

Abstract
Recent studies describe distinct DNA methylomes among phenotypic subclasses of neurons in the human brain, but variation in DNA methylation between common neuronal phenotypes distinguished by their function within distinct neural circuits remains an unexplored concept. Studies able to resolve epigenetic profiles at the level of microcircuits are needed to illuminate chromatin dynamics in the regulation of specific neuronal populations and circuits mediating normal and abnormal behaviors. The Illumina HumanMethylation450 BeadChip was used to assess genome-wide DNA methylation in stratum oriens GABAergic interneurons sampled by laser-microdissection from two discrete microcircuits along the trisynaptic pathway in postmortem human hippocampus from eight control, eight schizophrenia, and eight bipolar disorder subjects. Data were analysed using the minfi Bioconductor package in R software version 3.3.2. We identified 11 highly significant differentially methylated regions associated with a group of genes with high construct-validity, including multiple zinc finger of the cerebellum gene family members and WNT signaling factors. Genomic locations of differentially methylated regions were highly similar between diagnostic categories, with a greater number of differentially methylated individual cytosine residues between circuit locations in bipolar disorder cases than in schizophrenia or control (42, 7, and 7 differentially methylated positions, respectively). These findings identify distinct DNA methylomes among phenotypically similar populations of GABAergic interneurons functioning within separate hippocampal subfields. These data compliment recent studies describing diverse epigenotypes among separate neuronal subclasses, extending this concept to distinct epigenotypes within similar neuronal phenotypes from separate microcircuits within the human brain.
Introduction
Ongoing advances in technology for sampling and interrogating cells and tissues are refining long-standing concepts of cellular categorization. As the resolution and throughput of multiple - omics techniques improve, the field is developing new insight into the diversity of individual cells within specific subtypes of neurons (1,2). Chromatin regulation plays an established role in the generation and maintenance of cellular diversity in response to both programmed developmental events (3) and unique environmental influences (4). DNA methylation is one facet of chromatin regulation known to play a prominent role in CNS development and the pathogenesis of multiple psychiatric illnesses (5,6). Recent studies have demonstrated DNA methylation profiles to be distinct in separate subclasses of neurons in mouse and in human brain (7,8), and high-throughput single-cell DNA methylation sequencing has been used to identify novel neuronal populations within the human cortex (9). As we learn more about epigenomic mechanisms that distinguish neuronal subpopulations from one another, how those subpopulations are different or similar across separate circuit locations in health and disease is the next necessary question. A given neuronal subpopulation is likely not interchangeable from one circuit location to another, and distinct chromatin states among similar neuronal subpopulations functioning within separate circuits in the brain is an underexplored concept essential to our understanding of the mechanisms leading to dysfunction of specific circuits resulting in disease symptomatology.
The hippocampus is an attractive site for studying the regulation of neuronal subtype specific physiology across circuits due to its well-characterized and comparatively simple cytoarchitecture. The cornu ammonis (CA), or hippocampus proper, contains three cellular layers, the stratum oriens (SO), stratum pyramidale, and stratum radiatum, and the stratum lacunosum-moleculare containing fiber tracts and distal dendrites of pyramidal cells, which proceed through four subfields, CA4, CA3, CA2 (in this work considered together as CA3/2), and CA1 (Fig. 1). The neurons within this structure form a highly patterned cytoarchitecture organized along the trisynaptic pathway, a prototypical circuit composed of projections from the entorhinal cortex to the dentate gyrus, the dentate gyrus to sectors CA3/2, and sectors CA3/2 to sector CA1.
Figure 1.

Hippocampus cytoarchitecture and microdissection of stratum oriens tissue from CA3/2 and CA1. (A) The schematic depicts the cytoarchitecture of the human hippocampus, focusing on the neurons of SO and the projections of pyramidal neurons of SP. Within the SO, neurons are exclusively GABAergic and primarily fall into three subtypes: basket cells, with axons synapsing with pyramidal cell somata within SP; bistratified cells, with axons synapsing with pyramidal cell dendrites within layers SO and SR; and oriens-lacunosum-moleculare cells, with axons synapsing with pyramidal cell dendrites in the SLM. The organization of the trisynaptic pathway is depicted, and the circled numbers indicate the location of the three synapses within this prototypical hippocampal circuit. Synapse 1 occurs between projections from the entorhinal cortex layers II and III to the neurons of the dentate gyrus via the perforant pathway. Projections from the dentate gyrus (mossy fibers) to the pyramidal neurons of CA3 form synapse 2. Synapse 3 occurs between projections from CA3 pyramidal neurons (Schaffer collaterals) and pyramidal neurons of CA1. The major populations of SO GABAergic interneurons of CA3 and CA1 are the same; what distinguishes these neuronal populations is their functioning within separate microcircuits supporting the function of the second and third synapses within the trisynaptic pathway. Shown at bottom is a single Nissl-stained 30 µm section of postmortem human hippocampus before (B) and after (C) laser microdissection of tissue from stratum oriens of subfields CA3/2 and CA1. SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum; SLM, stratum lacunosum-moleculare; P, pyramidal neuron; BC, basket cell; BSC, bistratified cell; O-LM, oriens-lacunosum-moleculare cell; DG, dentate gyrus; EC, entorhinal cortex.
This work focused on the SO, a layer containing exclusively GABAergic interneurons (as well as glia and supportive cell populations) that form synapses with pyramidal neurons whose cell bodies reside within the stratum pyramidale. GABAergic interneurons within the SO fall into a small number of subclasses based on their connectivity, primarily basket cells, bistratified cells, and oriens-lacunosum-moleculare cells (10). While the neurons within separate subfields of the SO are not differentiated by typically assessed phenotypic markers such as connectivity, morphology, and calcium-binding peptide expression, they function within distinct microcircuits that support separate cognitive functions and are affected differently in health and disease. Activity within CA3 is implicated in direct encoding of associative memories (11), while CA1 operates as a comparator of input arriving from regions CA3 and entorhinal cortex layer III to discriminate between similar stimuli (12). In schizophrenia, imbalance between these opposing functions of pattern completion and pattern separation has been postulated to result in distorted perceptions and psychotic thought process (13).
We performed a genome-wide analysis of a previously described dataset to investigate the hypothesis that DNA methylation patterns differ between populations of neurons as closely related as GABAergic interneurons found within the SO of hippocampal subfield CA1 versus those of CA3/2. Further, we tested whether this mechanism acts differently across these sites in schizophrenia (SZ) and bipolar disorder (BD) as compared with healthy controls (CON). From a cohort of 24 postmortem human hippocampus tissue samples (eight cases from each diagnostic group), SO tissue was laser-microdissected from subfields CA3/2 or from CA1 separately, and in these regions, genome-wide DNA methylation was assessed using the Illumina HumanMethylation450 BeadChip (HM450, Illumina, San Diego, CA). Comparing CA1 to CA3/2 identified 11 highly significant differentially methylated regions (DMRegions) with large effect sizes associated with a set of ten genes with high construct-validity with multiple common themes including the WNT signaling pathway, zinc-finger transcription factors, divergently transcribed tandem gene pairs, and development. While the genomic locations of differentially methylated regions were highly similar between diagnoses, methylation differences were consistently of larger magnitude and more highly significant in BD as compared with SZ and CON cases. Additionally, there were a greater number of differentially methylated positions (DMPositions) comparing CA1 to CA3/2 in BD as compared with SZ and CON cases (42, 7, and 7 DMPositions, respectively).
