissue inhibitor of metalloproteinase 1; TIMP2: tissue inhibitor of metalloproteinase 2; COL I: collagen Iα1.
To confirm our results, we examined the levels of intracellular markers of cell activation (α-SMA and TGF-β receptor) and of extracellular levels of proteins involved in the matrix formation (collagen I) and remodelling (MMPs and TIMPs) in a time-dependent manner. Specifically, we examined intracellular markers and extracellular proteins after: a) 24 h incubation with retinol and palmitic acid followed by retinol depletion in retinol free medium for 12 h (Supplementary Material, Fig. S5A); b) 48 h incubation with retinol and palmitic acid followed by retinol depletion in retinol-free medium for 24 h (Supplementary Material, Fig. S5B). Consistently, we obtained similar results as in the experiment shown in Fig. 4 indicating that the differences observed are PNPLA3 mutation and not time dependent.
To test whether the release of retinol metabolites was responsible for the reduction in the secretion of MMP2, TIMP1 and TIMP2, we examined the levels of these secreted proteins in the medium of LX-2 cells overexpressing the wild type and the mutant protein in the absence of retinol and palmitic acid. We did not observe any difference in the intracellular (Supplementary Material, Fig. S4B) or extracellular protein levels (Fig. 4B) of any of the other markers examined.
To further examine the role of retinoids on MMP2, TIMP1 and TIMP2 secretion, we incubated cells overexpressing the PNPLA3 wild type protein with retinol, palmitic acid and increasing concentrations of liarozole hydrochloride, a potent inhibitor of intracellular retinol metabolism. We observed a dose-dependent increase in MMP2, TIMP1 and TIMP2 after incubation with liarozole hydrochloride (Fig. 5A). To further confirm the role of retinoids on changes of MMP2, TIMP1 and TIMP2 secretion, we examined the extracellular protein levels of LX-2 cells overexpressing the PNPLA3 148M mutant protein after incubation with increasing amount of all trans retinoic acid (ATRA), a potent retinoid acid and X receptor (RAR and RXR) agonist. We observed a dose-dependent reduction in extracellular protein levels of MMP2, TIMP1 and TIMP2 in cells overexpressing the PNPLA3 148M mutant protein (Fig. 5B).
Figure 5.

The PNPLA3 induced reduction in MMP2, TIMP1 and TIMP2 secretion is due to retinoids. (A) Liarozole hydrochloride induces an increase in the MMP2, TIMP1 and TIMP2 secretion in LX-2 cells stably overexpressing wild type PNPLA3. Left panels: Western blotting analysis of LX-2 cells stably over-expressing PNPLA3 148I incubated with regular medium plus retinol 10 µM, palmitic acid 100 µM and increasing Liarozole Hydrochloride (10 µM, 50 µM and 100 µM) for 24 h and then in retinol-palmitic acid free medium without FBS for 24 h. Right Panels: relative quantification of western blotting bands of the left panels. (B) All trans retinoic acid (ATRA) induces a reduction in the secretion of MMP2, TIMP1 and TIMP2 in LX-2 cells stably over-expressing PNPLA3 148M mutant protein. Right Panels: western blotting of MMP2, TIMP1 and TIMP2 in LX-2 cells stably over-expressing PNPLA3 148M incubated with regular medium plus retinol 10 µM, palmitic acid 100 µM and increasing ATRA (30 µM, 60 µM and 100 µM) for 24 h and then in retinol-palmitic acid free medium without FBS for 24 h. Right panels: relative quantification of western blotting band in the left panels. Albumin was used as loading control. MMP2: matrix metallopeptidase 2; TIMP1: tissue inhibitor of metalloproteinase 1; TIMP2: tissue inhibitor of metalloproteinase 2.
