

Abstract
Osteoarthritis (OA) is a common complex disease with high public health burden and no curative therapy. High bone mineral density (BMD) is associated with an increased risk of developing OA, suggesting a shared underlying biology. Here, we performed the first systematic overlap analysis of OA and BMD on a genome wide scale. We used summary statistics from the GEFOS consortium for lumbar spine (n = 31,800) and femoral neck (n = 32,961) BMD, and from the arcOGEN consortium for three OA phenotypes (hip, ncases=3,498; knee, ncases=3,266; hip and/or knee, ncases=7,410; ncontrols=11,009). Performing LD score regression we found a significant genetic correlation between the combined OA phenotype (hip and/or knee) and lumbar spine BMD (rg=0.18, P = 2.23 × 10−2), which may be driven by the presence of spinal osteophytes. We identified 143 variants with evidence for cross-phenotype association which we took forward for replication in independent large-scale OA datasets, and subsequent meta-analysis with arcOGEN for a total sample size of up to 23,425 cases and 236,814 controls. We found robustly replicating evidence for association with OA at rs12901071 (OR 1.08 95% CI 1.05–1.11, Pmeta=3.12 × 10−10), an intronic variant in the SMAD3 gene, which is known to play a role in bone remodeling and cartilage maintenance. We were able to confirm expression of SMAD3 in intact and degraded cartilage of the knee and hip. Our findings provide the first systematic evaluation of pleiotropy between OA and BMD, highlight genes with biological relevance to both traits, and establish a robust new OA genetic risk locus at SMAD3.
Introduction
Osteoarthritis (OA) is a degenerative disease of the joints affecting over 40% of people over 70 years (1). Hallmarks of OA include cartilage degradation, joint-space narrowing, formation of osteophytes within the joint and subchondral bone remodeling (2,3). Due to a lack of therapeutic options, the main treatment strategy consists of pain management and, in severe cases, joint replacement surgery (2). OA is a complex disorder with the 18 currently known risk loci accounting for approximately 11% of disease heritability (4). The biggest OA genome-wide association study (GWAS) published to date was conducted by the arcOGEN consortium in a two-stage design, culminating in a total discovery sample of 7,410 cases and 11,009 controls (5,6). Cases in the arcOGEN study had radiographic hip or knee OA (defined as a Kellgren Lawrence score ≥ 2), and approximately 80% had undergone total joint replacement surgery, indicating disease progression to a severe degree (6).
In addition to establishing bigger OA sample collections, studying OA in the context of related phenotypes may add to the currently short list of established loci. While most GWAS to date have investigated a single phenotype, it is estimated that around 5% of risk variants and 16% of genes identified through these studies have pleiotropic effects on multiple phenotypes (7). In recent years, the accumulation of large-scale genomic datasets has made it possible to study genetic pleiotropy by incorporating data from multiple epidemiologically linked traits. Leveraging available summary data of correlated traits to identify common genetic risk factors can help pinpoint causal pathways and refine our understanding of disease mechanisms. OA in itself is a heterogeneous disorder, with heritability varying depending on the affected joint. Of the 18 published risk loci to date, 6 and 7 are associated with knee OA only and hip OA only, respectively, while 5 are associated with both hip and knee OA (4). This further highlights how phenotypic variation is reflected by genetics, and demonstrates the need for strict phenotype definitions.
The link between bone mineral density (BMD) and OA was first reported in 1972 by Foss and Byers, who observed higher BMD in femoral heads excised during OA-related hip replacement surgery (8). Since then, a number of cross-sectional and longitudinal studies have found higher femoral neck (FN) and lumbar spine (LS) BMD, as well as total body BMD to be associated with incident OA at the hip, knee and other joint sites (9–12).
Findings with regards to the relationship between BMD and OA progression are less clear (13). Elevated bone turnover – usually a marker for decreased BMD – was reported in patients with progressive knee OA compared to patients with stable OA (14). Decreased baseline femoral neck BMD (FNBMD) has also been associated with knee OA progression (15,16). Conversely, data from the Rotterdam study showed a non-significant trend of higher odds of knee OA progression with increased lumbar spine BMD (LSBMD) (17), while another study found no link between knee OA progression and total body- or FNBMD (9).
The largest genetic studies on BMD to date have been carried out by the Genetic Factors for Osteoporosis (GEFOS) consortium: a large-scale GWAS in 2012 found 56 loci associated with BMD (18); more recently, a rare variant of large effect was identified combining whole-genome sequencing and GWAS imputation (19). Several biological mechanisms are implicated in both OA and BMD, such as bone remodeling, mesenchymal stem cell differentiation and inflammation (2,6,18,20). RUNX2, a key transcription factor regulating endochondral ossification and osteoblast differentiation (21,22), has been associated with both OA and BMD based on its proximity to genome-wide significant variants (6,18). The other locus with known GWAS hits for both traits is KLHL42 (or KLHDC5), although its biological relevance remains unclear (6,18).
In addition, Yerges-Armstrong and colleagues have previously shown nominal association of four BMD-linked single nucleotide polymorphisms (SNPs) with knee OA (23). However, despite the long-established epidemiologic link and shared biology, the genetic overlap of OA and BMD has not yet been assessed on a genome-wide level. Here, we present results from the first genome-wide analysis establishing shared genetic aetiology between OA and BMD.
Results
Known OA risk loci in BMD
Six loci associated with OA were nominally significant for FN- or LSBMD, including the SUPTH3/CDC5L locus, which is close to RUNX2, and KLHL42/PTHLH (Supplementary Material, Table S1).
Genome-wide correlation
We used linkage disequilibrium (LD) score regression to get an estimate of the genome-wide genetic correlation between OA and BMD. There was a significant correlation between combined OA and LSBMD (correlation = 0.18; P = 0.022), but not hip or knee OA and LSBMD or between FNBMD and any OA phenotype (Fig. 1).
Figure 1.

Genetic correlation between osteoarthritis (OA) and bone mineral density (BMD) as estimated by LD score regression. Rectangles show the correlation estimate (middle horizontal line) and standard errors (upper and lower bounds) of each comparison. Rectangles are coloured according to the strength of correlation. Significant correlation estimates are marked by an asterisk. LSBMD = lumbar spine BMD; FNBMD = femoral neck BMD.
