

Abstract
Approximately 50% of cystic fibrosis (CF) patients are heterozygous with a rare mutation on at least one allele. Several mutants exhibit functional defects, correctable by gating potentiators. Long-term exposure (≥24 h) to the only available potentiator drug, VX-770, leads to the biochemical and functional downregulation of F508del-CFTR both in immortalized and primary human airway cells, and possibly other CF mutants, attenuating its beneficial effect. Based on these considerations, we wanted to determine the effect of chronic VX-770 exposure on the functional and biochemical expression of rare CF processing/gating mutants in human airway epithelia. Expression of CFTR2 mutants was monitored in the human bronchial epithelial cell line (CFBE41o-) and in patient-derived conditionally reprogrammed bronchial and nasal epithelia by short-circuit current measurements, cell surface ELISA and immunoblotting in the absence or presence of CFTR modulators. The VX-770 half-maximal effective (EC50) concentration for G551D-CFTR activation was ∼0.63 μM in human nasal epithelia, implying that comparable concentration is required in the lung to attain clinical benefit. Five of the twelve rare CFTR2 mutants were susceptible to ∼20–70% downregulation by chronic VX-770 exposure with an IC50 of ∼1–20 nM and to destabilization by other investigational potentiators, thereby diminishing the primary functional gain of CFTR modulators. Thus, chronic exposure to VX-770 and preclinical potentiators can destabilize CFTR2 mutants in human airway epithelial models in a mutation and compound specific manner. This highlights the importance of selecting potentiator drugs with minimal destabilizing effects on CF mutants, advocating a precision medicine approach.
Introduction
Cystic fibrosis (CF) is the most common lethal genetic disease in the Caucasian population with an incidence of ∼1: 2500 (1,2). More than 2000 mutations have been identified in the CF transmembrane conductance regulator (CFTR) (3–5), a protein kinase A (PKA) activated chloride and bicarbonate selective anion channel. CFTR is expressed at the apical plasma membrane (PM) of secretory and resorptive epithelia of various organs, including the lung, intestine, pancreas and sweat gland (6,7). CF-causing mutations may interfere with CFTR transcription, splicing, translation, folding, trafficking, stability and channel function or a combination of these phenomena, manifesting in impaired transepithelial anion conduction and secondary water transport (8–11).
The most common CF-causing mutation, the deletion of phenylalanine residue 508 (F508del), is present in at least one allele of ∼90% of all CF patients (3), while ∼50% of patients have one or two CFTR alleles containing a rare mutation (hereafter referred to as CFTR2 mutation). The third most common mutation is G551D, with an incidence of ∼4% (2,12,13). G551D, an archetypal gating mutation, imposes a severe functional defect without influencing the channel processing or PM expression (14).
A recently approved drug, VX-770 or ivacaftor (marketed as Kalydeco), developed by Vertex Pharmaceuticals (Boston, Massachusetts, USA), profoundly ameliorates the clinical phenotype by augmenting the function of G551D-CFTR in patients carrying this mutation on at least one allele (12,15–17). Correction of the G551D-CFTR gating defect by ∼30%, as determined in cell culture models, (18,19) is sufficient to increase the predicted % forced expiratory volume in 1 s (FEV1%) by >10%, to reduce the exacerbation rate of lung infection and sputum P. aeruginosa density, and to slow down the long-term decay of lung function (15,17), the primary source of morbidity and mortality. VX-770 has been subsequently approved for a total of thirty two additional CFTR2 mutants displaying gating defects (19,20).
Based on observation made in CF bronchial epithelia (CFBE14o- designated as CFBE) and BHK cells heterologously overexpressing F508del-CFTR, we assume that the modest clinical efficacy of Orkambi (VX-770 combination with the folding corrector VX-809, also known as lumacaftor) in F508del patients can be explained by the limited improvement in F508del-CFTR folding by VX-809 (21,22). The modestly augmented folding, however, is reduced by chronic exposure of the CFBE to VX-770. Destabilization of F508del-CFTR both at the ER and at the cell surface was documented in CFBE (22,23), leading to reduced functional expression of F508del-CFTR as compared with the acute effect of VX-770 in both immortalized and patient-derived primary human bronchial epithelia (HBE) (22,23). Neither the WT nor the G551D-CFTR was sensitive to VX-770-mediated destabilization (22).
Though the biochemical destabilizing effect of VX-770 on the F508del and other mutants remains to be proven in patients, correlative evidence suggests that the in vitro model has predictive value for functional responsiveness of airway epithelia in vivo (24). The Orkambi rescued short circuit current (Isc) mediated by phosphorylated F508del-CFTR in monolayers of primary human nasal epithelia (HNE) is proportional with the improvement of the lung function (FEV1%) of individual patients (24). In analogy, attenuation of the F508del-CFTR activity by chronic over acute exposure to VX-770 would be consistent with proportionally reduced lung function improvement in patients with F508del-CFTR similar to other mutants susceptible to VX-770 downregulation.
Although some testing has been performed in Fischer Rat Thyroid (FRT) cells (19,25), the efficacy of VX-770 for most CFTR2 mutations in respiratory epithelia remains unknown. Considering that a significant fraction of the >2000 CFTR2 mutants have gating defects (13), we anticipate that robust functional correction will require administration of gating potentiators for several CFTR2 mutants. Using newly established conditionally reprogrammed human nasal epithelia as well as CFBE cell lines, we examined whether the observed biochemical and functional effect of VX-770 chronic exposure on F508del-CFTR represents an exception or is a general phenomenon that prevails in CFTR2 mutants.
Results
Conditionally reprogrammed human nasal epithelia (HNE) as a preclinical model for CF research
Conditional reprogramming (CR) has gained wide application to enhance the amplification and life span of primary epithelial cells, while largely preserving their characteristics (26,27) (Supplementary Material, Fig. S1A). In CF, this method was used to amplify human bronchial epithelia (HBE) (28) and alveolar type II cells (29), derived from lung samples collected during transplantations. However, the availability of lung tissues, especially those carrying rare mutations, is low, hampering CF and other lung disease research. To overcome this limitation, we collected HNE by nasal scraping (30) and used CR to amplify these cells. Upon differentiation, CR-HNE, similar to CR-HBE, formed pseudostratified epithelia expressing tight junction associated zonula occludens-1 (ZO-1), embedded with mucin (Muc5AC) producing goblet cells and acetylated tubulin (Ac. tub) expressing ciliated cells (Fig. 1A and B), with an overall transepithelial resistance of ∼500 Ω*cm2.
Figure 1.

