

Abstract
Mutations in the mitochondrial DNA polymerase, POLG, are associated with a variety of clinical presentations, ranging from early onset fatal brain disease in Alpers syndrome to chronic progressive external ophthalmoplegia. The majority of mutations are linked with disturbances of mitochondrial DNA (mtDNA) integrity and maintenance. On a molecular level, depending on their location within the enzyme, mutations either lead to mtDNA depletion or the accumulation of multiple mtDNA deletions, and in some cases these molecular changes can be correlated to the clinical presentation. We identified a patient with a dominant p.Y955H mutation in POLG, presenting with a severe, early-onset multi-systemic mitochondrial disease with bilateral sensorineural hearing loss, cataract, myopathy, and liver failure. Using a combination of disease models of Drosophila melanogaster and in vitro biochemistry analysis, we compare the molecular consequences of the p.Y955H mutation to the well-documented p.Y955C mutation. We demonstrate that both mutations affect mtDNA replication and display a dominant negative effect, with the p.Y955H allele resulting in a more severe polymerase dysfunction.
Introduction
The mitochondrial DNA polymerase γ (POLγ) is required for replication of the mitochondrial genome (mtDNA). The holoenzyme consists of the catalytic subunit POLγA, encoded by the POLG gene (MIM 174763), and by the dimeric processivity factor POLγB, encoded by the POLG2 gene (MIM 604983) (1). POLγA belongs to the family A polymerases, with an N-terminal 3′–5′ exonuclease domain, a central linker domain and a C-terminal polymerase domain (2). Replication of the mitochondrial genome is independent of the cell cycle with individual mtDNA molecules being randomly selected for replication, a phenomenon referred to as relaxed replication (3–6). The total mtDNA copy number, however, is maintained at a relatively constant level.
Defects in mtDNA replication or nucleotide metabolism can lead to rearrangements, deletions, point mutations, or depletion of mtDNA, often resulting in mitochondrial dysfunction and ultimately mitochondrial disease (7). Although several factors involved in mtDNA replication have been associated with mitochondrial diseases, mutations in POLG are most common with close to 230 different disease-causing mutations reported (8) [recently summarized in (9)]. The associated clinical symptoms can be quite variable, both with respect to disease onset and clinical presentation, and cause a number of different disease entities, such as Alpers syndrome (MIM 203700), mitochondrial neurogastrointestinal encephalopathy (MNGIE: MIM 613662), sensory ataxic neuropathy, dysarthria and ophthalmoparesis (SANDO: MIM 607459), spinocerebellar ataxia-epilepsy (SCAE: MIM 607459), and chronic progressive external ophthalmoplegia (CPEO: MIM 157640 and MIM 258450). On a molecular level, mutations in POLG lead to the accumulation of multiple mtDNA deletions or mtDNA depletion (5,9), which in turn cause reduced oxidative phosphorylation (OXPHOS).
CPEO is the most common mitochondrial myopathy, defined by a progressive bilateral ptosis and diffuse, symmetric reduction in ocular motility, often associated with additional symptoms, e.g. hearing loss and ataxia [reviewed in (10)]. About half of all CPEO cases are inherited and disease-causing mutations at seven different loci have so far been identified, including loci coding for the mitochondrial adenine nucleotide translocator 1 (SLC25A4: MIM 103220) (11), the mitochondrial DNA helicase, TWINKLE (TWNK: MIM 606075) (12) and POLγA (13). The p.Y955C mutation of human POLG results in adult onset autosomal dominant CPEO (13) as well as premature ovarian failure (14) and is associated with the accumulation of multiple mtDNA deletions in affected patients (13,15). Mutagenesis experiments identified Y955, together with residues R943, L947 and A957 of POLγA to be essential for nucleotide specificity, and processivity (16–21), with the Y955C mutation resulting in replicative stalling and formation of multiple mtDNA deletions (21).
We here identify an autosomal dominant p.Y955H mutation in POLG, leading to a severe multi-systemic mitochondrial disease with bilateral sensorineural hearing loss, cataract, myopathy and liver failure in a paediatric patient. Using disease models of Drosophila melanogaster (Dm) and in vitro characterization, we demonstrate that the p.Y955C and p.Y955H mutations both affect mtDNA replication and have a dominant negative effect on DNA synthesis. The p.Y955H mutation resulted in a more severe dysfunction than p.Y955C, in agreement with the clinical phenotype of the patient, which is unusually severe for a dominant POLG disease.
Results
Subjects
Subject 1 presented with progressive PEO and ovarian failure with infertility in adulthood. Measurements of mitochondrial OXPHOS function in a skeletal muscle biopsy showed a normal result (Fig. 1A and B), although morphological analysis revealed a high number of COX negative muscle fibres (Fig. 1C), as well as paracrystalline inclusions by electron microscopy (Supplementary Material, Fig. S1A), indicating mitochondrial dysfunction.
Figure 1.

Characterization of mitochondrial function in skeletal muscle. (A) Mitochondrial ATP production rate (MAPR) in fresh skeletal muscle mitochondria, isolated from subject 1, using six different substrate combinations as indicated. Results are presented as the ATP synthesis rate (units) per unit of CS activity (control n=11; age 0–5 years). (B) Respiratory chain enzyme activities of complexes I and III (NADH:cytochrome c reductase), complex I (NADH:coenzyme Q reductase), complexes II and III (succinate:cytochrome c reductase, SCR), complex IV (cytochrome c oxidase) and citrate synthase (CS) were determined. Results are presented as percentage of mean control (n=9; age 0–5 years) values. The range of control values is depicted as ± 2SD. (C) COX/SDH double staining of fresh-frozen skeletal muscle from subject 1. (D) MAPR as in (A) from subject 2. (E) Respiratory chain enzyme activities as in (B) from subject 2. (F) COX/SDH double staining of fresh-frozen skeletal muscle from subject 2. (G) Gomori-trichrome staining of fresh-frozen skeletal muscle from subject 2 revealing ragged-red fibres (RRF). (H) Electropherogram of POLγA from subject 1, 2 and control.