PNPLA3 is present in non-parenchymal hepatic sinusoidal cells
To examine the role of PNPLA3 wild type and mutant protein in human liver with steatosis, we used immunohistochemistry to assess the distribution of PNPLA3 in different liver cell types in hepatic specimens from patients homozygous for the wild type or mutant allele. PNPLA3 was present at higher levels in non-parenchymal sinusoidal cells of patients homozygous for the mutant allele (Fig. 6A–C).
Figure 6.

PNPLA3 is present in non-parenchymal hepatic sinusoidal cells. Immunohistochemistry (IHC) analysis of PNPLA3 was performed on liver specimens obtained from NAFLD patients carriers of the two different PNPLA3 genotypes (I148I panel A, M148M panel B). Non parenchymal cells, most likely HSCs localized in sinusoidal spaces, are indicated by the arrows. (C) Percentage of PNPLA3 positive cells, manually counted with ImageJ. The expression of PNPLA3 is higher in liver biopsies from M148M patients compared to I148I. (D) Liver sample characterized by fibrotic areas showing PNPLA3 cytoplasmic positivity in hepatocytes, but not in cells localized between fibrotic septa. Nuclei were contrasted by hematoxylin staining. Original magnification: 200X. n = 4 I148I PNPLA3 patients and n = 3 M148M PNPLA3 patients; *P < 0.05 compared to I148I genotype.
We finally examined the distribution of PNPLA3 in hepatic specimens from individuals with advanced liver disease and could not detect the protein in fibrotic strands (Fig. 6D).
Discussion
In this study, we examined the effect of the PNPLA3 protein and its I148M variant in HSC activation and the acquisition of the fibrogenic phenotype. We show that PNPLA3 is induced by TGF-β and that the PNPLA3 I148M mutation impairs retinol-palmitate release from primary human HSCs ex vivo. Moreover, upregulation of the PNPLA3 wild type but not the mutant protein results in reduced extracellular levels of proteins involved in fibrosis.
TGF-β is a potent inducer of HSC transactivation and fibrogenesis (23) whereas PDGF induces cell proliferation (24). We showed that TGF-β but not PDGF induced an upregulation of intracellular PNPLA3 levels in primary human HSCs, indicating the specificity of TGF-β in upregulating PNPLA3 levels. Similar to our previous results showing that upregulation of PNPLA3 by insulin induces the release of retinol from intracellular lipid droplets in human HSCs (12), here we showed that TGF-β induced a PNPLA3-mediated reduction in intracellular lipid droplets. These data suggest that PNPLA3 is upregulated during the process of HSC activation by fibrogenic stimuli.
To investigate the impact of PNPLA3 upregulation of the I148M mutation on the fibrogenic phenotype of HSCs, we generated a stable cell line of immortalized human HSCs overexpressing the wild type and the mutant protein. First, we examined intra and extracellular level of proteins involved in fibrosis in cells cultured with retinol. To confirm our results, we examined the effect of the PNPLA3 wild type and mutant protein on primary HSCs isolated from humans who were homozygous for either of the two genotypes. Cells expressing the PNPLA3 148M had higher intracellular neutral lipid content. This is the first demonstration that the PNPLA3 I148M mutation impairs retinol-palmitate release ex vivo from primary human HSCs. These data are consistent with previous data showing lower circulating free retinoids and higher retinoid levels in the liver in carriers of the PNPLA3 148M mutation (25,26).
Earlier results from both experimental models and human samples suggest that changes in expression of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) contribute to fibrosis remodelling (27). Specifically MMP2 expression increases in experimental fibrosis models and in human chronic liver disease (28–32). Moreover TIMP1 and TIMP2 expression increases dramatically in activated HSCs (33) and in the liver of rats treated with CCl4, a compound used to induce fibrosis (34). Here we showed that stable overexpression of the PNPLA3 wild type but not the mutant protein induced a reduction in extracellular levels of MMP2, TIMP1 and TIMP2 in the medium of HSCs incubated with retinol and palmitic acid. Our results suggest that upregulation of the PNPLA3 wild type protein prevents the release of proteins that increase liver fibrosis. The mutant protein is a loss of function variant and therefore the protective effect of PNPLA3 is hampered in HSCs carrying this mutation.
