

Abstract
The French-Canadian variant of Leigh Syndrome (LSFC) is an autosomal recessive oxidative phosphorylation (OXPHOS) disorder caused by a mutation in LRPPRC, coding for a protein involved in the stability of mitochondrially-encoded mRNAs. Low levels of LRPPRC are present in all patient tissues, but result in a disproportionately severe OXPHOS defect in the brain and liver, leading to unpredictable subacute metabolic crises. To investigate the impact of the OXPHOS defect in the liver, we analyzed the mitochondrial phenotype in mice harboring an hepatocyte-specific inactivation of Lrpprc. Loss of LRPPRC in the liver caused a generalized growth delay, and typical histological features of mitochondrial hepatopathy. At the molecular level, LRPPRC deficiency caused destabilization of polyadenylated mitochondrial mRNAs, altered mitochondrial ultrastructure, and a severe complex IV (CIV) and ATP synthase (CV) assembly defect. The impact of LRPPRC deficiency was not limited to OXPHOS, but also included impairment of long-chain fatty acid oxidation, a striking dysregulation of the mitochondrial permeability transition pore, and an unsuspected alteration of trans-membrane H2O2 diffusion, which was traced to the ATP synthase assembly defect, and to changes in the lipid composition of mitochondrial membranes. This study underscores the value of mitochondria phenotyping to uncover complex and unexpected mechanisms contributing to the pathophysiology of mitochondrial disorders.
Introduction
Leigh syndrome is the most common pediatric presentation of autosomal recessive oxidative phosphorylation (OXPHOS) diseases. This syndrome is an early onset, subacute neurodegenerative disorder accompanied by characteristic brain stem lesions, psychomotor regression, hypotonia, ataxia, and lactic acidosis (1–3). The so-called classical form of the syndrome can be caused by mutations in subunits of the pyruvate dehydrogenase (PDH) complex, or more frequently, by mutations in the structural subunits of OXPHOS complexes, or of factors required for their assembly, resulting in isolated or multi-complex deficiencies affecting Complex I, II, IV (CI, CII, CIV) and the ATP synthase (CV) (4,5).
In addition to classic LS, a variant known as Leigh Syndrome French Canadian (LSFC; OMIM #220111) has a characteristic pattern of tissue involvement, and a different clinical evolution, with acute metabolic crises usually leading to very early fatality (6–8). LSFC is caused by a mutation in LRPPRC, which codes for an RNA-binding protein involved in the stabilization of most mtDNA-encoded mRNAs (9–11). Most patients examined to date are homozygous for a single missense mutation, which results in low (i.e. less than 20% of normal levels) steady state amounts of LRPPRC protein in all tissues (9,12,13). This causes a pronounced complex IV (C-IV) deficiency in the liver and brain (20% of normal activity), a moderate C-IV deficiency in fibroblasts and heart (50% of normal activity) and a combined C-IV and complex I (C-I) defect in the skeletal muscle (40% of normal activity) (8,9,12,13). LSFC fibroblasts recapitulated several alterations in mitochondrial function including mitochondrial fragmentation, a decrease mitochondrial membrane potential, and reduced ADP-stimulated respiration (13,14). Consistent with these findings, a case-control prospective metabolic profiling study in LSFC patients revealed a metabolic signature of disrupted oxidative phosphorylation, which include markers reflecting changes in NAD+, lipid and amine metabolism (15). Additional mutations in the LRPPRC gene have recently been identified outside the French-Canadian population, resulting in similar multi-systemic and neurological phenotypes (13). The reasons underlying this spectrum of biochemical defects remain unclear, but is likely related to differences in the way mitochondrial mRNAs are handled in different tissues, and the ability of some cell types, but not others, to compensate for the absence of LRPPRC through adaptive changes in their mitochondrial translation machinery (12).
The aim of the present study was to examine the impact of LRPPRC deficiency on key aspects of the liver mitochondrial phenotype. We generated an hepatic knockout mouse model to investigate in detail the impact of LRPPRC deficiency on the phenotype of liver mitochondria. Our results reveal that loss of hepatic LRPPRC triggers a multi-faceted phenotypic remodelling that extends beyond OXPHOS impairment, and includes mitochondrial ultrastructure abnormalities, impaired lipid metabolism, dysregulation of the permeability transition pore, and changes in ROS dynamics, thus highlighting the complex pathogenesis of OXPHOS disorders.
Results
Loss of hepatic LRPPRC results in growth delay, and pronounced liver histopathological abnormalities
Homozygous knockout mice were viable, had a normal appearance and locomotor activity under normal cage bound conditions, but had reduced body weight at 5 weeks-old compared to littermate controls (Fig. 1A). After an overnight fast, only 2% (2 out of 98) of H-Lrpprc−/− mice were found lethargic in their cage due to hypoglycaemia (≤ 3.1 mmol/l), indicating a relatively preserved capacity to sustain hepatic glucose production. Of note, immunoblot analysis indicated the presence of residual amounts of LRPPRC (Fig. 2A), which is likely attributable to liver regeneration as previously observed in liver-specific COX10 knockout mice (16).
Figure 1.

General phenotype and liver histology in normal and liver-specific LRPPRC deficient mice. Panels A and B show mean body weight (n = 30), and ratio of liver to body weight (n = 16–17) in H-Lrpprc+/+ and H-Lrpprc−/− mice. Panel C shows representative images of H&E staining. Loss of lobular structure and dilated vessels (top), focal necrosis and infiltration of inflammatory cells (middle), and cholestasis (bottom) are visible in the H-Lrpprc−/− samples (arrows). Panel D shows the quantification of Oil Red’O staining intensity in individual hepatocytes from H-Lrpprc+/+ and H-Lrpprc−/− livers (n = 10). Panel E shows representative images of serial liver sections stained for COX (brown staining) and SDH (blue staining) activity. Panel F shows transmission electron micrographs from both mouse strains. Arrows point to large vacuolar structures and stacked cristae, which were commonly observed in H-Lrpprc−/−mitochondria. Difference between H-Lrpprc+/+ and H-Lrpprc−/− was assessed with a Student t-test: **P < 0.01; ***P < 0.001.
Figure 2.

Impact of LRPPRC deficiency on the OXPHOS system. Panel A and B: SDS-PAGE blots and densitometry analysis (n = 4–6 mice per group) showing the impact of LRPPRC deficiency on SLIRP and selected components of CI (NDUFA9), CII (SDHA), CIV (COX1), and outer membrane (Porin). SDH and Actin were used as loading controls. Panel C: Activity of OXPHOS (CI, CII, CIV) and TCA cycle (CS) enzymes in isolated liver mitochondria. Enzyme activity measured spectrophotometrically (n = 4–8 mice per experimental group). Panel D: BN-PAGE blot of OXPHOS complexes in normal and H-Lrpprc−/− mice. Antibodies to detect OXPHOS complexes were NDUFA9 (CI), SDHA (CII), UQCRC2 (CIII), COXIV (CIV) and ATP5A1 (CV). Data are representative of 4 independent experiments. Panel E: Expression of mitochondrial ribosomal subunits, and of selected mitochondrial and nuclear encoded transcripts in wild type and H-Lrpprc−/− mice. Data were obtained at 10 weeks of age (n = 3–5). Panel F: Polyadenylated tail length analysis for COX1, COX2 and ND3 mRNA. Data shows the proportion of mRNA 3’ end displaying PolyA short, >10 and >35 chain length in wild type and Lrpprc-knockout mice. Data were obtained at 10 weeks of age (3 experimental replicates per group, using pooled RNA from 3 WT and 3 KO mice). Difference between H-Lrpprc+/+ and H-Lrpprc−/− was assessed using one-way ANOVA: *P < 0.05; **P < 0.01, ***P < 0.001.
Livers of H-Lrpprc−/− mice displayed several macroscopic abnormalities. Liver mass was 25% greater than in control animals (Fig. 1B), liver lobes displayed scattered pale coalescing areas, characteristic of multifocal hepatic necrosis, and the gall bladder was severely swollen. Microscopically, the geometry of liver lobules was disrupted and numerous blood vessels were dilated (Fig. 1C). Cholestasis, focal necrosis, infiltration of inflammatory cells and microvesicular steatosis was also evident following H&E and Oil Red O staining (Fig. 1C and D). Consistent with a C-IV deficiency, a severe reduction of COX staining was present in liver sections from H-Lrpprc−/− mice. Furthermore, the normal peri-portal zonation of nuclear encoded CII (e.g. SDH) activity was lost in favour of a more homogeneous distribution across liver lobules (Fig. 1E).
Transmission electron microscopy showed the presence of notable abnormalities in mitochondria from H-Lrpprc−/− livers. In general, mitochondria appeared larger than in control mice, and many displayed altered cristae morphology characterized by loss of cristae ridges, and the presence of large vacuolar structures or patches of stacked cristae (Fig. 1F).
Loss of hepatic LRPPRC induces a multi-faceted bioenergetic phenotype
LRPPRC deficiency resulted in the reduction of the steady state levels of most mtDNA-encoded transcripts in mice at ten weeks of age, particularly COX1-3 and ATP6, while rRNA levels were normal (Fig. 2E). Furthermore, in H-Lrpprc−/− mice, a high proportion (70–91%) of transcripts for COX1, COX2 and ND3 were oligo adenylated with less than 10A’s, or lacked stop codons, and had short and variable chain lengths (Fig. 2F). However, there was no relationship between the severity of mRNA downregulation and the polyA tail length" based on ND3 analysis.