Subject 2 presented with a slowly progressive multi-systemic disorder at the age of 8 months, with weight loss, bilateral sensorineural hearing loss, bilateral cataract, myopathy, liver failure and feeding difficulties that required a percutaneous endoscopic gastrostomy. He had a normal intellectual capacity and development. Measurements of mitochondrial OXPHOS activity in a skeletal muscle biopsy was unremarkable at 10 months of age, but a reduced ATP production rate was determined in a second biopsy around 1 year later (Fig. 1D and E). Morphological analysis confirmed a mitochondrial dysfunction, exhibiting a high number of COX negative muscle fibres (Fig. 1F) and ragged-red fibres (Fig. 1G).
The p.Y955H mutation in POLγA is associated with early onset multi-systemic mitochondrial disease
Sequencing of the POLG locus in subject 1 revealed that the patient was heterozygous for the previously reported c.2864A>G, p.Y955C, mutation (13) (Fig. 1H). Southern blot analysis showed the accumulation of multiple mtDNA deletions (Supplementary Material, Fig. S1B), while mtDNA sequencing was unremarkable. Sequencing of POLG in subject 2 detected a previously unreported mutation, c.2863T>C, p.Y955H, additionally revealing a heterozygous p.Q1236H mutation, previously shown to be benign (22). Detailed allele-specific analysis revealed that the p.Y955H mutation was in cis with p.Q1236H on the paternal POLG allele but absent in the father (Table 1). The p.Y955H mutation had thus occurred de novo. Multiplex ligation-dependent probe amplification (MLPA) and cDNA analysis of POLG did not detect any other disease-causing mutation in the gene. Whole exome sequencing on DNA samples from subject 2, was performed as described previously (23,24), but revealed no other potentially disease-causing mutation involved in metabolic or mitochondrial diseases.
Table 1.
Nucleotide and amino acid changes in POLG in subject 2 and his parents
| Nucleotide | Protein | |
|---|---|---|
| Subject 2 | c.[2863T>C; 3708G>T];[=] | p.[Y955H; Q1236H];[=] |
| Father | c.[3708G>T];[=] | p.[Q1236H];[=] |
| Mother | c.[=];[=] | p.[=];[=] |
Previous reports suggested that mutations affecting position Y955 in human POLγA can lead to an increase in incorrect nucleotide incorporation, but cloning and sequencing of mtDNA from human muscle samples showed no difference in point mutation load, when compared with control samples (Supplementary Material, Table S1).
Mutations at p.Y873 are recessive lethal in Drosophila melanogaster
In order to understand the molecular and physiological consequences of the p.Y955H mutation we generated Drosophila melanogaster (Dm) models for both the p.Y955C and p.Y955H mutations (Y955 in human POLγA corresponds to Y873 in DmPOLγA). To this end we targeted the Dm tamas locus, coding for DmPOLγA, following a previously described procedure (25) (for details see materials and methods) (Supplementary Material, Fig. S2).
Flies heterozygous for the p.Y873C and p.Y873H mutations did not show any obvious phenotypic abnormalities. Eclosure rates were comparable to control flies (Fig. 2A), although flies heterozygous for p.Y873C mutation were developmentally delayed (Fig. 2B). Lifespans (Fig. 2C) were normal even after 15 generations of intercrossing (Supplementary Material, Fig. S3), whereas Dm lines homozygous for either of the two mutations were larval lethal at the third instar larval stage (Fig. 2D).
Figure 2.

Eclosion rates and life-spans in flies carrying the p.Y873C or p.Y873H mutations. (A) Eclosure rates, (B) life-span, (C) developmental time of F0 and F1 flies. (D) L3 larvae of control (wt/wt) and homozygous mutant (Y873C/Y873C and Y873H/Y873H) larvae at 4 days after egg laying.
The p.Y873H and p.Y873C mutations lead to mtDNA depletion in Drosophila melanogaster
The p.Y955C mutation causes multiple mtDNA deletions in humans (13,15) as observed in muscle samples from subject 1 (Supplementary Material, Fig. S1B). In contrast, no multiple deletions were observed in muscle samples from subject 2, carrying the p.Y955H mutation (Supplementary Material, Fig. S1B). Interestingly, flies carrying the p.Y873H or p.Y873C mutation did not present multiple deletions; neither as heterozygous flies (Fig. 3A) nor as homozygous larvae (Fig. 3B). However, qRT-PCR and Southern blot analysis revealed severe mtDNA depletion in L3 larvae homozygous for either of the two mutants, with a milder reduction in the heterozygous state (Fig. 3C and D). Heterozygous flies (p.Y873H or p.Y873C) showed an mtDNA depletion, which was somewhat more pronounced after 15 generations intercrossing (Fig. 3C).
Figure 3.

Mutations at p.Y873 lead to mtDNA depletion in flies. (A) Southern blot analysis of mtDNA of heterozygous mutant flies. (B) Long-range PCR of homozygous mutant and control larvae. Primers were situated as indicated. (C) Southern blot analysis of mtDNA from homozygous mutant larvae. (D) Relative mtDNA levels determined by qPCR in L3 larvae (L0), or flies intercrossed for 1 (F1) or 15 (F15) generations. TaqMan probes used as indicated. Error bars are ±SD (*P<0.05, **P<0.01, ***P<0.001, n=5).
POLγ:Y955H has lower affinity to DNA than WT POLγ or POLγ:Y955C
We next investigated the biochemical consequences of the p.Y955H mutation and for comparison we also analysed the previously characterized p.Y955C mutation (20,21). To this end, wild-type POLγA protein and mutant derivatives (POLγA:Y955C and POLγA:Y955H) were expressed and purified. DNA binding properties of all three POLγA versions were first assessed in the absence or presence of recombinant POLγB, using an electrophoretic mobility shift assay (EMSA). Both POLγA:Y955C and POLγA:Y955H bound to a primed DNA template independently of POLγB (Fig. 4A). However, the Kd (equilibrium dissociation constant) for binding to the template, was higher for POLγA:Y955H than for WT POLγA and POLγA:Y955C (Supplementary Material, Fig. S4).
Figure 4.