To investigate the mechanisms underlying the effect of PNPLA3 on the fibrogenic phenotype of HSCs we incubated cells without retinol and palmitic acid. We observed no differences in the extracellular protein levels of MMP2, TIMP1 and TIMP2 between the cells overexpressing the 148I wild type or the 148M mutant protein in the medium of cells incubated without retinol. These results suggest that the PNPLA3 effect on these markers is mediated by retinoids. To test this hypothesis, we incubated cells overexpressing the wild type with increasing amount of liarozole, a potent inhibitor of retinol metabolism. Increasing concentrations of liarozole induced a progressive increase in extracellular levels of MMP2, TIMP1 and TIMP2. Vice versa, incubation of the cells overexpressing the mutant protein with increasing amount of ATRA, a potent RAR/RXR agonist, induced a progressive reduction of extracellular levels of these enzymes. Thus, we recapitulated the wild type phenotype in cells carrying the mutant by incubating with a retinoid agonist, and the mutant phenotype in cells carrying the wild type protein by incubating with a retinoid antagonist. These results show that, in our model, the PNPLA3-mediated effect on proteins involved in fibrosis remodelling is mediated by retinoids.
We have previously shown that PNPLA3 is expressed and synthetized in hepatocytes, and that higher expression of the 148M mutant protein is associated with increased risk of liver damage related to nonalcoholic fatty liver disease, likely due to impairment of lipid droplet remodelling (12,35). Here, we showed that PNPLA3 was present at higher levels in non-parenchymal sinusoidal cells from humans who were homozygous for the mutant allele compared with the wild type PNPLA3. In contrast, PNPLA3 was not detectable in other cells of fibrotic strands during advanced human liver disease. These findings are consistent with the hypothesis that PNPLA3 mutant protein induces liver fibrosis by acting in HSCs and not in other fibrogenic cells involved in later stages of fibrosis. The detailed characterization of non-parenchymal PNPLA3-positive cells in human liver samples of healthy individuals and patients with liver diseases will require further studies evaluating by immunofluorescence the co-staining of PNPLA3 with α-SMA and other cell markers, which was not possible in our paraffin-embedded tissues.
It is generally accepted that retinol loss occurs after HSC activation, but it is not known whether this loss triggers, facilitates or increases the HSC activation. Our data suggest that retinol loss does not worsen the fibrogenic phenotype of HSCs. In fact, PNPLA3-mediated retinol release may rather be a protective mechanism against fibrosis involving regulation of HSC activation and extracellular matrix remodelling. The PNPLA3 I148M is a loss of function variant determining a reduced rate in the release of retinoids from lipid droplets impairing this feedback mechanism. On the basis of our results, we propose the following model: upon HSC activation by fibrogenic stimuli, upregulation of the wild type PNPLA3 protein counteracts fibrosis by reducing the release of enzymes that facilitate fibrosis in a process that is mediated by retinoids (Fig. 7). In carriers of the loss of function PNPLA3 mutant, the release of retinoids from lipid droplets is reduced, and thus the protection from fibrosis is lost (Fig. 7).
Figure 7.

Putative model of PNPLA3 in hepatic stellate cells. (A) PNPLA3 overexpression promotes release of retinoids which down-regulates in the nucleus the expression of MMP2, TIMP1, TIMP2. (B) When the 148M mutation occurs, the retinyl-palmitate lipase activity of PNPLA3 is lost, leading to retinol retention and subsequent increased secretion of pro-fibrotic factors and extracellular matrix deposition. LD: Lipid droplet; N: nucleus; MMP2: matrix metallopeptidase 2; TIMP1: tissue inhibitor of metalloproteinase 1; TIMP2: tissue inhibitor of metalloproteinase 2.
The main limitation of this work is that it does not account for the cross talk between different liver resident cell types. Further in vitro and in vivo studies are required to understand the mechanism linking PNPLA3 with liver fibrosis.