Extent of shared association signals
We found evidence for significant overlap of association signals at different P-value thresholds (Pt) between all three OA categories and LSBMD (permutation adjusted P-value (Pperm)<0.05) (Table 1).
Table 1.
SNP-based overlap analysis of OA and BMD. For each comparison, the total number of SNPs present in both datasets after LD-pruning is given, as well as the number of SNPs falling below each P-value threshold (Pt). Pperm = empirical overlap p-value obtained through permutation analysis
| Combined OA |
Hip OA |
Knee OA |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LSBMD |
FNBMD |
LSBMD |
FNBMD |
LSBMD |
FNBMD |
|||||||
| Total number of SNPs |
75,015 |
75,161 |
74,999 |
75,147 |
75,125 |
75,270 |
||||||
| Pt | SNPs | P_perm | SNPs | P_perm | SNPs | P_perm | SNPs | P_perm | SNPs | P_perm | SNPs | P_perm |
| 0.5 | 28,940 | 8.53 × 10−2 | 27,599 | 2.74 × 10−1 | 28,563 | 1.64 × 10−1 | 27065 | 7.33 × 10−1 | 28,910 | 7.95 × 10−1 | 27,534 | 5.84 × 10−2 |
| 0.1 | 2,633 | 7.92 × 10−2 | 2,119 | 5.41 × 10−1 | 2,513 | 1.46 × 10−2 | 2057 | 1.74 × 10−1 | 2,620 | 3.67 × 10−2 | 2,131 | 2.33 × 10−1 |
| 0.05 | 931 | 1.25 × 10−2 | 698 | 2.35 × 10−1 | 868 | 1.42 × 10−2 | 647 | 2.47 × 10−1 | 939 | 8.98 × 10−4 | 700 | 3.10 × 10−2 |
| 0.04 | 682 | 2.41 × 10−3 | 486 | 2.33 × 10−1 | 641 | 6.48 × 10−4 | 434 | 4.99 × 10−1 | 680 | 4.74 × 10−4 | 474 | 1.27 × 10−1 |
| 0.03 | 451 | 1.20 × 10−3 | 314 | 6.57 × 10−2 | 416 | 1.02 × 10−3 | 272 | 4.20 × 10−1 | 437 | 2.87 × 10−3 | 291 | 3.27 × 10−1 |
| 0.02 | 251 | 1.95 × 10−3 | 160 | 1.07 × 10−1 | 239 | 6.10 × 10−5 | 150 | 6.31 × 10−2 | 244 | 1.59 × 10−3 | 152 | 3.03 × 10−1 |
| 0.01 | 87 | 2.79 × 10−2 | 60 | 5.84 × 10−2 | 87 | 2.97 × 10−4 | 52 | 4.94 × 10−2 | 88 | 3.01 × 10−3 | 53 | 2.18 × 10−1 |
| 0.005 | 37 | 6.89 × 10−3 | 24 | 2.43 × 10−2 | 38 | 3.04 × 10−4 | 26 | 6.39 × 10−4 | 29 | 1.97 × 10−1 | 16 | 6.94 × 10−1 |
| 0.001 | 10 | 3.90 × 10−5 | 3 | 4.52 × 10−2 | 7 | 1.68 × 10−3 | 1 | 1.00 | 6 | 1.12 × 10−2 | 4 | 3.39 × 10−2 |
| 5 × 10−4 | 4 | 6.81 × 10−3 | 0 | 1.00 | 6 | 5.70 × 10−5 | 1 | 5.64 × 10−2 | 4 | 4.21 × 10−3 | 2 | 7.12 × 10−2 |
Analysis of the combined OA and LSBMD data resulted in significant overlap P-values at Pt=0.001 and 0.005, as well as at less stringent Pt. Four SNPs overlap at Pt=5×10−4 (rs17158899, rs4536164, rs11826287 and rs630765), one of which (rs11826287) is genome-wide significantly associated with FNBMD (P = 3.61×10−14) and maps to an intron in LRP5.
The highest overlap was observed between hip OA and LSBMD, with six SNPs overlapping at Pt=5×10−4 (Pperm=5.7×10−5). Two of these SNPs are genome-wide significantly associated with BMD in GEFOS (rs1524928, P = 5.29×10−9 and rs716255, P = 2.07×10−11). A significant overlap was also observed for Pt of 0.001, 0.005 and 0.01 in the hip OA-LSBMD comparison.
Compared to the hip OA and LSBMD analysis, overlap P-values for the knee OA and LSBMD comparison were at least one order of magnitude smaller. Four SNPs (rs7104420, rs9466056, rs881803 and rs4536164) overlapped at Pt=5×10−4 in this analysis (Pperm =4.21×10−3). Two of these, rs4536164 and rs9466056, fall within known BMD risk loci (18).
Overlap signal was much weaker for the OA and FNBMD comparisons, with only five Pt reaching statistical significance (Pt = 0.001 for knee OA, Pt = 0.005 and 0.01 for hip OA, and Pt = 0.001 and Pt = 0.005 for combined OA). The SNP overlapping at Pt=5×10−4 for hip OA and FNBMD (rs1524928) was also among the six SNPs identified in the hip OA and LSBMD analysis. Two SNPs overlapped at this Pt for knee OA, one being rs9466056 and the other rs1283614, which maps to an intron of the BMD locus MEF2C (24).
Evidence for colocalising regions
We employed a regional Bayesian colocalisation test that measures the posterior probabilities (PP) for each of four alternative hypotheses compared to one global null hypothesis (i.e. no associations in that region). We identified four independent genomic regions with a high posterior probability (PP) of harbouring one causal variant common to both traits analysed (PP for hypothesis 3 ≥ 0.9) (Table 2). The region containing the RPS6KA5 gene was identified by three comparisons (combined OA and LSBMD, hip OA and LSBMD, and hip OA and FNBMD). The most strongly associated SNPs in this region have genome-wide significant associations with increased BMD at both the LS and FN (rs1286147 and rs1286063, P < 5×10−8) and nominally significant associations with increased risk of combined and hip OA (rs1286077, P < 0.05); the three SNPs are in perfect LD (r2=1.00 for each pairwise combination).
Table 2.