In vitro DNA binding and polymerization properties of POLγA:Y955C and POLγA:Y955H. (A) Electrophoretic mobility shift assays showing that POLγA:Y955C and POLγA:Y955H bind DNA both in absence (lanes 5 and 8) and presence (lanes 6 and 9) of POLγB. Lanes 1, 4 and 7 contained no protein. Lower panel: schematic representation of the DNA template. The asterisk indicates the 32P label on the 5´ end of the 20-mer. (B) Coupled 3’–5’ exonuclease/polymerase assays show that both POLγA:Y955C and POLγA:Y955H required higher dNTP concentrations than WT POLγA to synthesize DNA (all experiments were performed in the presence of POLγB). Reactions were run on denaturing 15% PAGE. Below: scheme of the primer-extension reaction starting from the 20-meric primer to produce a 35-mer product.
Modifications at position Y955 are associated with difficulties of incorporating dATP
We previously demonstrated that the p.Y955C mutation leads to stalling at low dNTP concentrations. POLγA:Y955C is especially sensitive to low dATP concentrations, and the enzyme enters a polymerase/exonuclease idling mode at dATP insertion sites (21). To investigate if this is a general phenotype for mutations affecting Y955, we performed primer extension experiments, using two different primed DNA templates (Fig. 4B). In the absence of dNTPs, WT POLγA will use its 3′–5′ exonuclease activity to digest the labelled primer, but in the presence of dNTPs, the polymerase will initiate primer elongation. In this assay, POLγA:Y955H behaved as previously demonstrated for POLγA:Y955C and required higher dNTP concentrations to produce full-length products (Fig. 4B, upper panel). Increasing the number of thymines on the template strand, promoted stalling of both mutant polymerases (Fig. 4B, lower panel), consistent with preferred stalling at dATP insertion sites (Fig. 4B, compare upper and lower panel) (21).
POLγA:Y955H has a dominant negative effect on the replisome at low dNTP concentrations
In the presence of mtSSB and the TWINKLE helicase, WT POLγA in complex with POLγB is able to replicate a circular dsDNA template containing a preformed replication fork (Fig. 5A, lanes 1–4 and 13–16) (26). Neither POLγA:Y955H nor POLγA:Y955C could support DNA synthesis (Fig. 5A, lanes 5–8 and 17–20, respectively). In agreement with previous reports, POLγA:Y955C displayed a dominant negative effect on DNA synthesis in the presence of WT POLγA (Fig. 5A, lanes 21–24) (21). The dominant negative effect was less pronounced with POLγA:Y955H even when the mutant was added at a 3:1 molar ratio relative to WT POLγA (compare lanes 9–12 with lanes 21–24 in Fig. 5A). Postmitotic tissues contain lower dNTP concentrations than the 10 µM used here (27,28). We therefore repeated the experiment using 1 µM dNTP. At this concentration, POLγA:Y955H also displayed a dominant negative effect on WT POLγA activity (Fig. 5B, lanes 9–12). Furthermore, we also observed an accumulation of shorter-than-input length DNA products (Fig. 5B, lane 12), suggesting that POLγA:Y955H causes template degradation at lower dNTP concentrations.
Figure 5.

Rolling circle replication assay reveals dominant negative effect of POLγA:Y955C and POLγA:Y955H. (A) In vitro replication reactions performed at 10 µM dNTP and indicated POLγ versions (all experiments were performed in the presence of POLγB). Lanes 1–4, WT POLγA; lanes 5–8; POLγA:Y955H; lanes 9–12, WT POLγA and POLγA:Y955H; lanes 13–16, WT POLγA; lanes 17–20; POLγA:Y955C; lanes 21–24, WT and POLγA:Y955C. (B) As in (A) but at 1 µM dNTP.
POLγA:Y955C and POLγA:Y955H display reduced ligation efficiency
At the end of replication, DNA ends are ligated to produce a closed circular mtDNA molecule. We have previously shown that the 3′–5′ exonuclease and 5′–3′ polymerase activities of POLγA must be correctly balanced in order to support ligation (29). Since mutations affecting position Y955 shift the balance towards the exonuclease activity, we decided to monitor effects on ligation. To this end we performed coupled polymerase-ligation assays, using a template consisting of a radioactively 5′-labelled 60-mer hybridized to a circular 100-mer (Fig. 6A). On this template, DNA polymerase needs to extend the 3′ end to full circle and then terminate at the downstream 5′ end, in order to produce a ligateable nick. WT POLγA extended the 60-mer and provided ligateable ends already at 1 µM dNTP (Fig. 6B, lanes 2–4). The POLγA:Y955C mutant was also capable of generating ligateable full-length products, but it needed 100 times more dNTP, which reasons well with its lower dNTP affinity (Fig. 6B, lanes 5–7). The POLγA:Y955H mutant on the other hand had problems to fully extend the 60-mer even in the presence of 100 µM dNTP and were therefore unable to create ligateable ends (Fig. 6B, lanes 8–10). Here again, we observed that both mutants have increased exonuclease activity at low dNTP concentrations (see Fig. 6B, lanes 5 and 8). Next, we asked whether the presence of the mutants could affect the ability of the WT to create ligateable ends, which would reflect a typical situation in vivo. To answer this question, we performed time-dependent ligation assays at 1 µM dNTPs, where WT POLγA was mixed with either of the two mutants (Fig. 6C). At this dNTP concentration only the WT protein is able to extend the primer. Addition of POLγA:Y955C or POLγA:Y955H severely inhibited polymerization and the ability of WT POLγA to create ligateable nicks (Fig. 6C and D).
Figure 6.

Effects of POLγA:Y955C and POLγA:Y955H on ligation efficiency. (A) Schematic picture showing the template used in the coupled DNA synthesis-ligation assay. (B) Coupled DNA synthesis-ligation assay using different amounts of dNTPs (1, 10, or 100 µM) and indicated POLγ versions (all experiments were performed in the presence of POLγB). Lane l, input template: lanes 2–4, WT POLγA; lanes 5–7, POLγA:Y955C and lanes 8–10 POLγA:Y955H. (C) As in (B) but was performed as a time-course experiment (5, 15, 30 or 45 min) using 1 µM dNTPs and at a 1:1 ratio of indicated POLγ versions. Lanes 1, 6 and 11, input template; lanes 2–5, WT POLγA; lanes 7–10, WT POLγA and POLγA:Y955C; lanes 12–15, WT POLγA and POLγA:Y955H. (D) Ratio of ligated product compared with total DNA between different mixing-reactions of POLγ versions. Based on quantifications of band-intensity from reactions as shown in (C). Solid black bars are WT POLγA alone, grey bars are WT POLγA mixed with POLγA:Y955C and white bars represent WT POLγA mixed with POLγA:Y955H. Error-bars are standard deviations from three independent reactions. (E) Molecular model of Y955 and surrounding amino acids. Upper panel, WT POLγA; lower left panel, POLγA:Y955C; lower right panel, POLγA:Y955H.