In conclusion, we show a role for PNPLA3 in HSCs activation in response to fibrogenic stimuli. Moreover, our results indicate that PNPLA3-mediated retinol release may have a role in protecting against liver fibrosis by inducing the secretion of a specific signature of proteins that are involved in extracellular matrix remodelling.
Materials and Methods
Cell culture
Primary human hepatic stellate cells (pHSCs) cryopreserved after purification from human liver were purchased from 3H Biomedical. After thawing, cells were plated in T-75 flasks coated with poly-L-lysine and grown in SteCM medium containing foetal bovine serum (FBS) 2% for 5 passages. Then, retinol and palmitic acid were added to restore the quiescence state (36). Immortalized human HSCs (LX-2) were kindly provided by Professor Scott L. Friedman (Mount Sinai School of Medicine) (33). LX-2 cells were grown in high glucose DMEM containing FBS 10% in T-75 flasks. Cells were seeded in 24-well plates with cover slips for Oil Red O (ORO) staining or in 6-well plates and T-75 flasks for immunoblot. pHSCs were seeded in 6-well plates and 24-well plates with cover slips for 24 h. Then the cells were incubated with TGF-β 10 ng/ml (SigmaAldrich) or PDGF 10 ng/ml (SigmaAldrich) for 0, 4, 8 and 24 h. Western blotting was performed to assess the PNPLA3 levels and ORO staining was performed to measure intracellular lipid droplet content.
PNPLA3 knockdown
pHSCs were transfected with PNPLA3 small interfering RNA (siRNA) or negative control (scramble) siRNA (Ambion-Life Technologies by Thermo Fisher Scientific, Rockford, IL) with TurboFect Transfection Reagent (Thermo Scientific) according to the manufacturer’s instructions. The cells were transfected with 25 nM siRNA oligos in serum-free medium.
Stable cell line generation
Expression plasmids pcDNA 3.1 (30 µg for the T-75 plate) with a V5 fusion tag, containing the human 148I wild type or 148M mutant PNPLA3 cDNA were used to transfect LX-2 cells using TurboFect reagent according to the manufacturer’s protocol. An empty vector was used as a negative control. Forty-eight hours after transient transfection, LX-2 cells were treated with 4 mg/ml of geneticin-G418 (Life Technologies), to select resistant cells. After 15 days, six single colonies per each genotype were selected and expanded. An empty vector was used as a negative control. Western blotting was performed to determine PNPLA3 expression levels. A single cell clone for each genotype was used for all the experiments. Transfection efficiency was assessed using an anti-V5 antibody.
LX-2 cells stably expressing human V5-tagged 148I or 148M PNPLA3 were incubated with albumin (3 mg/ml), retinol (10 µM) and palmitic acid (100 µM) for: (a) 48 h; (b) 48 h plus 24 h with regular medium; (c) 24 h; (d) 24 h plus 12 h with regular medium; (e) 24 h plus 24 h with regular medium. ORO staining was performed for each time point.
Next, LX-2 stably expressing human V5-tagged 148I or 148M PNPLA3 were incubated with albumin (3 mg/ml), retinol (10 µM) and palmitic acid (100 µM) for: (a) 24 h plus 24 h with regular medium without FBS; (b) 24 h plus 12 h with regular medium without FBS; (c) 48 h plus 24 h with regular medium without FBS; (d) regular medium without retinol and palmitic acid for 24 h and then in regular medium without FBS for 24 h. Medium fractions and cells were collected and analysed by western blotting after each time point.
LX-2 cells stably expressing PNPLA3 148M were also incubated with albumin (3 mg/ml), retinol (10 µM), palmitic acid (100 µM) plus all trans retinoic acid (ATRA) (30 µM, 60 µM, 100 µM) for 24 h and then with regular medium without FBS for 24 h. Medium fractions were collected and analysed by western blotting.
