

Abstract
X-linked adrenoleukodystrophy (ALD) is a devastating inherited neurodegenerative disease caused by defects in the ABCD1 gene and affecting peripheral and central nervous system myelin. ABCD1 encodes a peroxisomal transmembrane protein required for very long chain fatty acid (VLCFA) metabolism. We show that zebrafish (Danio rerio) Abcd1 is highly conserved at the amino acid level with human ABCD1, and during development is expressed in homologous regions including the central nervous system and adrenal glands. We used TALENs to generate five zebrafish abcd1 mutant allele lines introducing premature stop codons in exon 1, as well as obtained an abcd1 allele from the Zebrafish Mutation Project carrying a point mutation in a splice donor site. Similar to patients with ALD, zebrafish abcd1 mutants have elevated VLCFA levels. Interestingly, we found that CNS development of the abcd1 mutants is disrupted, with hypomyelination in the spinal cord, abnormal patterning and decreased numbers of oligodendrocytes, and increased cell death. By day of life five abcd1 mutants demonstrate impaired motor function, and overall survival to adulthood of heterozygous and homozygous mutants is decreased. Expression of human ABCD1 in oligodendrocytes rescued apoptosis in the abcd1 mutant. In summary, we have established a zebrafish model of ALD that recapitulates key features of human disease pathology and which reveals novel features of underlying disease pathogenesis.
Introduction
Inherited leukodystrophies, a group of diseases impacting nervous system myelin, affect 1 in 7,500 live births and have a >30% mortality by age 8 years (1). Most leukodystrophies lack treatment, and their orphan status, relative rarity as individual diseases, and incomplete penetrance of phenotypes in animal models, have limited progress in understanding disease pathophysiology and in therapy development. X-linked adrenoleukodystrophy (ALD) is one of the first leukodystrophies in which the genetic defect was identified (2). ALD is caused by mutations of the X-linked ABCD1 gene, a peroxisomal transmembrane protein, which encodes an ATP-binding cassette transporter protein (3). One of the most unusual and poorly understood features of ALD is the phenotypic heterogeneity. The same mutation in ABCD1, even in siblings, can result in disease phenotypes ranging from unaffected, to endocrine-only (adrenal) disease, to lethal nervous system involvement. The most severe phenotype is a rapidly progressive demyelinating disease affecting school-aged boys (cerebral ALD); but the most common phenotype consists of a slowly progressive central and peripheral demyelination that affects males (4), and to a lesser extent female carriers, known as adrenomyeloneuropathy (AMN).
ALD is characterized by elevated levels and accumulation of very long chain fatty acids (VLCFAs) in serum and tissues. However, reduction of VLCFA levels does not improve outcomes (5). Mouse knock-outs of Abcd1 show a gradual elevation of VLCFAs and the slow progression of an AMN-like peripheral neuropathy over more than one year (6–8). Interestingly, the mouse mutants do not develop the rapidly progressive cerebral demyelination (9). ABCD1 also has other roles that may contribute to its disease effects. Studies from mouse and cell culture models show that loss of ABCD1 causes dysfunction of the inflammatory cascade including altered antioxidant signaling and abnormal endothelial function (10,11), and loss of ABCD1 is associated with a pro-inflammatory state (12,13). In addition, ABCD1 mutations are a rare cause of isolated primary adrenal insufficiency (14), and adrenal insufficiency is a known complication of ALD. However, whether ABCD1 has a developmental role that might predispose to subsequent disease progression has not been clear.
We report the development and characterization of a zebrafish (Danio rerio) model of ALD. Using transcription activator-like effector nucleases (TALENs) we generated five mutant alleles for abcd1, and we identified, validated, and characterized a sixth mutant allele from the Zebrafish Mutation Project (15). We show that the mutant abcd1 zebrafish have elevated VLCFA levels; and develop a motor impairment within the first week of life. The mutants have altered CNS development including reduced numbers of oligodendrocytes with altered patterning, hypomyelination, and increased apoptosis in the CNS. Our work reveals an unexpected role for abcd1 in early development, provides a small vertebrate model with early nervous system dysfunction useful for screening therapeutics, and suggests a developmental predisposition for the later progression of disease.
Results
Zebrafish Abcd1 shows amino acid conservation with human ABCD1 and is expressed in the nervous system and adrenal glands
We characterized the ABCD1 ortholog in zebrafish, abcd1 (ENSDARG00000074876). abcd1 is autosomal and is located on chromosome 8; its open-reading frame is 2341 base-pairs in length with a predicted protein size of 766 amino acids (Fig. 1A). Zebrafish Abcd1 has 70% amino acid identity with human and mouse ABCD1 with extensive conservation of key protein domains (Fig. 1B); most of the divergence is at the N- and C- termini (Fig. 1C). Zebrafish Abcd1 is the ortholog in the zebrafish genome most similar to mammalian ABCD1; by contrast, zebrafish Abcd2 is more similar to ABCD2 in mouse or human than to zebrafish Abcd1 (Fig. 1D).
Figure 1.

Overview of zebrafish Abcd1 protein. (A) Domain structure and size of Abcd1. (B) Cross-species amino acid conservation matrix (percentages) compared using the Clustal Omega method, species names are indicated; zebrafish (Danio rerio) is top row and has 70% amino acid identity with mouse and human ABCD1. (C) Zebrafish Abcd1 protein is highly conserved compared to human protein, except for the N- and C-termini. Degree of amino acid similarity is indicated by color, with red being identical, and blue being non-conserved, as visualized using PRALINE. (D) Phylogenetic cladogram of evolutionary relationships of Abcd1 and Abcd2 proteins between zebrafish and other species. Scale bar equals an evolutionary distance of 0.1 amino acid changes per position in the sequence (Phylodendron).
We evaluated the abcd1 expression during embryonic development. abcd1 expression is not visible until 48 hpf (hours post-fertilization), at which time expression is visible in the nervous system, floor plate, and pronephros/interrenal gland (the zebrafish homolog of the adrenal glands (16)) (Fig. 2A). By 72 hpf and persisting through 96 hpf expression is present in the central nervous system (CNS), floorplate, interrenal glands/pronephros, and gut. Double-labeling for abcd1 in Tg(olig2:dsRed) embryos revealed co-localization of abcd1 expression with olig2:dsRed-expressing cells in the brain and spinal cord (Fig. 2B and C), consistent with expression of abcd1 in progenitors of motor neurons and oligodendrocytes (17). abcd1 appears to be expressed in oligodendrocyte precursors, shown by visualizing co-expression of abcd1 with RFP in the spinal cord of Tg(sox10:mRFP) embryos (18) (Fig. 2D and E). Thus, our results suggest that abcd1 is expressed in oligodendrocyte precursors and in the interrenal gland.
Figure 2.