These data demonstrate that recently described neuronal subtype specific epigenotypes (7–9) also differ between common neuronal subtypes functioning within separate neuronal circuits. Our results offer insight into the depth of complexity involved in the regulation of cellular diversity within the human brain and identify genes of known and of unknown function as potentially important in the differences between phenotypically similar populations of GABAergic interneurons participating in distinct microcircuits within the human hippocampus. This work demonstrates the importance of pursuing microcircuit level resolution with physical microdissection techniques when sampling the brain, as traditional homogenization techniques and even emerging high-throughput single-cell technologies will not detect potentially important microcircuitry based differences such as those described here.
Results
Sample characterization
Tissue was sampled from SO of CA3/2 and from SO of CA1, locations containing exclusively GABAergic neurons functioning in support of the second and the third synapse of the trisynaptic circuit, respectively (Fig. 1). To demonstrate that DNA methylation differences between these samples is not due to different proportions of cellular subpopulations present in each location, we assessed gene-expression profiles from a previously reported microarray study that investigated equivalently microdissected tissue samples in a separate non-overlapping cohort of postmortem human hippocampus from 21 individuals (14). In this prior dataset, we investigated at each circuit location mRNA expression of genes commonly used as markers of cell phenotype, including ENO2, GFAP, GAD1 and GAD2, SLC17A7, SST, CCK, PVALB, CALB1, and CALB2, as normalized to the expression of the housekeeping gene GAPDH. Supplementary Material, Figure S1 shows that sampled tissues are not significantly different in the expression of any of these markers of neuronal and non-neuronal cellular phenotypes.
Differentially methylated regions
In each sample, DNA methylation was assessed across the genome at 485 512 CpG sites using the HM450. Methylation profiles were compared across CA3/2 and CA1 samples using the “bump hunting” approach (15) implemented with the minfi package (16) in R version 3.3.2. Our cohort is well balanced for potential confounds, including age, postmortem interval, and tissue pH (Table 1), and as all comparisons were done with perfect matching (the cases in the CA3/2 and CA1 groups are identical), demographic variables of the cohort were not further considered in our analyses.
Table 1.
Tissue cohort and demographic variables
| Diagnosis | Gender | Age | PMI | pH | COD | Medication exposures |
|---|---|---|---|---|---|---|
| CON | M | 57 | 22.3 | 6.60 | Cancer | Lorazepam |
| CON | F | 51 | 30.7 | 6.60 | PE | |
| CON | F | 71 | 22.0 | 6.13 | MI | |
| CON | F | 71 | 22.3 | 6.28 | Cancer | |
| CON | M | 88 | 11.1 | 6.37 | Cancer | |
| CON | M | 49 | 27.1 | 6.29 | MI | |
| CON | M | 75 | 20.3 | 6.41 | MI | |
| CON | F | 51 | 23.1 | 6.09 | CVA | |
| Mean | 4M|4F | 64.13 | 22.35 | 6.35 | ||
| SZ | F | 73 | 28.8 | 6.19 | Cancer | Clozapine, Lithium |
| SZ | M | 77 | 25.3 | 6.50 | PNA | Clozapine, Quetiapine, Valproate |
| SZ | F | 64 | 18.5 | 6.55 | Resp Fail | Lorazepam, Olanzapine |
| SZ | F | 73 | 24.0 | 6.05 | Cancer | Risperidone |
| SZ | M | 83 | 43.5 | 6.18 | Cancer | Fluphenazine |
| SZ | M | 56 | 20.0 | 6.24 | MI | Clozapine |
| SZ | F | 85 | 12.8 | 6.69 | MI | Fluphenazine, Haloperidol |
| SZ | M | 32 | 38.4 | 6.56 | Suicide | Risperidone, Valproate |
| Mean | 4M|4F | 67.88 | 26.42 | 6.37 | ||
| BD | F | 76 | 22.8 | 6.08 | MI | Lithium, Valproate |
| BD | M | 69 | 29.5 | 6.14 | PNA | Lithium |
| BD | M | 77 | 24.2 | 6.58 | Cancer | Diazepam, Lithium |
| BD | F | 51 | 30.1 | 6.45 | MI | Diazepam, Risperidone, Valproate |
| BD | M | 80 | 13.4 | 6.08 | PNA | Haloperidol, Lithium, Risperidone |
| BD | F | 76 | 19.9 | 6.52 | PNA | Fluphenazine, Lithium, Olanzapine |
| BD | F | 51 | 35.1 | 6.02 | PNA | Clozapine, Lithium, Valproate |
| BD | F | 66 | 25.0 | 6.27 | Suicide | Olanzapine, Valproate |
| Mean | 3M|5F | 68.25 | 24.98 | 6.27 | ||
| p | 0.86 | 0.82 | 0.58 | 0.46 |
Listed are the demographic variables for our assembled cohort. Sample groups were not significantly different for age, gender, PMI, or pH as assessed by ANOVA with p value listed in the bottom row. PMI, postmortem interval; COD, cause of death; CVA, cerebrovascular accident; PE, pulmonary embolism; MI, myocardial infarction; PNA, pneumonia
Pooled comparison of genome-wide DNA methylation patterns between hippocampal subfields in all 24 cases identified 11 DMRegions with FWER < 0.05 (Fig. 2A), six of which were relatively hypomethylated in CA3/2 versus CA1, and five hypermethylated (Table 2). Four of these DMRegions were found immediately downstream of genes within divergently transcribed tandem pairs, including three genes within the zinc finger of the cerebellum (ZIC) gene family (Fig. 3 - additional DMRegions are depicted in Supplementary Material, Fig. S2). As shown in Figure 4 and Supplementary Material, Figure S3, methylation measurements at sites within DMRegions are robustly different between circuit locations, with methylation measurements in CA1 and CA3/2 separating completely at most assessed CpG loci. Effect sizes measured by Cohen's d (difference in group means divided by the pooled standard deviation) at all 95 individual CpG sites found within significant DMRegions range from 0.68 to 6.33, with an average of 3.96.
Figure 2.