Discussion
Mitochondrial diseases form a highly diverse group, with a range of clinical symptoms and different ages of onset. They are predominantly monogenic disorders and recent advances in sequencing technologies have revolutionized the diagnosis of patients with mitochondrial diseases. Despite these advances, the correlation between genotype and phenotype remains difficult to predict and combined with the large heterogeneity of the human genome makes functional validation of novel disease variants essential. A complicating factor is that a biochemical defect is not always observed in patient samples (30–32). To date, almost 230 mutations have been identified in the human POLG gene alone (8), with both dominant and recessive inheritance patterns. A substantial number of mutations are also reported to only prompt clinical symptoms, when inherited in trans with other mutations as compound heterozygous, and it is thus important to fully characterise variants. We here used a combination of in vitro biochemistry, model organisms and patient investigations to demonstrate pathogenicity of a mutation in POLG, which causes a tyrosine to histidine change at position 955 in the human POLγA protein.
The tyrosine residue at position 955 of POLγA is essential for nucleotide incorporation into the growing DNA strand. An interaction between Y955 and E895 is required for the binding of the incoming nucleotide (Fig. 6E). The hydroxyl group of Y955 interacts with the carboxyl group of E895, which is located adjacent to the catalytic helix (residues 943–955). In T7 DNA polymerase this glutamate selectively stabilizes the incoming nucleotide. The polar contacts, rotomer conformation and steric influence of E895 likely contribute to the formation of a tight binding pocket. In addition, Y955 may aid catalysis via E895-mediated alignment of the catalytic helix towards the nucleotide.
The severity of the p.Y955C and p.Y955H mutations is due, in part, to the steric influence of Y955 in the active site (33). The large phenyl ring excludes solvent from the active site, while providing a tight binding pocket for the nucleotide. Removal of this phenyl ring results in a loss of favourable π–π stacking interactions between the incoming base and Y955 (negatively affecting nucleotide binding), which likely destabilises the active site. Differences in Kd were also observed between the p.Y955C and p.Y955H mutated versions of POLγ, with the latter having 5-fold weaker binding to a primed DNA template. This is not surprising given that the introduction of a polar histidine into an apolar buried cavity can negatively affect both protein stability and ligand binding (34–36). Replacement of Y955 with histidine is thus likely responsible for (i) loss of steric packing, (ii) loss of a stabilizing hydrogen bond with E895, (iii) loss of π-stacking interactions with Y951 and (iv) electrostatic repulsion within the binding cavity (Fig. 6E, lower left panel).
The p.Y955C mutation leads to reduced polymerase activity and replication stalling (16–21). Previous data also suggested that the p.Y955C mutation could cause increased mtDNA mutation loads (37–40), but this conclusion was not supported by data obtained here from human muscle.
The p.Y955C mutation is consistently associated with multiple deletions in mtDNA, but we could not reproduce this phenotype in flies harbouring the p.Y955C mutation. Neither did the p.Y955H mutation lead to multiple deletions in either human skeletal muscle samples or in the corresponding fly model. However, this is not surprising, since it has been shown that the number of deletion-molecules increases with age. The lack of deletion in the flies and in the p.Y955H patient could simply reflect the short lifespan of flies and the very young age of the patient. In humans, multiple mtDNA deletions do not become visible in Southern blots until ≈20 years of age (41). Nevertheless, we observed mtDNA depletion with both p.Y873C and p.Y873H, consistent with observations made in murine and yeast models of the p.Y955C mutation (18,37–40). Depletion is also consistent with results from the in vitro characterization of the mutants. Both POLγA:Y955C and POLγA:Y955H were unable to synthesise DNA and had a dominant negative effect on the DNA synthesis in the presence of WT POLγA. We favour a model in which the mutant proteins are competing with WT POLγA for the primer terminus, thereby slowing down replication fork progression (Fig. 7).
Figure 7.
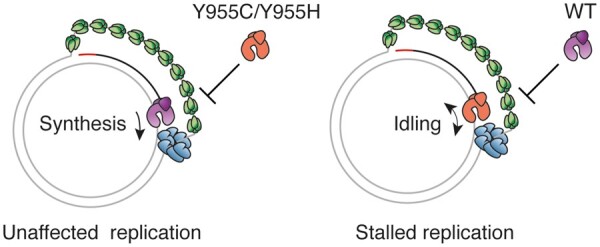
A schematic model for the effects of mutant POLγA:Y955C and POLγA:Y955H on replication fork progression. The mitochondrial replication machinery at the fork is dynamic and POLγ is regularly coming on and off the 3’ end of the newly synthesized DNA. When WT POLγ (purple) is present at the replication fork of a heterozygous patient, it prevents mutant POLγ (orange) from binding and mtDNA synthesis can therefore progress (left panel). Once mutant POLγ binds the primed template, it enters an idling mode (repeated cycles of incorporation and degradation at the primer terminus) and simultaneously blocks the WT protein from accessing the 3’ end (right panel). mtSSB is green and TWINKLE is blue.
As demonstrated here, the p.Y955C and p.Y955H mutations cause related, but distinct molecular phenotypes. Whereas POLγA:Y955H is unable to synthesise DNA, POLγA:Y955C has strongly impaired DNA synthesis activity that can be partly overcome at high dNTP concentrations. Furthermore, POLγA:Y955C has a stronger affinity for primed DNA templates, compared with POLγA:Y955H. Even if the molecular differences are subtle, they do lead to fundamentally different clinical presentations. Our data therefore further demonstrates the complexity of mitochondrial diseases and how difficult it is to predict clinical phenotypes based on molecular defects in these patients.
Materials and Methods
The Regional Ethics Committee at Karolinska Institutet approved the use of patient material in this study.
DNA and RNA isolation from patients
Genomic DNA from the patients was isolated from skeletal muscle, blood or fibroblasts, using QIAamp DNA Mini Kit (Qiagen). For RNA analysis, total RNA was isolated from fibroblasts using RNeasy Mini Kit (Qiagen).