Regions with strong evidence of pleiotropy. For each region the number of SNPs, start and stop position in basepairs (bp) and most strongly associated SNPs for OA and BMD are given. Chromosome coordinates are in hg19. Hypothesis 3 = one causal variant; hypothesis 4 = two distinct causal variants; PP = posterior probability
| Analysis | SNPs | Chr | Start (bp) | Stop (bp) | Top SNP BMD | Top SNP OA | PP | |
|---|---|---|---|---|---|---|---|---|
| Hypothesis 3 | combinedOA and LSBMD | 817 | chr14 | 91297823 | 93129850 | rs1286147; rs1286063 | rs1286077 | 0.95 |
| hipOA and FNBMD | 817 | chr14 | 91297823 | 93129850 | rs1286147 | rs1286077 | 0.98 | |
| hipOA and LSBMD | 817 | chr14 | 91297823 | 93129850 | rs1286147; rs1286063 | rs1286077 | 0.99 | |
| hipOA and LSBMD | 1242 | chr10 | 78708452 | 80875213 | rs7071206 | rs716255 | 0.92 | |
| hipOA and LSBMD | 531 | chr1 | 44974119 | 46897698 | rs7554123 | rs7545984 | 0.91 | |
| kneeOA and FNBMD | 1235 | chr6 | 19208477 | 21677746 | rs9466056 | rs9466056 | 0.99 | |
| Hypothesis 4 | hipOA and LSBMD | 268 | chr4 | 696848 | 1415698 | rs3755955 | rs3755920 | 0.97 |
| kneeOA and LSBMD | 382 | chr16 | 14464538 | 16152940 | rs4985155 | rs9935327 | 0.95 | |
| kneeOA and LSBMD | 1070 | chr6 | 150255029 | 151910904 | rs4869742 | rs9384514 | 0.90 |
Two further regions were identified in the hip OA and LSBMD analysis; one of these spans a known BMD locus close to the KCNMA1 gene, while the other does not contain genome-wide significant variants for either OA or BMD.
The region identified in the knee OA and FNBMD analysis contains one lead SNP for both traits, rs9466056, which is a known variant associated with high FN and LSBMD mapping to an intergenic region between CDKAL1 and SOX4.
We also identified three regions (Table 2) with a high PP of harbouring two distinct causal variants (PP for hypothesis 4 ≥ 0.9). All three of these contain a known BMD locus, with the top SNPs for LSBMD mapping to introns of IDUA, CCDC170 and PDXDC1. The top SNPs for knee and hip OA are nominally associated (P < 0.05) with these respective phenotypes in arcOGEN.
Gene and pathway analysis
Of the individual genes significantly associated (Q < 0.05) with at least one OA or BMD phenotype, SUPTH3, COL11A1, and APCDD1 overlapped between OA and BMD (Supplementary Material, Table S2). All three include variants that were identified in the SNP-wise overlap analysis and taken forward for replication.
There were no pathways significantly associated with any OA phenotype in any of the analyses. One of the CP pathways was associated with FNBMD (“basal cell carcinoma”, Q = 0.02) when allowing a 20 kilobase (kb) window around genes. Using GO annotations, a total of 33 unique pathways were associated with either BMD phenotype using strict or lenient gene definitions (Supplementary Material, Tables S3–S5), including several with direct biological relevance, such as “regulation of ossification” or “osteoblast development”.
Cross-phenotype meta-analysis
To search for potential novel associations not identified by single-trait GWAS, we performed a cross-phenotype meta-analysis between each pairwise combination of OA and BMD datasets. Using the CPASSOC method (25), we computed two statistics, Shom and Shet, which assume homogeneous and heterogeneous effects across studies, respectively. The quantile–quantile and Manhattan plots of these analyses are shown in Supplementary Material, Figures S1–S4. We identified 13 independent associations not previously reported for BMD or OA, which we followed up in the UK Biobank combined OA dataset (Supplementary Material, Table S10). One SNP, rs11164649, was nominally significant (P < 0.05). This SNP lies in an intron of the COL11A1 gene and is in strong LD (r2=0.92) with a variant (rs1903787) identified in the SNP-wise overlap analysis which was taken forward for replication.
Replication and meta-analysis for OA
We meta-analysed 143 SNPs identified in the colocalisation and/or P-value based overlap analysis in arcOGEN, UK Biobank and deCODE (Supplementary Material, Table S6). None of the SNPs taken forward are genome-wide significantly associated with arcOGEN. We found a significant excess of independent SNPs with the same direction of effect in all three meta-analysis cohorts among variants with Pmeta<0.05 (binomial sign test P = 7.75×10−11), as well as all independent SNPs included in the meta-analysis (binomial sign test P = 0.03).
Variants within several genes linked to bone, cartilage and extracellular matrix biology, including APCDD1, SUPTH3, COL11A1, NOTCH4, SEMA3A, LGR4, PTCH1 and RPS6KA5, were associated at Pmeta<0.05 (Supplementary Material, Table S6).
Two variants reached genome-wide significance in the meta-analysis across arcOGEN, deCODE and UK Biobank: rs12901071 (OR 1.08 95% CI 1.05–1.11, Pmeta=3.12×10−10) and rs10518707 (OR 1.07, 96% CI 1.03–1.09, Pmeta=2.15×10−8). Both are intronic variants in the SMAD3 gene (r2=0.645) and were identified in the SNP-wise overlap analysis of combined OA vs. LSBMD and hip OA vs. LSBMD, respectively.
Both new genome-wide significant SNPs for OA were imputed in the arcOGEN data (imputation info score > 0.95) and are nominally associated with combined OA (Supplementary Material, Table S6; Fig. S2). They are also nominally associated with increased LSBMD in GEFOS (rs12901071, P = 1.58×10−3 and rs10518707, P = 3.47×10−5) (Fig. 2), but not FNBMD (rs12901071, P = 3.72×10−1 and rs10518707, P = 2.46×10−1). SMAD3 is associated (P < 0.05) with LS and FNBMD, hip and combined OA in the gene analysis (Supplementary Material, Table S7), although this association only holds for LSBMD after using false discovery rate (FDR) correction (Q = 6.92×10−6).
Figure 2.