LX-2 cells stably expressing PNPLA3 148I wild type were incubated with albumin (3 mg/ml), retinol (10 µM), palmitic acid (100 µM) plus liarozole hydrochloride (10 µM, 50 µM, 100 µM) for 24 h and then with regular medium without FBS for 24 h. Medium fractions were collected and analysed by western blotting.
Human HSC isolation
Human HSCs were isolated from non-neoplastic human liver tissue obtained from explanted or resected livers of patients. All samples were negative for hepatitis B virus surface antigen (HBsAg) and for hepatitis C virus (HCV)-RNA. Samples from donors with variable steatosis and fibrosis grade and with no age restriction were included in the study. Informed written consent of each patient was obtained, according to legal and ethical rules established by the Ethics Committee of the Fondazione IRCCS Cà Granda, which has already approved the protocol. Liver tissues were stored in a Celsior preservation solution for a maximum of 8 h from the explantation at 4 °C.
Human HSCs were isolated by a multi-step ethylene glycol tetra-acetic acid (EGTA)/collagenase-pronase perfusion technique, through adequate micro cannulation of small blood vessels to ensure efficient perfusion of the tissue sample, followed by centrifugation on a density gradient with Optiprep to isolate human HSCs, taking advantage of their low density, as described before in a murine model (37).
Human HSCs isolated were cultured on plastic for a few passages (37). Cells were grown in DMEM medium in T-75 flasks. When confluent, cells were passaged using trypsin and seeded at a ratio of 1:3. Experiments were performed at the third passage of culture. Three independent lots of freshly isolated human HSCs carrying 148I/I and two carrying 148M/M PNPLA3 genotype, were used for experiments. Briefly, 4 h after isolation, primary human HSCs were treated with 10 µM retinol and 300 µM palmitic acid (Sigma Aldrich, St Louis, MO) for 24 h and then incubated with quiescence medium (DMEM 0.5% FBS) (wash-out). Lipid droplet content was visualized by ORO staining (12) and ORO positive area was quantified by ImageJ software in 10 random micrographs (magnification 200x) by calculating the ORO positive area as percentage of pixels above the threshold value with respect to the total pixels per area.
Oil red O (ORO) staining
The total area of ORO-stained lipid droplets was determined as described previously (38). Pictures were obtained using Axio KS 400 Imaging System and AxioVision 4.8 Software (Zeiss) at 100X magnification. ORO-stained area was quantified by BioPix or ImageJ software for the in vitro and ex vivo model respectively.
Immunoblot analysis
Cells were lysed in M-PER Mammalian Protein Extraction Reagent (Pierce, Thermo Fisher Scientific) containing complete protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Medium fractions were collected and concentrated 10X using VIVASPIN tubes (MWCO 10KDa). Immunoblot analysis was performed according to standard procedures. Bands were visualized using a Chemidoc XRS System and Image Lab Software (Bio-Rad).
The following antibodies were used: mouse anti-V5 (P/N 46-0705) by Invitrogen, rabbit anti-PNPLA3 (AV48936), rabbit anti-calnexin (C4731), rabbit anti-MMP14 (M5808), rabbit anti-MMP2 (HPA001939), mouse anti-MMP9 (SAB140274), mouse anti-TIMP2 (WH0007077M1), rabbit anti-collagen I (HPA011795), mouse anti-α-SMA (A5228), mouse anti-albumin (A6684) by Sigma-Aldrich, rabbit anti-TIMP1 (AB81282) and rabbit anti-TGF-β receptor (AB138248) by Abcam.
Statistical analysis
Data from in vitro experiments were analysed using Mann-Whitney non-parametric test. P-values of <0.05 were considered significant and indicated as * in figures. Bar graphs in figures show mean ± SD of three experiments unless otherwise specified.
Immunohistochemistry
Liver biopsies were fixed in 10% PBS buffered formalin and then embedded in paraffin within 24 h of formalin fixation. We performed immunohistochemical analysis to evaluate PNPLA3 expression in liver tissues from patients homozygous for the two different genotypes (148I, 148M), as previously described (35).