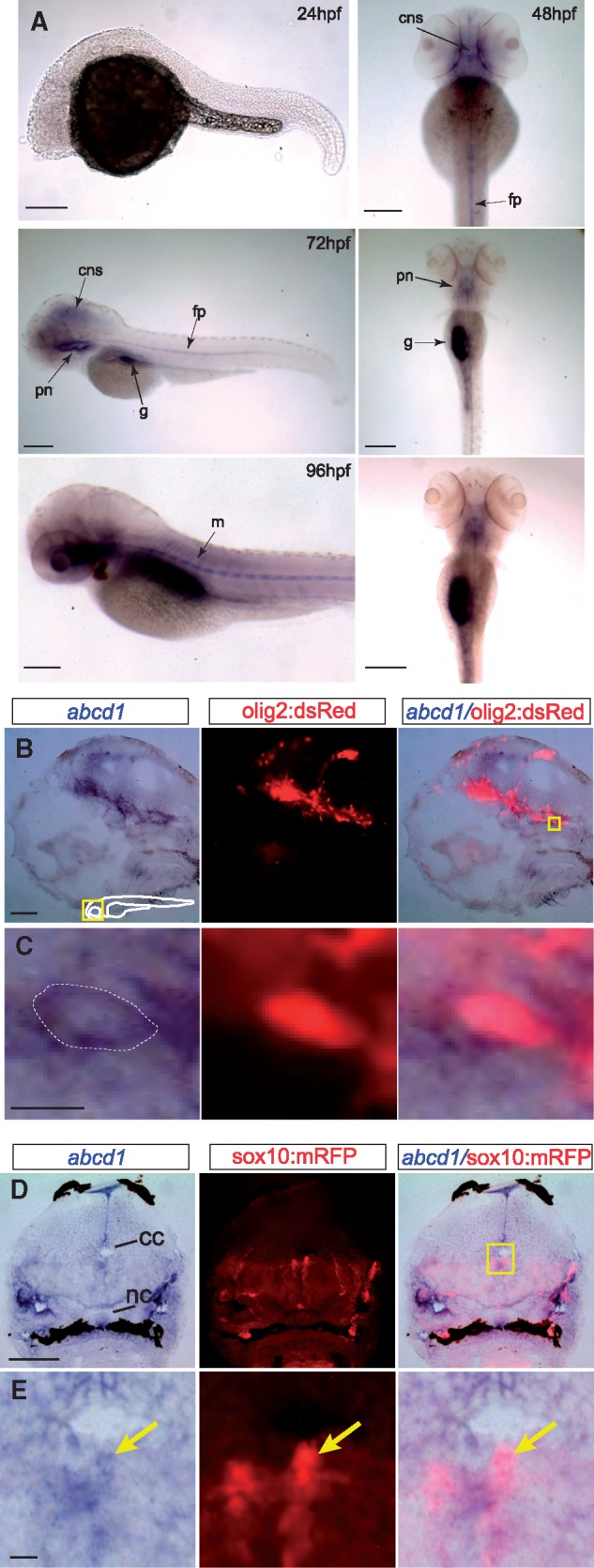
(A) Bright-field images of whole-mount embryos, in situs for abcd1, ages 24-96 hpf. At 24 hpf no abcd1 expression is seen. By 48 hpf expression is seen in the CNS and floorplate, and from 72-96 hpf additional expression becomes visible in the muscles, pronephros/adrenal glands, and gut. Left panel’s lateral views, dorsal to the top, rostral to the left; right panels, dorsal view, rostral to the top; scale bar 20 µm; cns, central nervous system; fp, floorplate; g, gut; m, muscles; pn, pronephros. (B,C) Section of 72 hpf Tg(olig2:dsRed) embryo double-stained for abcd1 in situ and α-dsRed. abcd1 is expressed in dsRed-labeled oligodendrocyte precursors in the brain. Area shown at higher resolution in C is the region boxed in far-right panel of B. Dotted line in C indicates the approximate boundary of cell membrane. Sagittal view in midline, rostral to the left, dorsal to top, scale bar 5 μm in B; 1 μm in C. (D,E) Cross-section of 72 hpf Tg(sox10:mRFP) embryo double-stained for abcd1 in situ and α-RFP in the spinal cord, dorsal to the top. abcd1 is expressed in dedicated oligodendrocyte precursors (arrow). Scale bar 10 μm in D, 1 μm in E; abbreviations: cc, central canal; nc, notochord.
TALEN generation of zebrafish mutant alleles and splice mutant allele
We generated zebrafish mutant alleles for abcd1. To do this we targeted a pair of transcription activator-like effector nuclease (TALEN) arms against sequence in exon 1 of abcd1. We also requested and obtained an abcd1 allele line from the Zebrafish Mutation Project (Fig. 3A and B) (15). Following TALEN mutagenesis, individual G0 fish were outcrossed to wild-type animals and offspring were screened by high-resolution melt analysis (HRMA) PCR (19). In addition to HRMA PCR genotyping, we confirmed genotypes by Sanger sequencing both in the G0 as well as in subsequent generations. We identified and isolated five distinct allele lines. The five alleles have mutations in exon 1 causing missense nucleotide changes with resultant amino acid changes, and resulting in premature stop codons occurring within 20 amino acids, all located within exon 1 (Fig. 3B). The abcd1 allele line abcd1sa509 obtained from the Zebrafish Mutation Project (referred to subsequently as sa509) is a point mutation (G > A) in the splice donor site of exon 9. Semi-quantitative RT-PCR showed that sa509 heterozygotes had decreased amount of abcd1 transcript, and that sa509 homozygotes had no detectable abcd1 transcript (Fig. 3C). In addition, PCR across the exon 9/10 boundary showed no evidence for the generation of alternative splice-forms (Fig. 3C). qRT-PCR for abcd1 in the zc90 TALEN mutant (abcd1zc90) revealed no change in transcript levels (Fig. 3D). Log2 fold changes were calculated relative to wild-type, using 28S rRNA as a standard, SEM for each in parenthesis, with two-way ANOVA with post-hoc Tukey HSD test performed. For +/+; zc90/+; and zc90/zc90 the log2 fold changes were 1.0 (0.27), 0.81 (0.19), and 1.22 (0.27). Overall morphology of the TALEN and sa509 heterozygous and homozygous animals appeared normal (images not shown). Both homozygous and heterozygous adults were generated from heterozygous in- crosses (see Fig. 7 for quantification of survival rates).
Figure 3.

(A) Sequence of TALEN arms for targeting of exon 1 in abcd1. (B) Schematic of zebrafish abcd1 exon/intron structure, and location of TALEN mutants and splice acceptor mutant (sa509, "sa"). TALEN mutant allele predicted amino acid sequences shown; and sa509 sequence change shown. (C) Semi-quantitative RT-PCR of wild-type compared to heterozygous and homozygous sa509 alleles. Arrowhead points to trace amount of PCR amplicon in heterozygote. (D) qRT-PCR shows no significant change in abcd1 transcript levels in zc90 allele. Error bars, SEM; ns, not significant. (E) Very-long chain fatty acid levels (VLCFAs) in sa509 heterozygotes and homozygotes, normalized to wild-type animals. * P < 0.05; ** P < 0.001, 2-way ANOVA.
Figure 7.

(A–F) abcd1sa509 allele. A, B) Kaplan-Meier survival curves for sa509 allele. Shown is composite data for three separate experimental replicates, n > 25 animals for each genotype for each replicate. 95% confidence intervals indicated by dotted lines. (A) Non-competitive survival analysis, in which each genotype (wild-type, homozygote, or heterozygote) was raised separately. (B) Competitive survival analysis, in which the three genotypes were raised together. There was a significant increase in mortality in sa509 mutants compared to wild-type (**, Mantel-Cox test P < 0.0001). (C) Activity traces show visualization of spontaneous behavior over 2’; with 6 dpf larvae in wild-type compared to heterozygous and homozygous sa509 mutants. (D) Scatter-plot comparison of spontaneous behavior response in wild-type, heterozygous, and homozygous sa509 6 dpf larvae. Each dot represents a separate animal; horizontal lines indicate the mean and standard error of the mean. * ANOVA, P = 0.19. (E) Activity traces show visualization of 1’ of swimming behavior after light flash; with 6 dpf larvae in wild-type compared to heterozygous and homozygous sa509 mutants. (F) Scatter-plot comparison of evoked behavior response in wild-type, heterozygous, and homozygous sa509 6 dpf larvae. Each dot represents a separate animal; horizontal lines indicate the mean and SEM. * ANOVA, P = 0.021. G-I: abcd1zc90 TALEN allele. (G) Competitive survival analysis for zc90. (H) Scatter-plot comparison of spontaneous behavior response (total distance moved) in wild-type, heterozygous, and homozygous zc90 6 dpf larvae. Each dot represents a separate animal; horizontal lines indicate the mean and standard error of the mean. ANOVA, P = 0.32. (I) Scatter-plot comparison of evoked behavior response (total distance swum) in wild-type, heterozygous, and homozygous zc90 6 dpf larvae. Each dot represents a separate animal; horizontal lines indicate the mean and SEM. ANOVA, P = 0.14.
Very-long chain fatty acid (VLCFA) levels are elevated in the mutant
Elevated very-long chain fatty acid (VLCFA) levels in tissues and serum are characteristic of human ABCD1 patients and carriers; in particular, elevations of C26:0 fatty acids. We tested VLCFA levels in sa509 heterozygotes and homozygotes using whole animal extracts performed at 7–8 days post-fertilization (dpf) (n = 20 embryos per replicate; 4 or more replicates performed for each genotype) (Fig. 3E). Compared to wild-type animals, sa509 heterozygotes and homozygotes had elevated C26:0 ratios (ratio (normalized to wild-type) 1.4 and 1.9, respectively; standard deviation (SD) 0.14 and 0.27, 2-way ANOVA P < 0.05 and 0.001); and sa509 homozygotes had an elevated C24:0 ratio (normalized ratio 1.2, SD 0.29, 2-way ANOVA P < 0.005). This result indicates that the zebrafish abcd1 mutant phenocopies a key biochemical marker of human ALD.