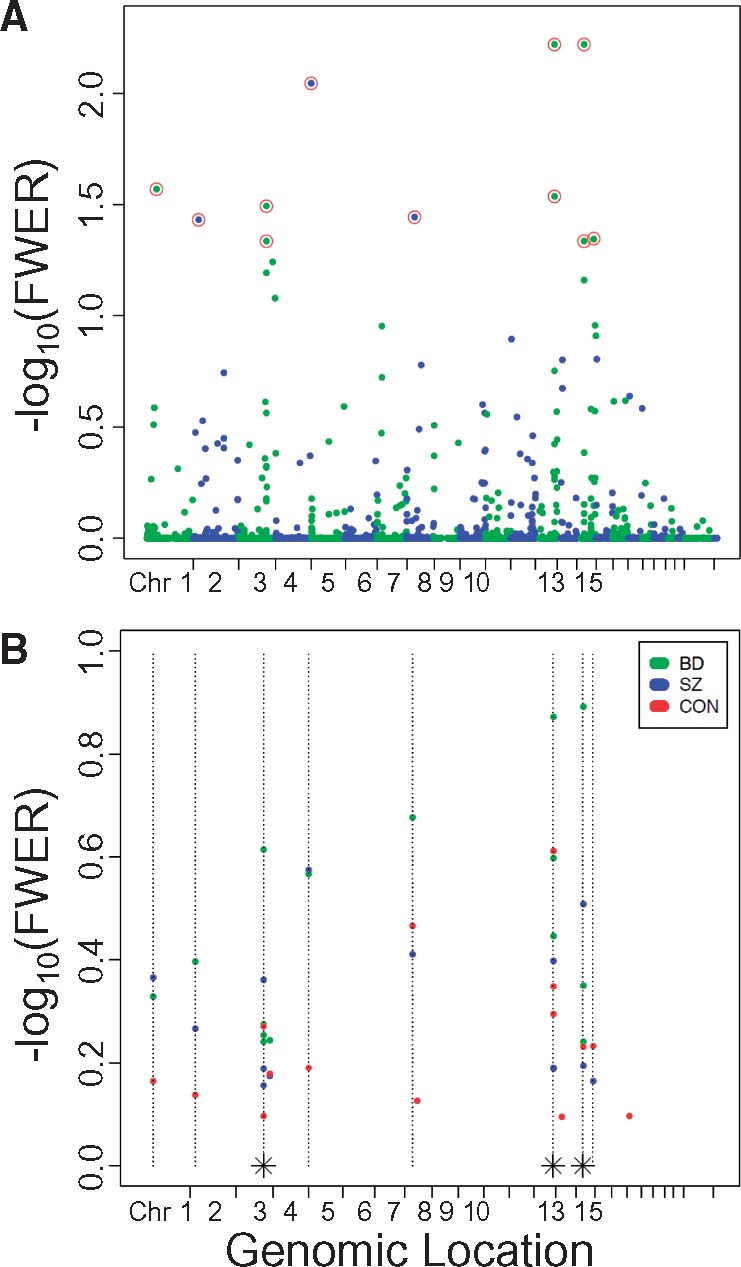
Manhattan plot of differentially methylated regions in CA3/2 versus CA1. (A) DMRegions identified by bump hunting analysis comparing all 24 CA3/2 cases to all 24 CA1 cases are plotted with genomic location indicated on the X-axis, and -log10(FWER) on the Y-axis. Colors alternate to identify chromosomal boundaries. Circled points indicate the 11 DMRegions with FWER less than 0.05. (B) DMRegions identified by bump hunting analysis comparing eight CA3/2 cases to eight CA1 cases in each separate diagnostic group are plotted with genomic location indicated on the X-axis, and -log10(FWER) on the Y-axis. Although no DMRegions were identified with FWER less than 0.05 in any single diagnosis, shown here are the 15 most significant DMRegions from analysis of each diagnosis. Dotted vertical lines indicate the genomic location of DMRegions identified in the pooled analysis of all diagnoses together. Dotted lines with an asterisk at their base indicate the location of two distinct DMRegions located too closely together to be resolved on this plot.
Table 2.
Differentially methylated regions and associated genes
| Genomic location | FWER | HM | Associated gene | DTTGP Member | Gene product | Protein function | DMR location |
|---|---|---|---|---|---|---|---|
| 13q32.3 | 0.006 | CA1 | ZIC2 | X | Zinc Finger of the Cerebellum 2 | Zinc Finger TF, | 2 kb Downstream |
| CNS Development, | |||||||
| WNT Signaling | |||||||
| 15q14 | 0.006 | CA1 | LOC145845/ MEIS2 | Long Non-Coding RNA | Non-Coding RNA | Third Intron, 10kb Downstream of MEIS2 | |
| 4q35.2 | 0.009 | CA2/3 | TRIML1 | X | Tripartite Motif Family Like-1 | Zinc Finger TF, CNS Development | 90 kb Downstream |
| 1p32.3 | 0.027 | CA1 | DMRTA2 | DoubleSex and Male Aberant-3 Related TF | CNS Development, Neuronal Differentiation, WNT Signaling | 7 kb Upstream of TSS | |
| 13q32.3 | 0.029 | CA1 | CLYBL | Citrate Lyase Beta Like | Mitochondrial B12 Metabolism | Ninth (Final) Exon | |
| 3q24 | 0.032 | CA2/3 | ZIC1 | X | Zinc Finger of the Cerebellum 1 | Zinc Finger TF, CNS Development, WNT Signaling | 6 kb Downstream |
| 8p11.21 | 0.036 | CA1 | SFRP1 | Secreted frizzled-related protein 1 | WNT Signaling | 0.6 kb Upstream of TSS | |
| 2p23.1 | 0.037 | CA1 | OTOF | Otoferlin Protein | Vesicle Membrane Fusion, Neurosensory Deafness | Upstream boundary exon 6 | |
| 15q26.1 | 0.045 | CA2/3 | MIR9-3HG | Long-Intergenic Non-coding RNA | microRNA9-3 host gene | Upstream boundary exon 2 | |
| 15q14 | 0.046 | CA2/3 | MEIS2 | TALE Homeobox Protein | Development | Fourth Intron | |
| 3q24 | 0.046 | CA2/3 | ZIC4 | X | Zinc Finger of the Cerebellum 4 | Zinc Finger TF, CNS Development, WNT Signaling | 4 kb Downstream |
| 13q32.3 | 0.176 | CA1 | ZIC5 | X | Zinc Finger of the Cerebellum 5 | Zinc Finger TF, CNS Development, WNT Signaling | 6.5 kb Downstream |
HM indicates which subfield is hypermethylated relative to the other at the indicated locus.
FWER, Family Wise Error Rate; DMR, Differentially Methylated Region; DTTGP, Divergently Transcribed Tandem Gene Pair.
Figure 3.
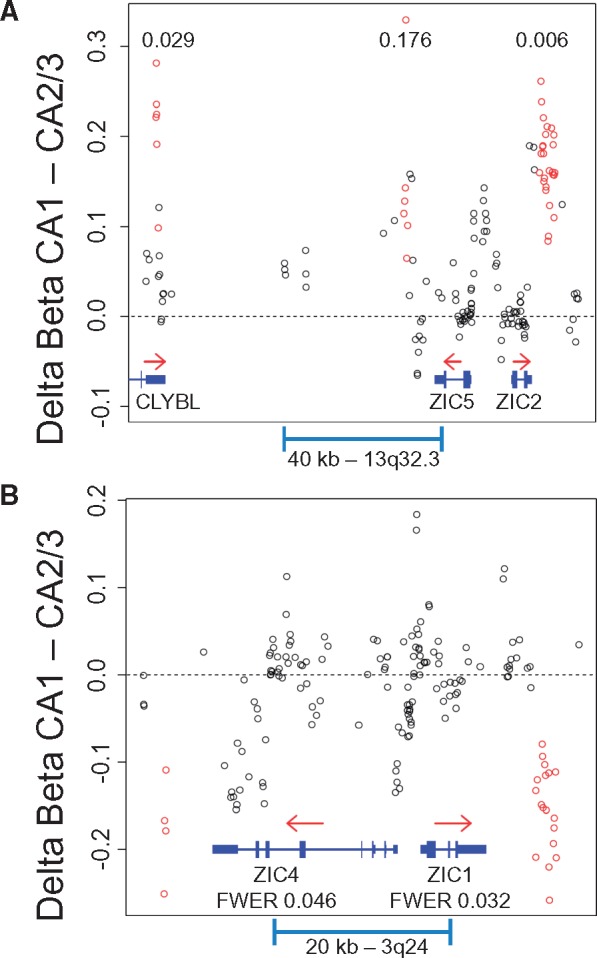
Differentially methylated regions associated with the ZIC gene family tandem gene pairs. DMRegions at the ZIC2 and ZIC5 locus on Chromosome 13 (A) and at the ZIC1 and ZIC4 locus on Chromosome 3 (B) are depicted with genomic location plotted on the X-axis, and delta beta value (CA1 - CA3/2) plotted on the Y-axis. Note that the DMRegion downstream of ZIC5 did not reach significance as defined by FWER < 0.05 (FWER 0.176, p value = 0.001). Open circles represent differences in averaged subfield methylation levels at individual CpG sites, with red circles found within identified DMRegions. Gene location and structure are depicted in blue, with red arrows indicating the direction of transcription. Numbers within plots indicate the FWER of the associated DMRegion.