Conditionally reprogrammed (CR) HNE and HBE have similar functional and morphological characteristics. (A,B) HBE and HNE with CFTRWT/WT genotype were expanded using CR, followed by differentiation on filter supports under ALI culture for ≥ 4 weeks. Representative laser confocal immunofluorescence micrographs are shown for HBE (A) and HNE (B). Epithelia were immunostained with goblet cell marker mucin5AC (green), ciliated cell marker acetylated tubulin (green) and tight junctional protein zonula occludens-1 (red). Nuclei were stained with DAPI. Top is the transverse (xz) plane and bottom is the frontal (xy) plane. (A) and (B, left) show the apical level; (A, insert) and (B, right) show the subapical level. Along the z-axis, the empty arrowheads and filled arrowheads indicate the apical and basal PM, respectively. Size bar is 10 µm. (C,D) Representative short-circuit current (Isc) traces for HBE (C) and HNE (D), with CFTRWT/WT genotype. After ENaC inhibition with amiloride (amil, 100 µM), WT-CFTR was stimulated by forskolin (fsk) titration and genistein (gen, 50 μM) or VX-770 (770, 10 µM), followed by specific inhibition of CFTR with CFTRinh-172 (172, 20 μM). Measurements were performed with equimolar chloride concentrations in both chambers (C) or in presence of a basolateral-to-apical chloride gradient (D). (E) Maximal forskolin response of HBE and HNE each from 5 CFTRWT/WT donors. (F) Dose-response to forskolin stimulation of HBE (EC50 = 0.22 µM), HNE (EC50 = 0.27 µM), each from 5 CFTRWT/WT donors and WT-CFTR overexpressing CFBE (EC50 = 0.13 µM). (G,H) HBE (G) and HNE (H) with CFTRΔF508/ΔF508 genotype were expanded and differentiated as described, followed by treatment with VX-809 (3 µM) for 24 h. Isc of ΔF508-CFTR was stimulated as in (C) and measurements were performed with equimolar chloride concentrations in both chambers (G) or in presence of a basolateral-to-apical chloride gradient (H). Bar graphs show maximal forskolin + potentiator responses of HBE and HNE from 3 CFTRΔF508/ΔF508 donors each as percentage of the respective WT controls. (I) HNE with CFTRG551D/Y1092X genotype were expanded and differentiated as described. Isc of G551D-CFTR was stimulated by forskolin (20 μM) and VX-770 titration, followed by CFTR inhibition. Measurements were performed with equimolar chloride concentrations in both chambers. (J) Dose-response to VX-770 stimulation of HNE (EC50 = 0.63 ±0.07 µM) with CFTRG551D/Y1092X genotype. Unless otherwise specified, all experiments are n = 3; error bars are SEM.
To assess the electrophysiological characteristics of CR-HNE, cells from five individuals with CFTRWT/WT genotype were collected and upon differentiation on filter supports, their CFTR function was determined by short-circuit current (Isc) measurement and compared with that of CR-HBE. While the PKA activator forskolin-stimulated CFTR Isc was ∼40% lower in CR-HNE than in CR-HBE (Fig. 1C–E), likely due to reduced CFTR expression in nasal epithelia, the cAMP sensitivity and fractional potentiator-independent current were similar between CR-HNE and CR-HBE (Fig. 1C, D and F). The functional phenotype of CR-HNE was stable for ∼10 passages (∼20 population doublings) as indicated by the comparable amiloride-inhibited ENaC and CFTR inhibitor 172 (CFTRinh-172)-inhibited current densities at passages 3, 6 and 10 (Supplementary Material, Fig. S1B and C). Furthermore, CR-HBE and CR-HNE each isolated from three patients with CFTRF508del/F508del genotype exhibited similar correction upon treatment with the folding corrector VX-809, resulting in CFTR function corresponding to 24.1 ±4.7% and 25.1 ±1.5% of the matching wild-type (WT) epithelia currents (Fig. 1G and H). Notable, the current values reported here are similar to those reported for VX-809 correction in primary HBE (22,31).
Potency of VX-770 in CR-HNE
Using the CR technique, we amplified HNE of a patient with CFTRG551D/Y1092X genotype to measure VX-770 potency of G551D-CFTR activation. The Y1092X mutant had negligible protein expression (Supplementary Material, Fig. S1D and E, Table S1), consequently, the VX-770 potentiated current could be attributed to the G551D mutant and yielded an EC50 of 0.63 µM (±0.07) (Fig. 1I and J), ∼2-fold higher than reported in primary HBE with CFTRG551D/F508del genotype (43). Due to slow activation of G551D-CFTR, subsequent additions of VX-770 were done before the current had reached steady-state. Owing to this logistical issue, we consider that the EC50 of 0.63 µM may be overestimated.
The profound clinical improvement of CF pathology seen in G551D-CFTR patients (15,17) implies that the EC50 of VX-770 is at least temporarily reached in the extracellular milieu of respiratory epithelia. This inference is supported by the steady-state serum concentration of VX-770 [∼ 2 μM (32)] as well as the >8 fold accumulation in the epithelial lining fluid of the lung compared with plasma concentration (33), also showing that plasma protein binding (34) is a minor determinant of in situ VX-770 concentration.
The EC50 of VX-770 was confirmed in the cystic fibrosis bronchial cell line CFBE41o- (CFBE) (35) harbouring the inducible expressed G551D-CFTR [Supplementary Material, Table S1, (36)]. Similar to HNE, the WT-, but not G551D-CFTR, was activated by forskolin in CFBE (Fig. 1F, Supplementary Material, Fig. S1F and G). Potentiation with VX-770 increased the WT current by ∼26% (Fig. 3E) but profoundly activated G551D-CFTR (Supplementary Material, Fig. S1F and G). Notably, the extent of VX-770 activated G551D current increased as a function of forskolin pre-treatment, indicating that the VX-770 efficacy depends on the level of channel phosphorylation (Supplementary Material, Fig. S1F and G). Likewise, forskolin-mediated channel phosphorylation, but not the presence of a 3HA-tag in the 4th extracellular loop (37), decreased the EC50 of VX-770-induced G551D-CFTR activation from 19.17 μM to 4.62 μM (Supplementary Material, Fig. S1H and I). As noted for the HBE CFTRG551D/Y1092X measurements, the EC50 values calculated for the G551D-CFTR activation in CFBE may also be also slightly overestimated (Supplementary Material, Fig. S1F and H). However, based on the similarity to the values calculated in Supplementary Material, Figure S1G and I, which were based on steady-state currents, we believe that our overestimation to be only slight.