Abnormal interrenal organ development in abcd1 mutants
Adrenocortical insufficiency is frequently observed in patients with ALD (20). We assayed development of the interrenal organ, the teleost equivalent to the tetrapod adrenal cortex (16), using visualization of steroidogenic acute regulatory protein (StAR) transcript expression (21). We found that while the interrenal organ was established with the same developmental timing in sa509 homozygotes as in wild-types, the size of the interrenal organ was ∼ 1.5x larger in wild-type animals (Fig. 4A–C) (72 hpf, wt vs. sa509 homozygotes, 49,147 vs. 32,157 pixels; SD 13,411 vs. 11,402, P < 0.0001; n = 20 animals each genotype). To test whether the function of the interrenal organ was affected, we measured cortisol levels in larvae and in adults. We found no significant differences in larval or adult cortisol levels by ANOVA including a post-hoc Tukey’s HSD (Fig. 4D and E) (larval, wt vs. sa509/+ vs. sa509/sa509, 1.2, 1.3, 1.5 pg cortisol/larva; SD 0.65, 0.50, 0.23, F2,6 = 1.09, P = 0.37; adults, wt vs. sa509/+ vs. sa509/sa509, 19.9, 16.6, 23.4 ng cortisol/g body weight; SD 7.2, 7.0, 4.8, F2,10 = 0.14, P = 0.87). Our results suggest that abcd1 is involved in adrenocortical development.
Figure 4.

Adrenal gland development is altered in abcd1 mutants (sa509/sa509). (A, B) Bright-field images of whole-mount embryos at 72 hpf, in situs for StAR labeling of interrenal gland. Lateral views, rostral to the left, dorsal top, scale bar 20 μm. (C) Area of the interrenal gland is decreased, area measured is shown in dotted yellow lines in A; box plot, y-axis pixels, P < 0.05. Cortisol levels are not significantly changed in larvae and adults. (D) Larval cortisol levels; E) Adult cortisol levels.
Altered myelin development in abcd1 mutants
To determine whether mutation of abcd1 affected development of the myelin, we analyzed major transcripts involved in myelin development. We performed quantitative reverse-transcription polymerase chain reaction (qRT-PCR) at 72 hpf in experimental triplicates, with n > 10 larvae in each group (Fig. 5A). Transcript levels of myelin basic protein (mbp), a protein component of the mature myelin sheath produced by oligodendrocytes, and of myelin protein zero (mpz), an oligodendrocyte-produced protein component of CNS compact myelin (22), were not significantly changed. However, levels of proteolipid protein 1a (plp1a), a major protein component of compact myelin, were significantly reduced in mutants (P < 0.05). The relative log2 fold change were calculated relative to wild-type, using 28S rRNA standard, SEM for each in parenthesis, with p values adjusted using Benjamini-Hochberg False Discovery Rate. The four genotypes were sa509/+; sa509/sa509; zc90/+; and zc90/zc90. For mbp, the log2 fold changes were 0.15 (0.33), 0.40 (0.30), 0.15 (0.41), and 0.33 (0.43). For mpz, the log2 fold changes were 0.03 (0.24), 0.07 (0.39), 0.04 (0.33), and 0.07 (0.27). For plp1a, the log2 fold changes were 0.01 (0.37), 0.01 (0.42), 0.01 (0.40), and 0.01 (0.44). These data suggest that myelin generation is impaired in abcd1 mutants.
Figure 5.

Decreased myelin protein transcripts and hypomyelination in abcd1 mutants. (A) qRT-PCR shows significant decrease in plp1a transcript levels. Error bars, SEM; * P < 0.05. (B) Schematic representation of a 5 dpf larval zebrafish; spinal cords were cut between segments 5 and 6 (red dashed line) and prepared for TEM. Axis shows the orientation of the embryo (A, anterior; D, dorsal). (C) Cross-section diagram of a 5 dpf zebrafish spinal cord, dorsal is up. The spinal cord is orange; ventral region used for quantification is boxed in red; notochord (n) in green. (D, E) Representative TEM images from the ventral spinal cord of wt and sa509/sa509 mutant larvae at 5 dpf. Myelinated axons are shaded green. (F) Quantification of the percent of myelinated axons in the ventral spinal cord of sa509/sa509 mutants compared with wt controls; ** P < 0.01.
We used transmission electron microscopy (TEM) to quantify the extent of myelination in the ventral spinal cord at 5 dpf (Fig. 5B and C). We found that the percentage of myelinated axons was reduced (wt vs. sa509 homozygotes, 8.8% vs. 3.8%, SD 4.2% vs. 3.1%, P = 0.0078; n = 4 animals each genotype) (Fig. 5D–F). These results show a developmental hypomyelination in abcd1 mutants.
abcd1 mutants have impaired oligodendrocyte patterning caused by elevated apoptosis
To determine the cause of the changes in early myelin development, we studied whether there were changes in oligodendrogliogenesis. Oligodendrocyte precursor cells (OPCs) can give rise to myelin-producing oligodendroglia (23,24). We used the transgenic line Tg(olig2:dsRed) to label OPCs and oligodendrocytes (25) in wild-type compared to heterozygous sa509 mutants. We noted a difference in the pattern and number of dsRed-expressing cells in the mutant brains (Fig. 6A and B; arrows). To quantify the change, we counted the number of dsRed+ cells in the spinal cord (Fig. 6C and D). We found a significant decrease in the total number of dsRed+ cell counts in the spinal cord of mutants (Fig. 6E) (sa509, wild-type versus heterozygous versus homozygous, n = 12, 15, 7 embryos each, average number of cells, 55, 41, and 38, SEM 4.3, 4.0, and 4.1, one-way ANOVA F2, 39 = 4.2 P = 0.023; post-hoc Tukey’s HSD, wild-type versus heterozygous p= 0.015; P = 0.007 for wild-type versus homozygous).
Figure 6.

Abnormal OPC development. Confocal z-stack projections, 72 hpf, scale bar 50 μm. (A–D) Representative images of Tg(olig2:dsRed) embryos, immunohistochemistry for α-dsRed, shows decreased number of olig2+ cells in the brain and spinal cord and altered patterning (arrows), quantified in (E) * P < 0.05. A-B, dorsal views of the brain, rostral to the top; (C,D), lateral views of spinal cord, rostral to the left, dorsal to the top. F) TUNEL counts in the brain, area of quantification shown in G). At 72 hpf there was a significant increase in TUNEL counts in sa509 mutants, ** P < 0.001. (G, H) Confocal images of TUNEL labeling, lateral views of the head, rostral to the left, dorsal to top. Yellow box indicates region for quantification. I) In sa509 mutants there was no significant co-localization of TUNEL+ labeling in dsRed (olig2+) cells at 72 hpf.
Because we observed this change in the amount and in the distribution of olig2:dsRed labeling, we tested whether there was an increase in apoptosis. We observed a significant increase in apoptosis in the brains of sa509 but not zc90 mutants at 72 hpf: sa509, wt versus heterozygous versus homozygous, cell counts 11, 27, 52, SEM 1.2, 1.4, 4.2, F2,33 = 112.7, post-hoc Tukey’s HSD P < 0.001; zc90, wt versus heterozygous versus homozygous, 19, 18, 21, SEM 2.0, 2.6, 4.3, ANOVA F2,55 = 1.48, P > 0.05 (Fig. 6F–H). To determine if OPCs were particularly vulnerable to apoptosis, we determined whether the percentage of TUNEL-positive olig2-positive cells was increased at 72 hpf. We found that there was not a difference in percentages of apoptosis in olig2-positive cells at 72 hpf in sa509, wild-type versus heterozygous versus homozygous, n = 14, 13, 7 embryos each, average percentage 2.8, 3.5, 2.3, SEM 0.2, 0.4, and 0.3, F2, 31 = 2.8 P = 0.077. Taken together, our findings show that early OPC patterning and numbers are affected by mutation in abcd1.
abcd1 mutants demonstrate reduced survival and impaired motor swimming response
Although the abcd1 TALEN and sa509 alleles generated both heterozygous and homozygous animals surviving to adulthood, we noted a reduction in expected numbers of sa509 animals. We found that when different genotypes (wild-type, heterozygotes, homozygotes) were reared separately that there was no difference in overall survival for sa509 (Fig. 7A). However, when all three genotypes were raised together, heterozygous and homozygous animals died at statistically significant increased rates compared to wild-types (Mantel-Cox log rank test, P < 0.0001) (Fig. 7B), suggesting a potential competitive disadvantage in the mutants. There was no statistically significant difference in survival between heterozygous and homozygous sa509 mutants. The zc90 allele did not have a significant difference in survival between wild-type, heterozygotes, and homozygotes (Fig. 7G).