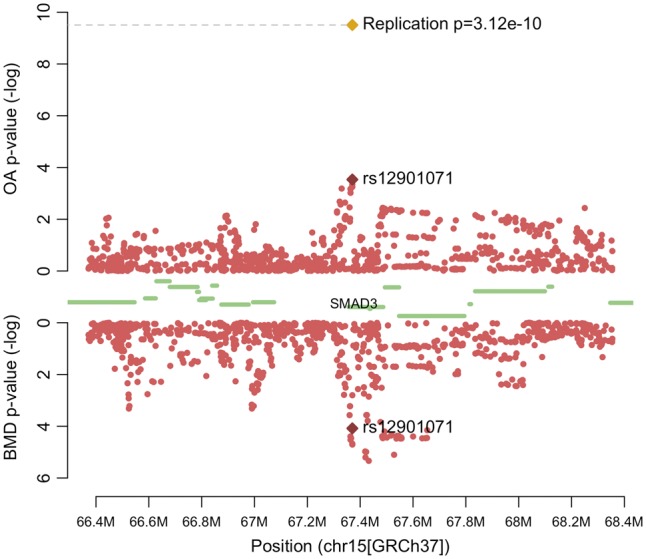
Regional association plot of SMAD3. The -log(p-values) of SNPs in the arcOGEN combined osteoarthritis (OA) data (top) and GEFOS lumbar spine bone mineral density (LSBMD) data (bottom) are plotted against their chromosomal position. The meta-analysis p-value of rs12901071 is plotted as a golden diamond. Protein coding genes are represented by green bars.
In the colocalisation analysis, the region in which both top SNPs reside (chr15:67,095,629-69,017,421) has a PP of containing a single pleiotropic variant associated with hip OA and LSBMD (hypothesis 3) of 0.88.
Functional follow-up of SMAD3
Using RNA sequencing data, we confirmed the expression of SMAD3 in low-grade degenerate articular cartilage of 12 knee and 9 hip OA patients undergoing total joint replacement (26) (Supplementary Material, Fig. S5). SMAD3 is among the 30% most expressed genes in the knee articular cartilage samples, and among the 15% most expressed genes in the hip articular cartilage samples.
Discussion
The analysis of shared genetic aetiology across epidemiologically linked traits can enhance power to identify disease variants and shed light into the biological mechanisms underpinning these associations. We conducted the first genome-wide overlap analysis of BMD and OA using summary statistics from the two largest GWAS of these two traits.
We identified 143 variants with evidence for association with OA and BMD. Many of these reside in or near biologically relevant genes, two of which (KLHL42/KLHDC5 and SUPT3H/RUNX2) are established loci for both traits (6,18). Variants in three loci (SUPTH3, APCDD1, and COL11A1) were also significantly associated with at least one OA and BMD phenotype in the gene analysis. APCDD1 is an inhibitor of WNT signaling (27), which is implicated in both OA and BMD. COL11A1 encodes collagen type 11, an important component of cartilage and bone, and has been associated with OA in a candidate gene meta-analysis (28). Other examples include the LGR4 gene, in which a rare variant in the Icelandic population has been associated with low BMD and osteoporotic fractures (29); and SEMA3A, which affects bone remodeling in rats (30).
We identified novel genome-wide significant associations at two intronic SNPs in SMAD3, and confirm expression of this gene in primary chondrocytes from articular cartilage of OA patients undergoing total joint replacement surgery. Activated SMAD3 acts downstream of TGF-β, repressing osteoblast differentiation and the production of bone matrix (31,32). It also represses the cartilage-degrading enzyme matrix metalloproteinase 13 in chondrocytes (32). Missense mutations in a conserved protein domain of SMAD3 have been linked to aneurysm-osteoarthritis syndrome, a congenital disorder characterised by arterial aneurysms, heart abnormalities and early-onset OA (33).
Due to its role in bone and cartilage biology, SMAD3 has been previously assessed in a candidate gene study of hip and knee OA (34). Despite their small sample size (number of cases < 400), the investigators found nominal associations (P < 0.05) for both OA phenotypes in their discovery, which were further strengthened in a meta-analysis (hip OA P = 4×10−4; knee OA P = 7.5×10−6). Notably, their top signal (rs12901499) maps to the same locus as our lead SNP (r2=0.645).
More recently, two studies have shown SMAD3 expression to be correlated with the genotype at a 3’UTR SNP (35), and to be significantly higher in cartilage from OA patients compared to healthy controls (36). The authors postulate that this could be a compensatory mechanism to counteract existing cartilage damage, or that SMAD3 expression levels outside a narrow range have detrimental effects.
We observed a stronger overlap between LSBMD and OA. The fact that only the correlation between combined OA and LSBMD was significant could be due to the bigger sample size in this OA dataset compared to the hip or knee OA data. While the FN- and LSBMD datasets are very similar in size, the knee OA and hip OA datasets each contain approximately half the number of cases compared to the combined OA dataset. This difference in power might at least partly explain why the genetic correlation estimates for joint-specific OA and LSBMD did not achieve statistical significance. Epidemiological data from the Chingford study have shown increased baseline BMD to be associated with incident radiographic knee OA, with the mean increase in LSBMD being approximately twice as high as the increase in FNBMD (10,15). Incident knee OA was also linked to higher baseline LSBMD, but not FNBMD, in the Baltimore Longitudinal Study of Ageing (37). The reasons for this differential association of FN- and LSBMD with OA remain unclear. One possible explanation could be the comorbidity of knee and spinal OA, characterised by spinal osteophytes, which could lead to increased LSBMD measurements. However, in one study, adjustment for the presence of osteophytes at the lumbar spine did not change the strength of association between OA and LSBMD (10). Damage to the spine accumulates over time and can lead to changes such as breakdown of the invertebral discs, scoliosis and osteochondrosis. This process is also referred to as degenerative disc disease (DDD). Although the association between DDD and LSBMD remains inconclusive (38–41), it is known that the presence of degenerative features can increase LSBMD measurements obtained via dual X-ray absorptiometry (42). While this may have contributed to the observed association between LSBMD and OA, we found genetic correlations of a similar magnitude between OA and skull, as well as total body BMD measurements in a paediatric cohort (43) (Supplementary Material, Table S11; Supplementary Methods). As DDD and related features such as osteophytes are unlikely to be present in young individuals, these results suggest that the correlation between OA and LSBMD is not purely artefactual.