Molecular analysis in patients
The entire mtDNA sequence from the patients was determined from total DNA isolated from skeletal muscle, while the POLG gene was sequenced from total DNA isolated from the patient’s blood. MtDNA was amplified by PCR in 28 overlapping M13-tailed fragments as described before (42). The coding exons 2–23 of POLG were amplified by PCR using M13-tagged intronic primers. Subsequent sequencing of all PCR products was carried out with M13 primers, using the BigDye version 3.1 sequencing kit (Applied Biosystems) on a 3130xl Genetic Analyser (Applied Biosystems). MtDNA sequence data were compared with the revised Cambridge reference sequence for human mtDNA (GenBank NC_012920.1) and for POLG to the reference sequence NM_002693.2. MLPA analysis, using kit P010 (MRC Holland, Amsterdam), for detection of deletions or duplications of each exon of the POLG gene was performed according to the manufacturer’s protocol as described earlier (43). RT-PCR was performed on isolated RNA, using GeneAmp RNA PCR kit (Applied Biosystems) and exons 2–23 of POLG cDNA were amplified by PCR in 10 overlapping fragments. The size and amount of the PCR products was compared with a control sample in 2% agarose gel. Sequencing of the PCR products from cDNA was performed as described for genomic DNA above. Southern blot analysis was done using aliquots of 0.2 µg total DNA isolated from skeletal muscle, which were digested with the restriction enzyme PvuII (New England Biolabs) and fractionated by electrophoresis in 0.5% agarose gels. The DNA was then transferred to Hybond XL nylon filter (GE Healthcare) by capillary blotting under standard procedures. The filter was hybridized with an equimolar mix of radiolabelled mtDNA probes corresponding to nucleotides 1–12 640, as described previously (44).
Expression and purification of recombinant human proteins
Mutated versions of POLγA were constructed using the QuikChange Lightning Site-directed mutagenesis kit according to the provided protocol (Agilent Technologies) and confirmed by sequencing (Eurofins MWG Operon). Recombinant baculoviruses coding for TWINKLE, mitochondrial single-stranded DNA-binding protein (mtSSB), WT POLγA, POLγA:Y955C, POLγA:Y955H and POLγB were expressed in Sf9 cells and purified as previously described (26).
EMSA and Kd determination
The affinity between POLγ (POLγA in complex with POLγB) and a primer template was monitored using an EMSA as previously described (25). Each experiment was repeated at least three times. Band intensities representing unbound and bound DNA were quantified using Fujifilm Multi Gauge V3.1 software. The fraction of DNA bound in each reaction was plotted versus the concentration of POLγ. Data were fitted with the binding equation (Fraction bound = (MaxB × [POLγ])/(MaxB + [POLγ]) using EXCELs Add-in ‘Solver’ to perform non-linear regression and obtain values for Kd (as the value corresponding to the midpoint of MaxB) and using MaxB set to 1 (the fraction bound at which the data plateaus).
Coupled 3′–5′ exonuclease/polymerase assays
A 20-mer (5′-CGG TCG AGT CTA GAG GAG CC-3′) labelled at the 5′ end with [γ-32P] ATP was annealed to a 35-mer oligonucleotide containing a poly-dT stretch, 5′-TTT TTT TTT TAT CCG GGC TCC TCT AGA CTC GAC CG-3′, or an oligonucleotide without a poly-dT stretch (5′-GAC AAC CAG CAG CCG GGC TCC TCT AGA CTC GAC CG-3′, as illustrated in Fig. 4B. These two templates were used as substrates to investigate DNA polymerization and 3′–5′ exonuclease activity as previously described (21).
In vitro rolling circle DNA replication
The reaction mixtures (20 μl) contained 10 fmol of rolling circle template and reactions were performed as described previously except that lower dNTP concentrations (1 µM or 10 µM) were used. POLγ (75 fmol WT POLγA in complex with 300 fmol POLγB, 225 fmol POLγA:Y955C in complex with 675 fmol POLγB, or 225 fmol POLγA:Y955H in complex with 675 fmol POLγB) were added as indicated in the figure legends (26).
Coupled DNA synthesis-ligation assay
The synthesis-ligation assay was adapted from (29). Here a circular DNA template was used instead of a linear (Fig. 4a). The closed circular template was constructed by circularizing the 100 nt oligonucleotide (5′-GAG GGG TAT GTG GCC ACA GCA CTT AAA CAC ATC TCT GCC AAA CCC AAA AAC AAA GAA CCC TAA CAC CAG CCT AAC CAG ATT TCA AAT TTC ATA CCC CTA T-3′) with CircLigase TMssDNA ligase (Epicentre), followed by annealing to a 32P-labelled 60-mer (5′-TCT GGT TAG GCT GGT GTT AGG GTT CTT TGT TTT TGG GTT TGG CAG AGA TGT GTT TAA GTG-3′). The reactions were performed in a volume of 20 µl containing 20 mM Tris–HCl (pH 7.5), 1 mM DTT, 0.1 mg/ml BSA, 10 mM MgCl2, 0.5 mM ATP, 4 units of T4 DNA ligase, 10 fmol circular DNA substrate, indicated POLγ version, and varying amounts of dNTPs as specified in the figure legends. In the mixing experiments, 75 fmol WT POLγA, POLγA:Y955H, or POLγA:Y955C in complex with 300 fmol POLγB were pre-incubated in the mixture on ice for 10 min before an additional 75 fmol of WT POLγA in complex with 300 fmol POLγB was added to each reaction. Reactions were incubated for 30 min if nothing else is indicated in the figure legends and terminated with 2× stop buffer (Formamide with 10 mM EDTA, 0.025% bromophenol blue, 0.025% xylene cyanol). Samples were run in 7 M urea/10% polyacrylamide gels and visualized with a PhosphorImager or autoradiography.
Drosophila stocks and maintenance
All genomically engineered fly strains were constantly backcrossed into a white Dahomey Wolbachia-free (wDahT) WT strain. All fly stocks were maintained at 25 °C on a 12:12 h light/dark cycle with 60% humidity and fed on a sugar/yeast/agar (SYA) diet (25).