We next tested whether there was a functional effect on motor behavior in the sa509 mutants that might contribute to the competitive survival disadvantage (Fig. 7C–F). First, we analyzed motor behavior in larvae at 6 dpf. Animals were adapted for 30’, and then recorded for 2’. For evoked behavior, animals were similarly adapted, and then a single white light flash was provided for stimulus and behavior was recorded for 1’. For spontaneous swimming there was no significant difference (wt, heterozygous, homozygous; total distance 235, 177, 190 mm; standard error of the mean (SEM) 25, 22, 24; n = 23, 22, 19 larvae; ANOVA P = 0.19) (Fig. 7C and D). Evoked behavior was significantly different in heterozygous and homozygous animals: total distance swam was an average of 166 mm in wild-type larvae compared to 137 mm and 119 mm in heterozygotes and in homozygotes (SEM 66, 45, and 58; n = 24, 20, and 23 larvae; ANOVA p 0.021). Post-hoc analysis with Tukey’s HSD test was significant between wild-type and homozygous mutants (P = 0.018) but not for any other comparisons. Evoked behavior thus showed a greater difference in response between the different genotypes (Fig. 7E and F) than spontaneous behavior. Other parameters did not have statistically significant differences: percentage of time spent moving (wt, homozygous; percentage 3.5, 4.6; SEM 0.6; ANOVA P = 0.23); and high-velocity percent of time (wt, homozygous; percentage 3.4, 4.4; SEM 0.8; ANOVA P = 0.37). Thus, we found that both heterozygous and homozygous sa509 animals had impaired motor swimming at 6 dpf.
We also tested the motor behavior of zc90 mutants (Fig. 7H and I). We analyzed motor behavior in larvae at 6 dpf using the same measures of spontaneous and evoked behavior. For spontaneous swimming there was no significant difference in time spent moving, high-velocity percent of time, or total distance swum (Fig. 7H) (for total distance swum: wt, heterozygous, homozygous; mean total distance 310, 286, 225 mm; SEM 37, 27, 35; n = 18, 33, 11 larvae; ANOVA P = 0.32). Evoked behavior was significantly different for time spent moving (ANOVA P = 0.04; post-hoc Tukey’s HSD P = 0.048 for wt vs heterozygous). However, other parameters were not significant, including high-velocity percent of time (data not shown) and total distance swum (Fig. 7I) (wt, heterozygous, homozygous; mean total distance 241, 180, 161 mm; SEM 37, 17, 28; ANOVA P = 0.14). Our data thus demonstrate that impairments in movement are a consistent feature of the zebrafish abcd1 mutants across different alleles.
We then tested adult motor behavior. We analyzed adult sa509 homozygous mutants to determine if a motor deficit was present as compared to wild-type siblings. In analysis of spontaneous swimming (n = 11 each genotype), we found significant differences in the distance swum, velocity, and time spent moving (distance, wt versus sa509; 3138 cm, 2160 cm; SEM 303, 146; P < 0.05; velocity, 5.1 cm/s, 3.5 cm/s; SEM 0.51, 0.24; P < 0.05; time spent moving, 569 s, 485 s; SEM 47.3, 26.3; P < 0.05) (Fig. 8). There were no differences of thigmotaxis (444 s, 380 s; SEM 21, 32), acceleration (1291 cm/s, 548 cm/s; SEM 488, 209), mean mobility (28%, 25%; SEM 2.8, 2.6), or frequency of mobility (1604 s, 1658 s; SEM 77, 95). Mobility is defined as movement of any part of the animal’s body axis without movement of the center point of the animal (e.g. no overall displacement of the animal). Overall, these results show that abcd1 mutants display early quantifiable defects in survival and motor activity that were also present in the adult animals.
Figure 8.

Adult sa509 homozygous mutants show abnormal swimming behavior. 10’ spontaneous swimming behavior box-plot analyses; * P < 0.05. sa509 mutants show evidence of impaired movement, as indicated by significant decreases in distance, velocity, and time spent moving. Thigmotaxis (as well as acceleration, mean mobility, and frequency of mobility) were not significantly different between wild-type siblings and sa509 mutants.
Human ABCD1 Rescue in sa509 Mutants
We tested whether transient expression of human ABCD1 cDNA in oligodendrocytes could reduce phenotypes of the sa509 mutants. We injected full-length ABCD1 expressed under control of the sox10 promoter into sa509 homozygous mutant offspring. We saw reduced apoptosis in embryos expressing the human ABCD1 construct compared to non-injected mutants (Fig. 9A). Quantification revealed a statistically significant decrease in apoptosis (uninjected versus injected homozygotes, n = 9 embryos each, cell counts 16.9 vs 2.8, SEM 2.5 and 1.4, P < 0.0001, Student’s t-test) (Fig. 9B). Further, we found that human ABCD1 rescued the number and patterning of oligodendrocytes in sa509 heterozygotes (Fig. 9C). Finally, in the analysis of motor swimming behavior, we found that expression of human ABCD1 improved the evoked swimming deficit in sa509 mutants (n > 45 animals each group, 35.2 vs 46.5 mm, SEM 2.6, 3.4, P < 0.05) (Fig. 9D). These results suggest that human and zebrafish ABCD1 perform functionally similar roles, and demonstrate that expression of ABCD1 in oligodendrocytes is sufficient to rescue several of the mutant phenotypes.
Figure 9.

Expression of human ABCD1 cDNA in oligodendrocytes rescues apoptosis in sa509 mutants. (A-A’’) Confocal z-stack images of ABCD1/GFP expression in Tg(olig2:dsRed) embryo injected with sox10:ABCD1-2A-GFP plasmid. (B, C) Confocal z-stack images of TUNEL and DAPI staining in 30 hpf embryos, showing decrease in apoptosis in embryos injected with sox10:ABCD1-2A-GFP. Rostral to left, dorsal top; area for quantification in B shown as yellow box; scale bar 50 μm. (D) Quantification of apoptosis, P < 0.0001, Student’s t-test, n = 9 embryos each. (E, F) Confocal z-stack images of α-dsRed and DAPI staining in 72 hpf embryos, arrows point to regions with normalized oligodendrocyte pattern and numbers in embryos injected with sox10:ABCD1-2A-GFP. Dorsal views, rostral top, scale bar 50 μm. (G) Quantification of evoked behavior response, P < 0.05.
Discussion
We have characterized a small vertebrate model for ALD, by generating mutations in the zebrafish gene abcd1. The zebrafish abcd1 mutant alleles recapitulate key elements of human ALD, including elevated VLCFA levels, altered myelination, and evidence of nervous system dysfunction including altered motor behavior. The motor phenotypes are apparent within the first week of life and could be used for screening of therapeutic compounds. Importantly, our data reveal a heretofore unrecognized developmental defect in oligodendrocyte generation and patterning, and requirement for abcd1 in oligodendrocytes, which may be important for understanding ALD pathophysiology and the predisposition for disease progression.
The ABCD1 gene encodes a peroxisomal ATP-binding cassette protein transporter, and ABCD1 appears necessary for very-long chain fatty acid metabolism. ALD is characterized by elevated VLCFAs in the serum as well as accumulation in tissues (26,27). However, several studies have found that levels of VLCFA β-oxidation are not affected in ABCD1 mouse mutants (28,29), and correction of VLCFA levels does not appear to prevent or reverse disease progression (5,30). Nonetheless the presence of elevated VLCFAs remains a reliable diagnostic marker for mutations in ABCD1, and which we noted in our zebrafish mutant by day of life seven. Both the heterozygous and homozygous sa509 zebrafish had VLCFA elevation. In human, male ALD patients VLCFA levels are increased roughly 4-fold over baseline (31), and we observed a 2-fold increase over baseline, although differences in sample source (human serum vs. whole tissue) and methodology of sample preparation are limitations in interpreting this result.
In zebrafish, we noted that abcd1 RNA expression is seen by 48 hpf in the floor plate, which regulates neuron and oligodendrocyte patterning in the CNS (31–33). Consistent with this floor plate expression, loss of abcd1 was associated with multiple defects in the development of the CNS, including abnormal axon tract placement, altered oligodendrocyte patterning with reductions in oligodendrocyte numbers, and impaired myelination. Because the oligodendrocyte progenitor population also gives rise to motor neurons (34), our results offer a potential insight into the susceptibility for motor defects in patients with the AMN form of ALD, which has altered motor neuron function (35). Future studies could examine if numbers and patterning of motor neurons is intact in abcd1 mutants. Zebrafish abcd1 is also expressed in the interrenal/pronephros, the zebrafish homolog of the adrenal glands (16), similar to human ABCD1, which is associated with Addison’s disease and adrenal insufficiency. We observed a defect in interrenal gland development, but no change in cortisol levels. However, in humans the adrenal insufficiency is only present in some patients, and can manifest at any age from young childhood to middle age, with rates ranging from ∼1% in females to eventually > 80% in males (36).