Figure 3.

Functional analysis of CFTR2 mutants. (A) Representative Isc traces of CFBE cells expressing the indicated CFTR2 mutants. Cells grown on filter supports were incubated for 24 h with DMSO (red traces) or VX-809 (3 μM, blue traces). Isc of mutant CFTR was stimulated with forskolin (20 μM) and increasing concentrations of genistein until saturation, or a single high concentration of genistein (gen, 100 μM), followed by inhibition of CFTR with CFTRinh-172 (20 μM). Measurements were performed in the presence of a basolateral-to-apical chloride gradient after basolateral permeabilization with amphotericin B (100 μM). (B) Quantification of currents in (A); forskolin was measured relative to initial baseline; genistein was measured relative to fsk plateau. (C) Normalization of data from B showing fold increase of Isc in the presence of forskolin plus genistein relative to that in the presence of forskolin alone. Dotted line is relative genistein response of WT-CFTR. (D) Quantification of CFTR2 Isc potentiated with VX-770 (3 μM). Forskolin was measured relative to initial baseline; VX-770 was measured relative to fsk. (E) Normalization of data from D as described in C to display VX-770 effect relative to forskolin. Dotted line is relative VX-770 response of WT-CFTR. All experiments are n = 3; error bars are SEM.
Taken together, these results warrant the examination of the chronic effect of VX-770 in the concentration range of 1 nM to 1 μM and the use of the CFBE cells as a model to determine the biochemical and functional stability of CFTR2 mutants upon prolonged potentiator exposure.
Biochemical characterization of selected CFTR2 mutants
We selected twelve mutants which exhibited variable extent of processing and gating defects in the FRT heterologous expression system (10,25). These mutations are distributed over the four major CFTR domains, the membrane spanning domains 1 and 2 (MSD1–2), and the nucleotide binding domains 1 and 2 (NBD1–2) (Fig. 2A, Supplementary Material, Fig. S2, Table S1) (38). CFBE cells were transduced with lentiviral particles, encoding the 3HA-tagged CFTR2 mutants.
Figure 2.

Biochemical analysis of selected CFTR2 processing/gating mutants shows different levels of protein biogenesis. (A) Electron cryo-microscopy model (38) of human CFTR showing the positions of the selected CFTR2 mutations in red. (B) Immunoblot of CFBE expressing inducible CFTR mutants with an extracellular 3HA tag under the control of the TetON doxycycline (dox) regulated transactivator with or without (-) dox (500 ng/ml) induced expression for 3 days. The cells were incubated for 24 h with DMSO or VX-809 (3 μM). CFTR was visualized with anti-HA antibody, and anti–Na+/K+-ATPase antibody served as loading control. The empty arrowheads show the mature, complex glycosylated CFTR (C-band), and the filled arrowhead show the immature, core glycosylated protein (B-band). (C) PM density measurements of CFTR2 mutant expression by cell-surface ELISA, with and without 24 h correction with VX-809 (3 μM). The PM density is expressed as % of WT DMSO treated cells. The expression was normalized to viability, determined by AlamarBlue assay and to CFTR mRNA expression, measured by RT-qPCR, to account for differences in cell seeding concentration and viral transduction/induction efficiency, respectively. (D) PM density, as shown in (B) correlated (R2 = 0.8593, P < 0.0001) to the mature protein (C-band) of CFTR mutants (F508del (red), CFTR2 mutants (blue)) expressed in FRT cells, published by Van Goor. (25). All experiments are n = 3; error bars are SEM.
Cellular expression of CFTR2 mutants, determined by immunoblotting, showed that the mature, complex-glycosylated (band C) form exhibited mutation-dependent variable biogenesis in CFBE cells (Fig. 2B, Supplementary Material, S3A and B). Despite substantial mRNA expression (Supplementary Material, Fig. S3C), E92K, R347P, S492F, V520F, R560T, D614G, L1077P, M1101K of the CFTR2 mutants yielded low or negligible levels of band C, whereas others displayed moderate (S1235R, N1303K) or WT-like protein expression (S341P, R347H) (Fig. 2B, Supplementary Material, S3A and B) (22).
Similar results were obtained when the PM expression of CFTR2 mutants was determined by cell surface ELISA (39) (Fig. 2C, Supplementary Material, S3D). The PM densities determined in CFBE cells correlated with the published complex-glycosylated protein expression in FRT cells (25), indicating that, unlike corrector efficacy (40), the biogenesis of CFTR2 mutants was largely independent of the expression system (Fig. 2D). This inference was further supported by the correlation of CFTR2 mutant PM expression between CFBE and BHK cells and between FRT protein expression (25) and BHK PM density (Supplementary Material, Fig. S3E).
Importantly, VX-809 increased both band C abundance and PM expression of E92K, S341P, S492F, D614G, L1077P, M1101K and S1235R mutants by >2-fold (Fig. 2B and C). In contrast, baseline and VX-809 rescued expression of V520F- and R560T-CFTR were marginal and these mutants were removed from further investigation.
Assessment of the gating defect severity of CFTR2 mutants
Measurement of the CFTR2 channel activity by Isc showed that all mutants except V520F, R560T and M1101K, were able to confer anion transport upon phosphorylation that was further increased by acute addition of the potentiator genistein (Fig. 3A and B, Supplementary Material, S3F). Depending on the mutation, VX-809 increased the CFTR mediated Isc by ∼0.5–6 fold (Fig. 3B).
To quantify the severity of the gating defect of the mutants, we calculated the fold increase of Isc in the presence of forskolin plus genistein relative to that in the presence of forskolin alone (Fig. 3C). The WT-CFTR has low sensitivity to genistein, as indicated by the ∼0.1 fold increase of Isc in the presence of genistein, confirming previous observations (41). In contrast, most mutants, with the exception of S341P and M1101K, were stimulated by >0.1 fold by genistein, indicating a gating defect. The activation induced by genistein was generally similar upon VX-809 treatment, suggesting that the gating defect is not attenuated by processing correction (Fig. 3C).
Acute potentiation of the phosphorylated channel by VX-770 was similar to that of genistein (Fig. 3D and E). The E92K, D614G, S1235R mutants, however, exhibited greater, while others (e.g. L1077P) displayed attenuated fractional potentiation with VX-770, as compared with genistein. These observations suggest that CFTR2 mutants differ in their susceptibility to structurally distinct gating potentiators, with important therapeutic implications.