Generation of genomically engineered DmPOLγA flies
Human POLG sequence (NP_002684.1) was aligned to the Drosophila melanogaster homolog (tamas; NP_476821.1) and the fly-equivalent position for p.Y955 was identified at position p.Y873. Genetically modified flies were generated essentially as described previously (25,45). We previously cloned the Tamas locus into the fly-specific targeting vector pGEattB-GMR (25), which was here modified by site-directed mutagenesis to introduce the p.Y873C and p.Y873H mutations. Mutant variants of the tamas gene were then injected into the embryos of DmPolγA (Tamas) KO founder embryos (25), expressing ϕ31 integrase by the in-house Drosophila transgenic core facility. Positive flies were selected by eye colour and confirmed by Southern blot/sequencing.
Life-span determination
Newly eclosed adult flies were mated for 2 days before they were sorted for lifespan analyses. Two hundred females per genotype were used at a density of 10 flies per vial. Flies were transferred to new vials every 2–3 days and dead flies were counted.
Hatching and eclosure rates
Fly development was assessed as follows. Flies were allowed to lay eggs on grape juice agar plates for 2 h. One hundred eggs per genotype were individually picked and placed into vials with SYA food. The number of eclosed flies was scored every 12 h. At least five biological replicates were done for each genotype.
DNA isolation and southern blot analysis from flies
For relative mtDNA copy number determination, total DNA extractions were prepared from L3 larvae using DNeasy Blood and Tissue Kit (Qiagen). Five biological replicates, each with 10 larvae, were prepared for each genotype. Quantification of mtDNA levels was done using SYBR-Green qPCR analyses and primers targeting the mitochondrial encoded genes 12S, COX1 and CytB, and normalized to the nuclear encoded histone gene. All data were normalized against wild-type levels.
For Southern blot analysis, total DNA was extracted from 20 to 30 L3 larvae, either using the DNeasy Blood and Tissue kit (Qiagen), or by homogenizing samples with a tissue grinder in 400 µl of buffer A (100 mM Tris–HCl, pH 7.5; 100 mM EDTA; 100 mM NaCl and 0.5% SDS). After incubation at 65 °C for 30 min, 800 µl of freshly prepared Buffer B (4 µl of 5 M KOAc and 10 µl of 6 M lithium chloride) was added and samples were left on ice for 120 min. After incubation, samples were centrifuged at 12 000g for 15 min and supernatant was transferred into a new tube. About 540 µl of isopropanol was added to the supernatant and samples were further centrifuged at 12 000g for 15 min. Pellets were washed with 70% ethanol, dried and resuspended in 100 µl nuclease-free water containing 20 mg/ml RNase A. After incubation at 37 °C for 1 h, samples were stored at +4 °C.
Approximately 1–3 µg of total DNA was cut using either XhoI or StyI restriction endonuclease (NEB). Digestions were run on 0.8% agarose gels, and blotted to Hybond-N membrane (Amersham Bioscience). COXI or ND6-CytB (positions 10169–11169) were used as probes, and signals were visualized by autoradiography. 32P-labelling of Southern probes was done according to manufacturer’s instructions (Prime-IT II Random Prime Labelling Kit, Agilent).
Long-Range PCR was performed using primers: Dmel_Long 1: 5’AAT TAA GCT ACT GGG TTC ATA CCC C3’, Dmel_Long 2: 5’GCT CTA AAA TAT GTA CAC ATC GCC C3’, Dmel_Long 3: 5’TTT GAA GCA GCT GCA TGA TAT TGA C3’ and Dmel_Long 4: 5’AGA GTT TGA CAT TGA AGA TGT TAT GGA G3’.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank all affected individuals and their families for their participation and for providing important samples for the present research study.
Conflict of Interest statement. None declared.
Funding
This study was supported by the Swedish Research Council [A.W. (VR521-2012-2571), A.We. (VR521-2013-2341) and M.F. (VR521-2013-3621)]; Stockholm County Council [A.W. (K0176-2012) and A.We. (20140053)]; Swedish Foundation for Strategic Research [A.W. (ICA 12-0017)]; Knut & Alice Wallenberg Foundation [A.W. and A.We. (KAW 2013.0026) and M.F. (KAW 2011 and KAW 2014]); The Swedish Cancer Foundation [M.F. (CAN 2016/816]; European Research Council [M.F.]; The Swedish Brain Foundation [A.We. (FO2015-0146)]. A.W. is a Ragnar Söderberg fellow in Medicine (M77/13). Funding to pay the Open Access publication charges for this article was provided by the Swedish Research Council (VR521-2012-2571).
Contributor Information
Triinu Siibak, Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, University of Gothenburg, Gothenburg SE-405 30, Sweden
Paula Clemente, Max Planck Institute Biology of Ageing - Karolinska Institutet Laboratory, Division of Metabolic Diseases, Department of Laboratory Medicine, Karolinska Institutet, Stockholm, SE-171 77, Sweden, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm SE-171 77, Sweden,
Ana Bratic, Department of Mitochondrial Biology, Max Planck Institute for Biology of Ageing, Cologne D-50931, Germany,.
Helene Bruhn, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm SE-171 77, Sweden,; Centre for Inherited Metabolic Diseases, Karolinska University Hospital, Stockholm SE-171 76, Sweden,
Timo E.S. Kauppila, Department of Mitochondrial Biology, Max Planck Institute for Biology of Ageing, Cologne D-50931, Germany,
Bertil Macao, Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, University of Gothenburg, Gothenburg SE-405 30, Sweden.
Florian A. Rosenberger, Max Planck Institute Biology of Ageing - Karolinska Institutet Laboratory, Division of Metabolic Diseases, Department of Laboratory Medicine, Karolinska Institutet, Stockholm, SE-171 77, Sweden, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm SE-171 77, Sweden,
Nicole Lesko, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm SE-171 77, Sweden,; Centre for Inherited Metabolic Diseases, Karolinska University Hospital, Stockholm SE-171 76, Sweden,
Rolf Wibom, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm SE-171 77, Sweden,; Centre for Inherited Metabolic Diseases, Karolinska University Hospital, Stockholm SE-171 76, Sweden,
Karin Naess, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm SE-171 77, Sweden,; Centre for Inherited Metabolic Diseases, Karolinska University Hospital, Stockholm SE-171 76, Sweden,
Inger Nennesmo, Department of Pathology, Karolinska University Hospital, SE-171 77 Stockholm, Sweden.