Figure 4.
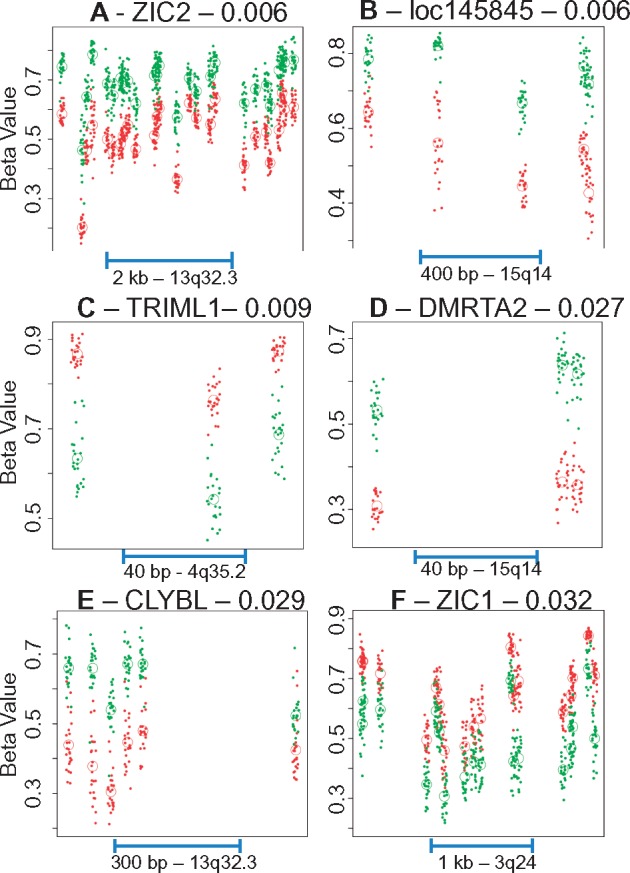
Methylation profiles of individual differentially methylated regions. Each plot depicts the methylation profiles of all 24 cases at a single identified genome-wide significant DMRegion plotted as a function of genomic location. Each point represents the beta value at a single CpG in a single case, with green points indicating CA1 measurements and red points indicating CA3/2 measurements. Open circles indicate the average beta value at a single CpG for all cases in that subfield. Genomic location and length of scale bar are indicated at the base of each plot. Similar plots for the remaining five significant DMRs, as well as ZIC5, are included in Supplementary Material, Figure S3.
As revealed by cluster analysis, methylation profiles differ more across hippocampal subfields than across diagnoses (Fig. 5). Comparison of the eight CA3/2 samples to the eight CA1 samples within each diagnostic group identified zero DMRegions with FWER < 0.05. However, as shown in Figure 2B, the 15 most significant DMRegions in each diagnostic group show a high degree of overlap with the locations of DMRegions identified in the pooled analysis. DMRegions within individual groups were highly similar in location and in rank order to each other and to DMRegions identified in pooled analysis of all 24 cases. All of the 11 significant pooled comparison DMRegions (with the exception of TRIML1) were found within the top 14 DMRegions in analyses of each individual diagnosis. Despite this high degree of similarity in methylation profiles among diagnostic groups, the BD group contained more DMRegions with uncorrected P-value less than 0.01 than the SZ and CON groups (22, 9, and 6 DMRegions, respectively; chi-squared = 11.73, df = 2, P = 0.0028).
Figure 5.

Cluster dendrogram depicting the relatedness of the six sample groups in our dataset. Methylation profiles differ more across hippocampal subfields than they do across diagnostic categories. Note the similarity within each hippocampal subfield, with psychiatric subjects clustering together and away from control subjects.
With the same approach described under “Sample Characterization,” we checked for differential expression between SO of CA1 and CA3/2 of genes associated with DMRegions in the pooled analysis within the previously reported gene expression dataset. Of the 11 DMRegion-associated genes, only five (MEIS2, OTOF, SFRP1, ZIC1, and ZIC4) are assessed by probes present on the microarray used in the previous gene expression study (14), and none of these five genes were found to be differentially expressed in that dataset (Supplementary Material, Fig. S4).
Differentially methylated positions
In addition to the bump hunting analysis, we investigated differences between hippocampal subfields at the level of individual CpG sites by performing paired Student's t-test between DNA methylation measurements from CA3/2 and CA1 at each of the 485 512 assessed CpG sites and applying Bonferroni correction. In diagnosis-specific analyses, we observed 7, 7, and 42 significantly differentially methylated CpG sites (DMPositions – Fig. 6B) in the CON, SZ, and BD groups, respectively (χ2 = 44, df = 2, P-value = 3.2e-10). While many of the significant DMPositions in individual diagnoses fall within the genomic locations of DMRegions, there is little overlap between the specific CpGs identified as DMPositions in each diagnosis, with zero DMPositions common to all three diagnoses, and just one DMPosition shared by the BD and CON groups (cg25334575).
Figure 6.
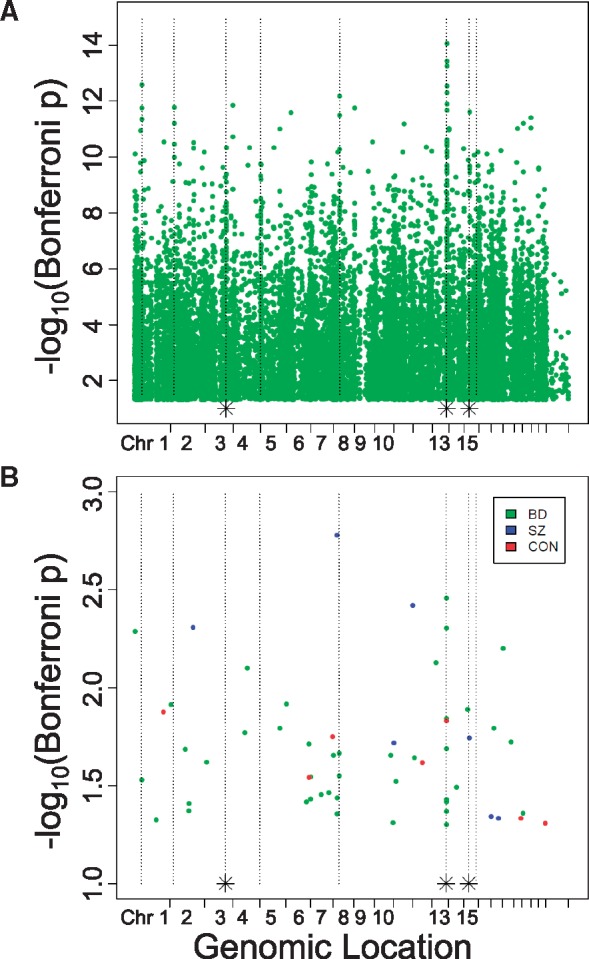
Differentially methylated positions in CA3/2 versus CA1. DMPositions with Bonferroni-corrected P-value less than 0.05 identified by single-probe analysis in all 24 cases together (A) and in separate diagnostic categories (B) are plotted with genomic location indicated on the X-axis, and -log10(Bonferroni-corrected P-value) on the Y-axis. As in Figure 2b, dotted vertical lines indicate the genomic location of DMRegions identified in the pooled analysis of all diagnoses together, and dotted lines with an asterisk at their base indicate the location of two distinct DMRegions located too closely together to be resolved on this plot.