In five weeks old mice, SDS-PAGE experiments indicated a near complete absence of LRPPRC in whole liver extracts (Fig. 2A), which was accompanied by a drastic reduction of the LRPPRC binding partner SLIRP (Fig. 2B). The abundance of the mitochondria-encoded CIV subunit COX1 was also significantly reduced as compared to control mice, while the abundance of CI (NDUFA9) and CII (SDHA) subunits were normal (Fig. 2A and B). Measurement of enzyme activities and BN-PAGE analysis confirmed a severe (>80%) decrease in the activity (Fig. 2C), and assembly of CIV (Fig. 2D). At this age, the activity of the TCA-cycle enzyme CS, and the amounts of assembled CI, CII, and CIII were similar to controls (Fig. 2C and D). However, at ten weeks of age, the amount of all three complexes was higher in H-Lrpprc−/− mice, suggesting a compensatory upregulation of other electron transport chain complexes over time (Supplementary Fig. S1).
We also investigated whether or not in addition to a CIV deficiency, mitochondria from H-Lrpprc−/− mice also displayed abnormalities at the level of the phosphorylation machinery, particularly the ATPsynthase (CV). As shown in Fig. 3A, CV activity was similar in the two experimental groups. However, an important loss of sensitivity to inhibition by oligomycin was noted in H-Lrpprc−/− samples, suggesting a CV defect. BN-PAGE analysis in DDM-solubilized mitochondrial extracts indicated a reduction in the amount of assembled CV monomers in H-Lrpprc−/− samples (Fig. 3B). Moreover, lower molecular weight ATPα immuno-reactive bands were visible, suggesting an accumulation of sub-assembled CV (Fig. 3B). CN-PAGE and in-gel activity measurements were also performed in digitonin-solubilized mitochondria to resolve CV dimers (Fig. 3C). These experiments confirmed the assembly defect, and further indicated an important reduction in amount of assembled CV dimers in H-Lrpprc−/− mice.
Figure 3.

Impact of LRPPRC deficiency on ATP synthase (CV) activity and assembly. Panel A: Enzyme activity measured spectrophotometrically in mitochondrial extracts in absence and presence of the CV inhibitor Oligomycin (1.2 µM). Data are expressed as fold changes vs wild type values (n = 6–8). Panel B: BN-PAGE blot of DDM-solubilized mitochondria from wild type and H-Lrpprc−/− mice. Membranes were probed with anti-ATPα Mono: CV monomers, Sub: Sub-assembled CV complexes. Panel C: Supramolecular assembly of CV revealed by in gel activity measurement. CN-PAGE was performed in duplicates for each sample. The first gel was used for in gel activity measurements (right lanes for each sample), while the other gel was stained with coomassie blue. Experiments were performed following extraction with DDM, which fully dissociates CV into monomers or with digitonin to preserve dimeric, and oligomeric CV complexes. Blots are representative of at least 3 independent experiments. Significantly different from the H-Lrpprc+/+ group: **P < 0.01.
Respirometry studies were performed to examine the bioenergetic consequences of these OXPHOS defects. While subtle differences in baseline state 2 respiration were apparent, the most striking abnormality was a 40–60% reduction of maximal ADP-stimulated respiration in the presence of CI or CII substrates (Fig. 4A). This reduction was attributable to the ATP synthase defect, rather than to loss of CIV activity, since mitochondria from control and H-Lrpprc−/− mice had similar maximal respiration rates following uncoupling with CCCP (Fig. 4A). Since the activity of CIV measured in solubilized extracts was reduced by 80% in H-Lrpprc−/− mitochondria (Fig. 3A), the surprizing capacity of residual CIV to support normal rates of respiration was investigated by monitoring CIV activity in its native membrane environment using TMPD/Ascorbate as a respiratory substrate (Fig. 4B). Under those conditions, CIV activity of H-Lprrpc−/− mitochondria was only 30% lower than in controls (P = 0.07), indicating that the detergent extraction step used for the spectrophotometric assessment of CIV activity amplifies the defect in H-Lrpprc−/− mitochondria. Interestingly, CIV was significantly more sensitive to cyanide (Fig. 4C) in H-Lrpprc−/− mitochondria compared to controls, indicating a greater vulnerability to inhibition.
Figure 4.

Impact of LRPPRC deficiency on the mitochondrial bioenergetics phenotype. Panel A: Baseline state 2, state 3 (1 mM ADP), and CCCP (0.03 µM)-uncoupled respiration in liver mitochondria energized with CI (glutamate/malate [GM: 5/2.5 mM]), CII (Succinate 5 mM) substrates, or following stimulation of long-chain fatty acid oxidation (palmitoyl-CoA [20μM]) (n = 5–9). Panel B: Maximal state 3 respiration in isolated liver mitochondria energized with CIV substrates (ascorbate/TMPD: 9mM/0.9mM) (n = 5). Panel C: Titration of CIV-driven (Ascorbate/TMPD 9/0.9 mM) state 3 respiration with potassium cyanide (KCN) in wild type and H-Lrpprc−/− mitochondria. Best fit and 95% confidence intervals are shown for each dataset (n = 5).
Mitochondria from H-Lrpprc−/− mice also showed a 50% reduction in ADP-stimulated respiration in the presence of palmitoyl-CoA (Fig. 4A). Because the respiration rates observed with palmitoyl-CoA, are less than half those achieved with CII substrates, this result suggest that in addition to causing OXPHOS defects, Lrpprc deficiency has a direct impact on mitochondrial fatty acid oxidation capacity.
The ATP synthase assembly defect associated with LRPPRC deficiency alters the mitochondrial permeability transition pore
Recent studies suggest that ATP synthase dimers may constitute the structural core of the PTP (17,18). In the presence of increased Ca2+ concentrations and Pi, which are key triggers of pore opening, Ca2+ is suggested to displace Mg2+ from catalytic sites on the F1 sector. Under this condition, CV would adopt a conformation that induces pore formation at the interface between the two CV monomers. In this model, the OSCP subunit, located on the lateral stalk of CV is suggested to act as a negative modulator of PTP opening, presumably by limiting access of the Ca2+ binding sites on the F1 ATPase (17,18). Importantly, this negative modulation is suggested to be relieved when Cyclophilin-D (CypD), a key pore-sensitizing protein present in the mitochondrial matrix, is recruited to OSCP (17,18).
Given the CV assembly defect observed in H-Lrpprc−/− mice, Ca2+ retention capacity (CRC) experiments were performed in the presence of Pi to determine whether pore regulation was altered. As shown in Figure 5A and D, CRC was nearly two-fold greater in mitochondria from H-Lprrpc−/− mice, indicating a striking resistance to PTP opening. CRC was also performed in the presence of Mg2+, ADP, and oligomycin, to delineate the nature of this resistance. Under this condition, CRC was increased by 2.3–2.5 fold in both experimental groups, indicating normal sensitivity in H-Lrpprc−/− mitochondria to ligands of the catalytic F1 sector, and the OSCP subunit (Fig. 5C and D). Furthermore, OSCP levels in the whole mitochondrial lysate and extracts used for CN-PAGE were similar between the two experimental groups (Fig. 5F). CRC experiments were also performed in the presence of Cyclosporin-A (CsA), which delays PTP opening by preventing recruitment of CypD to OSCP (17,18). As expected, CsA increased resistance to Ca2+-induced PTP opening by more than 2.5 fold in control mice. In contrast, mitochondria from H-Lrpprc−/− mice were largely insensitive to CsA (Fig. 5B and D). Lack of sensitivity to CsA is normally observed in cells that express low levels of CypD (19,20), and in tissues from CypD-KO mice (21). However, this was not the case in H-Lrpprc−/− mice since total mitochondrial CypD content was twofold higher than in controls (Fig. 5E). Together, these data indicate that in H-Lrpprc−/− mitochondria the CV assembly defect leads to intrinsic resistance to pore opening, through a loss of regulation by CypD.
Figure 5.

Impact of LRPPRC deficiency on the permeability transition pore: Panel A-C: Representative tracings of mitochondrial calcium retention capacity (CRC) in isolated liver mitochondria exposed to consecutive pulses of calcium (2.5 μmole/mg protein per pulse). All experiments were performed in the presence of Succinate (5 mM), Rotenone (1 µM) and Pi (10 mM). In panel B and C, the incubation buffer was respectively supplemented with Cyclosporin-A (1µM), or a combination of ADP (12 µM), MgCl2 (0.6 mM) and Oligomycin (27 µM). Panel D: Average Calcium Retention Capacity observed in the three experimental conditions described in A-C (n = 8–11). Panel E: Immunoblot and densitometric analysis of Cyclophilin-D (CypD) content in isolated liver mitochondria from wild type and H-Lrpprc−/− mitochondria. Mitochondrial lysates from wild-type and CypD deficient Ppif−/− mice were run as controls control (n = 6). Panel F: Immunoblot and densitometric analysis of OSCP content in whole mitochondrial lysates and OXPHOS extracts obtained following digitonin treatment. Significantly different from wild type mice: **P < 0.01, ***P < 0.001; Significantly different from control condition: Φ: P < 0.001.
Hepatic LRPPRC deficiency induces pronounced changes in mitochondrial H2O2 dynamics
An important factor believed to underlie cellular dysfunction in genetic mitochondrial diseases is enhanced production of mitochondrial ROS, which can disrupt normal ROS-dependent cellular signal transduction, and trigger oxidative damage to cellular components (22,23). To address whether LRPPRC deficiency had an impact on mitochondrial ROS dynamics, net mitochondrial H2O2 release was measured in respiring mitochondria. Strikingly, mitochondria from H-Lrpprc−/− mice released tenfold less H2O2 compared to controls in conditions where superoxide production is promoted through reverse electron flow (i.e. during state 2 respiration with succinate ± glutamate/malate) (Fig. 6A). To investigate factors underlying this striking difference, mitochondrial membrane potential, which is an important determinant of superoxide production, was measured. As shown in Figure 6B, membrane potential was reduced by 2–3 mV in H-Lrpprc−/−, which can account for no more than ∼7% of the difference in H2O2 release observed vs wild type (24,25).
Figure 6.