VX-770 reduces the biochemical and functional expression of some CFTR2 mutants
To assess the effect of long-term VX-770 exposure on channel expression, CFBE expressing CFTR2 mutants were incubated with increasing concentration (1–1000 nM) of VX-770 for 24 h, in the presence or absence of VX-809. Immunoblot analysis showed reduced expression of the complex-glycosylated CFTR2 form of D614G, L1077P, and S1235R at 10 nM VX-770 while others (e.g. N1303K) were not affected (Fig. 4A, Supplementary Material, S4A). Cell surface ELISA confirmed these results, documenting reduced PM expression of E92K-, S341P-, D614G-, L1077P- and S1235R-CFTR by chronic VX-770 exposure in a concentration dependent manner, resulting in a mutation-specific 20–75% downregulation (Fig. 4B and C). Importantly, VX-809 was unable to considerably rescue VX-770-induced CFTR2 downregulation (Fig. 4C and D), matching observations on F508del-CFTR (22,23).
Figure 4.

Extended exposure to VX-770 leads to downregulation of a subset of CFTR mutants in a dose-dependent manner. (A) CFBE cells expressing the indicated CFTR mutants were incubated for 24 h with VX-809 (3 μM) and increasing concentrations of VX-770 and subsequently lysed to collect protein samples. Protein was detected by immunoblotting with anti-HA antibody. Na+/K+ ATPase was used as loading control. The empty arrowheads show the mature, complex glycosylated CFTR protein (C-band), and the filled arrowhead show the immature, core glycosylated protein (B-band). (B) PM density of CFTR2 mutants expressed in CFBE cells. Cells were treated with VX-770 for 24 h in the presence or absence of 3 µM VX-809, and the values, normalized with cell viability, are expressed as percentage of non–VX-770-treated controls (n = 3). (C) Quantification of remaining CFTR2 PM density, shown in B, after chronic treatment with 100 nM VX-770, as percent of DMSO control. Half maximal inhibitory concentration (IC50) of VX-770 on PM expression of CFTR2 mutants, calculated on the basis of the measurements shown in panel B. Experiments in B, C and D are n = 3; error bars are SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
The consequence of chronic VX-770 on the function of the CFTR2 mutants was evaluated next. Current measurements showed a mutation-specific susceptibility to chronic VX-770-induced downregulation, resulting in 0–50% reduction in relative Isc (Fig. 5A–F). The current reduction of the L1077P-CFTR activity was partially reversed in the presence of VX-809 (Fig. 5F). Similar to the biochemical results (Fig. 4C and D), the most susceptible and resistant mutants to VX-770 were L1077P and N1303K, respectively. Surprisingly, the VX-809 corrected E92K-CFTR was less affected by VX-770-mediated downregulation at the functional level, and non-affected in the absence of correction (Fig. 5A and F). The short circuit current profiles differ between the VX-770 treated and DMSO treated controls, due to the pre-existence of the potentiator in the Ussing chamber at the time of activation (Fig. 5A–E and G).
Figure 5.

Long-term treatment with VX-770 leads to CFTR2 mutant functional downregulation in CFBE and HNE. (A–E) Representative Isc traces of CFBE cells expressing the indicated CFTR2 mutants under the control of the TetON inducible promoter. Polarized CFBE were incubated for 24 h with DMSO or VX-770 (100 nM, top), VX-809 (3 μM) or VX-809 + VX-770 combination (bottom). Isc of mutant CFTR was stimulated with forskolin (20 μM) and VX-770 (3 μM) and measurements were performed in the presence of a basolateral-to-apical chloride gradient after basolateral permeabilization with Amphotericin B (100 μM). (F) Quantification of maximal currents expressed as % of DMSO control for each mutant. The currents were measured as the difference between maximal induced by VX-770 and the baseline current after CFTRinh-172 addition. (G) HNE with CFTRF508del/P67L genotype were expanded and differentiated as described, followed by treatment with VX-809 (3 µM) and VX-770 (1 μM) for 24 h. Isc of CFTR was stimulated by forskolin (20 μM) and VX-770 (10 μM), followed by CFTR inhibition with CFTRinh-172 (20 μM). Measurements were performed in the presence of a basolateral-to-apical chloride gradient. (H) Quantification of maximal currents expressed as % of control. All experiments are n = 3; error bars are SEM; ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
We reported previously the susceptibility of the P67L mutant to chronic VX-770 exposure in CFBE resulting in a 32% and 43% downregulation of the channel function and PM density, respectively (22). Here we isolated HNE from a CF patient with CFTRF508del/P67L genotype. Upon VX-809 treatment, these cells exhibit a maximal current corresponding to 84.4% of the WT, i.e. >3.5 times higher than homozygous F508del HNE. Chronic VX-770 exposure resulted in a 34% current decrease (Fig. 5G and H), further supporting the notion that results obtained in the CFBE cell model (Supplementary Material, Fig. S4B) parallel the results in HNE.
Potentiators have CFTR2 mutation-specific destabilizing effects
The destabilizing effect on F508del-CFTR is not restricted to VX-770, but is a common phenomenon of many investigational potentiators (22). To test whether VX-770 susceptible CFTR2 mutants would also be sensitive to destabilization by other potentiators, we measured their PM density upon chronic exposure to seven potentiators from the CFFT Inc. panel (22). The PM densities of E92K and L1077P were attenuated by all tested potentiators, albeit to different extents (Fig. 6A). In contrast, S1235R was resistant to downregulation by P1 and P8, indicating mutation specific potentiator susceptibility (Fig. 6A).
Figure 6.

Other classes of potentiators have widely varying effects on CFTR2 mutant expression. (A) PM densities of CFTR2 mutants expressed in CFBE cells after 24 h treatment with potentiators from the CFFT panel that downregulate F508del expression (22), with and without VX-809 (3 μM). Potentiators were used at the following concentrations: P2, P3—3 µM; P8—20 µM; P1, P4, P7–30 µM, P6–100 µM. The PM densities were normalized for cell viability and are expressed as % of DMSO control (dotted line). (B) Cell surface expression of CFBE cells expressing a subset of CFTR2 mutants performed after 24 h incubation with P5, P10, P12, A04 or H02 (10 μM), five potentiators that do not downregulate F508del-CFTR (42). The dotted line represents the expression in DMSO treated cells. (C) Immunoblots of CFBE cells expressing the indicated CFTR mutants after incubation for 24 h with P12, A04 or H02 (10 μM), with and without VX-809 (3 μM). CFTR was detected with anti-HA antibody, Na+/K+ ATPase served as loading control. The empty arrowheads show the mature, complex glycosylated CFTR protein (C-band), and the filled arrowhead show the immature, core glycosylated protein (B-band). All experiments are n = 3; error bars are SEM.