Pooled analysis of all 24 cases using the paired Student's t-test to assess each CpG locus individually identified 16 089 sites, or 3.3% of all assessed sites, as DMPositions with Bonferroni-corrected P-value < 0.05. These sites are diffusely distributed across the genome (Fig. 6A), but the most significantly different sites again show a large amount of overlap with locations of significant DMRegions.
To investigate the potential influence of the disparate medication exposures present in the SZ, BD, and CON groups on the differences in the numbers of CA1 vs. CA3/2 DMPositions observed in each group, we repeated the DMPosition analysis in the seven patients with exposure to lithium, 6 patients with exposure to valproate, and 9 patients with exposure to dibenzodiazepine type antipsychotics. The number of DMPositions identified with Bonferroni-corrected P-value <0.05 from each medication exposed group was compared with the number identified by an equivalent analysis of a randomly selected group of subjects without the exposure. For each medication class the number of DMPositions identified in the exposed subjects was comparable to the number identified in the unexposed subjects (lithium exposed 5, lithium unexposed 3; dibenzodiazepine antipsychotic exposed 56, dibenzodiazepine antipsychotic unexposed 61; valproate exposed 1; valproate unexposed 1).
Distribution of DMRegions and DMpositions among functional genomic elements
We investigated the overlap of DMRegions with annotated functional genomic elements by assessing the number of DMRegion probes located within each class of element, normalized to the total number of DMRegion probes genome-wide (Fig. 7). A similar calculation was performed for probes identified as DMPositions, and for all probes present on the HM450. If DMRegions and DMPositions are distributed randomly across the genome, we would expect the ratios observed for each genomic element to be similar between all HM450 probes and probes associated with DMRegions or DMPositions. What we found however indicates that sites of significant methylation change are enriched and depleted within specific classes of functional genomic elements. DMPositions are enriched at enhancer elements and gene bodies, while they are depleted from CpG islands. DMRegions are enriched within CpG islands and shores, highly enriched within previously annotated DMRegions, and depleted from gene bodies.
Figure 7.
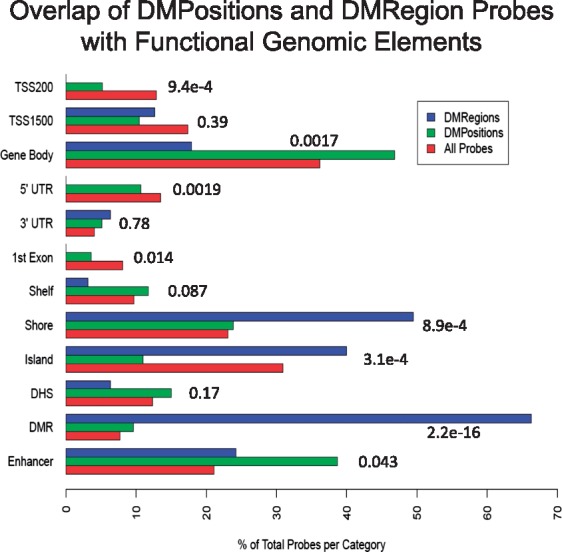
Overlap of differentially methylated locations with functional genomic elements. Barplot of the percentage of the total number of probes within the categories of DMRegion probes and all HM450 probes that overlap with the indicated functional genomic elements. TSS200, within 200 bp of the transcription start site; TSS1500, within 1500 bp of the transcription start site; Shelf, CpG island shelf; Shore, CpG island shore; Island, CpG island; DHS, DNAse hypersenstive site; DMR, previously annotated differentially methylated region.
Pyrosequencing validation
In the same layer- and subfield-specific DNA samples used for the HM450 analysis, we performed bisulfite pyrosequencing measurements of DNA methylation levels within the most significant (ZIC2-associated) and the least significant (ZIC4-associated) DMRegions from the bump hunting analysis to validate our microarray findings. These assays measured methylation levels at four contiguous CpG sites within the ZIC2-associated DMRegion, and five contiguous CpG sites within the ZIC4 DMRegion. Methylation at all nine of these CpG sites was highly significantly different between CA1 and CA3/2 (uncorrected P-values between 6e-12 and 1e-19 – Supplementary Material, Fig. S5).
Discussion
To our knowledge, this study is the first genome-wide investigation of methylation patterns in phenotypically similar populations of neurons sampled from multiple circuit locations within a single structure of the human brain. The majority of prior epigenetic postmortem human brain analyses have been performed using homogenized samples of whole cortex. Bioinformatic approaches have been developed to deconvolute signal from the many distinct cell types present within such a sample (17,18), and such approaches are necessary to control for differences in the cellular composition of samples across comparator groups, and to identify which cell-type may be the host of a significant finding. These approaches are limited, however, in that they cannot address the increased risk of type II statistical error present in studies of cell-specific phenomena such as DNA methylation performed with highly heterogeneous samples, and they cannot localize findings to common cellular subtypes at one circuit location but not another within the larger structure. For these reasons, more advanced methods for physically fractionating sampled cell types and circuit locations [laser microdissection (5,19), fluorescence-activated cell or nuclear sorting (7,20), genetic labeling of neuronal subpopulations in animal models (8), microfluidics (21,22)] are necessary to advance our understanding of the regulation of neuronal diversity and the functional distinctions that occur across cell types and neuronal circuits in both health and disease.
Essential in the analysis of postmortem human brain is rigorous control for the many potential confounding factors present in an assembled cohort. Our study design mitigates this problem by comparing two hippocampal subregions microdissected from each individual case, resulting in a perfectly balanced comparison and sidestepping the confounding influence of factors such as the age, gender, agonal state, and lifetime exposures unique to each case. Nevertheless, the cohort employed in this study was well balanced across diagnostic groups with respect to age, gender, and postmortem interval (Table 1). Exposure to psychotropic medications is also perfectly balanced in each CA1 versus CA3/2 comparison, but is not balanced between the control and patient groups. The issue of chronic medication exposure as a confounding factor in DNA methylation studies of psychiatric illness is difficult to rigorously control for multiple ethical and practical reasons. The ideal study would assess a large cohort of medication naive patients, but this is difficult to achieve, especially for a study requiring access to brain tissue. Model systems have been used to measure epigenetic effects of medication exposure in rodent, non-human primate, or cell culture models (23), with findings often specific to the medication administered and the genomic loci assessed. Statistical methods employed to control for medication effects in human studies are to include medication or other environmental exposures (i.e. smoking or drugs of abuse) as a covariate in regression analysis where appropriate (24), to reanalyse data grouping subjects on exposure status rather than diagnostic categorization (5,25), or to investigate the impact of surrogate variables (i.e. principal components), thought to encompass unmeasured potential confounds including medication effects, on DNA methylation variability (26). In our cohort circuitry specific DMRegions are highly similar between medication exposed patient groups and the unexposed control subjects, leading us to conclude that medication is not a driving factor in these methylation differences. To further investigate the potential influence of medication exposure on differential methylation across hippocampal subfields in our cohort, we analysed DMPositions in groups of patients with exposure to three types of medication, lithium, which has been shown to influence DNA methylation in a gene-specific manner in human neuroblastoma cells (27), valproate, which is known to influence chromatin structure through its activity as a histone deacetylase inhibitor (28), and dibenzodiazepine type antipsychotic medications (including clozapine, olanzapine, and quetiapine) which have been shown in animal studies to alter DNA methylation patterns while other antipsychotic medications (including haldol and risperidone) did not (23). The number of significant DMPositions identified in each medication-exposed group was comparable to that observed in a randomly selected group of subjects of equal size without the exposure, again supporting our conclusion that medication exposure is not a primary factor in the differences observed between the SZ, BD, and CON groups.