Anna Wedell, Max Planck Institute Biology of Ageing - Karolinska Institutet Laboratory, Division of Metabolic Diseases, Department of Laboratory Medicine, Karolinska Institutet, Stockholm, SE-171 77, Sweden,; Centre for Inherited Metabolic Diseases, Karolinska University Hospital, Stockholm SE-171 76, Sweden, Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm SE-171 76, Sweden
Bradley Peter, Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, University of Gothenburg, Gothenburg SE-405 30, Sweden.
Christoph Freyer, Max Planck Institute Biology of Ageing - Karolinska Institutet Laboratory, Division of Metabolic Diseases, Department of Laboratory Medicine, Karolinska Institutet, Stockholm, SE-171 77, Sweden,; Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm SE-171 77, Sweden, Centre for Inherited Metabolic Diseases, Karolinska University Hospital, Stockholm SE-171 76, Sweden,
Maria Falkenberg, Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, University of Gothenburg, Gothenburg SE-405 30, Sweden.
Anna Wredenberg, Max Planck Institute Biology of Ageing - Karolinska Institutet Laboratory, Division of Metabolic Diseases, Department of Laboratory Medicine, Karolinska Institutet, Stockholm, SE-171 77, Sweden,; Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm SE-171 77, Sweden, Centre for Inherited Metabolic Diseases, Karolinska University Hospital, Stockholm SE-171 76, Sweden,
References
- 1. Lee Y.-S., Kennedy W.D., Yin Y.W. (2009) Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell, 139, 312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Longley M.J., Nguyen D., Kunkel T.A., Copeland W.C. (2001) The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem., 276, 38555–38562. [DOI] [PubMed] [Google Scholar]
- 3. Birky C.W. (1994) Relaxed and stringent genomes: why cytoplasmic genes don‘t obey Mendel’s laws. J. Hered., 85, 355–365. [Google Scholar]
- 4. Clayton D.A. (1992) Transcription and replication of animal mitochondrial DNAs. Int. Rev. Cytol., 141, 217–232. [DOI] [PubMed] [Google Scholar]
- 5. Falkenberg M., Larsson N.-G., Gustafsson C.M. (2007) DNA replication and transcription in mammalian mitochondria. Annu. Rev. Biochem., 76, 679–699. [DOI] [PubMed] [Google Scholar]
- 6. Gustafsson C.M., Falkenberg M., Larsson N.-G. (2016) Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem., 85, 133–160. [DOI] [PubMed] [Google Scholar]
- 7. Zeviani M., Di Donato S. (2004) Mitochondrial disorders. Brain, 127, 2153–2172. [DOI] [PubMed] [Google Scholar]
- 8.Human DNA polymerase gamma mutation database human DNA polymerase gamma mutation database. https://tools.niehs.nih.gov/polg/.
- 9. Young M.J., Copeland W.C. (2016) Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev., 38, 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McClelland C., Manousakis G., Lee M.S. (2016) Progressive external ophthalmoplegia. Curr. Neurol. Neurosci. Rep., 16, 53.. [DOI] [PubMed] [Google Scholar]
- 11. Kaukonen J., Juselius J.K., Tiranti V., Kyttälä A., Zeviani M., Comi G.P., Keränen S., Peltonen L., Suomalainen A. (2000) Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science, 289, 782–785. [DOI] [PubMed] [Google Scholar]
- 12. Spelbrink J.N., Li F.Y., Tiranti V., Nikali K., Yuan Q.P., Tariq M., Wanrooij S., Garrido N., Comi G., Morandi L. et al. (2001) Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet., 28, 223–231. [DOI] [PubMed] [Google Scholar]
- 13. Van Goethem G., Dermaut B., Löfgren A., Martin J.J., Van Broeckhoven C. (2001) Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet., 28, 211–212. [DOI] [PubMed] [Google Scholar]
- 14. Pagnamenta A.T., Taanman J.-W., Wilson C.J., Anderson N.E., Marotta R., Duncan A.J., Bitner-Glindzicz M., Taylor R.W., Laskowski A., Thorburn D.R. et al. (2006) Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum. Reprod., 21, 2467–2473. [DOI] [PubMed] [Google Scholar]
- 15. Van Goethem G., Martin J.J., Löfgren A. (1997) Unusual presentation and clinical variability in Belgian pedigrees with progressive external ophthalmoplegia and multiple deletions of mitochondrial DNA. Eur. J. Neurol., 4, 476–484. [Google Scholar]
- 16. Lim S.E., Ponamarev M.V., Longley M.J., Copeland W.C. (2003) Structural determinants in human DNA polymerase gamma account for mitochondrial toxicity from nucleoside analogs. J. Mol. Biol., 329, 45–57. [DOI] [PubMed] [Google Scholar]
- 17. Graziewicz M.A., Longley M.J., Bienstock R.J., Zeviani M., Copeland W.C. (2004) Structure-function defects of human mitochondrial DNA polymerase in autosomal dominant progressive external ophthalmoplegia. Nat. Struct. Mol. Biol., 11, 770–776. [DOI] [PubMed] [Google Scholar]
- 18. Lewis W., Day B.J., Kohler J.J., Hosseini S.H., Chan S.S.L., Green E.C., Haase C.P., Keebaugh E.S., Long R., Ludaway T. et al. (2007) Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab. Invest., 87, 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Graziewicz M.A., Bienstock R.J., Copeland W.C. (2007) The DNA polymerase gamma Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7,8-dihydro-8-oxo-2'-deoxyguanosine. Hum. Mol. Genet., 16, 2729–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Estep P.A., Johnson K.A. (2011) Effect of the Y955C mutation on mitochondrial DNA polymerase nucleotide incorporation efficiency and fidelity. Biochemistry, 50, 6376–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Atanassova N., Fusté J.M., Wanrooij S., Macao B., Goffart S., Bäckström S., Farge G., Khvorostov I., Larsson N.-G., Spelbrink J.N. et al. (2011) Sequence-specific stalling of DNA polymerase γ and the effects of mutations causing progressive ophthalmoplegia. Hum. Mol. Genet., 20, 1212–1223. [DOI] [PubMed] [Google Scholar]
- 22. Luoma P.T., Luo N., Löscher W.N., Farr C.L., Horváth R., Wanschitz J., Kiechl S., Kaguni L.S., Suomalainen A. (2005) Functional defects due to spacer-region mutations of human mitochondrial DNA polymerase in a family with an ataxia-myopathy syndrome. Hum. Mol. Genet., 14, 1907–1920. [DOI] [PubMed] [Google Scholar]