Briefly, de-paraffinated sections were re-hydrated in alcohol and endogenous peroxidase activity was blocked with methanol/10% hydrogen peroxide. After unmasking with the proper antigen retrieval and treating with blocking solutions, the slides were incubated overnight at 4 °C with PNPLA3 antibody (Abcam, ab188856) 1:75. After rinsing, slides were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody and developed with 3-3-diaminobenzidine (DAB). The slides were analysed with the Leica DMD108 microscope (Leica) connected to a digital camera (Leica). PNPLA3 quantification was performed in 10 random non-overlapping fields per slide (200X magnification) by manually counting the number of cells localized in sinusoidal spaces with the software ImageJ. The results are expressed as percentage of positive cells localized in sinusoidal space per field.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank Dr Rosie Perkins for editing the manuscript and Professor Scott L. Friedman for providing LX-2 cells.
Conflict of Interest statement. None declared.
Funding
This work was supported by the Swedish Research Council [Vetenskapsrådet (VR), 254439006], the Swedish Heart Lung Foundation [244439007], the Swedish federal government funding under the Agreement on Medical Training and Medical Research (ALF) [76290], the Novo Nordisk Foundation Grant for Excellence in Endocrinology [244439012], the Swedish Diabetes Foundation [DIA 2014-052] (S.R.), the Wilhelm and Martina Lundgren Science Fund (P.P., B.M.M, R.M.M. and S.R.), the Nilsson-Ehle funds from the Fysiografiska Sällskapet in Lund (R.M.M.), the Ricerca Corrente Fondazione Ca’ Granda IRCCS Policlinico of Milan, Associazione Malattie Metaboliche del Fegato ONLUS, and the Fondazione Policlinico – INGM Molecular Medicine grant 2014-2016, My First AIRC Grant project code 16888 (L.V.), the co-financed grant from the European Commission, the European Social Fund and Calabria Region (S.M.L.). Funding to pay the Open Access publication charges for this article was provided by the Swedish Research Council.
References
- 1. Li J., Chen K., Li S., Feng J., Liu T., Wang F., Zhang R., Xu S., Zhou Y., Zhou S., et al. (2016) Protective effect of fucoidan from Fucus vesiculosus on liver fibrosis via the TGF-β1/Smad pathway-mediated inhibition of extracellular matrix and autophagy. Drug Des. Devel. Ther., 10, 619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedman S.L. (2008) Hepatic fibrosis – overview. Toxicology, 254, 120–129. [DOI] [PubMed] [Google Scholar]
- 3. Kocabayoglu P., Friedman S.L. (2013) Cellular basis of hepatic fibrosis and its role in inflammation and cancer. Front. Biosci. (Schol Ed), 5, 217–230. [DOI] [PubMed] [Google Scholar]
- 4. Senoo H., Yoshikawa K., Morii M., Miura M., Imai K., Mezaki Y. (2010) Hepatic stellate cell (vitamin A-storing cell) and its relative–past, present and future. Cell Biol. Int., 34, 1247–1272. [DOI] [PubMed] [Google Scholar]
- 5. Bataller R., Brenner D.A. (2005) Liver fibrosis. J. Clin. Invest., 115, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marra F. (2002) Chemokines in liver inflammation and fibrosis. Front. Biosci., 7, d1899–1d1914. [DOI] [PubMed] [Google Scholar]
- 7. Nakatsukasa H., Nagy P., Evarts R.P., Hsia C.C., Marsden E., Thorgeirsson S.S. (1990) Cellular distribution of transforming growth factor-beta 1 and procollagen types I, III, and IV transcripts in carbon tetrachloride-induced rat liver fibrosis. J. Clin. Invest., 85, 1833–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castilla A., Prieto J., Fausto N. (1991) Transforming growth factors beta 1 and alpha in chronic liver disease. Effects of interferon alfa therapy. N. Engl J. Med., 324, 933–940. [DOI] [PubMed] [Google Scholar]