Limitations of this study include the sample size, which is limited by the labor and time intensive task of laser microdissection. This is most problematic in the analyses of individual diagnostic groups where no DMRegions met our stringent measure of genome-wide significance and follow-up studies of larger cohorts may identify circuitry specific DMRegions that are specific to patient or control populations. Additional limitations are intrinsic to the methodology, including the bias introduced by any microarray technology that can only investigate genomic regions targeted by the array, and the inability of any bisulfate conversion based assay of DNA methylation to differentiate 5-methylcytosine from 5-hydroxymethylcytosine. Furthermore, the HM450 array targets only cytosine residues within CpG dinucleotides, while non-CpG cytosine methylation may be more informative for neuronal physiology (9,29). Also, while gene expression analysis from a separate non-overlapping cohort did not detect gene expression changes in DMRegion associated genes, this postmortem human brain analysis is unable to investigate causal links to other potential functional outcomes of the observed methylation differences.
The neuronal populations sampled from CA3/2 and from CA1 are highly similar and are differentiated primarily by their functioning within discrete microcircuits along the trisynaptic pathway. While phenotypic characteristics including expression of a number of mRNAs commonly used as phenotypic markers of GABAergic interneuronal subtypes (Supplementary Material, Fig. S1) are not distinct between CA3/2 and CA1 on a population basis, subtle differences in GABAergic populations identified by patterns of connectivity in SO of these subfields have been described (back-projection neurons specific to CA1 SO, horizontal trilaminar cells specific to CA1 SO) (10). Considering the robust effect sizes and the fact that subfield-specific GABAergic neuron subtypes are greatly outnumbered by neuronal populations common to CA3/2 and CA1 (10), we propose that the observed subfield-specific methylation profiles are present in common GABAergic subtypes distinguished not by phenotype but by the unique environment of each hippocampal subfield.
The data presented here identify significant differences in DNA methylation profiles observed in SO of CA3/2 as compared with SO of CA1. While there are interesting differences in methylation profiles between SZ, BD, and CON at specific circuit locations (5), cluster analysis demonstrates that methylation profiles differ more between hippocampal subfields than between diagnostic categories (Fig. 5). An analysis of methylation differences between hippocampal circuit locations within individual diagnostic groups identified multiple DMPositions significant after Bonferroni correction (Fig. 6), and DMPositions in all diagnoses often overlap with a discrete number of genomic loci corresponding to DMRegions described below. Beyond these similarities between diagnoses, the BD group was found to have significantly more DMPositions (Fig. 6B, P-value = 3.2e-10) than the other two groups. While interpretation of this finding is uncertain, it suggests that phenomena driving methylation differences between hippocampal subfields may be more prominent in BD as compared with both CON and SZ. Additionally, while many DMPositions are found near the genomic sites of DMRegions, there is little overlap at the level specific CpG loci, with just one common DMPosition shared between the BD and CON groups.
The bump hunting algorithm implemented within the minfi package has been used by multiple groups to identify regions of methylation change expected to be of greater biological significance as compared with differentially methylated single CpG sites (15,30). Bump hunting analysis of our dataset identifies 11 DMRegions with FWER < 0.05, and large effect sizes an order of magnitude beyond those most often reported in the literature assessing DNA methylation change (average effect size is 3.96 for assessed CpGs within DMRegions). In this genome-wide unbiased comparison, there is no reason to assume a priori that these DMRegions would be associated with genes of common function. Interestingly, however, these DMRegions are associated with a group of ten genes with high construct-validity, as most are known to play roles relevant to the development and function of the central nervous system (CNS). Four of these genes encode zinc-finger transcription factors, including three members of a single gene family, the zinc finger of the cerebellum genes (ZIC1, ZIC2, ZIC4 - a non-significant DMRegion with FWER 0.176 was also associated with ZIC5). These four zinc finger protein-associated DMRegions are found in a similar configuration downstream of genes within divergently transcribed tandem gene pairs. While none of the five DMRegion associated genes that were assessed in a prior gene-expression study showed differential expression between SO regions CA1 and CA3/2 (Supplementary Material, Fig. S4), there is a growing literature describing multiple genomic functions of DNA methylation beyond direct regulation of transcription (31).
The ZIC gene family contains five members in two divergently transcribed tandem gene pairs, ZIC1 and ZIC4 on chromosome 3, and ZIC2 and ZIC5 on chromosome 13, plus ZIC3 as a singleton on the X chromosome. This unusual arrangement is conserved over large evolutionarily distances similar to other gene families critical for developmental patterning (32). Within the ZIC gene family’s two tandem gene pairs, DMRegions were found between two and six kb downstream of the 3’ end of each gene (Fig. 3). Interestingly, within each pair the direction of methylation change is consistent, but inconsistent across pairs. The ZIC genes encode zinc-finger transcription factors involved with neuroectoderm differentiation and specification of neuronal cell fate with complex patterns of expression across time and location. Mutations in ZIC genes lead to varied developmental malformations and lack of specific structures within the CNS, with an extreme example being the association of loss of ZIC2 with holoprosencephaly, a failure of the forebrain to separate into distinct left and right hemispheres (33). During development, ZIC genes are involved in preventing the premature exit of neuronal precursors from the cell cycle (34). ZIC genes continue to be expressed in adult neurons, where they function in the coordination of mature neuronal gene expression patterns through action at neuronal enhancer elements regulated through chromatin dynamics (35). ZIC genes are known to interact with the WNT signaling pathway that regulates many functions within the developing CNS, including cell proliferation and specification, neuronal migration and polarity, axon pathfinding and branching, and synapse assembly (36). In the adult brain, ZIC genes continue to be expressed and interact with the WNT signaling pathway in the regulation of synaptic structure and function, both pre- and post-synaptically (37).
In addition to the ZIC genes, SFRP1 and DMRTA2 also play important roles in modulation of the WNT signaling pathway in the developing and adult brain. SFRP1 encodes the secreted frizzled-related protein 1, a biphasic modulator of the WNT signaling pathway (38). The DMRTA2 gene encodes the DMRT like family A2 protein, a transcription factor that promotes neurogenesis in the developing brain and continues to be expressed in adult hippocampus selectively in neurons (39). In mice, the loss of DMRTA2 leads to complete agenesis of the hippocampus (40). DMRTA2 has been shown to be involved in the development of the early telencephalon through the formation of the cortical hem and to support the maintenance of neural progenitors through WNT pathway activity (40).