A major area of uncertainty surrounding human ALD is the phenotypic heterogeneity noted with the same gene mutation, such that even family members carrying the same gene change can have different phenotypes ranging from adult onset adrenomyeloneuropathy (AMN) to pediatric onset rapidly progressive cerebral ALD (3). Our observation that numbers of oligodendrocyte progenitor cells (OPCs) and oligodendrocytes are reduced suggests a potential mechanism for the susceptibility for AMN. The reduced numbers make any additional loss of oligodendrocytes and motor axons more problematic for the animal. Since impaired peroxisomal function has been associated with increased susceptibility to neuroinflammation (37), including in ABCD1 mutants (38,39), specific triggers of inflammation such as trauma (40), or slowly progressive neuroinflammatory-mediated loss of oligodendrocytes, could cause further loss of oligodendrocytes. At some threshold the number of oligodendrocytes will no longer be able to maintain normal myelin structure and function.
Another area of investigation regards the dosage requirement for ABCD1. In humans, ABCD1 is located on the X-chromosome, and affected males as well as heterozygous females develop neurological features. It is not known in humans whether the phenotype reflects a partial compensation, for example by the closely-related ABCD2; and/or whether heterozygous females are affected by X-inactivation of the non-mutant ABCD1 allele. Our zebrafish abcd1 model displays phenotypes in both heterozygotes and homozygotes. We think our results suggest that the phenotype in the zebrafish model reflects a dose-dependent requirement for Abcd1; particularly since both the splice-blocking and the exon 1 TALEN mutants display similar phenotypes.
The reduction of oligodendrocytes in abcd1 mutants is correlated with increased apoptosis in early development. While loss of ABCD1 has been noted to impair energy production (41), loss of oligodendrocytes had not been examined. Future studies can examine whether the increased apoptosis is secondary to impaired bioenergetics, or instead due to an unrecognized developmental role. In adult patients peripheral neuropathy is the most common phenotype (adrenomyeloneuropathy- AMN), and appears to consist of a "dying-back" axonopathy (4). Future studies in the zebrafish ALD model can examine the extent of peripheral axon and myelin loss.
The absence of interventions to prevent or slow disease progression in AMN, or to prevent or slow conversion to cerebral ALD, makes identification of new therapeutics of high relevance. The zebrafish ALD mutants demonstrate motor swimming defects by day six of life, when the larvae are still small enough to be tested in 96-well plates. While the current motor assay has significant phenotypic overlap between the different genotypes, with refinements the zebrafish ALD model might provide the basis for a drug compound screen. We have also found that adult mutants have impaired swimming behavior. The adult swimming behavior is quantifiable but is not obvious on casual inspection by eye. However, we have not serially followed adult individual animals over time to determine whether there is disease progression.
Drug screens in zebrafish have been used to identify target pathways as well as therapeutic compounds for human diseases (42–45). Since zebrafish have conservation of many of the genetic pathways for myelin generation, and our own work demonstrates conservation of abcd1 expression and function, mechanisms of action for compounds identified in zebrafish might be relevant for mammals as well. This is supported by our data showing that human ABCD1 expression in zebrafish can rescue the increased apoptosis and behavioral phenotype seen in sa509 mutants. Finally, the zebrafish model offers the opportunity to investigate the pathogenesis of conversion to rapidly progressive cerebral ALD, since we note early death in a subset of mutant animals. Future work can test the role and biochemical pathways played by environmental triggers or genetic background effects.
Materials and Methods
Ethics statement
Zebrafish experiments were performed in strict accordance with guidelines from the University of Utah Institutional Animal Care and Use Committee (IACUC), regulated under federal law (the Animal Welfare Act and Public Health Services Regulation Act) by the U.S. Department of Agriculture (USDA) and the Office of Laboratory Animal Welfare at the NIH, and accredited by the Association for Assessment and Accreditation of Laboratory Care International (AAALAC).
Fish stocks and embryo raising
Adult fish were bred according to standard methods. Embryos were raised at 28.5 °C in E3 embryo medium and staged by time and morphology (46). For in situ staining and immunohistochemistry, embryos were fixed in 4% paraformaldehyde (PFA) in PBS overnight (O/N) at 4 °C, washed briefly in PBS with 0.1% Tween-20, dehydrated, and stored in 100% MeOH at −20 °C until use.
Transgenic fish lines and alleles used in this paper were the following: Tg(olig2:dsRed)vu19 (23); Tg(mbp:egfp-caax)zc95. Injection of DNA constructs and generation of stable transgenic lines was performed essentially as described (47). Lines are available upon request.
Sequence analysis
Human and zebrafish ABCD1 amino acid sequences were compared using Clustal Omega and aligned using PRALINE (48,49); phylogenetic tree analysis was performed with Phylodendron (http://iubio.bio.indiana.edu/treeapp/treeprint-form.html; date last accessed July 04, 2017).
TALEN plasmid construction and mutant generation and screening
Design and construction of TALENs targeting the D. rerio abcd1 gene (Ensembl(Zv9):ENSDARG00000074876) was performed by the University of Utah Mutation Generation and Detection Core as previously described (50,51). Potential TALEN target sites were identified using the TAL Effector Nucleotide Targeter 2.0 program (https://tale-nt.cac.cornell.edu; date last accessed July 04, 2017). TALEN sites were selected using the design parameters of: 1) spacer length: 14–18bp; 2) TALE repeat array length of 15–21 and 3) applying all additional options that restrict target choice (52). The TAL Effector Nucleotide Targeter 2.0 program’s Target Finder and Paired Target Finder, and a BLAST analysis, were used to ensure that highly similar left and right binding sites in close proximity to each other were not present at other regions in the D. rerio genome. The TALEN Golden Gate Kit (Addgene #1000000024) was used to produce RVD repeat array sequences that were cloned into pCS2TAL3-DDD (Addgene, #48637) and pCS2TAL3-RRR (Addgene, #48636) expression vectors to generate left and right TALENs, respectively. acbd1 TALEN plasmids were linearized with NotI and 5’-capped mRNA was generated (mMESSAGE mMACHINE SP6 Transcription Kit, Ambion) and purified (MEGAclear Kit, Ambion). 50–250 pg of equal amounts of each TALEN arm was injected into one-cell stage embryos and scored for dysmorphology and ability to induce mutations at the abcd1 locus, as assayed using high-resolution melt analysis (HRMA) PCR. Potential G0 founder mutant alleles were out-crossed to wild-type fish.
HRMA PCR
PCR and high-resolution melt analysis (HRMA) was used for genotyping following published conditions with LightScanner Master Mix (Biofire Defense, Inc.) (19,53). For the zc90 allele in exon 1, we used the following primers and conditions: (forward primer)
5’- GTGGCTCATCTGTATTCAAACCT -3’ and (reverse primer) 5’- CAGCCGTTTTAATGAGCGTGTA -3’; 94˚C for 2 min (m), followed by 30 cycles of 94˚C for 15 s (s), 62˚C for 15s, 72˚C for 15s, and a final extension of 72˚C for 2m. Following the PCR, DNAs were run on a 3.5% super fine resolution agarose gel in 1XTBE to differentiate wild type (79bp) from mutant (67bp) or heterozygote (79bp and 67bp) products. For the other TALEN alleles in exon 1, we used the following primers and conditions: (forward primer)
5’-AGGTTGGCAAACCCTGACCA-3’ and (reverse primer) 5’-GTGTTGGCGCCTTTGGATTC-3’; 94˚C for 3 min (m), followed by 45 cycles of 94˚C for 30 s (s), 60˚C for 30s, 72˚C for 30s, and a final extension of 72˚C for 10m. For sa509 genotyping we did PCR with the following primers and conditions: (forward primer) 5’- TTACTCACCGTCCTTCTCTC-3’ and (reverse primer) 5’-TCATACGATTCAAAACCCAC -3’; 95˚C for 2 m, followed by 30 cycles of 95˚C for 15s, 68.1˚C for 15s, followed by a final denaturation and annealing step of 95˚C for 30s, 25˚C for 15s, in an optically transparent plate with a mineral oil overlay. We then performed HRMA in order to differentiate the sa509 melt-curves.