Our analyses showed a greater degree of overlap between hip OA and both BMD measurements than between knee OA and BMD. Hip OA is estimated to have a higher heritability than knee OA (44), with environmental risk factors such as physical activity and BMI more strongly associated with the latter (45).
The analyses outlined here present the first comprehensive evaluation of genetic overlap between BMD and radiographic OA. Our results lend further support to the hypothesis of common genetic factors underlying these two traits, and establish SMAD3 as a genome-wide significant risk locus for OA with a potential pleiotropic effect on BMD.
Our work exemplifies the potential to uncover new disease risk loci by combining data of epidemiologically linked traits. Recent efforts in the development of statistical methods to detect pleiotropy, as well as the establishment of cross-disorder working groups (46) and of biobank collections (47), have made it possible to systematically assess pleiotropy in human disease genetics. Methods combining univariate summary statistics of different traits – such as the colocalisation analysis employed here (48) – often do not require a locus to be genome-wide significantly associated with any of the individual studies to detect a cross-phenotype association. Hence, they can increase power to identify associated variants or regions without the need to collect larger sample sizes (49).
There is a stark difference in sample size and, consequently, statistical power between the arcOGEN and GEFOS GWAS datasets. Larger datasets where phenotype information for both OA and BMD is available in the same individuals will be necessary to further disentangle the extent of shared genetics between them. Pinpointing the common biological pathways of these two complex traits will provide insight into the underlying mechanisms of OA, facilitating the identification of novel targets or drug repurposing opportunities for its treatment.
Materials and Methods
Datasets
All analyses outlined here were conducted using summary association statistics from the arcOGEN (6) and GEFOS consortia (18). The arcOGEN data comprised three OA phenotypes: knee OA, hip OA, and knee and/or hip OA (combined OA). A detailed description of the contributing studies and phenotype definitions can be found in (18). Briefly, OA case status was determined radiographically as a Kellgren-Lawrence grade score ≥ 2. Most cases included in arcOGEN had progressed to a severe disease endpoint, as evident from the fact that 80% had undergone total joint replacement surgery.
For the P-value-based overlap analysis as well as the gene and pathway analysis, we excluded samples from London-based cohorts (TwinsUK and Chingford Study) from the arcOGEN datasets to avoid overlap with GEFOS. After exclusion of 714 samples, we carried out genome-wide association analyses on the arcOGEN dataset for each of the three phenotype groups using the “–method score” option in SNPTEST v2.5 (50). The full arcOGEN dataset was used for all other analyses described.
The BMD data consisted of meta-analysis summary statistics for FN and LSBMD (18).
For replication, we used summary statistics from two OA GWAS: the UK Biobank (47) and the deCODE study. Details on case definitions in UK Biobank and deCODE can be found in the Supplementary Methods.
Sample sizes for each dataset are outlined in Supplementary Material, Table S8.
Genome-wide correlation
We performed LD score regression analysis (51) on each pairwise combination between the BMD and OA datasets, using pre-computed LD scores based on the European sample of the 1000 Genomes Project (52). We accounted for sample overlap between each pair of datasets through tetrachoric correlation of the Z-scores of SNPs present in both studies.
Assessment of shared association signals
For each pairwise combination between the two BMD and three OA phenotypes we assessed the extent of shared signals at different p-value cutoffs, following the approach described by Elliott and colleagues (53). Briefly, we filtered both datasets to a common set of SNPs on which p-value-informed LD pruning was performed. To this end, SNPs were sorted based on their association with OA, and, starting with the lowest p-value, any SNP in LD with the index SNP (r2>0.05) was removed. The next SNP was then considered, and so on.
The extent of shared association signals between OA and BMD was assessed by counting the number of overlapping variants above and below ten different p-value thresholds (Pt: 0.5, 0.1, 0.05, 0.04, 0.03, 0.02, 0.01, 0.005, 0.001, 5 × 10−5). To test for significance of overlap, a chi-squared test on the resulting 2×2 contingency tables was performed at each Pt.
Empirical overlap p-values were obtained by repeating the chi-squared test after randomly permuting the GEFOS p-values. This was done 1,000,000 times to obtain a null distribution of overlap p-values against which the original overlap p-value could be compared.
Colocalisation analysis
We employed a Bayesian colocalisation method to search for genomic regions harbouring cross-phenotype associations between OA and BMD (48). Briefly, the algorithm uses Z-scores and standard errors from two association studies to generate posterior probabilities for each of five hypotheses:
H0: the region contains no variants associated with trait 1 or trait 2
H1: the region contains one variant associated with trait 1
H2: the region contains one variant associated with trait 2
H3: the region contains one variant associated with both trait 1 and trait 2
H4: the region contains one variant associated with trait 1 and a second variant associated with trait 2
Genomic regions were defined according to approximately independent LD-blocks (54). Analogous to LD score regression, we used the tetrachoric correlation of the Z-scores of SNPs present in datasets as an estimate of sample overlap.
Gene and pathway analysis
Gene- and pathway analyses were performed on each OA and BMD dataset using MAGMA (55). First, SNPs are assigned to genes, which are tested for their association with the phenotype. Results from this step are then combined into pathway-based association statistics.
For the gene analysis, we grouped variants into genes using SNP locations from dbSNP version 135 and NCBI 37.3 gene definitions. We performed this step twice, once annotating SNPs to a gene only if they fell within the gene’s transcription start and stop site, and once including SNPs that fell within a 20 kilobase window of the gene.
We ran two separate pathway analyses, one using the Molecular Signatures Database canonical pathways collection (56), comprising 1,329 manually curated gene-sets from nine databases (Supplementary Material, Table S9), and one using 6,166 gene-sets from the Gene Ontology pathway database (57).
Significance was defined using a 5% FDR equivalent to a Q-value of 0.05 for both the gene and pathway analyses (58).