Next, potentiators that do not downregulate F508del-CFTR expression upon chronic exposure, including P5 and P10 (22), P12, a more potent P5 analog (42), as well as A04 and H02, two recently identified potentiators of novel chemical classes (42), were tested for their effect on CFTR2 mutants. Some of these potentiators downregulated the PM expression of particular mutants (Fig. 6B); L1077P-CFTR was not affected by chronic P5, P10 and P12 treatment but showed decreased PM density upon long-term treatment with A04, a potentiator which preserved the expression of all other tested mutants (Fig. 6B). Immunoblot analysis confirmed the cell surface ELISA data for the P12, A04 and H02 potentiators, demonstrating the mutation-specific downregulation of the respective complex-glycosylated CFTR2 mutants (Fig. 6C and F).
Comparison of the PM densities after chronic treatment with downregulating and non-downregulating potentiators between mutants showed no significant correlation (Supplementary Material, Tables S2 and S3), suggesting the necessity to empirically select the best potentiator for each CFTR2 mutant.
Empirical selection of CFTR2 mutation-specific potentiators
To demonstrate the functional advantage of selecting mutation specific potentiators lacking destabilizing effects, we performed current measurements in CFBE expressing the three CFTR2 mutants E92K, L1077P and S1235R. Consistent with the biochemical data, the least destabilizing potentiators for E92K, L1077P and S1235R mutations were A04 (Fig. 7A), P12 (Fig. 7B), and P12 or A04 (Fig. 7C), respectively. These potentiators elicited a current comparable to that recorded by acute VX-770 exposure, indicating that they do not downregulate their respective CFTR2 mutant and are able to potentiate the Isc to a level equivalent to VX-770. On the basis of these observations, we suggest a precision medicine approach to screen gating mutants against a panel of structurally distinct potentiators when they become available in order to select the most efficacious activators which do not exhibit adverse effects.
Figure 7.
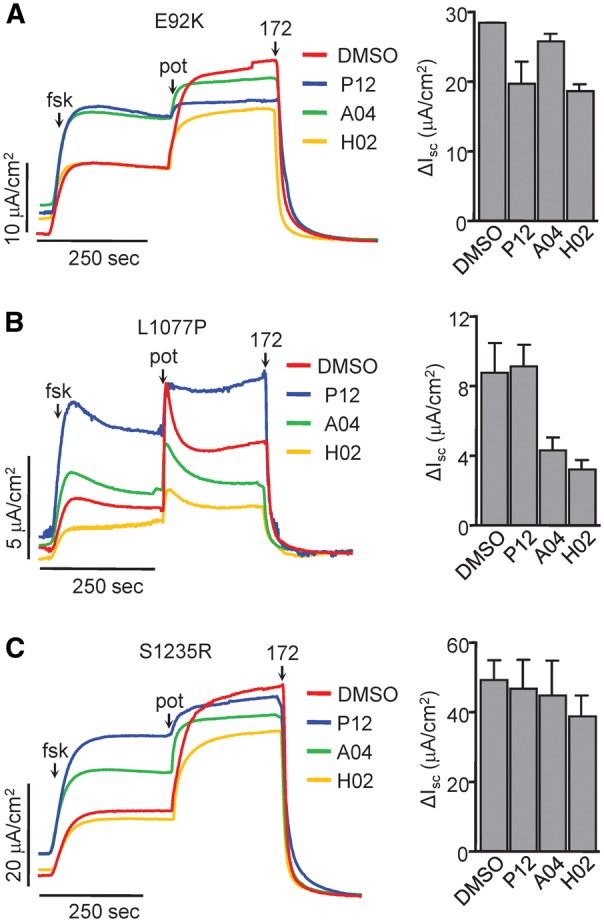
Identification of non-downregulating potentiators with efficacy similar to VX-770. (A–C,left) Representative Isc traces of polarized CFBE cells expressing the indicated CFTR2 mutants. The cells were incubated for 24 h with VX-809 (3 μM, red traces) with and without 10 μM P12 (blue), A04 (green) or H02 (yellow). Isc of mutant CFTR was stimulated with forskolin (20 μM) and 10 μM of the respective long-term treatment potentiator. Non-potentiator pre-treated cells were stimulated with 3 μM VX-770. CFTR was inhibited with CFTRinh-172 (20 μM). Measurements were performed in the presence of a basolateral-to-apical chloride gradient after basolateral permeabilization with amphotericin B (100 μM). (A–C, right) Quantification of maximal currents performed similarly as in Fig. 5 F. All experiments are n = 2, error bars are SEM.
Discussion
Despite a considerable amount of effort from both academia and industry, VX-770 remains the only gating potentiator drug which has been FDA approved for clinical therapy of thirty three rare CF-causing mutations (20,43,44). In an attempt to better estimate the drug concentration, which likely prevails in the lungs of CF patients undergoing VX-770 therapy, we determined the dose response of G551D-CFTR to VX-770 in primary HNE, expressing the channel endogenously. To overcome the limitation of sample availability, we used primary HNE instead of HBE cells and introduced conditional reprogramming to expand HNE cell population, while preserving their morphological and electrophysiological characteristics.
We found that the EC50 of VX-770 in HNE cells (0.63 ±0.07 μM) was comparable to that found previously in HBE (0.23 ±0.20 μM) (45), a relevant observation when trying to extrapolate from in vitro studies to the VX-770 pharmacodynamics in the CF lung. The EC50 values of VX-770 are nearly two orders of magnitude higher than the recently proposed free plasma concentration (1.5–8.5 nM) of VX-770 (34) and exceed by 20–40 fold the IC50 of VX-770-mediated F508del-CFTR destabilization (22). Meanwhile, the EC50 of VX-770 is below the minimal plasma concentration in adults (1.97 ±1.19 μM or 774 ±468 ng/ml) and in epithelial lining fluid (16 μM), reported by the European Medicines Agency (33). Cholon and coworkers also showed that the VX-770 concentration is ∼3-fold higher in HBE cells as compared with its extracellular level (23). Collectively, these represent compelling evidences that the free plasma concentration of VX-770 is a minor determinant of the respective tissue concentration, with the examples of lungs, adrenal glands and lymphoid nodes showing profound accumulation (33). Clinical studies have shown ∼10% improvement in the predicted FEV1% in heterozygous G551D-CFTR patients undergoing VX-770 treatment (15,17). This implies that the VX-770 concentration in the G551D-CFTR heterozygous lung must reach, and likely surpasses, the EC50 (∼0.2–0.6 µM). Taken together, these data support the notion that CFTR channels at the apical surface of airway epithelia are exposed to at least hundreds of nanomolar concentration of VX-770.