Impact of LRPPRC deficiency on mitochondrial H2O2 dynamics: Panel A: Net H2O2 release from mitochondria under state 2 and state 3 (1 mM ADP) conditions in the presence of substrates for CI + CII (G/M/Succ: 5/2.5/5 mM) (n = 8–16). In some experiments (n = 3), net H2O2 release was measured in the presence of succinate alone (Succ: 5 mM) and rotenone (1 µM) was added to confirm that H2O2 release occurred mainly through reverse electron backflow to CI. Panel B: Mitochondrial membrane potential measured on respiring isolated liver mitochondria energized with complex I + II (G/M/Succ: 5/2.5/5 mM) or complex II (succinate 5mM) substrates. (n = 6). Panel C: Immunoblot and densitometric analysis of SOD2 and catalase content in mitochondria from wild type and H-Lrpprc−/− mice (n = 6). Panel D: Kinetics of scavenging of an exogenous H2O2 load (3 nmoles) by mitochondria energized with CI substrates (Gutamate/Malate: 50/20 µM) (n = 6). Panel E: Net H2O2 release from mitochondria under state 2 conditions in the presence of substrates for CI + CII (G/M/Succ: 5/2.5/5 mM). Following baseline measurements, digitonin was progressively added and changes in H2O2 release were monitored (n = 6). Insets in panel E shows progressive inhibition of respiration in H-Lrpprc+/+(black tracing) H-Lrpprc−/− (grey tracing) in response to progressive permeabilization with digitonin. Panel F: H2O2 production measured in sub-mitochondrial particles (SMP) in the presence of succinate (5 mM) alone, or with Antimycin-A (AA: 2 µM) (n = 4). Panel G: Immunoblot and densitometric analysis of AQP8 content in mitochondria from wild type and H-Lrpprc−/− mitochondria (n = 6). Significantly different from wild type mice: *P < 0.05, **P < 0.01, ***P < 0.001.
To determine whether enhanced ROS scavenging was responsible for the dramatic reduction of mitochondrial H2O2 release in H-Lrpprc−/− mice, the capacity to scavenge a pulse of exogenous H2O2 was also measured directly in respiring mitochondria. This scavenging rate represents the sum of the rate of all mitochondrial H2O2 consuming systems. Surprisingly, mitochondria from H-Lrpprc−/− mice were completely unable to scavenge exogenous H2O2 (Fig. 6C). The level of key antioxidant enzymes was therefore measured. The expression of SOD2 was similar in the two groups, while the levels of catalase were modestly reduced, suggesting no major collapse of the antioxidant machinery (Fig. 6D). This observation was overall compatible with the results of the H2O2 release experiment, since a collapse of antioxidant systems would have caused an increase in H2O2 release, not a drastic reduction as observed. Together, the lack of endogenous H2O2 emission and exogenous H2O2 scavenging pointed to a major barrier hindering H2O2 diffusion in and out of mitochondria in Lrpprc-deficient mice.
To address this possibility, respiring mitochondria were titrated with increasing concentration of digitonin (0.01 to 0.03%) to achieve progressive membrane permeabilization. As shown in Figure 6E, addition of digitonin to H-Lrpprc−/− mitochondria progressively raised the amount of H2O2 released in the media to the level observed in non-permeabilized mitochondria from control mice, suggesting trapping of H2O2 in mitochondria from H-Lrpprc−/− mice. To test this hypothesis, H2O2 production was measured in inside-out sub-mitochondrial, where respiratory chain complexes generate H2O2 directly in the incubation medium, bypassing the normal diffusion step across the lipid bilayer. In this condition, H2O2 release was actually higher in H-Lrpprc−/− compared to control (Fig. 6F), confirming that LRPPRC deficiency induced a strong reduction of mitochondrial permeability to H2O2.
To investigate the underlying mechanism, the expression of aquaporin-8 channels, which are known to facilitate the diffusional transport of H2O2 across the inner mitochondrial membrane (26,27) was measured. AQP-8 content was 35% higher in H-Lrpprc−/− mice, indicating that changes in AQP8 expression were not responsible for the altered H2O2 diffusion (Fig. 6G). Non-targeted lipidomic analysis was also performed to examine potential changes in the membrane lipid composition. In H-Lrpprc−/− mitochondria, 65 features reached the established significance threshold (FC > 2 and FDR < 5%, corresponding to a p value of 0.01) (Fig. 7A). From these features, thirty-three distinct lipids were identified in H-Lrpprc−/− mitochondria of which 14 were upregulated and 18 downregulated. Interestingly, increased lipids were essentially glycerophospholipids mostly represented by phosphatidylcholines (PC) and few phosphatidylethanolamine (PE), while decreased lipid species were more diversified. The latter included in particular 7 triglycerides, 2 diglycerides, 6 sphingomyelins, as well as cholesterol and cholesterol sulfate (Fig. 7B). Together, these results indicated that loss of LRPPRC triggers broad changes in the lipid composition of mitochondrial membranes, which likely underlie the impairment of H2O2 diffusion in and out of mitochondria.
Figure 7.
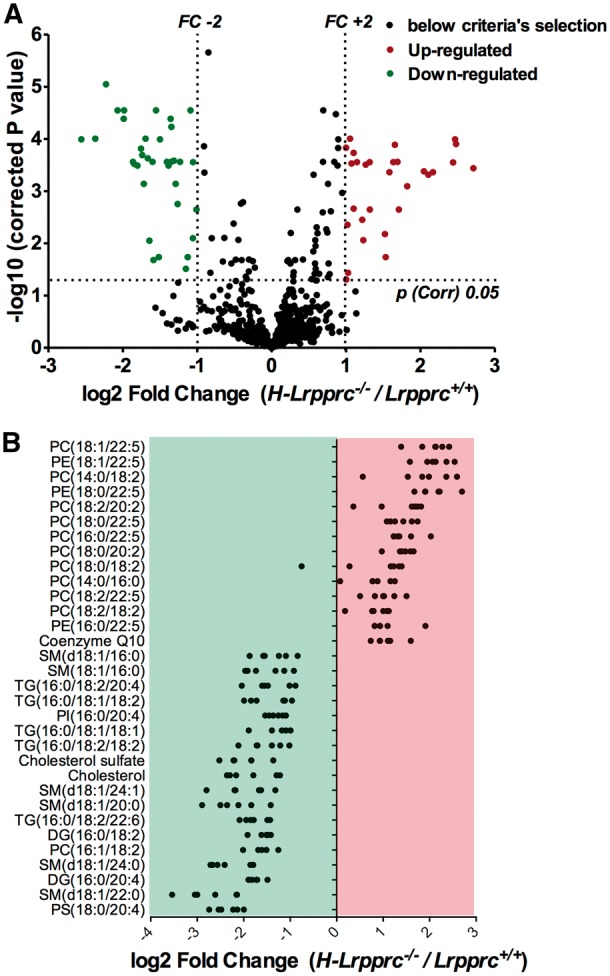
Impact of LRPPRC deficiency on mitochondrial membrane lipid composition: Panel A: Volcano plot showing entities that were identified as differentially expressed, either up or downregulated, in H-Lrpprc−/− mitochondria according to the following selection criteria: fold change > 2 and corrected p value < 0.05. Panel B: Individual lipid species found to be significantly up or down-regulated on H-Lrpprc−/− mitochondria. Each dot represents a Log2-transformed ratio of an individual H-Lprrpc−/− mitochondria relative to the mean value obtained in the wild type group (n = 6 per group).
Discussion
Our results reveal that specific loss of LRPPRC in the liver induces a generalized growth delay, and typical histological features of severe mitochondrial hepatopathy. At the molecular level, LRPPRC deficiency causes destabilization of polyadenylated mitochondrial mRNAs, resulting in a severe CIV and CV assembly defect that compromises OXPHOS capacity. Importantly, our data show that the impact of LRPPRC deficiency is not limited to OXPHOS, but also includes impairment of long-chain fatty acid oxidation, a striking dysregulation of the mitochondrial permeability transition pore, and an unsuspected alteration of trans-membrane H2O2 diffusion. Dysregulation of the PTP and changes in mito-cellular H2O2 exchanges can be traced to the ATP synthase assembly defect, and to changes in the lipid composition of mitochondrial membranes, revealing novel intricacies of mitochondrial OXPHOS deficiencies that have potential clinical impact.
Bioenergetics phenotype of hepatic LRPPRC deficiency
Previous studies have shown that CIV activity is reduced by 80% in liver samples from LSFC patients (8,12), which is expected to have severe bioenergetic consequences. Yet, some patients survive several years, with no signs of chronic liver dysfunction despite the presence of steatosis (6). Our data suggest that this may be explained in part by the fact that biochemical measurement of CIV activity following detergent extraction exaggerates the actual severity of the enzyme defect. Indeed, when measured in intact mitochondria, our data show that CIV activity was only reduced by 30% in LRPPRC-deficient livers, as opposed to 80% when measured following detergent extraction. This significant residual activity was sufficient to sustain normal respiration with substrates feeding the ETC at the level of CI or CII, albeit at a higher percentage of maximal CIV capacity, which likely put cells at greater risk of bioenergetic crisis in the presence of factors that negatively modulate CIV activity in vivo (14).
Sensitivity of CIV to inhibition in the presence of detergents has been reported previously in cells from Surf1 patients (28), although the underlying mechanisms were not established. One factor that likely plays a role is membrane lipids, which are known to be crucial for proper assembly, and function of ETC complexes (29,30). For CIV in particular, mass spectrometry coupled with X-ray structure analysis has identified thirteen lipids including CL, PCs, PEs, PGs and TGs that are integral to the complex, and likely play a structural and functional role (31). Interestingly, one intrinsic PC [PC(16:1/18:2)], and three TGs [TG(16:0/18:1/18:1); TG(16:0/18:1/18:2) believed to stabilize the dimeric state of CIV, and the assembly between the three core subunits (COX1-3), and the nuclear encoded subunits were significantly reduced in H-Lrpprc−/− mitochondria. These results suggest that the reduced abundance in key structural lipids could increase the lability of CIV in LRPPRC-deficient mitochondria, making the enzyme more vulnerable to inhibition in the presence of detergents. An interesting question that remains to be investigated is whether the reduced abundance of these lipids is simply a consequence of the lower level of assembled CIV. However, the fact that several other lipids are also modified in H-Lrpprc−/− mitochondria suggests that it is part of a more global remodelling of membrane lipid composition.