Immunohistochemistry and in situ hybridization
Immunohistochemistry was performed as previously described (47; 38]. Antibodies used were: mouse anti-acetylated tubulin 1:250 (Sigma), mouse monoclonal anti-GFP 1:250 (Millipore), chicken anti-GFP 1:1000 (Aves Labs), mouse anti-HuC/D 1:400 (ThermoFisher), rabbit anti-dsRed 1:250 (Clontech), Cy-3 anti-rabbit 1:400 (Millipore), Alexa 488 donkey anti-mouse 1:400, Alexa 488 donkey anti-chicken 1:400, Alexa 555 rabbit anti-goat 1:400 (ThermoFisher), and 4',6-diamidino-2-phenylindole (DAPI).
We tested several different commercial antibodies against ABCD1 by whole-mount immunohistochemistry and on western blots; as well as generating and testing a rabbit polyclonal antibodies against a zebrafish 17-mer polypeptide. None of the antibodies tested gave a specific band. Antibodies against human ABCD1 that were tested by whole-mount immunohistochemistry and on western blots were Abcam clone 4E2 ab180485; Acris 11159-1-A; and USBio 209323. We also generated and tested rabbit polyclonal antibodies against a zebrafish Abcd1 protein 17-mer, amino acids 751-766 (CTERDVQSAGKEKDLME).
In situ hybridization
The antisense digoxigenin-labeled RNA probe for zebrafish abcd1 (ENSDARG00000074876) was prepared by generation of an amplicon from exon 1 (primers, forward 5’- CTGAAGAAACCCCTGGTGAA -3’; reverse, 5’-CTTTCCTCCTGGCTTCCTCT -3’). Following gel purification, the fragment was cloned into a TOPO TA dual promoter PCR4 TOPO vector (Invitrogen) and was sequenced to confirm identity. The construct was linearized with BamHI, and digoxigenin-labeled antisense mRNA probe was generated by T7 in vitro transcription (DIG-RNA labeling kit, Roche 11-175-025-910). Whole-mount in situ hybridization was performed as described (54).
In situ hybridization combined with immunohistochemistry on embryonic sections
In situ hybridization was performed as described above. After in situ was completed, embryos were sequentially transferred to 5% for 3 h, 15% sucrose overnight, and 30% sucrose overnight (sucrose was prepared in 1X PBS). Embryos were then embedded in OCT with a dry ice ethanol bath (dry ice submerged in 100% ethanol) and stored at −20˚C. 20 μm sections were cut for immunostaining. Sections were blocked for 30 min in blocking solution (1% BSA. 0.1% fish skin gelatin (Sigma G7765), 0.1% Tween-20 in PBS). The sections were then incubated with primary antibody, rabbit anti-dsRed, for 1 h at room temperature. After removal of the primary antibody with three washes in PBS, the second antibody, donkey anti-rabbit Alexa Fluor 555, was added for 30 min at room temperature. Sections were then washed three times in 1x PBS and mounted in Fluoromount G (SouthernBiotech).
Cloning
Expression clones were built using the Tol2 kit and recombination reactions with Gateway (ThermoFisher) plasmids (55). The identity of the constructs was confirmed by restriction enzyme digests and by sequencing both strands (for coding sequences) or by partial end-sequencing (for enhancers). Specific plasmids used for cloning were p5E-mbp; p5E-sox10; pME-basEGFP-caax (middle entry clone with EGFP-caax preceded by minimal promoter); pME-ABCD1 (human); p3E-2A-EGFP; p3E-pA; into pDestTol2pA2 (53). p5E-mbp was generated by cloning the mbp enhancer (20,56) using primers forward 5’- GTCGACCAGATGCTGAGATGTGACTACTGCAAATGA -3’ and reverse 5’- GGATCCGTTGATCTGTTCAGTGGTCTACAGTCTGGA -3’.
Very long chain fatty acid analysis
Samples were transferred to 13 ml culture tubes with PTFA-lined caps and 1.5 ml of 3% HCl in MeOH containing n-heptadecanoic acid (0.06 mg/ml) was added. Samples were vortexed for 30 s then placed into a sand bath heated at 80 °C for 16 h. The resulting methanolysis reaction products were extracted with hexanes (2 × 1 ml), transferred to Eppendorf tubes, briefly evaporated in a speedvac, then resuspended in 30 µl EtOAc and transferred to GC/MS vials with insert. A pooled QC sample was generated by taking small aliquots from each reaction vial. A blank process sample was also made. GC/MS analyses are conducted using an HP6890 instrument interfaced with an MSD-HP5973 detector and equipped with a Zebron ZB-5MSi Guardian (30 m × 0.25 mm ID, 0.25 µm film thickness; Phenomenex) column and an HP7682 injector. Helium was used as a carrier gas with a 10:1 split ratio at an injection volume of 1 µl. The injector temperature was 250 °C. The oven temperature gradient was: 95 °C held for 1.5 min increased at a rate of 40 °C/min to 118 °C, held for 1 min, increased at a rate of 5 °C/min to 250 °C, increased at a rate of 25 °C/min to 330 °C and held for 12.3 min. MS spectra were obtained in CI mode (methane gas) from a range of m/z 45 - 400. The MS quad temperature was 150 °C, MS source temperature was 230 °C, solvent cut time was 4 min, and then scanned at 16 scans/s. Supelco's FAMES 37 component mixture was used as a standard.
Microscopy and image analysis
Image acquisition and analysis were performed as described previously (47). Immunostained embryos were transferred step-wise into 80% glycerol/20% PBST, mounted on a glass slide with a #0 coverslip placed over a well-made using electrical tape, and imaged on a confocal microscope. Confocal stacks were projected in ImageJ, and images composed with Adobe Photoshop and Illustrator.
olig2:dsRed quantification
Confocal images of Tg(olig2:dsRed) embryos immunostained for dsRed and for DAPI were obtained, and quantification was performed in ImageJ by compiling a maximum intensity projection of 30 z-slices, step-size 1.14 μm, into a single image; a 300 × 150 pixel box was drawn with the rostral edge at the hindbrain boundary, and the total pixel intensity was calculated.
TUNEL quantification
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) was performed on whole-mount larvae (ApopTag Fluorescein in Situ Apoptosis Detection Kit; Millipore) as described (57). Confocal imaging was performed and quantification was performed in ImageJ by compiling a sum projection of 75 slices (step size 1.14 μm) into a single z-stack image. For counts at 72 hpf a 340×170 pixel box was drawn in the telencephalon at with the ventral edge place on the dorsal edge of the eye and the rostral edge at the nose; at 30 hpf and 48 hpf a 450×300 pixel box was drawn around the entire brain. Thresholding was applied from a minimum of 125 to a maximum of 255; and the "Analyze Particles" function was used for quantification.
Semi-quantitative RT-PCR
A semi-quantitative PCR reaction for abcd1 was performed on 72 hpf embryos. Primers for abcd1 were the same as used to generate the in situ probe. polyA RNA was quantified for each sample, and equal amounts were used in each PCR. Total RNA was extracted using Trizol (Invitrogen), purified (Qiagen RNeasy mini-columns), and reverse-transcribed (SuperscriptTM III First-strand Invitrogen kit, 18080-051) as previously described (53).
Survival analysis
Three separate experimental replicates were performed for each analysis with n > 25 animals of each genotype for each experimental replicate. Survival was checked no less than every 3rd day. Animals were raised at a density of 25 animals per tank. For competitive survival analysis the different genotypes were raised together; on a daily basis tanks were checked, and dead animals were removed and genotyped by PCR. Statistical analysis was performed in GraphPad Prism 6.
Cortisol assays
Larval cortisol preparation
7 dpf larvae were immediately anesthetized with Tricaine. 20 larvae per 1.7 ml tube were suspended in 200 μl 10 mM Tris pH 8.0 on ice and homogenized with a pellet pestle. Tubes were spun for 10 min at maximum speed in a microfuge at 4 °C. The entire supernatant was removed to a new tube and kept on ice or at −80 °C before processing over columns.
Strata-X columns (cat#8E-S100-UGB, Phenomenex) were conditioned with 1 ml MeOH and then equilibrated with 1 ml water. The columns were then washed with 1 ml water and 1 ml 10% MeOH in water. Finally, the samples were eluted with 2 × 500 mcL 2% formic acid in ethyl acetate/isopropanol (85:15), dried down in a speedvac, and reconstituted in 100 μl of ELISA buffer from a cortisol ELISA kit (Cayman Chemical Company, cat #500530). The samples were further diluted 1:10 in ELISA buffer and a cortisol ELISA was performed in triplicate, according to the manufacturer’s directions.