Cross-phenotype meta-analysis
We used a multi-trait extension to meta-analysis to search for novel associations in each pairwise combination of arcOGEN and GEFOS datasets (25). The method, CPASSOC, requires only summary statistics and generates two test statistics: the first, Shom, assumes homogeneous effects across studies and is equivalent to performing an inverse variance weighted meta-analysis if no sample overlap between the studies exists. The second, Shet, is more powerful if effects are heterogeneous between studies. Both statistics require the specification of a correlation matrix between the included studies. To calculate this, we followed the approach by Park et al. (59), using all independent SNPs (r2 < 0.2) present in both the OA and BMD dataset that were not associated with either trait (−1.96 > Z score < 1.96), and taking the Pearson’s correlation of their Z-scores (25).
We performed an in-silico lookup in the UK Biobank hip and/or knee OA data of top SNPs with P < 5×10−8 in any of the CPASSOC analyses that did not fall into known OA or BMD loci.
Replication and meta-analysis for OA
We took forward a total of 143 SNPs for in silico replication. This set comprises the two most strongly associated variants (one for each trait) in each region from the Bayesian colocalisation test, as well as all variants overlapping at Pt=0.005 in the SNP-based overlap analyses. We used the METAL (60) software package to perform inverse variance weighted meta-analysis of these SNPs in relation to hip and/or knee OA using summary statistics from the arcOGEN combined OA dataset (including London samples), the UK Biobank (47) and the deCODE (61) study.
Functional follow-up of SMAD3
Samples were obtained and processed as in Ref. 25. Briefly, articular cartilage was obtained from 12 patients undergoing total joint replacement for knee OA, and 9 patients for hip OA. Cartilage was graded using the OARSI cartilage classification system (62,63).
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
This study utilised genotype data from arcOGEN (http://www.arcogen.org.uk/) funded by a special purpose grant from Arthritis Research UK (grant 18030).
The population controls in arcOGEN were from the 1958 British Birth Cohort collection funded by the Medical Research Council (grant G0000934) and The Wellcome Trust (grant 068545) and from the UK Blood Services Collection of Common Controls funded by The Wellcome Trust. This research utilizes resources provided by the Type 1 Diabetes Genetics Consortium, a collaborative clinical study sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), National Human Genome Research Institute (NHGRI), National Institute of Child Health and Human Development (NICHD), and Juvenile Diabetes Research Foundation International (JDRF) and supported by U01 DK062418. F.R. and K.T. received funding from the Netherlands Organization for Health Research and Development (ZonMw VIDI 460 016.136.367). K.T. received funding from the mobility stimuli plan of the European Union 461 Erasmus Mundus Action 2: ERAWEB programme.
R.A.B received funding from the National Institute for Health Research (Cambridge Biomedical Research Centre).
This study makes use of data generated by the Wellcome Trust Case-Control Consortium (the 1958 British Birth Cohort collection and the UK Blood Services Collection). A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk.
We would like to acknowledge the Cambridge Biomedical Research Center Human Research Tissue Bank for storing the hip gene expression and methylation replication samples.
This research has been conducted using the UK Biobank Resource under Application Number 9979.
Conflict of Interest statement. U.S., U.T. and K.S. are employed by deCODE Genetics/Amgen. All other authors declare no competing interests.
Funding
This work was funded by the Wellcome Trust (WT098051). Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
References
- 1. Zhang Y., Jordan J.M. (2010) Epidemiology of osteoarthritis. Clin. Geriatr. Med., 26, 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glyn-Jones S., Palmer A.J.R., Agricola R., Price A.J., Vincent T.L., Weinans H., Carr A.J. (2015) Osteoarthritis. Lancet, 386, 376–387. [DOI] [PubMed] [Google Scholar]
- 3. Hochberg M.C., Yerges-Armstrong L., Yau M., Mitchell B.D. (2013) Genetic epidemiology of osteoarthritis: recent developments and future directions. Curr. Opin. Rheumatol., 25, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zengini E., Finan C., Wilkinson J.M. (2016) The genetic epidemiological landscape of hip and knee osteoarthritis: Where are we now and where are we going?. J. Rheumatol., 43, 260–266. [DOI] [PubMed] [Google Scholar]
- 5. Panoutsopoulou K., Southam L., Elliott K.S., Wrayner N., Zhai G., Beazley C., Thorleifsson G., Arden N.K., Carr A., Chapman K.. et al. (2011) Insights into the genetic architecture of osteoarthritis from stage 1 of the arcOGEN study. Ann. Rheum. Dis., 70, 864–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zeggini E., Panoutsopoulou K., Southam L., Rayner N.W., Day-Williams A.G., Lopes M.C., Boraska V., Esko T., Evangelou E., Hofman A.. et al. (2012) Identification of new susceptibility loci for osteoarthritis (arcOGEN): A genome-wide association study. Lancet, 380, 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sivakumaran S., Agakov F., Theodoratou E., Prendergast J.G., Zgaga L., Manolio T., Rudan I., McKeigue P., Wilson J.F., Campbell H. (2011) Abundant pleiotropy in human complex diseases and traits. Am. J. Hum. Genet., 89, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Foss M.V., Byers P.D. (1972) Bone density, osteoarthrosis of the hip, and fracture of the upper end of the femur. Ann. Rheum. Dis., 31, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nevitt M.C., Zhang Y., Javaid M.K., Neogi T., Curtis J.R., Niu J., McCulloch C.E., Segal N.A., Felson D.T. (2010) High systemic bone mineral density increases the risk of incident knee OA and joint space narrowing, but not radiographic progression of existing knee OA: the MOST study. Ann. Rheum. Dis., 69, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hart D.J., Mootoosamy I., Doyle D.V., Spector T.D. (1994) The relationship between osteoarthritis and osteoporosis in the general population: the Chingford Study. Ann. Rheum. Dis., 53, 158–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hannan M.T., Anderson J.J., Zhang Y., Levy D., Felson D.T. (1993) Bone mineral density and knee osteoarthritis in elderly men and women. the framingham study. Arthritis Rheum., 36, 1671–1680. [DOI] [PubMed] [Google Scholar]