Results from our study also show that the impact of LRPPRC-deficiency in the mouse liver is not limited to CIV but also includes a CV assembly defect that was largely responsible for the impairment of OXPHOS capacity we observed. While a similar CV defect was reported following Cre-mediated inactivation of Lrpprc in the mouse heart (32), recent results from our group indicate that this abnormality is likely absent in the liver and heart from LSFC patients. Indeed, the only defect observed is a severe (80%) reduction in CIV assembly in the liver, and a more moderate (50%) reduction in the heart (12) with no signs of other OXPHOS complexes being affected. Absence of CV defect was also recently reported in patients with a mutation of LRPPRC that is distinct from the one causing LSFC (13). This difference could be explained by the presence of greater residual amounts of LRPPRC in patient tissues compared to knockout mouse tissues, which would be sufficient to allow proper assembly of CV. However, we consider this possibility to be unlikely since in both organs is either undetectable, or extremely low (<10%) (12). Moreover, when the level of LRPPRC is further decreased in patient fibroblasts using siRNAs, it is the assembly of all OXPHOS complexes, not just CV, that is compromised (9) Therefore, the CV assembly defect reported in the liver and heart likely represents a species-specific response to LRPPRC deficiency.
In addition to affecting OXPHOS directly, our data show that LRPPRC deficiency impairs mitochondrial long-chain acyl-CoA oxidation which is consistent with our recent metabolic profiling of plasma from LSFC patients (15). Indeed, we identified elevated circulating acylcarnitines irrespective of chain length suggesting the existence of multiple defects in mitochondrial fatty acid β-oxidation. Because oxidation of long-chain fatty acid is controlled by steps located upstream of the ETC (33,34), this impairment is unlikely to be explained by the reduction of OXPHOS capacity per se. Although we did not pursue this further, previous studies indicate that knockdown of Lrpprc in primary hepatocytes does not lower the expression of key genes involved in mitochondrial fatty acid uptake and β-oxidation, despite reducing overall cellular fatty acid uptake, and oxidation (35,36). It is therefore likely that post-translational changes secondary to the OXPHOS defect, or broader changes in lipid metabolism revealed by our lipidomic analysis, underlie the reduced fatty acid uptake/oxidation capacity and drive steatosis in the Lrpprc-deficient liver.
PTP dysregulation in LRPPRC-deficient mitochondria
The PTP, located in the mitochondrial inner membrane, plays an important role in the physiological regulation of ROS production, mitochondrial Ca2+ release, and cellular differentiation, as well as pathological activation of cell death (see (37) for recent review). Despite being well characterized functionally, the molecular identity of the PTP continues to be a subject of debate. Several molecular candidates have been proposed including ANT (38), VDAC (39) and the Pi carrier (40). However, none of these putative pore components has survived experimental scrutiny. Recently, studies in yeast, and mammalian cell models have provided strong evidence that CV acts as the core constituent of the PTP (18,41,42). Our study, by showing a striking dysregulation of the PTP in a mouse model of primary CV defect, provides support for this model. Interestingly, in fibroblasts from LSFC patients, we previously reported an enhanced sensitivity to PTP opening in the presence of Ca2+(14), which may be due to the absence of CV assembly defects in patient cells.
The molecular mechanism by which CV shifts from an ATP synthetizing unit to a high conductance non-specific pore remains largely unknown (17,43). One model proposes that the PTP forms at the interface of CV dimers (17,44). At this level, several subunits of the Fo sector (a, e, f, g and A6L) bind together, and stabilize the central c-ring of the Fo sector, and the lateral stalk of the F-ATPase (45–48), which contributes to the formation of 86°-angled CV dimers that define areas of high membrane curvature at the level of cristae ridges (49–51). In the presence of increased Ca2+ concentrations, Ca2+ is suggested to displace Mg2+ from catalytic sites on the F1 sector. Under this condition, CV would adopt a conformation that induces pore formation at the interface between the two CV monomers (17,44). The exact mechanism of pore formation currently remains unclear. The c-ring was initially proposed as the pore-forming channel (42), but recent studies strongly argue against this possibility (52). Subunits of the Fo sector that are required for the stabilization of CV dimers (53) however remain serious candidates (17). Results showing that knockout of subunits e and g induces a strong resistance to PTP opening argue in favour of this model (41). Irrespective of the exact mechanism of pore formation, studies show that CV is regulated by binding of CypD to the OSCP subunit of the lateral stalk, which induces a conformational change that facilitates pore formation (18,54).
Our data show that the CV assembly defect associated with the loss of LRPPRC induces a marked resistance to PTP opening that can be explained by a loss of control of PTP sensitization by CypD. This effect is not related to reduced CypD expression, as the protein level is upregulated twofold in the absence of LRPPRC. Since OSCP levels were comparable across the two experimental groups, and that OSCP is present in CV monomers and dimers following cardiac inactivation of Lrpprc (32), it is also unlikely that lack of CypD binding sites on CV is responsible for pore desensitization in LRPPRC deficient mitochondria. One possibility is that post-translational modifications of OSCP, such as a reduction of K40 acetylation, could impair CypD recruitment, as recently shown (55). Alternately, loss of mitochondrially-encoded subunits a (ATP6) and A6L (ATP8) (Fig. 2A and (32)) and potentially other associated Fo subunits may lead to alterations in the assembly between the Fo sector, and the peripheral stalk which prevent CypD binding from exerting its pore-promoting conformational changes. More detailed structure-function studies are required to elucidate the precise mechanisms involved.
Altered ROS dynamics in LRPPRC-deficient mitochondria
Oxidative stress is considered a significant contributor to the pathogenesis of OXPHOS defects (22,23). Mitochondrial ROS production, and its consequences on signalling pathways, and mitochondrial/cellular injury is generally viewed as the result of a disequilibrium between the production of ROS by dysfunctional ETC complexes, and their elimination by various mitochondrial antioxidant systems. However, our results show the importance of considering mitochondrial membranes as potential diffusion barriers that can affect mito-cellular ROS dynamics in pathological states. Indeed, we show that lack of LRPPRC drastically reduced the capacity of mitochondria to release and take-up H2O2. This effect occurred in the absence of major remodelling of key antioxidant enzymes (Catalase and SOD2) and was abolished by progressive permeabilization with small amounts of digitonin. Furthermore, mitochondrial and H2O2 production was higher in LRPPRC-deficient mice when measured in inside-out submitochondrial particles, suggesting that reduced membrane permeability to H2O2 in LRPPRC-deficient livers confined ROS to the mitochondrial compartment.
Although H2O2 is generally considered to diffuse freely across membranes, increasing evidence suggest that this process may be regulated. One of the mechanisms involves diffusion-facilitating channels of the aquaporin family, particularly AQP8 isoforms, which are expressed in mitochondrial membranes, where they are thought to assist diffusion of water, NH3, and H2O2 (26,27). Knockdown of AQP8 was shown to inhibit mitochondrial H2O2 release (27), and cause mitochondrial dysfunctions including oxidative stress, respiratory impairments, and loss of membrane potential (26,27). Based on these results, lower expression of AQP8 might have been expected in H-Lrpprc−/− mitochondria; however, our results indicate that AQP8 expression was in fact higher in the absence of LRPPRC, indicating that this is not the primary mechanism responsible for the impaired diffusion of H2O2. In fact, our results would suggest that in the absence of LRPPRC AQP8 expression may be upregulated in an attempt to compensate the H2O2 diffusion deficit.
In view of our results, changes in the lipid composition of membranes in LRPPRC-deficient mitochondria appear as the main factor responsible for the impaired diffusion of H2O2. Although the precise mechanism remains unclear, one possibility is that loss of membrane cholesterol, which is already present at low levels in mitochondrial membranes (56), contributes to the H2O2 diffusion impairment. Lipid composition and their arrangement in the bilayer, as well as physicochemical parameters of lipids surrounding cholesterol molecules, might have an impact on lipid packing density and thickness which could eventually lead to a packing defect. In H-Lrpprc−/− mitochondria, the significant decrease in cholesterol as well as the increase amount in PC-containing polyunsaturated FA highlighted perturbations on mitochondrial membranes physical properties thereby promoting increased membrane fluidity, decreased thickness and major packing defects (57,58), which could interfere with H2O2 diffusion. Support for a role of cholesterol and its lipid interactome in affecting trans-membrane H2O2 diffusion comes from studies in yeast showing that downregulation of ergosterol (the main sterol in in fungi and protozoa) during the stationary phase reduces H2O2 diffusion into cells, while upregulation of ergosterol content during exponential growth facilitates H2O2 diffusion out of cells (59,60). Studies in yeast also showed that gain of function mutation of the ergosterol synthesis pathway enhanced H2O2 permeability (61). Together, these data have led to the suggestion that in yeast, modulation of ergosterol content is part of a cellular strategy to manage oxidative stress in the face of pronounced metabolic transitions (59–61). Interestingly, in mammalian erythrocytes, upregulation of membrane cholesterol content was found to decrease the trans-membrane diffusion of oxygen (62), which is compatible with a role of cholesterol in facilitating the diffusion of small molecules. As for cholesterol sulphate, it is suggested to play a role in the stabilization of erythrocyte membrane (63), but whether its downregulation could alter diffusion is unknown. It should however be noted that under normal healthy conditions, mitochondrial membranes may not constitute a major diffusion barrier, since in our experiments progressive permeabilization of mitochondrial membranes with digitonin did not reveal evidence of H2O2 trapping in wild-type mitochondria. Pathological loss of cholesterol and specific changes in the lipid composition of mitochondrial membranes may be required to observe a diffusion limitation.