- 9. Pinzani M., Gesualdo L., Sabbah G.M., Abboud H.E. (1989) Effects of platelet-derived growth factor and other polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J. Clin. Invest., 84, 1786–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pingitore P., Pirazzi C., Mancina R.M., Motta B.M., Indiveri C., Pujia A., Montalcini T., Hedfalk K., Romeo S. (2014) Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta, 1841, 574–580. [DOI] [PubMed] [Google Scholar]
- 11. He S., McPhaul C., Li J.Z., Garuti R., Kinch L., Grishin N.V., Cohen J.C., Hobbs H.H. (2010) A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem., 285, 6706–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pirazzi C., Valenti L., Motta B.M., Pingitore P., Hedfalk K., Mancina R.M., Burza M.A., Indiveri C., Ferro Y., Montalcini T., et al. (2014) PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet., 23, 4077–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Romeo S., Kozlitina J., Xing C., Pertsemlidis A., Cox D., Pennacchio L.A., Boerwinkle E., Cohen J.C., Hobbs H.H. (2008) Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet., 40, 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Romeo S., Sentinelli F., Dash S., Yeo G.S., Savage D.B., Leonetti F., Capoccia D., Incani M., Maglio C., Iacovino M., et al. (2010) Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int. J. Obes. (Lond), 34, 190–194. [DOI] [PubMed] [Google Scholar]
- 15. Pirazzi C., Adiels M., Burza M.A., Mancina R.M., Levin M., Ståhlman M., Taskinen M.R., Orho-Melander M., Perman J., Pujia A., et al. (2012) Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol., 57, 1276–1282. [DOI] [PubMed] [Google Scholar]
- 16. Sookoian S., Pirola C.J. (2011) Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology, 53, 1883–1894. [DOI] [PubMed] [Google Scholar]
- 17. Valenti L., Al-Serri A., Daly A.K., Galmozzi E., Rametta R., Dongiovanni P., Nobili V., Mozzi E., Roviaro G., Vanni E., et al. (2010) Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology, 51, 1209–1217. [DOI] [PubMed] [Google Scholar]
- 18. Burza M.A., Pirazzi C., Maglio C., Sjöholm K., Mancina R.M., Svensson P.A., Jacobson P., Adiels M., Baroni M.G., Borén J., et al. (2012) PNPLA3 I148M (rs738409) genetic variant is associated with hepatocellular carcinoma in obese individuals. Dig. Liver Dis., 44, 1037–1041. [DOI] [PubMed] [Google Scholar]
- 19. Valenti L., Dongiovanni P., Ginanni Corradini S., Burza M.A., Romeo S. (2013) PNPLA3 I148M variant and hepatocellular carcinoma: a common genetic variant for a rare disease. Dig. Liver Dis., 45, 619–624. [DOI] [PubMed] [Google Scholar]
- 20. Dongiovanni P., Donati B., Fares R., Lombardi R., Mancina R.M., Romeo S., Valenti L. (2013) PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol., 19, 6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang P., Chandra V., Rastinejad F. (2014) Retinoic acid actions through mammalian nuclear receptors. Chem. Rev., 114, 233–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okuno M., Kojima S., Akita K., Matsushima-Nishiwaki R., Adachi S., Sano T., Takano Y., Takai K., Obora A., Yasuda I., et al. (2002) Retinoids in liver fibrosis and cancer. Front. Biosci., 7, d204–2d218. [DOI] [PubMed] [Google Scholar]