- 23. Stranneheim H., Engvall M., Naess K., Lesko N., Larsson P., Dahlberg M., Andeer R., Wredenberg A., Freyer C., Barbaro M. et al. (2014) Rapid pulsed whole genome sequencing for comprehensive acute diagnostics of inborn errors of metabolism. BMC Genomics, 15, 1090.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Freyer C., Stranneheim H., Naess K., Mourier A., Felser A., Maffezzini C., Lesko N., Bruhn H., Engvall M., Wibom R. et al. (2015) Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet., 52, 779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bratic A., Kauppila T.E.S., Macao B., Grönke S., Siibak T., Stewart J.B., Baggio F., Dols J., Partridge L., Falkenberg M. et al. (2015) Complementation between polymerase- and exonuclease-deficient mitochondrial DNA polymerase mutants in genomically engineered flies. Nat. Commun., 6, 8808.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korhonen J.A., Pham X.H., Pellegrini M., Falkenberg M. (2004) Reconstitution of a minimal mtDNA replisome in vitro. EMBO J., 23, 2423–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song S., Pursell Z.F., Copeland W.C., Longley M.J., Kunkel T.A., Mathews C.K. (2005) DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc. Natl. Acad. Sci. USA, 102, 4990–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gandhi V.V., Samuels D.C. (2011) A review comparing deoxyribonucleoside triphosphate (dNTP) concentrations in the mitochondrial and cytoplasmic compartments of normal and transformed cells. Nucleosides Nucleotides Nucleic Acids, 30, 317–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Macao B., Uhler J.P., Siibak T., Zhu X., Shi Y., Sheng W., Olsson M., Stewart J.B., Gustafsson C.M., Falkenberg M. (2015) The exonuclease activity of DNA polymerase γ is required for ligation during mitochondrial DNA replication. Nat. Commun., 6, 7303.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zeviani M., Moraes C.T., DiMauro S., Nakase H., Bonilla E., Schon E.A., Rowland L.P. (1988) Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology, 38, 1339–1346. [DOI] [PubMed] [Google Scholar]
- 31. Kurt B., Jaeken J., Van Hove J., Lagae L., Löfgren A., Everman D.B., Jayakar P., Naini A., Wierenga K.J., Van Goethem G., Copland W.C., DiMauro S. (2010) A novel POLG gene mutation in 4 children with Alpers-like hepatocerebral syndromes. Arch Neurol., 67, 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Uusimaa J., Gowda V., McShane A., Smith C., Evans J., Shrier A., Narasimhan M., O’Rourke A., Rajabally Y., Hedderly T., Cowan F., Fratter C. et al. (2013) Prospective study of POLG mutations presenting in children with intractable epilepsy: prevalence and clinical features. Epilepsia, 54, 1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kool E.T. (2002) Active site tightness and substrate fit in DNA replication. Annu. Rev. Biochem., 71, 191–219. [DOI] [PubMed] [Google Scholar]
- 34. Shoichet B.K., Baase W.A., Kuroki R., Matthews B.W. (1995) A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. USA, 92, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merski M., Shoichet B.K. (2012) Engineering a model protein cavity to catalyze the Kemp elimination. Proc. Natl. Acad. Sci. USA, 109, 16179–16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Merski M., Shoichet B.K. (2013) The impact of introducing a histidine into an apolar cavity site on docking and ligand recognition. J. Med. Chem., 56, 2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stuart G.R., Santos J.H., Strand M.K., van Houten B., Copeland W.C. (2006) Mitochondrial and nuclear DNA defects in Saccharomyces cerevisiae with mutations in DNA polymerase gamma associated with progressive external ophthalmoplegia. Hum. Mol. Genet., 15, 363–374. [DOI] [PubMed] [Google Scholar]
- 38. Baruffini E., Lodi T., Dallabona C., Puglisi A., Zeviani M., Ferrero I. (2006) Genetic and chemical rescue of the Saccharomyces cerevisiae phenotype induced by mitochondrial DNA polymerase mutations associated with progressive external ophthalmoplegia in humans. Hum. Mol. Genet., 15, 2846–2855. [DOI] [PubMed] [Google Scholar]
- 39. Ponamarev M.V., Longley M.J., Nguyen D., Kunkel T.A., Copeland W.C. (2002) Active site mutation in DNA polymerase gamma associated with progressive external ophthalmoplegia causes error-prone DNA synthesis. J. Biol. Chem., 277, 15225–15228. [DOI] [PubMed] [Google Scholar]
- 40. Qian Y., Kachroo A.H., Yellman C.M., Marcotte E.M., Johnson K.A. (2014) Yeast cells expressing the human mitochondrial DNA polymerase reveal correlations between polymerase fidelity and human disease progression. J. Biol. Chem., 289, 5970–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tyynismaa H., Mjosund K.P., Wanrooij S., Lappalainen I., Ylikallio E., Jalanko A., Spelbrink J.N., Paetau A., Suomalainen A. (2005) Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA, 102, 17687–17692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taylor R.W., Taylor G.A., Durham S.E., Turnbull D.M. (2001) The determination of complete human mitochondrial DNA sequences in single cells: implications for the study of somatic mitochondrial DNA point mutations. Nucleic Acids Res., 29, E74–E74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Naess K., Barbaro M., Bruhn H., Wibom R., Nennesmo I., von Döbeln U., Larsson N.-G., Nemeth A., Lesko N. (2012) Complete deletion of a POLG1 allele in a patient with Alpers syndrome. JIMD Rep., 4, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Larsson N.-G., Holme E., Kristiansson B., Oldfors A., Tulinius M. (1990) Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediatr Res., 28, 131–136. [DOI] [PubMed] [Google Scholar]
- 45. Huang J., Zhou W., Dong W., Watson A.M., Hong Y. (2009) From the Cover: Directed, efficient, and versatile modifications of the Drosophila genome by genomic engineering. Proc. Natl. Acad. Sci. USA, 106, 8284–8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.