In summary, our mouse model of hepatic LRPPRC deficiency reveals that genetic OXPHOS defects can be accompanied by broad and surprising phenotypic alterations that cannot easily be predicted based on knowledge of the causal genetic defect. From a clinical perspective, our results also indicate that the investigation of PTP dysregulation is warranted in patients harbouring CV assembly defects. Furthermore, the substantial remodeling of the lipid composition of mitochondrial membranes in our mouse model suggests that membrane lipidomics should be performed in patients with genetic OXPHOS defects as this might assist diagnostic and have significant impact on mitochondrial functions and the disease phenotype.
Materials and Methods
Animal care and generation of conditional Lrpprc knockout mice
All experiments on animals were approved by the Université de Montréal Institutional Animal Care Committee and conducted according to the directives of the Canadian Council on Animal Care. Mice were maintained in ventilated cage racks by groups of 4–6 mice. All mice were kept on a regular 12–12h light-dark cycle, and had access to food and water ad libitum. To disrupt LRPPRC expression in the liver, the Lrpprc knock-out mice line Lrpprctm1a(K°MP)Wtsiproduced in C57BL/6N embryonic stem (ES) cells was acquired from KOMP repository (University of California, California). The mutated locus was transmitted through the germline to obtain heterozygous Lrpprc+/l°x-ne° animals. These animals were then bred with flp producing animal B6(C3)-Tg(Pgk1-FLPo)10Sykr/J (The Jackson Laboratory) in order to excise the neomycin resistance cassette, recreating a LRPPRC protein producing locus having lox sites in intron 3 and 5. The resulting Lrpprc+/l°xP mice were then mated to get homozygous Lrpprc l°xP/l°xP mice and only animals that were exempt of the flp allele were kept. To achieve liver specific inactivation of Lrpprc, Lrpprc l°xP/l°xP mice were crossed with B6.Cg-Tg(Alb-cre)21Mgn/J (The Jackson Laboratory) mice producing the Cre recombinase under the control of the albumin promoter and bred to homozygous state, Hep-Crecre/cre. These mice were then bred with the Lrpprc l°xP/l°xP to generate double homozygous mice Hep-Lrpprcl°xP/l°xP;cre/cre. However, in order to simultaneously generate homozygous knockout and wild type littermate controls, Hep-Lrpprcl°xP/l°xP;cre/0 individuals were inter-crossed.
All animals were genotyped at weaning via tail biopsies (20 mg/ml), which were incubated at 55 °C for 4 h in buffer (50 mM Tris, 30 mM EDTA, and 0.25% SDS) containing (1,000 μg/ml) proteinase K (Sigma, St. Louis, MO). After digestion, the samples were diluted 1:250 in DNAse/RNAse free water along with 15 mg chelex-resin (Bio-Rad Laboratories, Hercules, CA) and heated at 95 °C for 5 min. They were then analyzed in duplicate by quantitative qPCR with 2X Platinum SYBR Green qPCR Supermix-UDG, according to the manufacturer's specifications (Invitrogen Life Technologies, Carlsbad, CA). Cycling was achieved in a MX3005p cycler (Stratagene, Mississauga, Ontario, Canada) with the following conditions: 95 °C for 10 min and 40 cycles of 95 °C for 30 s, 47 °C for 45 s, and 72 °C for 45 s. The primers for qPCR were designed to target the Cre gene cassette (reverse: CCAGCTTGCATGATCTCC; forward: CGCTAAGGATGACTCTGG), and the corresponding PCR signal was expressed relative to cyclophilin A (Ppia) genomic DNA (reverse: GCCGCCAGTGCCATTATG; forward: CCGATGACGAGCCCTTGG). Lox allele containing LRPPRC was detected by end-point PCR. The primers for PCR were designed to target the lox containing neighbouring sequences located in the third LRPPRC intron (reverse: ATGAGTTCGATTCCCAGCAAC; forward: CGTAGGCAGTATCCACAC). The lox-containing PCR amplicon is 474 bp while the endogenous amplicon is 386 bp. Animals used (males and females) for experiments were euthanized by cervical dislocation at 5 or 10 weeks of age following an overnight fast. Unless indicated otherwise, results presented were obtained using 5 weeks-old mice.
Histology
Haematoxylin/Eosin. For a general assessment of histopathology, livers were rapidly excised and fixed overnight in 10% formalin. Samples were embedded in paraffin and 5μm thick sections were stained with Haematoxylin and Eosin.
Oil Red’O. To quantify steatosis, livers were snap frozen in liquid nitrogen for Oil red’O staining. 10μm thick sections were fixed in 10% neutral buffered formalin for 5 min, washed 4 times in distilled water and stained for 30 min at room temperature with either Oil Red’O (working solution: 0,5% Oil Red’O in propylene glycol) or propylene glycol. Following thorough rinsing with distilled water, slides were mounted using Pro-long Gold anti-fade reagent with DAPI (Invitrogen) and examined by confocal microscopy. Lipid droplet number and morphology in individual hepatocytes was quantified using ImageJ (NIH).
COX/SDH activity. Cytochrome c oxidase (COX) and succinate dehydrogenase (SDH) activity staining was adapted from (64). Serial cryostat sections were cut 10μm thick, brought to room temperature and were left to air dry for 1 hour. The first section of each sample was incubated for COX activity staining (in mM: 3.75 DAB, 0.1 Cytochrome C, pinch of catalase in 0.1 NaPi buffer, pH 7,0) for 30 min at 37 °C. The second section was incubated for SDH activity staining (in mM: 130 Na- Succinate, 0,2 PMS, 0.1 Sodium Azide, 1.5 NBT in 0.1 NaPi buffer, pH 7,0) for 30 min at 37 °C. The third section was incubated for COX activity staining buffer first, washed with distilled water and incubated for SDH activity staining buffer to achieve double staining. Sections were washed with distilled water and mounted with glycerol gelatine. Slides were examined by light microscopy.
Transmission electron microscopy
Following anaesthesia (8% chloral hydrate; 600 mg/kg), mice were perfused with 10ml PBS and 5ml 2,5% glutaraldehyde via the vena cava. Livers were excised, sliced and fixed over night at 4 °C in 2,5% glutaraldehyde in phosphate buffer. After sample preparation, 90–100 nm thick sections were mounted onto a 200 mesh copper grid (Electron Microscopy Sciences) and imaged with a FEI Tecnai 12 120 kV transmission electron microscope equipped with an AMT XR80C 8 megapixel CCD camera as previously described (65).
Preparation of isolated liver mitochondria
Liver mitochondria were isolated as described by Debray (2) with slight modifications. Following cervical dislocation, livers were rapidly excised and transferred to cold isolation buffer (in mM: 300 sucrose, 10 Tris-HCl, 1 EGTA-Tris Base; pH 7,2). The tissue was minced using scissors and 4mm2 pieces were allowed to sediment. The decanted suspension was transferred in 15ml Potter-Elvehjem homogeniser and homogenised with 5 strokes, the pestle rotating at 500rpm. The homogenate was centrifuged 10 min at 1000Xg, 4 °C and the supernatant was transferred in a clean tube and re-centrifuged at the same speed. The resulting supernatant was transferred using a syringe attached to a metal filling cannula and centrifuged 10 min at 8000Xg, 4 °C. The supernatant was aspirated and the pellet was re-suspended in suspension buffer (in mM: 300 sucrose, 10 Tris-HCl, 0.05 EGTA-Tris Base; pH 7,2). The suspension was centrifuged 10 min at 8000Xg, 4 °C; and the last two steps repeated twice. The final pellet was re-suspended in 300μl suspension buffer and kept on ice until use.
Preparation of sub-mitochondrial particles
Submitochondrial particles were prepared as in the previously published literature (3–5). Frozen isolated liver mitochondria were thawed on ice and centrifuged for 10 min at 8000Xg, 4 °C. The pellet was re-suspended in sonication buffer (in mM: 250 sucrose, 10 K-phosphate, 10 Tris-HCl, 2 EGTA, 2 MgCl2; pH 7,4) and sonicated 5×15 s using a Branson sonicator at 50% amplitude. The suspension was centrifuged 10 min at 10 000Xg, 4 °C; the supernatant was transferred and centrifuged 60 min at 100 000Xg, 4 °C in a swinging-bucket rotor. The final pellet was re-suspended in a small volume (25μl or less) of suspension buffer (in mM: 1 EDTA, 1 MgCl2, 75 sodium phosphate; pH 7,4) and kept on ice until use.
Mitochondrial functions
Respirometry. Respiration was measured using Clark-type electrodes at 23 °C under continuous stirring based on previous protocols (66). Mitochondria (0.3 mg prot./ml) were suspended in respiration buffer (in mM: 10 KCl, 5 K2HPO4, 10 MOPS, 9 Pi, 2,5 MgCl2, 1mg/ml BSA; pH 7,4). Following baseline recording, respiration was measured following sequential additions: i) glutamate + malate (5:2.5 mM) for Complex-I (CI) or succinate (5 mM) for complex-II (CII), ii) ADP (1 mM), and iii) and carbonyl cyanide m-chlorophenylhydrazone (CCCP: 0.03 µM). To measure to oxidation of long-chain fatty acids, respiration was measured following supplementation with palmitoyl-carnitine (20 µM) in the presence of malate (2.5 mM) and ADP (1 mM). To measure the activity of complex-IV (CIV) under native conditions, maximal ADP stimulated respiration was measured in the presence of TMPD/Ascorbate (5/0.3 mM), and titrated with increasing concentrations of potassium cyanide, which allowed to correct respiration values for the auto-oxidation of TMPD/Ascorbate.