Of the DMRegion associated genes not explicitly related to the WNT signaling pathway, MEIS2, TRIML1, and MIR9–3HG are also important regulators of early development. MEIS2 encodes the Meis homeobox 2 protein, a highly conserved TALE homeobox protein and transcriptional regulator essential for development (41). MEIS2 contains a subfield-specific DMRegion within its fourth intron. This gene is known to produce a number of transcript variants encoding distinct isoforms of the protein, and a role for methylation of intronic sequences has been proposed in the regulation of alternative splicing (31). The TRIML1 gene, like the ZIC genes, encodes a zinc-finger protein and is found in a divergently transcribed tandem gene pair. TRIML1 encodes tripartite motif family-like 1, a RING (Really Interesting New Gene) finger domain protein and E3 ubiquitin ligase important for early embryo development (42). TRIML1 is expressed in adult brain, where it is found in particularly high levels in neurons within the hippocampus and is not detected in glia (39). MIR9–3HG is the MIR9–3 host gene found 10 kb downstream of the MIR9–3 gene and the two are likely co-regulated (43). MIR9–3 is one of three genomic loci that encode microRNA9, which is expressed in both neurons and glia but is specific to brain, is involved in neuronal differentiation and patterning of the developing brain (44), and is regulated by the cAMP response element binding protein (CREBBP) (45).
Among this group of DMRegion associated genes, only two, CLYBL and OTOF, do not fit neatly into the categories of WNT signaling modulators and regulators of development. CLYBL encodes the citrate lyase beta-like enzyme, a component of the mitochondrial B12 pathway found in mitochondria of all mammalian tissues, including both neurons and glia of adult hippocampus (39). Finally, the OTOF gene is associated with neurosensory nonsyndromic recessive deafness (46). OTOF is expressed in adult hippocampus selectively in neurons, and the otoferlin protein is thought to be involved with vesicle membrane fusion. OTOF produces multiple transcript variants (47), and the OTOF-associated DMRegion straddles the 3’ boundary of the sixth exon.
This work demonstrates robust and specific differences in the DNA methylomes of populations of neurons that are phenotypically highly similar and are differentiated by the circuitry in which they are embedded, suggesting a role for DNA methylation in the regulation of neuronal genomic processes in response to the unique cellular environment found at separate circuit locations within the human brain. A striking number of the genes associated with DMRegions in our analysis perform roles in CNS development, continue to be expressed in adult hippocampus, and are expressed specifically in neurons. Association of DMRegions with three out of five ZIC gene family members, as well as DMRTA2 and SFRP1, implicates the WNT signaling pathway in these phenomena. Finally, this work demonstrates the necessity of physically sampling neuronal circuit locations individually, as even advanced cell-sorting methodologies will overlook circuit specific epigenomic distinctions between phenotypically similar cellular populations when applied to larger brain structures such as whole hippocampus or whole cortex.
Materials and Methods
Cohort description
A cohort of 24 postmortem human hippocampus samples containing eight healthy controls, eight schizophrenia, and eight bipolar disorder cases matched for age, gender, postmortem interval, and pH was obtained from the Harvard Brain Tissue Resource Center, as previously described (5). All cases were obtained by family referral; none was referred by a medical examiner's office. Cases with documentation of illicit drug abuse were excluded.
On arrival to the facility, hippocampal tissue blocks were dissected from fresh brain at the level of the pulvinar thalami along the rostro-caudal axis of the hippocampus. Tissue blocks were lightly fixed in ice-cold 0.1% formalin in 0.1M phosphate buffer (pH 7.2) for 90 minutes before cryoprotection with 30% sucrose in phosphate buffer overnight. Tissue blocks were then embedded in optimum cutting temperature compound (Sakura Finetek, Torrance, CA) and stored at −80 °C.
Tissue processing and arrays
Frozen postmortem human hippocampus was sectioned to 30 µm in a Microm HM560 cryostat at -20 °C and mounted on polyethylene terephthalate frame slides (Leica, Wetzlar, Germany). Mounted tissue sections were processed through a graded series of ice-cold acetone and ethanol, hydrated in phosphate-buffered saline, stained with cresyl violet, and dehydrated through ascending concentrations of ethanol. The slides were air dried, and SO tissue was dissected from region CA3/2 or CA1 from approximately 60 sections per case using a Leica LMD6500 laser microdissection system (Fig. 1). DNA was extracted from collected tissue using the QIAamp DNA Micro Kit (Qiagen, Hilden, Germany) and stored at −80 °C until further use.
For each sample, 500 ng of genomic DNA was bisulfite modified using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA) with modified parameters recommended by Illumina. The bisulfite-modified DNA was analysed using the HM450 per the manufacturer's instructions, with samples randomly distributed to 48 arrays across four slides.
Sample characterization
A previously reported microarray experiment investigated gene-expression patterns in equivalently microdissected postmortem human hippocampus in a non-overlapping cohort of 21 cases (14). This existing dataset was interrogated to demonstrate that the cellular populations present in SO do not differ between subfields CA3/2 and CA1 as assessed by mRNA expression for genes commonly used to characterize cellular phenotypes pertinent to GABAergic interneurons. Comparisons were made using two-tailed paired t-tests in 21 CA1 samples versus 21 CA3/2 samples. Genes examined include GFAP, ENO2, GAD1, GAD2, CCK, SST, PVALB, SLC17A7, CALB1, and CALB2, and gene expression was normalized to expression of the housekeeping gene GAPDH (Supplementary Material, Fig. S1).
Data analysis
All statistical analyses were performed using R software version 3.3.2. Raw intensity (idat) files were analysed with the minfi Bioconductor package (16), and data were normalized by stratified quantile normalization. Internal control probes were investigated and no samples were identified as outliers, and no samples were excluded from analysis. M values, the log ratios of percent methylation measurements, were extracted and used for all analyses, and data are presented graphically using the more intuitive beta values (48), which represent percent methylation measurements. As each group in the comparison (CA3/2 versus CA1) contains tissue sampled from an identical set of cases, potential confounds are equally distributed across groups and were not considered further in the assessment of differences across subfields. Additionally, while some HM450 probes have been shown to be influenced by the genotype of underlying single-nucleotide polymorphisms (SNP) and these probes are sometimes excluded from analysis, we did not exclude these probes as genotype is identical between the CA3/2 CA1 pair from each individual case. Probes known to have an SNP at their target CpG (SNP probes) were present within the set of probes associated with DMPositions at lower frequency than among all probes on the array (4.0% versus 4.3%), and no SNP probes were associated with CpG sites within DMRegions (49). DMPositions were identified using two-tailed paired Student's t-test to compare subfields at each individual HM450 probe in both individual diagnostic groups (eight vs eight samples in each group) and in all samples together (24 vs 24 samples), with Bonferroni-corrected P-value < 0.05. To investigate the influence of specific classes of medication on methylation differences between subfields, we compared the output of this DMPosition analysis of array-wide CA3/2 versus CA1 methylation measurements in patients with exposure to lithium, valproate, or dibenzidiazepine type antipsychotic medication, and in a randomly selected group of cases of equal number without exposure to each medication class. DMRegions were identified with the bump hunter algorithm within the minfi package based on 1000 permutations with family wise error rate (FWER) < 0.05, and this analysis was again performed both in each eight case diagnostic group and in all 24 cases pooled together.