Adult cortisol preparation
Adult male fish were immediately euthanized in ice, frozen in liquid nitrogen, and ground to powder with a mortar and pestle. The powder was placed in a 1.7 ml tube and weighed prior to addition 200 μl 10 mM Tris pH 8.0, followed by vortexing. Extracts were spun for 10 min at maximum speed at 4 °C, and the supernatant removed to a new tube and kept on ice or at −80 °C before processing over columns and performing the ELISA as for larvae, above.
Behavior analysis
Larval behavior analysis was performed on 6 dpf larvae in 96-well square bottom plates (Krackeler Scientific) using a video analysis software program (Noldus EthoVision). For spontaneous behavior, animals were transferred at 5 dpf to the 96-well plate and kept at 28.5˚C overnight. At 6 dpf the plate was placed on the video imaging system and animals were allowed to adapt for 30’, and then recording was performed in three separate epochs of 2’ each. For evoked behavior, animals were similarly adapted, and then a single white light flash was provided for stimulus and behavior was recorded for 1’.
Adult behavior analysis was performed on adults (age > 6 months) in 7.5 × 9.5 cm (inner dimensions) rectangular glass dishes. Adults were transferred to the behavior analysis room on the morning of testing in their home tanks, allowed to acclimatize for 1 h, and then transferred individually to the testing dish. The animal and testing dish was placed in the recording arena and 30’ of acclimatization occurred, followed by 10’ of recording of spontaneous behavior.
Statistical analysis
Statistical analyses were performed using Prism6 software (GraphPad). Student’s t-test was used for two-way comparisons; comparisons between three or more groups was performed with ANOVA with post-hoc Tukey’s HSD between individual means.
Acknowledgements
We thank T. Dahlem and the University of Utah Mutation Generation and Detection Core; M. Hobbs and the Centralized Zebrafish Animal Resource (CZAR) at the University of Utah, supported in part by NIH grant #1G20OD018369-01; J. Cox and the University of Utah Metabolomics core, supported in part by 1S10OD016232-01, 1S10OD021505-01 and 1U54DK110858-01; and the University of Utah Fluorescence Microscopy Core Facility, supported in part by NCRR Shared Equipment Grant # 1S10RR024761-01. We thank F. Eichler for sharing the human ABCD1 cDNA; S. Ryu for sharing the star probe; and S. Rosenthal for assistance with survival analysis.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health [DP2 MH100008], European Leukodystrophy Association [2015-006I1] and Bray Chair in Child Neurology Research.
References
- 1. Bonkowsky J.L., Nelson C., Kingston J.L., Filloux F.M., Mundorff M.B., Srivastava R. (2010) The burden of inherited leukodystrophies in children. Neurology, 75, 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mosser J., Douar A.M., Sarde C.O., Kioschis P., Feil R., Moser H., Poustka A.M., Mandel J.L., Aubourg P. (1993) Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature, 361, 726–730. [DOI] [PubMed] [Google Scholar]
- 3. Berger J., Forss-Petter S., Eichler F.S. (2014) Pathophysiology of X-linked adrenoleukodystrophy. Biochimie, 98, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Powers J.M., DeCiero D.P., Ito M., Moser A.B., Moser H.W. (2000) Adrenomyeloneuropathy: a neuropathologic review featuring its noninflammatory myelopathy. J. Neuropathol. Exp. Neurol., 59, 89–102. [DOI] [PubMed] [Google Scholar]
- 5. Aubourg P., Adamsbaum C., Lavallard-Rousseau M.C., Rocchiccioli F., Cartier N., Jambaque I., Jakobezak C., Lemaitre A., Boureau F., Wolf C.. et al. (1993) A two-year trial of oleic and erucic acids ("Lorenzo's oil") as treatment for adrenomyeloneuropathy. N. Engl. J. Med., 329, 745–752. [DOI] [PubMed] [Google Scholar]
- 6. Forss-Petter S., Werner H., Berger J., Lassmann H., Molzer B., Schwab M.H., Bernheimer H., Zimmermann F., Nave K.A. (1997) Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J. Neurosci. Res., 50, 829–843. [DOI] [PubMed] [Google Scholar]
- 7. Kobayashi T., Shinnoh N., Kondo A., Yamada T. (1997) Adrenoleukodystrophy protein-deficient mice represent abnormality of very long chain fatty acid metabolism. Biochem. Biophys. Res. Commun., 232, 631–636. [DOI] [PubMed] [Google Scholar]
- 8. Lu J.F., Lawler A.M., Watkins P.A., Powers J.M., Moser A.B., Moser H.W., Smith K.D. (1997) A mouse model for X-linked adrenoleukodystrophy. Proc. Natl. Acad. Sci. U. S. A, 94, 9366–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pujol A., Hindelang C., Callizot N., Bartsch U., Schachner M., Mandel J.L. (2002) Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum. Mol. Genet., 11, 499–505. [DOI] [PubMed] [Google Scholar]
- 10. Musolino P.L., Gong Y., Snyder J.M., Jimenez S., Lok J., Lo E.H., Moser A.B., Grabowski E.F., Frosch M.P., Eichler F.S. (2015) Brain endothelial dysfunction in cerebral adrenoleukodystrophy. Brain, 138, 3206–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruiz M., Jove M., Schluter A., Casasnovas C., Villarroya F., Guilera C., Ortega F.J., Naudi A., Pamplona R., Gimeno R.. et al. (2015) Altered glycolipid and glycerophospholipid signaling drive inflammatory cascades in adrenomyeloneuropathy. Hum. Mol. Genet., 24, 6861–6876. [DOI] [PubMed] [Google Scholar]
- 12. Schluter A., Espinosa L., Fourcade S., Galino J., Lopez E., Ilieva E., Morato L., Asheuer M., Cook T., McLaren A.. et al. (2012) Functional genomic analysis unravels a metabolic-inflammatory interplay in adrenoleukodystrophy. Hum. Mol. Genet., 21, 1062–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singh J., Khan M., Singh I. (2009) Silencing of Abcd1 and Abcd2 genes sensitizes astrocytes for inflammation: implication for X-adrenoleukodystrophy. J. Lipid Res, 50, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guran T., Buonocore F., Saka N., Ozbek M.N., Aycan Z., Bereket A., Bas F., Darcan S., Bideci A., Guven A.. et al. (2016) Rare causes of primary adrenal insufficiency: genetic and clinical characterization of a large nationwide cohort. J. Clin. Endocrinol. Metab., 101, 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kettleborough R.N., Busch-Nentwich E.M., Harvey S.A., Dooley C.M., de Bruijn E., van Eeden F., Sealy I., White R.J., Herd C., Nijman I.J.. et al. (2013) A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature, 496, 494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsu H.J., Lin G., Chung B.C. (2003) Parallel early development of zebrafish interrenal glands and pronephros: differential control by wt1 and ff1b. Development, 130, 2107–2116. [DOI] [PubMed] [Google Scholar]
- 17. Ravanelli A.M., Appel B. (2015) Motor neurons and oligodendrocytes arise from distinct cell lineages by progenitor recruitment. Genes Dev., 29, 2504–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park H.C., Boyce J., Shin J., Appel B. (2005) Oligodendrocyte specification in zebrafish requires notch-regulated cyclin-dependent kinase inhibitor function. J. Neurosci., 25, 6836–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xing L., Quist T.S., Stevenson T.J., Dahlem T.J., Bonkowsky J.L. (2014) Rapid and efficient zebrafish genotyping using PCR with high-resolution melt analysis. J. Vis. Exp., 84, e51138.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Engelen M., Kemp S., de Visser M., van Geel B.M., Wanders R.J., Aubourg P., Poll-The B.T. (2012) X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis., 7, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gutierrez-Triana J.A., Herget U., Castillo-Ramirez L.A., Lutz M., Yeh C.M., De Marco R.J., Ryu S. (2015) Manipulation of Interrenal Cell Function in Developing Zebrafish Using Genetically Targeted Ablation and an Optogenetic Tool. Endocrinology, 156, 3394–3401. [DOI] [PubMed] [Google Scholar]