Ca2-induced opening of the permeability transition pore (PTP). Mitochondria (0.5 mg prot./ml) were incubated in a sucrose buffer (in mM: 250 sucrose, 0.005 EGTA-Tris base, 10 Tris-MOPS; pH 7.55) containing succinate (5 mM), rotenone (1 μM) and Pi (10 mM). Changes in extra-mitochondrial calcium concentration were monitored fluorimetrically (Hitachi, F4500 spectrofluorometer) using Calcium-green 5N (1 μM, ex–em: 505–535 nm) as described previously (67). Residual calcium concentration was adjusted to the same level at the beginning of every experiment by adding a small amount of EGTA. Calcium pulses (2.5 µmol/mg protein) were added at 2 min intervals until a Ca2+-induced Ca2+ release was observed. In some experiments, the following PTP inhibitors were added prior to Ca2+ pulses: Cyclosporin-A (1 µM), MgCl2 + ADP + Oligomycin (1.2, 0.6, and 0.00127 mM respectively). In all experiments, Calcium Retention Capacity (CRC) was taken as the total amount of Ca2+ accumulated by mitochondria prior to the Ca2+ pulse triggering Ca2+ release.
Mitochondrial membrane potential. Mitochondria (0.25mg prot./ml) were incubated at room temperature in a K-MES buffer (in mM: 110 K-MES, 35 KCl, 1 EGTA, 5 K2HPO4, 3 MgCl2, 0.5mg/ml BSA) supplemented with the potentiometric probe Rhodamine 123 (0.2 µM, ex-em: 503–525 nm) and either glutamate:malate (5: 2.5 mM) or succinate (5 mM). Membrane potential (ΔΨ) was calculated from changes in the concentration of extra-mitochondrial Rhodamine 123 as per (68) using the equation ΔΨ=59 × log [Rhodamine]in/[Rhodamine]out. To calculate the intra-mitochondrial concentration of Rhodamine, the amount of Rhodamine taken-up by mitochondria was determined by measuring changes in fluorescence following addition of respiratory substrates. A matrix distribution volume of 1 µl/mg protein, and a non-specific binding of rhodamine to mitochondria of 30% was assumed in all groups (68).
Mitochondrial H2O2 release and membrane permeabilization. Mitochondria (0.2 mg prot./ml) were incubated at room temperature in a K-MES buffer (in mM: 110 K-MES, 35 KCl, 1 EGTA, 3 MgCl2,10 K2HPO4, 0.5mg/ml BSA; pH 7,55) supplemented with the H2O2-sensitive probe Amplex red (1.5μM; ex-em: 563–587nm) and horseradish peroxidase (1.2U/ml), as previoulsly described (69). Net mitochondrial H2O2 release was measured: i) under state 2 conditions in the presence of succinate (5 mM) alone or with glutamate-malate (5:2.5 mM) to elicit reverse electron flow, ii) under state 3 conditions following the addition of ADP (1 mM) and iii) under state 2 following inhibition of reverse electron flow with rotenone (1 µM). To assess mitochondrial H2O2 trapping, net H2O2 release was first measured under state 2 conditions and membranes were subsequently permeabilized by adding increasing pulses of digitonin (0.01% per pulse) at regular interval. An increase in H2O2 emission above basal levels was used as an indicator of H2O2 entrapment.
H2O2 Scavenging. H2O2 scavenging was measured fluorimetrically in a 96 well plate reader using a protocol modified from (69). Briefly, mitochondrial aliquots (0.1 mg prot/ml) were distributed in 7 wells in K-MES buffer supplemented with glutamate (50 µM) and malate (20 µM). A pulse of 3 nmoles of H2O2 was added to the first well at t = 0 s, and to each subsequent well at 20 s intervals. At t = 100 s, 100 µl of K-MES buffer supplemented with 10 μM Amplex Red, and 0.5U/ml horseradish peroxidase was added to all wells, and fluorescence corresponding to the amount un unscavenged H2O2 remaining in each well was immediately measured in end-point measurement mode. In all experiments a control well containing no mitochondria was used as the baseline control.
Immunoblotting
SDS-PAGE. Whole liver extracts or isolated mitochondria were extracted in 1% lauryl maltoside/PBS, and 10, 20 or 50 µg of protein per sample were loaded and run on 12 or 15% polyacrylamide gels, then transferred to PVDF membrane and used for the detection of LRPPRC (1/1000; in house), SLIRP (1/50; in house), COX I (1/2000; Abcam #ab14705), NDUFA9 (1/1000; Abcam #ab14713), SDHA (1/1000; Abcam #ab14715), SOD2 (1/2000; Abcam #ab16956), CoreII (1/100; in house), Catalase (1/1000; Abcam #ab52477), Cyclophilin-D (1/1000; Pierce Thermos-Fisher #PA1-028), AQP8 (1/1000; Abcam #203682), OSCP (1/1000; Santa Cruz #365162), Porin (1/2000; Abcam #14734), and Actin (1/1000; Genescript #A00702). LRPPRC polyclonal antibodies were prepared by immunizing rabbits with peptides of 22 amino acids corresponding the sequence CEPPESFEFYAQQLRKLRENSS (antibody 295–313; Zymed Laboratories, San Francisco, CA). The density of immunoreactive bands was determined using ImageJ. Unless indicated in the figure legend, all results were normalised by ponceau staining of the membranes.
Clear-Native-PAGE and in gel ATPase activity. OXPHOS complexes were extracted from isolated mitochondria using either Dodecylmaltoside (DDM: 2%) or digitonin (2g digitonin/1g mitochondrial protein) in extraction buffer (in mM: 30 HEPES, 150 K-acetate, 2 6-aminocaproic acid, 1 EDTA, 12% glycerol; 10μl buffer/100μg protein). The suspension was incubated on ice for 30 min and centrifuged for 45 min at 20 400Xg at 4 °C. The supernatant was supplemented with 750μM 6-aminocaproic acid, and separated by Clear Native (CN)-PAGE on 3–12% Bis-Tris Novex gels (Life Technologies). Half of gel was stained with coomasie blue. The other half of the gel was incubated in an ATP synthase activity buffer (in mM: 50 glycine, 5 MgCl2, 50 HEPES, 30 CaCl2, 10 ATP; pH 7,8) overnight at 4 °C under agitation to resolve native ATP synthase complexes.
Blue-Native-PAGE. Mitochondria extracts were solubilised in 1% lauryl maltoside, 10 µg of protein was loaded on 6–15% polyacrylamide gradient gel and then transferred to nitrocellulose membrane. Individual structural subunits of complexes I (NDUFA9; 1/1000; Abcam #ab14713), II (SDHA; 1/1000; Abcam #ab14715), III (CoreII; 1/100; in house), IV (COX I; 1/2000; Abcam #ab14705) and V (ATP5a; 1/5000; Abcam #ab14748) were detected.
Enzyme activities
Activities of CI (NADH-CoQ reductase), CII (succinate dehydrogenase), CIV (cytochrome oxidase), CV (ATP synthase) and citrate synthase (CS) were measured spectrophotometrically in a plate reader using standard coupled enzyme assays adapted from (66) and from (70) for CV. Activities were expressed in mU.min−1.mg mitochondrial prot−1. To facilitate comparisons, all enzyme activities were subsequently expressed as fold difference vs H-Lrpprc+/+.
Mitochondrial polyadenylation tail length assay (MPAT)
The MPAT assay was performed as per (71). A universal linker DNA oligonucleotide 5’-phospho-ATG TGA GAT CAT GCA CAG TCA TA-3’-NH2 was ligated to the 3’ termini of total RNA (2.5 μg) by T4 RNA ligase (New England Biolabs) at 37 °C for 3h. The ligated RNA was subjected to phenol/chloroform extraction and ammonium acetate/ethanol precipitation and then amplified using one-step RT-PCR kit (QIAGEN) with anti-linker primer (5’ TAT GAC TGT GCA TGA TCT CAC AT 3’). A first round of PCR (35 cycles) was applied using a gene-specific upper primer and the anti-linker primer, followed by a second round of 10-cycle PCR using a gene-specific lower primer and the anti-linker primer. Half of the reaction product was resolved by 10% polyacrylamide gel electrophoresis in TBE buffer, dyed with SYBR green (Invitrogen) and visualized by PhosphorImage, and the remaining half was cloned in TOPO TA cloning vector (Invitrogen) and a large number of clones were subjected to Sanger sequencing.
Mitochondrial membrane lipid composition using LC-MS analysis
Procedures for lipid extraction and analysis were adapted as previously described (72,73). Briefly, lipids were extracted from isolated mitochondria (200 µg protein), which had been spiked with six internal standards. Samples (equivalent to 2.6 µg of protein extract) were injected onto a 1290 Infinity HPLC coupled with a 6530 accurate mass QTOF (Agilent, Santa Clara, USA) via a dual electrospray ion source. Lipids were eluted on a Zorbax Eclipse plus C18, 2.1 × 100 mm, 1.8µm (Agilent, Santa Clara, USA) kept at 40 °C with a gradient of 83 min and were analysed in both negative and positive scan mode. Each feature or entity characterized by a specific mass and retention time, were identified using Mass Hunter B.07.00 software (Agilent, Santa Clara, USA). A frequency filter of 100% was applied and signal intensities were normalized using Quantile algorithm with Mass Profiler Pro. software (MPP, Agilent, Santa Clara, USA). MS entities that discriminated the two conditions based on a false discovery rate of 5% and a fold change >2, were subjected to tandem mass spectrometry for lipid identification. Note that for cholesterol, identification was also confirmed by LC-MS analysis of the corresponding standard.