- 23. Dooley S., ten Dijke P. (2012) TGF-β in progression of liver disease. Cell Tissue Res., 347, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kinnman N., Goria O., Wendum D., Gendron M.C., Rey C., Poupon R., Housset C. (2001) Hepatic stellate cell proliferation is an early platelet-derived growth factor-mediated cellular event in rat cholestatic liver injury. Lab. Invest., 81, 1709–1716. [DOI] [PubMed] [Google Scholar]
- 25. Mondul A., Mancina R.M., Merlo A., Dongiovanni P., Rametta R., Montalcini T., Valenti L., Albanes D., Romeo S. (2015) PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity. J. Nutr., 145, 1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kovarova M., Königsrainer I., Königsrainer A., Machicao F., Häring H.U., Schleicher E., Peter A. (2015) The Genetic Variant I148M in PNPLA3 Is Associated With Increased Hepatic Retinyl-Palmitate Storage in Humans. J. Clin. Endocrinol. Metab., 100, E1568–15E1574. [DOI] [PubMed] [Google Scholar]
- 27. Iredale J.P. (2001) Hepatic stellate cell behavior during resolution of liver injury. Semin. Liver Dis., 21, 427–436. [DOI] [PubMed] [Google Scholar]
- 28. Préaux A.M., Mallat A., Nhieu J.T., D'Ortho M.P., Hembry R.M., Mavier P. (1999) Matrix metalloproteinase-2 activation in human hepatic fibrosis regulation by cell-matrix interactions. Hepatology, 30, 944–950. [DOI] [PubMed] [Google Scholar]
- 29. Benyon R.C., Iredale J.P., Goddard S., Winwood P.J., Arthur M.J. (1996) Expression of tissue inhibitor of metalloproteinases 1 and 2 is increased in fibrotic human liver. Gastroenterology, 110, 821–831. [DOI] [PubMed] [Google Scholar]
- 30. Takahara T., Furui K., Funaki J., Nakayama Y., Itoh H., Miyabayashi C., Sato H., Seiki M., Ooshima A., Watanabe A. (1995) Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology, 21, 787–795. [PubMed] [Google Scholar]
- 31. Takahara T., Furui K., Yata Y., Jin B., Zhang L.P., Nambu S., Sato H., Seiki M., Watanabe A. (1997) Dual expression of matrix metalloproteinase-2 and membrane-type 1-matrix metalloproteinase in fibrotic human livers. Hepatology, 26, 1521–1529. [DOI] [PubMed] [Google Scholar]
- 32. Milani S., Herbst H., Schuppan D., Grappone C., Pellegrini G., Pinzani M., Casini A., Calabró A., Ciancio G., Stefanini F. (1994) Differential expression of matrix-metalloproteinase-1 and -2 genes in normal and fibrotic human liver. Am. J. Pathol., 144, 528–537. [PMC free article] [PubMed] [Google Scholar]
- 33. Xu L., Hui A.Y., Albanis E., Arthur M.J., O'Byrne S.M., Blaner W.S., Mukherjee P., Friedman S.L., Eng F.J. (2005) Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut, 54, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peng J., Li X., Feng Q., Chen L., Xu L., Hu Y. (2013) Anti-fibrotic effect of Cordyceps sinensis polysaccharide: Inhibiting HSC activation, TGF-β1/Smad signalling, MMPs and TIMPs. Exp. Biol. Med. (Maywood), 238, 668–677. [DOI] [PubMed] [Google Scholar]
- 35. Donati B., Motta B.M., Pingitore P., Meroni M., Pietrelli A., Alisi A., Petta S., Xing C., Dongiovanni P., del Menico B., et al. (2016) The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology, 63, 787–798. [DOI] [PubMed] [Google Scholar]
- 36. Yoneda A., Sakai-Sawada K., Niitsu Y., Tamura Y. (2016) Vitamin A and insulin are required for the maintenance of hepatic stellate cell quiescence. Exp. Cell Res., 341, 8–17. [DOI] [PubMed] [Google Scholar]
- 37. Mederacke I., Dapito D.H., Affò S., Uchinami H., Schwabe R.F. (2015) High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc., 10, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nicoletti A., Kaveri S., Caligiuri G., Bariéty J., Hansson G.K. (1998) Immunoglobulin treatment reduces atherosclerosis in apo E knockout mice. J. Clin. Invest., 102, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.