Recent in vitro studies of VX-770 effect on F508del-CFTR expressing cell lines and patient-derived primary cells have shown that extended exposure to VX-770 leads to biochemical and functional destabilization of F508del-CFTR, causing decreased biogenesis and cell surface half-life with accelerated lysosomal delivery of the mutant (22,23). Thus the primary effect of improved channel gating by VX-770 is partly offset by the decreased folding and stability of F508del-CFTR, causing submaximal improvement in the PM ion conduction in in vitro models (46). While we do not have direct evidence that a similar phenomenon prevails in CF patients, the following consideration suggests that chronic VX-770 exposure may attenuate the PM expression of F508del-CFTR and, by analogy, CFTR2 mutants as well in vivo. The 2–4% increase in the predicted FEV1% seen in the Orkambi (VX-809 and VX-770) clinical trials (47–49) represents an unexpectedly modest clinical improvement, as compared with the documented ∼25% WT activity of Orkambi treated F508del HBE and HNE but with acute VX-770 stimulation (31). In contrast, comparable activation of G551D-by VX-770 alone (∼15–30% of WT), causes markedly improved clinical phenotype of patients with >10% increased FEV1%. Since F508del CFTR functional rescue by Orkambi in primary HBE and HNE correlates with the responsiveness of the lung function (24), Orkambi treatment should coincide with a substantial clinical improvement as observed in G551D patients (17,45) as consequence of restoring the mutant activity to ∼25% of the WT (31). One possible explanation of this discrepancy is that the preclinical studies (31) did not take into account F508del-CFTR downregulation by chronic VX-770 exposure, while the G551D mutant is resistant to VX-770 mediated downregulation (22,23).
These observations prompted us to measure the long-term effect of VX-770 both in presence and absence of VX-809 on a variety of CFTR2 mutants and seek for a possible pattern in the downregulation propensity of susceptible mutations (e.g. domain-specific clustering). We identified that 5 of the 12 tested CFTR2 gating mutants were susceptible to chronic VX-770-induced downregulation, in addition to two previously identified mutants (P67L- and R170G-CFTR, Supplementary Material, Fig. S4B and C) (22). The mutations, P67L, E92K, R170G, S341P, D614G, L1077P, and S1235R, which render CFTR sensitive to VX-770-mediated destabilization are located in all four major domains of CFTR. VX-770 sensitivity did not correlate with the PM expression, function, fractional potentiator sensitivity or VX-809 correctability of mutants (data not shown), thus at least for this set of parameters the results do not allow any prediction as to VX-770 mutation susceptibility. Importantly, some of the gating potentiators that were described not to interfere with F508del-CFTR functional expression in human airway epithelia (42), downregulated some CFTR2 mutants in a mutation-specific manner. In contrast, other CFTR2 mutants, similarly to G551D, were resistant to destabilization by gating potentiators (22). There was no significant correlation between the extent of downregulation and potentiator effect on various mutants, reinforcing the unpredictable nature of the potentiator effect in relation to specific mutations. On the other hand, we were able to identify at least one investigational potentiator without chronic destabilizing effect for E92K, L1077P and S1235R mutants, with similar potentiation efficacy as acute VX-770. Therefore, we argue that these results warrant the implementation of precision medicine to optimize functional rescue of CFTR2 gating defects for individual mutants.
As shown for one patient with CFTRF508del/P67L genotype, it is feasible to isolate primary HNE cells for assessing the acute and chronic potentiator and corrector effects on CFTR activity in differentiated molayers at ALI. Chronic exposure of heterozygous P67L HNE to VX-770 decreased the CFTR transport function by ∼34%, similar to that (32%) in CFBE overexpressing P67L as compared with acute VX-770 application, supporting the authenticity of the CFBE model.
Upon the clinical availability of additional potentiator drugs, we propose to test their chronic effect on each gating mutant to identify potentiators which augment CFTR function without compromising biochemical and functional stability in conditionally reprogrammed primary HNE. Such an approach could improve clinical outcome by identifying those patients who are responsive to a given treatment and by finding the best available drug combination in a mutant-specific manner.
Materials and Methods
Cell lines
CFBE cells expressing WT- and ΔF508-CFTR have been described before (22). Most CFTR2 (RefSeq NM_000492.3) mutants (except S341P-CFTR) were engineered using by subcloning the mutations from pBI plasmids, kindly provided by P. Thomas into pNUT or pLVX-Tight-Puro expression plasmids, containing CFTR with a 3HA-tag in the fourth extracellular loop as described (21,37). Stably expressing BHK-21 cell lines were generated as before (21). Stable epithelial cell lines were produced by lentiviral transduction of CF bronchial epithelial cells (CFBE41o-, designated as CFBE) (35) with inducible CFTR mutants as described (36). To avoid clonal bias, pools of >50–100 clones were selected and expanded for subsequent biochemical and functional studies.
Primary nasal epithelium collection, conditional reprogramming and differentiation
The isolation of human nasal epithelia (HNE) from healthy and CF human subjects under the protocol and consent form approved by the McGill MUHC Research Ethics Board (14–234-BMB) was performed as described (30). Following outgrowth of the explants, HNE cells were conditionally reprogrammed according to the protocol developed by Liu and coworkers (27). Following amplification, the cells were differentiated on Snapwell filter supports as previously described (50). HBE cells were isolated from bronchi obtained following lung transplantation for diseases other than CF according to protocol approved by the Human Research Protection Program Institutional Review Board of UCSF (No 10–02253) or were purchased from the Cystic Fibrosis Translational Research center (CFTRc), McGill University.
Biochemical and functional assays
CFTR short-circuit current (Isc) (36,51), PM density measurements (52), immunofluorescence (36) and quantitative RT-qPCR (36) were performed as described previously. Additional details on these methods are provided in the online data supplement.
Statistical analysis
Results are presented as mean ± SEM for the indicated number of experiments. Unless otherwise specified, statistical analysis was performed by two-tailed Student's t-test with the means of at least three independent experiments and the 95% confidence level was considered significant. Dose-response plots were fitted with a Hill function and EC50 was calculated using the GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, California, USA).