Statistical analyses
Unless indicated, data are presented as mean ± SEM. Unpaired student’s t-test or a one-way analysis of variance (ANOVA) followed by Bonferroni multiple comparison post hoc analysis was used to determine significant difference between two or among multiple groups, respectively. Statistically significant differences were considered for P < 0.05. All analyses were performed on Prism 6.0 for Mac OS-X. For lipidomics analysis, data were analyzed with Mass Profiler Professional (MPP) using unpaired student’s t-test followed by Benjamini Hochberg correction and statistical significance was set at a false discovery rate of 5%; data are depicted as a volcano plot.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
{kind=link}
Acknowledgements
We would like to thank Mélanie Brunette, François Brisebois, Nicolas Morin and Nicolas Sgarioto for technical assistance. This work was supported by the Canadian Institutes of Health Research (CIHR; Grant #102168 to the CIHR Emerging Team in Leigh Syndrome French Canadian (LSFC): Translating gene discovery into treatments for patients (awarded to CDR, YB, JDR, and EAS), the Lactic Acidosis Association (AAL) and Fondation du Grand Défi Pierre Lavoie. YB is a Ottawa University Chair in Integrative Mitochondrial Biology. JDR holds the Canada Research Chair in Genetics and Genomic Medicine. AC is the recipient of a graduate scholarship from Fondation du Grand Défi Pierre Lavoie.
Members of the LSFC consortium at the time this study was initiated were in alphabetical order: Bruc G. Allen, Claudine Beauchamp, Chantal Bemeur, Yan Burelle, Guy Charron, Lise Coderre, Alexanne Cuillerier, Christine Des Rosiers, Sonia Deschênes, François Labarthe, Jeanine Landry, Catherine Laprise, Geneviève Lavallée, Pierre Lavoie, Sylvie Lesage, Bruno Maranda, Charles Morin, Yvette Mukazena, John D. Rioux, Marie-Eve Rivard, Eric A. Shoubridge, Jessica Tardif, Julie Thopson-Legault, Nancy Tremblay, Vanessa Tremblay-Vaillancourt, Luc Vachon, and Josée Villeneuve.
Conflict of Interest statement. None declared.
Funding
Canadian Institutes of Health Research (CIHR; Grant #102168 to JDR, YB, EAS, CDR for the CIHR Emerging Team in Leigh Syndrome French Canadian (LSFC): Translating gene discovery into treatments for patients.
References
- 1. Leigh D. (1951) Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatr., 14, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Debray F.-G., Lambert M., Mitchell G.A. (2008) Disorders of mitochondrial function. Curr. Opin. Pediatr., 20, 471–482. [DOI] [PubMed] [Google Scholar]
- 3. Dimauro S., Schon E.A. (2008) Mitochondrial Disorders in the Nervous System. Annu. Rev. Neurosci., 31, 91–123. [DOI] [PubMed] [Google Scholar]
- 4. Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M.. et al. (1998) SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet., 20, 337–343. [DOI] [PubMed] [Google Scholar]
- 5. Rahman S., Blok R.B., Dahl H.H., Danks D.M., Kirby D.M., Chow C.W., Christodoulou J., Thorburn D.R. (1996) Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann. Neurol., 39, 343–351. [DOI] [PubMed] [Google Scholar]
- 6. Debray F.-G., Morin C., Janvier A., Villeneuve J., Maranda B., Laframboise R., Lacroix J., Decarie J.-C., Robitaille Y., Lambert M.. et al. (2011) LRPPRC mutations cause a phenotypically distinct form of Leigh syndrome with cytochrome c oxidase deficiency. J. Med. Genet., 48, 183–189. [DOI] [PubMed] [Google Scholar]
- 7. Morin C., Mitchell G., Larochelle J., Lambert M., Ogier H., Robinson B.H., De Braekeleer M. (1993) Clinical, metabolic, and genetic aspects of cytochrome C oxidase deficiency in Saguenay-Lac-Saint-Jean. Am. J. Hum. Genet., 53, 488–496. [PMC free article] [PubMed] [Google Scholar]
- 8. Merante F., Petrova-Benedict R., MacKay N., Mitchell G., Lambert M., Morin C., De Braekeleer M., Laframboise R., Gagné R., Robinson B.H. (1993) A biochemically distinct form of cytochrome oxidase (COX) deficiency in the Saguenay-Lac-Saint-Jean region of Quebec. Am. J. Hum. Genet., 53, 481–487. [PMC free article] [PubMed] [Google Scholar]
- 9. Sasarman F., Brunel-Guitton C., Antonicka H., Wai T., Shoubridge E.A.. LSFC Consortium (2010) LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol. Biol. Cell, 21, 1315–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruzzenente B., Metodiev M.D., Wredenberg A., Bratic A., Park C.B., Camara Y., Milenkovic D., Zickermann V., Wibom R., Hultenby K.. et al. (2011) LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. embo J., 31, 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mootha V.K., Lepage P., Miller K., Bunkenborg J., Reich M., Hjerrild M., Delmonte T., Villeneuve A., Sladek R., Xu F.. et al. (2003) Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl Acad. Sci. U S A, 100, 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sasarman F., Nishimura T., Antonicka H., Weraarpachai W., Shoubridge E.A.. LSFC Consortium (2015) Tissue-specific responses to the LRPPRC founder mutation in French Canadian Leigh Syndrome. Hum. Mol. Genet., 24, 480–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oláhová M., Hardy S.A., Hall J., Yarham J.W., Haack T.B., Wilson W.C., Alston C.L., He L., Aznauryan E., Brown R.M.. et al. (2015) LRPPRCmutations cause early-onset multisystem mitochondrial disease outside of the French-Canadian population. Brain, 138, 3503–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burelle Y., Bemeur C., Rivard M.-E., Thompson Legault J., Boucher G. LSFC Consortium Morin C., Coderre L., RosiersDes C. (2015) Mitochondrial vulnerability and increased susceptibility to nutrient-induced cytotoxicity in fibroblasts from leigh syndrome French canadian patients. PLoS One, 10, e0120767.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Legault J.T., Strittmatter L., Tardif J., Sharma R., Tremblay-Vaillancourt V., Aubut C., Boucher G., Clish C.B., Cyr D., Daneault C.. et al. (2015) A metabolic signature of mitochondrial dysfunction revealed through a monogenic form of Leigh syndrome. CellReports, 13, 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diaz F., Garcia S., Hernandez D., Regev A., Rebelo A., Oca-Cossio J., Moraes C.T. (2008) Pathophysiology and fate of hepatocytes in a mouse model of mitochondrial hepatopathies. Gut., 57, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bernardi P., Di Lisa F., Fogolari F., Lippe G. (2015) From ATP to PTP and back: a dual function for the mitochondrial ATP synthase. Circ. Res., 116, 1850–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giorgio V., von Stockum S., Antoniel M., Fabbro A., Fogolari F., Forte M., Glick G.D., Petronilli V., Zoratti M., Szabò I.. et al. (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl Acad. Sci. U S A, 110, 5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eliseev R.A., Filippov G., Velos J., VanWinkle B., Goldman A., Rosier R.N., Gunter T.E. (2007) Role of cyclophilin D in the resistance of brain mitochondria to the permeability transition. Neurobiol. Aging, 28, 1532–1542. [DOI] [PubMed] [Google Scholar]
- 20. Li B., Chauvin C., De Paulis D., De Oliveira F., Gharib A., Vial G., Lablanche S., Leverve X., Bernardi P., Ovize M.. et al. (2012) Inhibition of complex I regulates the mitochondrial permeability transition through a phosphate-sensitive inhibitory site masked by cyclophilin D. Biochim. Biophys. Acta Bioenerg., 1817, 1628–1634. [DOI] [PubMed] [Google Scholar]
- 21. Daussin F.N., Godin R., Ascah A., Deschenes S., Burelle Y. (2011) Cyclophilin-D is dispensable for atrophy and mitochondrial apoptotic signalling in denervated muscle. J. Physiol. (Lond.), 589, 855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verkaart S., Koopman W.J.H., van Emst-de Vries S.E., Nijtmans L.G.J., van den Heuvel L.W.P.J., Smeitink J.A.M., Willems P.H.G.M. (2007) Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim. Biophys. Acta, 1772, 373–381. [DOI] [PubMed] [Google Scholar]
- 23. Distelmaier F., Visch H.-J., Smeitink J.A.M., Mayatepek E., Koopman W.J.H., Willems P.H.G.M. (2009) The antioxidant Trolox restores mitochondrial membrane potential and Ca2+-stimulated ATP production in human complex I deficiency. J. Mol. Med., 87, 515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Starkov A.A., Fiskum G. (2003) Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem., 86, 1101–1107. [DOI] [PubMed] [Google Scholar]
- 25. Korshunov S.S., Skulachev V.P., Starkov A.A. (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett., 416, 15–18. [DOI] [PubMed] [Google Scholar]
- 26. Ikaga R., Namekata I., Kotiadis V.N., Ogawa H., Duchen M.R., Tanaka H., Iida-Tanaka N. (2015) Knockdown of aquaporin-8 induces mitochondrial dysfunction in 3T3-L1 cells. Biochem. Biophy. Rep., 4, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marchissio M.J., Francés D.E.A., Carnovale C.E., Marinelli R.A. (2012) Mitochondrial aquaporin-8 knockdown in human hepatoma HepG2 cells causes ROS-induced mitochondrial depolarization and loss of viability. Toxicol. Appl. Pharmacol., 264, 246–254. [DOI] [PubMed] [Google Scholar]
- 28. Pecina P., Capková M., Chowdhury S.K.R., Drahota Z., Dubot A., Vojtísková A., Hansíková H., Houst'ková H., Zeman J., Godinot C.. et al. (2003) Functional alteration of cytochrome c oxidase by SURF1 mutations in Leigh syndrome. Biochim. Biophys. Acta Mol. Bas. Dis., 1639, 53–63. [DOI] [PubMed] [Google Scholar]