- 12. Hardcastle S. a., Dieppe P., Gregson C.L., Smith G.D., Tobias J.H. (2015) Osteoarthritis and bone mineral density : are strong bones bad for joints ?. Bonekey Rep., 4, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hardcastle S. a., Dieppe P., Gregson C.L., Arden N.K., Spector T.D., Hart D.J., Edwards M.H., Dennison E.M., Cooper C., Sayers A.. et al. (2015) Individuals with high bone mass have an increased prevalence of radiographic knee osteoarthritis. Bone, 71, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bettica P., Cline G., Hart D.J., Meyer J., Spector T.D. (2002) Evidence for increased bone resorption in patients with progressive knee osteoarthritis: Longitudinal results from the Chingford study. Arthritis Rheum., 46, 3178–3184. [DOI] [PubMed] [Google Scholar]
- 15. Hart D.J., Cronin C., Daniels M. (2002) The relationship of bone density and fracture to incedent and progressive radiographic osteoarthritis of the knee. Arthritis Rheum., 46, 92–99. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y., Hannan M.T., Chaisson C.E., McAlindon T.E., Evans S.R., Aliabadi P., Levy D., Felson D.T. (2000) Bone mineral density and risk of incident and progressive radiographic knee osteoarthritis in women: the Framingham Study. J. Rheumatol., 27, 1032–1037. [PubMed] [Google Scholar]
- 17. Bergink A.P., Uitterlinden A.G., Van Leeuwen J.P.T.M., Hofman A., Verhaar J.A.N., Pols H.A.P. (2005) Bone mineral density and vertebral fracture history are associated with incident and progressive radiographic knee osteoarthritis in elderly men and women: the Rotterdam Study. Bone, 37, 446–456. [DOI] [PubMed] [Google Scholar]
- 18. Estrada K., Styrkarsdottir U., Evangelou E., Hsu Y.H., Duncan E.L., Ntzani E.E., Oei L., Albagha O.M.E., Amin N., Kemp J.P.. et al. (2012) Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet., 44, 494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng H., Forgetta V., Hsu Y., Estrada K., Rosello‐Diez A., Leo P.J., Dahia C.L., Park‐Min K.H., Tobias J.H., Kooperberg C.. et al. (2015) Whole‐genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature, 526, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geusens P.P., van den Bergh J.P. (2016) Osteoporosis and osteoarthritis: shared mechanisms and epidemiology. Curr. Opin. Rheumatol., 28, 97–103. [DOI] [PubMed] [Google Scholar]
- 21. Balooch G., Balooch M., Nalla R.K., Schilling S., Filvaroff E.H., Marshall G.W., Marshall S.J., Ritchie R.O., Derynck R., Alliston T. (2005) TGF-beta regulates the mechanical properties and composition of bone matrix. Proc. Natl. Acad. Sci. U. S. A, 102, 18813–18818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Panoutsopoulou K., Zeggini E. (2013) Advances in osteoarthritis genetics. J. Med. Genet., 50, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yerges-Armstrong L.M., Yau M.S., Liu Y., Krishnan S., Renner J.B., Eaton C.B., Kwoh C.K., Nevitt M.C., Duggan D.J., Mitchell B.D.. et al. (2014) Association analysis of BMD-associated SNPs with knee osteoarthritis. J. Bone Miner. Res., 29, 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rivadeneira F., Styrkársdottir U., Estrada K., Halldórsson B.V., Hsu Y.-H., Richards J.B., Zillikens M.C., Kavvoura F.K., Amin N., Aulchenko Y.S.. et al. (2009) Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat. Genet., 41, 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu X., Feng T., Tayo B.O., Liang J., Young J.H., Franceschini N., Smith J. a., Yanek L.R., Sun Y.V., Edwards T.L.. et al. (2015) Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am. J. Hum. Genet., 96, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steinberg J., Ritchie G.R.S., Roumeliotis T.I., Jayasuriya R.L., Brooks R.A., Binch A.L.A., Shah K.M., Coyle R., Pardo M., Le Maitre C.L.. et al. (2016) Integrative epigenomics, transcriptomics and proteomics of patient chondrocytes reveal genes and pathways involved in osteoarthritis. bioRxiv, https://doi.org/10.1101/038067. [DOI] [PMC free article] [PubMed]
- 27. Shimomura Y., Agalliu D., Vonica A., Luria V., Wajid M., Baumer A., Belli S., Petukhova L., Schinzel A., Brivanlou A.H.. et al. (2010) APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature, 464, 1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodriguez-Fontenla C., Calaza M., Evangelou E., Valdes A.M., Arden N., Blanco F.J., Carr A., Chapman K., Deloukas P., Doherty M.. et al. (2014) Assessment of osteoarthritis candidate genes in a meta-analysis of nine genome-wide association studies. Arthritis Rheumatol., 66, 940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Styrkarsdottir U., Thorleifsson G., Sulem P., Gudbjartsson D.F., Sigurdsson A., Jonasdottir A., Jonasdottir A., Oddsson A., Helgason A., Magnusson O.T.. et al. (2013) Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature, 497, 517–520. [DOI] [PubMed] [Google Scholar]
- 30. Fukuda T., Takeda S., Xu R., Ochi H., Sunamura S., Sato T., Shibata S., Yoshida Y., Gu Z., Kimura A.. et al. (2013) Sema3A regulates bone-mass accrual through sensory innervations. Nature, 497, 490–493. [DOI] [PubMed] [Google Scholar]
- 31. Alliston T., Choy L., Ducy P., Karsenty G., Derynck R., Akiyoshi S., Inoue H., Hanai J., Kusanagi K., Nemoto N.. et al. (2001) TGF-beta-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. embo J., 20, 2254–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen C.G., Thuillier D., Chin E.N., Alliston T. (2012) Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum., 64, 3278–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van de Laar I.M.B.H., Oldenburg R.A., Pals G., Roos-Hesselink J.W., de Graaf B.M., Verhagen J.M.A., Hoedemaekers Y.M., Willemsen R., Severijnen L.-A., Venselaar H.. et al. (2011) Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet., 43, 121–126. [DOI] [PubMed] [Google Scholar]
- 34. Valdes A.M., Spector T.D., Tamm A., Kisand K., Doherty S.A., Dennison E.M., Mangino M., Tamm A., Kerna I., Hart D.J.. et al. (2010) Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheum., 62, 2347–2352. [DOI] [PubMed] [Google Scholar]