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank P. Thomas (UT Southwestern Medical Center at Dallas) for providing vectors encoding CFTR2 mutants, D. Gruenert (University of California–San Francisco) for the parental CFBE41o- cell line; W.E. Finkbeiner for supplying HBE cells; A. Verkman (University of California–San Francisco), R.J. Bridges (Rosalind Franklin University of Medicine and Science) and Cystic Fibrosis Foundation Therapeutics (CFFT) Inc. for the CFTR potentiators.
Conflict of Interest statement. None declared.
Funding
Canadian Institutes of Health Research [MOP-142221], National Institute of Diabetes & Digestive & Kidney Diseases [5R01DK075302] and Cystic Fibrosis Canada. RGA is a recipient of the Fonds de recherche Santé Québec Doctoral training Scholarship. AS is a recipient of the CF Canada Fellowship. GLL is a Canada Research Chair. WE Finkbeiner, Department of Pathology, University of California, San Francisco (UCSF) provided support for some of the cell culture studies and was funded by the National Institute of Health grant [DK072517]; and Cystic Fibrosis Foundation Research and Translational Core Center grant [VERKMA15R0].
References
- 1. Rowe S.M., Miller S., Sorscher E.J. (2005) Cystic fibrosis. N. Engl. J. Med., 352, 1992–2001. [DOI] [PubMed] [Google Scholar]
- 2. Cutting G.R. (2015) Cystic fibrosis genetics: from molecular understanding to clinical application. Nat. Rev. Genet., 16, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Riordan J.R. (2008) CFTR function and prospects for therapy. Annu. Rev. Biochem., 77, 701–726. [DOI] [PubMed] [Google Scholar]
- 4. CFTR2 Database. Available: http://www.cftr2.org; date last accessed September 27, 2017.
- 5. Cystic Fibrosis Mutation Database. Available: http://www.genet.sickkids.on.ca/app; date last accessed September 27, 2017.
- 6. Denning G.M., Ostedgaard L.S., Cheng S.H., Smith A.E., Welsh M.J. (1992) Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J. Clin. Invest., 89, 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crawford I., Maloney P.C., Zeitlin P.L., Guggino W.B., Hyde S.C., Turley H., Gatter K.C., Harris A., Higgins C.F. (1991) Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc. Natl. Acad. Sci. USA, 88, 9262–9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sosnay P.R., Siklosi K.R., Van Goor F., Kaniecki K., Yu H., Sharma N., Ramalho A.S., Amaral M.D., Dorfman R., Zielenski J. (2013) Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet., 45, 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lukacs G.L., Verkman A.S. (2012) CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol. Med., 18, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Veit G., Avramescu R.G., Chiang A.N., Houck S.A., Cai Z., Peters K.W., Hong J.S., Pollard H.B., Guggino W.B., Balch W.E.. et al. (2016) From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol. Biol. Cell, 27, 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Welsh M.J., Smith A.E. (1993) Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell, 73, 1251–1254. [DOI] [PubMed] [Google Scholar]
- 12. Harrison M.J., Murphy D.M., Plant B.J. (2013) Ivacaftor in a G551D Homozygote with Cystic Fibrosis. N. Eng. J. Med., 369, 1280–1282. [DOI] [PubMed] [Google Scholar]
- 13. Bobadilla J.L., Macek M. Jr., Fine J.P., Farrell P.M. (2002) Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum. Mut., 19, 575–606. [DOI] [PubMed] [Google Scholar]
- 14. Xu Z., Pissarra L.S., Farinha C.M., Liu J., Cai Z., Thibodeau P.H., Amaral M.D., Sheppard D.N. (2014) Revertant mutants modify, but do not rescue, the gating defect of the cystic fibrosis mutant G551D-CFTR. J. Physiol., 592, 1931–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramsey B.W., Davies J., McElvaney N.G., Tullis E., Bell S.C., Dřevínek P., Griese M., McKone E.F., Wainwright C.E., Konstan M.W.. et al. (2011) A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med., 365, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hisert K.B., Heltshe S.L., Pope C., Jorth P., Wu X., Edwards R.M., Radey M., Accurso F.J., Wolter D.J., Cooke G.. et al. (2017) Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med., 195, 1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Accurso F.J., Rowe S.M., Clancy J.P., Boyle M.P., Dunitz J.M., Durie P.R., Sagel S.D., Hornick D.B., Konstan M.W., Donaldson S.H.. et al. (2010) Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med., 363, 1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Char J.E., Wolfe M.H., Cho H.J., Park I.H., Jeong J.H., Frisbee E., Dunn C., Davies Z., Milla C., Moss R.B.. et al. (2014) A little CFTR goes a long way: CFTR-dependent sweat secretion from G551D and R117H-5T cystic fibrosis subjects taking ivacaftor. PLoS One, 9, e88564.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu H., Burton B., Huang C.J., Worley J., Cao D., Johnson J.P. Jr., Urrutia A., Joubran J., Seepersaud S., Sussky K.. et al. (2012) Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros., 11, 237–245. [DOI] [PubMed] [Google Scholar]
- 20.FDA Approves KALYDECO® (ivacaftor) for More Than 900 People Ages 2 and Older with Cystic Fibrosis Who Have Certain Residual Function Mutations. Press release http://investors.vrtx.com/releasedetail.cfm? ReleaseID=1026864 (17 May 2017); date last accessed September 27, 2017.
- 21. Okiyoneda T., Veit G., Dekkers J.F., Bagdany M., Soya N., Xu H., Roldan A., Verkman A.S., Kurth M., Simon A.. et al. (2013) Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol., 9, 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Veit G., Avramescu R.G., Perdomo D., Phuan P.W., Bagdany M., Apaja P.M., Borot F., Szollosi D., Wu Y.S., Finkbeiner W.E.. et al. (2014) Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci. Transl. Med., 6, 246ra297.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cholon D.M., Quinney N.L., Fulcher M.L., Esther C.R. Jr., Das J., Dokholyan N.V., Randell S.H., Boucher R.C., Gentzsch M. (2014) Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci. Transl. Med., 6, 246ra296.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pranke I.M., Hatton A., Simonin J., Jais J.P., Le Pimpec-Barthes F., Carsin A., Bonnette P., Fayon M., Stremler-Le Bel N., Grenet D.. et al. (2017) Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep., 7, 7375.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Goor F., Yu H., Burton B., Hoffman B.J. (2014) Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros., 13, 29–36. [DOI] [PubMed] [Google Scholar]
- 26. Liu X., Krawczyk E., Suprynowicz F.A., Palechor-Ceron N., Yuan H., Dakic A., Simic V., Zheng Y.L., Sripadhan P., Chen C.. et al. (2017) Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc., 12, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu X., Ory V., Chapman S., Yuan H., Albanese C., Kallakury B., Timofeeva O.A., Nealon C., Dakic A., Simic V.. et al. (2012) ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol., 180, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gentzsch M., Boyles S.E., Cheluvaraju C., Chaudhry I.G., Quinney N.L., Cho C., Dang H., Liu X., Schlegel R., Randell S.H. (2017) Pharmacological rescue of conditionally reprogrammed cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol., 56, 568–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bove P.F., Dang H., Cheluvaraju C., Jones L.C., Liu X., O’Neal W.K., Randell S.H., Schlegel R., Boucher R.C. (2014) Breaking the in vitro alveolar type II cell proliferation barrier while retaining ion transport properties. Am. J. Respir. Cell Mol. Biol., 50, 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muller L., Brighton L.E., Carson J.L., Fischer W.A. 2nd, Jaspers I. (2013) Culturing of human nasal epithelial cells at the air liquid interface. J. Vis. Exp., 80, e50646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Goor F., Hadida S., Grootenhuis P.D.J., Burton B., Stack J.H., Straley K.S., Decker C.J., Miller M., McCartney J., Olson E.R.. et al. (2011) Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl Acad. Sci. USA, 108, 18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Access data FDA: 203188Orig1s000. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203188Orig1s000OtherRedt.pdf; date last accessed September 27, 2017.