- 29. Baker C.D., Basu Ball W., Pryce E.N., Gohil V.M. (2016) Specific requirements of nonbilayer phospholipids in mitochondrial respiratory chain function and formation. Mol. Biol. Cell, 27, 2161–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Böttinger L., Horvath S.E., Kleinschroth T., Hunte C., Daum G., Pfanner N., Becker T. (2012) Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratorychain supercomplexes. J. Mol. Biol., 423, 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shinzawa-Itoh K., Aoyama H., Muramoto K., Terada H., Kurauchi T., Tadehara Y., Yamasaki A., Sugimura T., Kurono S., Tsujimoto K.. et al. (2007) Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. embo J., 26, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mourier A., Ruzzenente B., Brandt T., Kuhlbrandt W., Larsson N.G. (2014) Loss of LRPPRC causes ATP synthase deficiency. Hum. Mol. Genet., 10.1093/hmg/ddt652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spurway T.D., Sherratt H.A., Pogson C.I., Agius L. (1997) The flux control coefficient of carnitine palmitoyltransferase I on palmitate beta-oxidation in rat hepatocyte cultures. Biochem. J., 323 (Pt 1), 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fritzen A.J., Grunnet N., Quistorff B. (2007) Flux control analysis of mitochondrial oxidative phosphorylation in rat skeletal muscle: pyruvate and palmitoyl-carnitine as substrates give different control patterns. Eur. J. Appl. Physiol., 101, 679–689. [DOI] [PubMed] [Google Scholar]
- 35. Lei S., Sun R.-Z., Wang D., Gong M.-Z., Su X.-P., Yi F., Peng Z.-W. (2016) Increased hepatic fatty acids uptake and oxidation by LRPPRC-driven oxidative phosphorylation reduces blood lipid levels. Front. Physiol., 7, 287.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu L., Sanosaka M., Lei S., Bestwick M.L., Frey J.H., Surovtseva Y.V., Shadel G.S., Cooper M.P. (2011) LRP130 protein remodels mitochondria and stimulates fatty acid oxidation. J. Biol. Chem., 286, 41253–41264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kwong J.Q., Molkentin J.D. (2015) Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab., 21, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Woodfield K., Brdiczka D., Halestrap A. (1998) Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J., 336 (Pt 2), 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szabò I., Zoratti M. (1993) The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Lett., 330, 201–205. [DOI] [PubMed] [Google Scholar]
- 40. Leung A., Varanyuwatana P., Halestrap A. (2008) The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem., 283, 26312–26323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carraro M., Giorgio V., Šileikytė J., Sartori G., Forte M., Lippe G., Zoratti M., Szabò I., Bernardi P. (2014) Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J. Biol. Chem., 289, 15980–15985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alavian K.N., Beutner G., Lazrove E., Sacchetti S., Park H.A., Licznerski P., Li H., Nabili P., Hockensmith K., Graham M.. et al. (2014) An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl Acad. Sci. U S A, 111, 10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Solesio M.E., Elustondo P.A., Zakharian E., Pavlov E.V. (2016) Inorganic polyphosphate (polyP) as an activator and structural component of the mitochondrial permeability transition pore. Biochem. Soc. Trans., 44, 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bernardi P. (2013) The mitochondrial permeability transition pore: a mystery solved? Front. Physiol., 10, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spannagel C., Vaillier J., Arselin G., Graves P.V., Grandier-Vazeille X., Velours J. (1998) Evidence of a subunit 4 (subunit b) dimer in favor of the proximity of ATP synthase complexes in yeast inner mitochondrial membrane. Biochim. Biophys. Acta, 1414, 260–264. [DOI] [PubMed] [Google Scholar]
- 46. Everard-Gigot V., Dunn C.D., Dolan B.M., Brunner S., Jensen R.E., Stuart R.A. (2005) Functional analysis of subunit e of the F1Fo-ATP synthase of the yeast Saccharomyces cerevisiae: importance of the N-terminal membrane anchor region. Eukaryotic Cell, 4, 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bisetto E., Picotti P., Giorgio V., Alverdi V., Mavelli I., Lippe G. (2008) Functional and stoichiometric analysis of subunit e in bovine heart mitochondrial F0F1ATP synthase. J. Bioenerg. Biomembr., 40, 257–267. [DOI] [PubMed] [Google Scholar]
- 48. Bustos D.M. (2005) The modification of the conserved GXXXG motif of the membrane-spanning segment of subunit g destabilizes the supramolecular species of yeast ATP synthase. J. Biol. Chem., 280, 29004–29010. [DOI] [PubMed] [Google Scholar]
- 49. Davies K.M., Strauss M., Daum B., Kief J.H., Osiewacz H.D., Rycovska A., Zickermann V., Kühlbrandt W. (2011) Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl Acad. Sci. U S A, 108, 14121–14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Davies K.M., Anselmi C., Wittig I., Faraldo-Gómez J.D., Kühlbrandt W. (2012) Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci U S A, 109, 13602–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Strauss M., Hofhaus G., Schröder R.R., Kühlbrandt W. (2008) Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. embo J., 27, 1154–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. He J., Ford H.C., Carroll J., Ding S., Fearnley I.M., Walker J.E. (2017) Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl Acad. Sci. U S A, 114, 3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wittig I., Meyer B., Heide H., Steger M., Bleier L., Wumaier Z., Karas M., Schägger H. (2010) Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochim. Biophys. Acta., 1797, 1004–1011. [DOI] [PubMed] [Google Scholar]
- 54. Giorgio V., Bisetto E., Soriano M.E., Dabbeni-Sala F., Basso E., Petronilli V., Forte M.A., Bernardi P., Lippe G. (2009) Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem., 284, 33982–33988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee C.F., Chavez J.D., Garcia-Menendez L., Choi Y., Roe N.D., Chiao Y.A., Edgar J.S., Goo Y.A., Goodlett D.R., Bruce J.E.. et al. (2016) Normalization of NAD+ redox balance as a therapy for heart failure. Circulation, 134, 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Horvath S.E., Daum G. (2013) Lipids of mitochondria. Prog. Lipid Res., 52, 590–614. [DOI] [PubMed] [Google Scholar]
- 57. Bigay J., Antonny B. (2012) Curvature, lipid packing, and electrostatics of membrane organelles: defining cellular territories in determining specificity. Developmental Cell, 23, 886–895. [DOI] [PubMed] [Google Scholar]
- 58. Holthuis J.C.M., Menon A.K. (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature, 510, 48–57. [DOI] [PubMed] [Google Scholar]
- 59. Sousa-Lopes A., Antunes F., Cyrne L., Marinho H.S. (2004) Decreased cellular permeability to H2O2 protects Saccharomyces cerevisiae cells in stationary phase against oxidative stress. FEBS Lett., 578, 152–156. [DOI] [PubMed] [Google Scholar]
- 60. Folmer V., Pedroso N., Matias A.C., Lopes S.C.D.N., Antunes F., Cyrne L., Marinho H.S. (2008) H2O2 induces rapid biophysical and permeability changes in the plasma membrane of Saccharomyces cerevisiae. Biochim. Biophys. Acta, 1778, 1141–1147. [DOI] [PubMed] [Google Scholar]
- 61. Branco M.R. (2003) Decrease of H2O2 Plasma membrane permeability during adaptation to H2O2 in Saccharomyces cerevisiae. J. Biol. Chem., 279, 6501–6506. [DOI] [PubMed] [Google Scholar]
- 62. Dumas D., Muller S., Gouin F., Baros F., Viriot M.L., Stoltz J.F. (1997) Membrane fluidity and oxygen diffusion in cholesterol-enriched erythrocyte membrane. Arch. Biochem. Biophys., 341, 34–39. [DOI] [PubMed] [Google Scholar]
- 63. Strott C.A. (2003) Cholesterol sulfate in human physiology: what's it all about? J. Lipid Res., 44, 1268–1278. [DOI] [PubMed] [Google Scholar]
- 64. Picard M., Azuelos I., Jung B., Giordano C., Matecki S., Hussain S.N.A., White K., Li T., Liang F., Benedetti A.. et al. (2015) Mechanical ventilation triggers abnormal mitochondrial dynamics and morphology in the diaphragm. J. Appl. Physiol., 118, 1161–1171. 10.1152/japplphysiol.00873.2014. [DOI] [PubMed] [Google Scholar]
- 65. Cadete V.J.J., Deschenes S., Cuillerier A., Brisebois F., Sugiura A., Vincent A., Turnbull D., Picard M., McBride H.M., Burelle Y. (2016) Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. (Lond.), 594, 5343–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Marcil M., Ascah A., Matas J., Belanger S., Deschepper C.F., Burelle Y. (2006) Compensated volume overload increases the vulnerability of heart mitochondria without affecting their functions in the absence of stress. J. Mol. Cell Cardiol., 41, 998–1009. [DOI] [PubMed] [Google Scholar]
- 67. Matas J., Young N., Bourcier-Lucas C., Ascah A., Marcil M., Deschepper C.F., Burelle Y. (2009) Increased expression and intramitochondrial translocation of cyclophilin-D associates with increased vulnerability of the permeability transition pore to stress-induced opening during compensated ventricular hypertrophy. J. Mol. Cell Cardiol., 46, 420–430. [DOI] [PubMed] [Google Scholar]
- 68. Emaus R.K., Grunwald R., Lemasters J.J. (1986) Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim. Biophys. Acta, 850, 436–448. [DOI] [PubMed] [Google Scholar]
- 69. Godin R., Daussin F., Matecki S., Li T., Petrof B.J., Burelle Y. (2012) Peroxisome proliferator-activated receptor coactivator 1- gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. J. Physiol. (Lond.), 590, 5487–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pon L.A., Schon E.A. (2017) Mitochondria 2nd edn. Academic Press, 944 p, ISBN-13: 978-0125441735.
- 71. Temperley R.J., Seneca S.H., Tonska K., Bartnik E., Bindoff L.A., Lightowlers R.N., Chrzanowska-Lightowlers Z.M.A. (2003) Investigation of a pathogenic mtDNA microdeletion reveals a translation-dependent deadenylation decay pathway in human mitochondria. Hum. Mol. Genet., 12, 2341–2348. [DOI] [PubMed] [Google Scholar]
- 72. Matyash V., Liebisch G., Kurzchalia T.V., Shevchenko A., Schwudke D. (2008) Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res., 49, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sandra K., Pereira Ados S., Vanhoenacker G., David F., Sandra P. (2010) Comprehensive blood plasma lipidomics by liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr A., 1217, 4087–4099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.