- 35. Raine E.V.A., Reynard L.N., van de Laar I.M.B.H., Bertoli-Avella A.M., Loughlin J. (2014) Identification and analysis of a SMAD3 cis-acting eQTL operating in primary osteoarthritis and in the aneurysms and osteoarthritis syndrome. Osteoarthr. Cartil., 22, 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aref-Eshghi E., Liu M., Razavi-Lopez S.B., Hirasawa K., Harper P.E., Martin G., Furey A., Green R., Sun G., Rahman P.. et al. (2016) SMAD3 is upregulated in human osteoarthritic cartilage independent of the promoter DNA methylation. J. Rheumatol., 43, 388–394. [DOI] [PubMed] [Google Scholar]
- 37. Hochberg M.C., Lethbridge-Cejku M., Tobin J.D. (2004) Bone mineral density and osteoarthritis: Data from the Baltimore Longitudinal Study of Aging. Osteoarthr. Cartil., 12, 45–48. [DOI] [PubMed] [Google Scholar]
- 38. Harada A., Okuizumi H., Miyagi N., Genda E. (1998) Correlation between bone mineral density and intervertebral disc degeneration. Spine (Phila. Pa. 1976), 23, 857–861. [DOI] [PubMed] [Google Scholar]
- 39. Grams A.E., Rehwald R., Bartsch A., Honold S., Freyschlag C.F., Knoflach M., Gizewski E.R., Glodny B. (2016) Correlation between degenerative spine disease and bone marrow density: a retrospective investigation. BMC Med. Imaging, 16, 17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pye S.R., Reid D.M., Adams J.E., Silman A.J., O’Neill T.W. (2006) Radiographic features of lumbar disc degeneration and bone mineral density in men and women. Ann. Rheum. Dis., 65, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miyakoshi N., Itoi E., Murai H., Wakabayashi I., Ito H., Minato T. (2003) Inverse relation between osteoporosis and spondylosis in postmenopausal women as evaluated by bone mineral density and semiquantitative scoring of spinal degeneration. Spine (Phila. Pa. 1976), 28, 492–495. [DOI] [PubMed] [Google Scholar]
- 42. Rand T., Seidl G., Kainberger F., Resch A., Hittmair K., Schneider B., Glüer C.C., Imhof H. (1997) Impact of spinal degenerative changes on the evaluation of bone mineral density with dual energy X-ray absorptiometry (DXA). Calcif. Tissue Int., 60, 430–433. [DOI] [PubMed] [Google Scholar]
- 43. Kemp J.P., Medina-Gomez C., Estrada K., St Pourcain B., Heppe D.H.M., Warrington N.M., Oei L., Ring S.M., Kruithof C.J., Timpson N.J.. et al. (2014) Phenotypic dissection of bone mineral density reveals skeletal site specificity and facilitates the identification of novel loci in the genetic regulation of bone mass attainment. PLoS Genet., 10, e1004423.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Valdes A.M., Spector T.D. (2011) Genetic epidemiology of hip and knee osteoarthritis. Nat. Rev. Rheumatol., 7, 23–32. [DOI] [PubMed] [Google Scholar]
- 45. Blagojevic M., Jinks C., Jeffery A., Jordan K.P. (2010) Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis. Osteoarthr. Cartil., 18, 24–33. [DOI] [PubMed] [Google Scholar]
- 46. Smoller J.W. (2013) Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet, 381, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sudlow C., Gallacher J., Allen N., Beral V., Burton P., Danesh J., Downey P., Elliott P., Green J., Landray M.. et al. (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med., 12, e1001779.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pickrell J., Berisa T., Segurel L., Tung J.Y., Hinds D. (2015) Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet., 48, 709–717. [DOI] [PMC free article] [PubMed]
- 49. Solovieff N., Cotsapas C., Lee P.H., Purcell S.M., Smoller J.W. (2013) Pleiotropy in complex traits: challenges and strategies. Nat. Rev. Genet., 14, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marchini J., Howie B., Myers S., McVean G., Donnelly P. (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet., 39, 906–913. [DOI] [PubMed] [Google Scholar]
- 51. Bulik-Sullivan B., Finucane H.K., Anttila V., Gusev A., Day F.R., Loh P.-R. ReproGen Consortium, Psychiatric Genomics Consortium, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3 Duncan L.. et al. (2015) An atlas of genetic correlations across human diseases and traits. Nat. Genet., 47, 1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bulik-Sullivan B.K., Loh P.-R., Finucane H.K., Ripke S., Yang J. Schizophrenia Working Group of the Psychiatric Genomics Consortium Patterson N., Daly M.J., Price A.L., Neale B.M. (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., advance on, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Elliott K.S., Chapman K., Day-Williams A., Panoutsopoulou K., Southam L., Lindgren C.M., Arden N., Aslam N., Birrell F., Carluke I.. et al. (2013) Evaluation of the genetic overlap between osteoarthritis with body mass index and height using genome-wide association scan data. Ann. Rheum. Dis., 72, 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berisa T., Pickrell J.K. (2016) Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics, 32, 283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. de Leeuw C.A., Mooij J.M., Heskes T., Posthuma D. (2015) MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol., 11, e1004219.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.. et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci., 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T.. et al. (2000) Gene ontology: tool for the unification of biology. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B, 57, 289–300. [Google Scholar]
- 59. Park H., Li X., Song Y.E., He K.Y., Zhu X. (2016) Multivariate analysis of anthropometric traits using summary statistics of genome-wide association studies from GIANT Consortium. PLoS One, 11, e0163912.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Willer C.J., Li Y., Abecasis G.R. (2010) METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gudbjartsson D.F., Helgason H., Gudjonsson S.A., Zink F., Oddson A., Gylfason A., Besenbacher S., Magnusson G., Halldorsson B.V., Hjartarson E.. et al. (2015) Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet., 47, 435–444. [DOI] [PubMed] [Google Scholar]
- 62. Mankin H.J., Dorfman H., Lippiello L., Zarins A. (1971) Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips: II. correlation of morphology with biochemical and metabolic data. J. Bone Jt. Surg., 53, 523–537. [PubMed] [Google Scholar]
- 63. Pearson R.G., Kurien T., Shu K.S.S., Scammell B.E. (2011) Histopathology grading systems for characterisation of human knee osteoarthritis – reproducibility, variability, reliability, correlation, and validity. Osteoarthr. Cartil., 19, 324–331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.