- 33. European Medicines Agency. Assessment report: Kalydeco. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002494/WC500130766.pdf; date last accessed September 27, 2017.
- 34. Matthes E., Goepp J., Carlile G.W., Luo Y., Dejgaard K., Billet A., Robert R., Thomas D.Y., Hanrahan J.W. (2016) Low free drug concentration prevents inhibition of F508del CFTR functional expression by the potentiator VX-770 (ivacaftor). Br. J. Pharmacol., 173, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ehrhardt C., Collnot E.M., Baldes C., Becker U., Laue M., Kim K.J., Lehr C.M. (2006) Towards an in vitro model of cystic fibrosis small airway epithelium: characterisation of the human bronchial epithelial cell line CFBE41o. Cell Tissue Res., 323, 405–415. [DOI] [PubMed] [Google Scholar]
- 36. Veit G., Bossard F., Goepp J., Verkman A.S., Galietta L.J., Hanrahan J.W., Lukacs G.L. (2012) Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Mol. Biol. Cell, 23, 4188–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zerangue N., Schwappach B., Jan Y.N., Jan L.Y. (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron, 22, 537–548. [DOI] [PubMed] [Google Scholar]
- 38. Liu F., Zhang Z., Csanady L., Gadsby D.C., Chen J. (2017) Molecular Structure of the Human CFTR Ion Channel. Cell, 169, 85–95.e88. [DOI] [PubMed] [Google Scholar]
- 39. Peters K.W., Okiyoneda T., Balch W.E., Braakman I., Brodsky J.L., Guggino W.B., Penland C.M., Pollard H.B., Sorscher E.J., Skach W.R.. et al. (2011) CFTR Folding Consortium: Methods available for studies of CFTR folding and correction. Methods Mol. Biol, 742, 335–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pedemonte N., Tomati V., Sondo E., Galietta L.J. (2010) Influence of cell background on pharmacological rescue of mutant CFTR. Am. J. Physiol. Cell Physiol., 298, C866–C874. [DOI] [PubMed] [Google Scholar]
- 41. Hwang T.C., Wang F., Yang I.C., Reenstra W.W. (1997) Genistein potentiates wild-type and delta F508-CFTR channel activity. Am. J. Physiol., 273, C988–C998. [DOI] [PubMed] [Google Scholar]
- 42. Phuan P.W., Veit G., Tan J.A., Finkbeiner W.E., Lukacs G.L., Verkman A.S. (2015) Potentiators of defective DeltaF508-CFTR gating that do not interfere with corrector action. Mol. Pharmacol., 88, 791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vertex Receives U.S. Food and Drug Administration Approval of KALYDECO® (ivacaftor) for Children with Cystic Fibrosis Ages 2 to 5 who have Specific Mutations in the CFTR Gene. Press release http://investors.vrtx.com/releasedetail.cfm? releaseid=902211 (18 March 2015); date last accessed September 27, 2017.
- 44.Vertex Receives Two EU Approvals for KALYDECO® (ivacaftor) for People with Cystic Fibrosis. Press release http://investors.vrtx.com/releasedetail.cfm? releaseid=943300 (18 November 2015); date last accessed September 27, 2017.
- 45. Van Goor F., Hadida S., Grootenhuis P.D., Burton B., Cao D., Neuberger T., Turnbull A., Singh A., Joubran J., Hazlewood A.. et al. (2009) Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl Acad. Sci. USA, 106, 18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mall M.A., Sheppard D.N. (2014) Chronic ivacaftor treatment: getting F508del-CFTR into more trouble? J. Cyst. Fibros., 13, 605–607. [DOI] [PubMed] [Google Scholar]
- 47.Two 24-Week Phase 3 Studies of Lumacaftor in Combination with Ivacaftor Met Primary Endpoint with Statistically Significant Improvements in Lung Function (FEV1) in People with Cystic Fibrosis who have Two Copies of the F508del Mutation. Press release http://investors.vrtx.com/releasedetail.cfm? releaseid=856185 (24 June 2014); date last accessed September 27, 2017.
- 48. Boyle M.P., Bell S.C., Konstan M.W., McColley S.A., Rowe S.M., Rietschel E., Huang X., Waltz D., Patel N.R., Rodman D. (2014) A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir. Med., 2, 527–538. [DOI] [PubMed] [Google Scholar]
- 49. Wainwright C.E., Elborn J.S., Ramsey B.W., Marigowda G., Huang X., Cipolli M., Colombo C., Davies J.C., De Boeck K., Flume P.A.. et al. (2015) Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl J. Med., 373, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Neuberger T., Burton B., Clark H., Van Goor F. (2011) Use of primary cultures of human bronchial epithelial cells isolated from cystic fibrosis patients for the pre-clinical testing of CFTR modulators. Methods Mol. Biol., 741, 39–54. [DOI] [PubMed] [Google Scholar]
- 51. Namkung W., Finkbeiner W.E., Verkman A.S. (2010) CFTR-adenylyl cyclase I association responsible for UTP activation of CFTR in well-differentiated primary human bronchial cell cultures. Mol. Biol. Cell, 21, 2639–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Glozman R., Okiyoneda T., Mulvihill C.M., Rini J.M., Barriere H., Lukacs G.L. (2009) N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J. Cell Biol., 184, 847–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.