- 22. Bai Q., Sun M., Stolz D.B., Burton E.A. (2011) Major isoform of zebrafish P0 is a 23.5 kDa myelin glycoprotein expressed in selected white matter tracts of the central nervous system. J. Comp. Neurol., 519, 1580–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Almeida R.G., Czopka T., Ffrench-Constant C., Lyons D.A. (2011) Individual axons regulate the myelinating potential of single oligodendrocytes in vivo. Development, 138, 4443–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simons M., Nave K.A. (2015) Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol., 8, a020479.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kucenas S., Takada N., Park H.C., Woodruff E., Broadie K., Appel B. (2008) CNS-derived glia ensheath peripheral nerves and mediate motor root development. Nat. Neurosci., 11, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rasmussen M., Moser A.B., Borel J., Khangoora S., Moser H.W. (1994) Brain, liver, and adipose tissue erucic and very long chain fatty acid levels in adrenoleukodystrophy patients treated with glyceryl trierucate and trioleate oils (Lorenzo's oil). Neurochem. Res., 19, 1073–1082. [DOI] [PubMed] [Google Scholar]
- 27. Moser H.W., Moser A.B., Frayer K.K., Chen W., Schulman J.D., O'Neill B.P., Kishimoto Y. (1981) Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids. Neurology, 31, 1241–1249. [DOI] [PubMed] [Google Scholar]
- 28. Oezen I., Rossmanith W., Forss-Petter S., Kemp S., Voigtlander T., Moser-Thier K., Wanders R.J., Bittner R.E., Berger J. (2005) Accumulation of very long-chain fatty acids does not affect mitochondrial function in adrenoleukodystrophy protein deficiency. Hum. Mol. Genet., 14, 1127–1137. [DOI] [PubMed] [Google Scholar]
- 29. McGuinness M.C., Lu J.F., Zhang H.P., Dong G.X., Heinzer A.K., Watkins P.A., Powers J., Smith K.D. (2003) Role of ALDP (ABCD1) and mitochondria in X-linked adrenoleukodystrophy. Mol. Cell. Biol., 23, 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Geel B.M., Assies J., Haverkort E.B., Koelman J.H., Verbeeten B. Jr., Wanders R.J., Barth P.G. (1999) Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy despite treatment with "Lorenzo's oil". J. Neurol. Neurosurg. Psychiatry, 67, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valianpour F., Selhorst J.J., van Lint L.E., van Gennip A.H., Wanders R.J., Kemp S. (2003) Analysis of very long-chain fatty acids using electrospray ionization mass spectrometry. Mol. Genet. Metab., 79, 189–196. [DOI] [PubMed] [Google Scholar]
- 32. Placzek M., Briscoe J. (2005) The floor plate: multiple cells, multiple signals. Nat. Rev. Neurosci., 6, 230–240. [DOI] [PubMed] [Google Scholar]
- 33. Richardson W.D., Pringle N.P., Yu W.P., Hall A.C. (1997) Origins of spinal cord oligodendrocytes: possible developmental and evolutionary relationships with motor neurons. Dev. Neurosci., 19, 58–68. [DOI] [PubMed] [Google Scholar]
- 34. Fu H., Qi Y., Tan M., Cai J., Takebayashi H., Nakafuku M., Richardson W., Qiu M. (2002) Dual origin of spinal oligodendrocyte progenitors and evidence for the cooperative role of Olig2 and Nkx2.2 in the control of oligodendrocyte differentiation. Development, 129, 681–693. [DOI] [PubMed] [Google Scholar]
- 35. Masur H., Zierz S., Ludolph A.C., Elger C.E., Palm D.G. (1990) Corticomotoneuronal latencies after non-invasive stimulation of the motor cortex in adrenomyeloneuropathy and adrenoleukodystrophy: results in patients and their relatives. Funct. Neurol., 5, 55–59. [PubMed] [Google Scholar]
- 36. Burtman E., Regelmann M.O. (2016) Endocrine Dysfunction in X-Linked Adrenoleukodystrophy. Endocrinol. Metab. Clin. North Am., 45, 295–309. [DOI] [PubMed] [Google Scholar]
- 37. Kassmann C.M., Lappe-Siefke C., Baes M., Brugger B., Mildner A., Werner H.B., Natt O., Michaelis T., Prinz M., Frahm J.. et al. (2007) Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat. Genet., 39, 969–976. [DOI] [PubMed] [Google Scholar]
- 38. Fourcade S., Lopez-Erauskin J., Galino J., Duval C., Naudi A., Jove M., Kemp S., Villarroya F., Ferrer I., Pamplona R.. et al. (2008) Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum. Mol. Genet., 17, 1762–1773. [DOI] [PubMed] [Google Scholar]
- 39. Kemp S., Wanders R. (2010) Biochemical aspects of X-linked adrenoleukodystrophy. Brain Pathol., 20, 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raymond G.V., Seidman R., Monteith T.S., Kolodny E., Sathe S., Mahmood A., Powers J.M. (2010) Head trauma can initiate the onset of adreno-leukodystrophy. J. Neurol. Sci., 290, 70–74. [DOI] [PubMed] [Google Scholar]
- 41. Kruska N., Schonfeld P., Pujol A., Reiser G. (2015) Astrocytes and mitochondria from adrenoleukodystrophy protein (ABCD1)-deficient mice reveal that the adrenoleukodystrophy-associated very long-chain fatty acids target several cellular energy-dependent functions. Biochim. Biophys. Acta, 1852, 925–936. [DOI] [PubMed] [Google Scholar]
- 42. Baraban S.C., Dinday M.T., Hortopan G.A. (2013) Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Comm., 4, 2410.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peal D.S., Lynch S.N., Milan D.J. (2011) Patterning and development of the atrioventricular canal in zebrafish. J. Cardiovasc. Transl. Res., 4, 720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laggner C., Kokel D., Setola V., Tolia A., Lin H., Irwin J.J., Keiser M.J., Cheung C.Y., Minor D.L. Jr., Roth B.L.. et al. (2011) Chemical informatics and target identification in a zebrafish phenotypic screen. Nat. Chem. Biol., 8, 144–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ridges S., Heaton W.L., Joshi D., Choi H., Eiring A., Batchelor L., Choudhry P., Manos E.J., Sofla H., Sanati A.. et al. (2012) Zebrafish screen identifies novel compound with selective toxicity against leukemia. Blood, 119, 5621–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn., 203, 253–310. [DOI] [PubMed] [Google Scholar]
- 47. Bonkowsky J.L., Wang X., Fujimoto E., Lee J.E., Chien C.B., Dorsky R.I. (2008) Domain-specific regulation of foxP2 CNS expression by lef1. BMC Dev. Biol., 8, 103.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J.. et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol., 7, 539.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simossis V.A., Heringa J. (2005) PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res., 33, W289–W294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dahlem T.J., Hoshijima K., Jurynec M.J., Gunther D., Starker C.G., Locke A.S., Weis A.M., Voytas D.F., Grunwald D.J. (2012) Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet., 8, e1002861.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu R., Wallace J., Dahlem T.J., Grunwald D.J., O'Connell R.M. (2013) Targeting human microRNA genes using engineered Tal-effector nucleases (TALENs). PloS One, 8, e63074.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Doyle E.L., Booher N.J., Standage D.S., Voytas D.F., Brendel V.P., Vandyk J.K., Bogdanove A.J. (2012) TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res., 40, W117–W122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xing L., Hoshijima K., Grunwald D.J., Fujimoto E., Quist T.S., Sneddon J., Chien C.B., Stevenson T.J., Bonkowsky J.L. (2012) Zebrafish foxP2 zinc finger nuclease mutant has normal axon pathfinding. PloS One, 7, e43968.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bonkowsky J.L., Chien C.B. (2005) Molecular cloning and developmental expression of foxP2 in zebrafish. Dev. Dyn., 234, 740–746. [DOI] [PubMed] [Google Scholar]
- 55. Kwan K.M., Fujimoto E., Grabher C., Mangum B.D., Hardy M.E., Campbell D.S., Parant J.M., Yost H.J., Kanki J.P., Chien C.B. (2007) The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn., 236, 3088–3099. [DOI] [PubMed] [Google Scholar]
- 56. Jung S.H., Kim S., Chung A.Y., Kim H.T., So J.H., Ryu J., Park H.C., Kim C.H. (2010) Visualization of myelination in GFP-transgenic zebrafish. Dev. Dyn., 239, 592–597. [DOI] [PubMed] [Google Scholar]
- 57. Lambert A.M., Bonkowsky J.L., Masino M.A. (2012) The conserved dopaminergic diencephalospinal tract mediates vertebrate locomotor development in zebrafish larvae. J. Neurosci., 32, 13488–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]