DMRegion validation by bisulfite pyrosequencing
Methylation measurements were validated using pyrosequencing at multiple CpG sites within separate DMRegions associated with the ZIC2 and ZIC4 genes. These sites were chosen because they are associated with the highly interesting ZIC genes, and also because they are the most and the least significant DMRegions that met the criteria of FWER < 0.05. Regions of interest were amplified using 15 ng of bisulfite-modified genomic DNA from each sample with the Qiagen Pyromark PCR kit using the following primers designed using MethPrimer (50): ZIC2 forward primer—5'- GTT AAG GTT GTG GGT TAT TAG GT -3'; ZIC2 reverse primer—5'-/5BiosG/AAA TAT CAC CTC TTC AAA AAC TC -3'; ZIC4 forward primer—5'- AGG AGT TTT ATT TAG ATT GTA GGT G -3'; ZIC4 reverse primer—5'-/5BiosG/CCA AAC CTT ACC CTT CCT AAA AA -3'. PCR conditions were as follows: 95 °C for 15 minutes, (94 °C for 30 seconds, 56 °C for 30 seconds, 72 °C for 30 seconds) for 45 cycles, 72 °C for 10 minutes, 4 °C hold. In both assays the forward primer was used as the sequencing primer. The amplified template was sequenced using a Pyromark MD96 instrument per the manufacturer’s protocol, and methylation measurements were assessed for significant differences between subfields using two-tailed paired Student’s t-test comparing 24 CA1 samples to 24 CA3/2 samples.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors thank Jill Ruzicka (HelloPaper.co) for assistance with generating figures.
Conflict of Interest statement. None declared.
Funding
National Institute of Mental Health (grant numbers MH077175, MH/NS077550) to F.M.B., Maria Lorenz Pope Fellowship and the Pope-Hintz Fellowship from McLean Hospital, a NARSAD Young Investigator Award from the Brain and Behavior Research Foundation, the MD/PhD Fellowship from the American Psychiatric Association and Pfizer Pharmaceuticals, and a Dupont-Warren Fellowship from Harvard Medical School (W.B.R.).
References
- 1. Tanay A., Regev A. (2017) Scaling single-cell genomics from phenomenology to mechanism. Nature, 541, 331–338. 10.1038/nature21350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanes J.R., Masland R.H. (2015) The types of retinal ganglion cells: current status and implications for neuronal classification. Annu. Rev. Neurosci., 38, 221–246. [DOI] [PubMed] [Google Scholar]
- 3. Hirabayashi Y., Gotoh Y. (2010) Epigenetic control of neural precursor cell fate during development. Nat. Rev. Neurosci., 11, 377–388. [DOI] [PubMed] [Google Scholar]
- 4. Sng J., Meaney M.J. (2009) Environmental regulation of the neural epigenome. Epigenomics, 1, 131–151. 10.2217/epi.09.21 [DOI] [PubMed] [Google Scholar]
- 5. Ruzicka W.B., Subburaju S., Benes F.M. (2015) Circuit- and diagnosis-specific DNA methylation changes at gamma-aminobutyric acid-related genes in postmortem human hippocampus in schizophrenia and bipolar disorder. JAMA Psychiatry, 72, 541–551. 10.1001/jamapsychiatry.2015.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nestler E.J., Pena C.J., Kundakovic M., Mitchell A., Akbarian S. (2016) Epigenetic basis of mental illness. Neuroscientist, 22, 447–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kozlenkov A., Wang M., Roussos P., Rudchenko S., Barbu M., Bibikova M., Klotzle B., Dwork A.J., Zhang B., Hurd Y.L.. et al. (2016) Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res., 44, 2593–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mo A., Mukamel E.A., Davis F.P., Luo C., Henry G.L., Picard S., Urich M.A., Nery J.R., Sejnowski T.J., Lister R.. et al. (2015) Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron. 86, 1369–1384. 10.1016/j.neuron.2015.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luo C., Keown C.L., Kurihara L., Zhou J., He Y., Li J., Castanon R., Lucero J., Nery J.R., Sandoval J.P.. et al. (2017) Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science (New York, N.Y.), 357, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Freund T.F., Buzsaki G. (1996) Interneurons of the hippocampus. Hippocampus. 6, 347–470. [DOI] [PubMed] [Google Scholar]
- 11. Leutgeb S., Leutgeb J.K. (2007) Pattern separation, pattern completion, and new neuronal codes within a continuous CA3 map. Learn. Memory, 14, 745–757. [DOI] [PubMed] [Google Scholar]
- 12. Chen J., Olsen R.K., Preston A.R., Glover G.H., Wagner A.D. (2011) Associative retrieval processes in the human medial temporal lobe: hippocampal retrieval success and CA1 mismatch detection. Learn. Mem., 18, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li W., Ghose S., Gleason K., Begovic A., Perez J., Bartko J., Russo S., Wagner A.D., Selemon L., Tamminga C.A. (2015) Synaptic proteins in the hippocampus indicative of increased neuronal activity in CA3 in schizophrenia. Am. J. Psychiatry, 172, 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benes F.M., Lim B., Matzilevich D., Walsh J.P., Subburaju S., Minns M. (2007) Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc. Natl. Acad. Sci. U. S. A, 104, 10164–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaffe A.E., Murakami P., Lee H., Leek J.T., Fallin M.D., Feinberg A.P., Irizarry R.A. (2012) Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol., 41, 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics (Oxford), 30, 1363–1369. England), [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guintivano J., Aryee M.J., Kaminsky Z.A. (2013) A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics, 8, 290–302. 10.4161/epi.23924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Houseman E.A., Accomando W.P., Koestler D.C., Christensen B.C., Marsit C.J., Nelson H.H., Wiencke J.K., Kelsey K.T. (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics, 13, 86–2105. 2113-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ruzicka W.B., Zhubi A., Veldic M., Grayson D.R., Costa E., Guidotti A. (2007) Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol. Psychiatry, 12, 385–397. [DOI] [PubMed] [Google Scholar]
- 20. Shulha H.P., Crisci J.L., Reshetov D., Tushir J.S., Cheung I., Bharadwaj R., Chou H.J., Houston I.B., Peter C.J., Mitchell A.C.. et al. (2012) Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol., 10, e1001427.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M.. et al. (2015) Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161, 1202–1214. 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rotem A., Ram O., Shoresh N., Sperling R.A., Goren A., Weitz D.A., Bernstein B.E. (2015) Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol., 33, 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grayson D.R., Guidotti A. (2013) The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology, 38, 138–166. 10.1038/npp.2012.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jaffe A.E., Gao Y., Deep-Soboslay A., Tao R., Hyde T.M., Weinberger D.R., Kleinman J.E. (2016) Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci., 19, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Montano C., Taub M.A., Jaffe A., Briem E., Feinberg J.I., Trygvadottir R., Idrizi A., Runarsson A., Berndsen B., Gur R.C.. et al. (2016) Association of DNA methylation differences with schizophrenia in an epigenome-wide association study. JAMA Psychiatry, 73, 506–514. 10.1001/jamapsychiatry.2016.0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hannon E., Dempster E., Viana J., Burrage J., Smith A.R., Macdonald R., St Clair D., Mustard C., Breen G., Therman S.. et al. (2016) An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol., 17, 176–016. 1041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Asai T., Bundo M., Sugawara H., Sunaga F., Ueda J., Tanaka G., Ishigooka J., Kasai K., Kato T., Iwamoto K. (2013) Effect of mood stabilizers on DNA methylation in human neuroblastoma cells. Int. J. Neuropsychopharmacol., 16, 2285–2294. [DOI] [PubMed] [Google Scholar]
- 28. Phiel C.J., Zhang F., Huang E.Y., Guenther M.G., Lazar M.A., Klein P.S. (2001) Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem., 276, 36734–36741. [DOI] [PubMed] [Google Scholar]
- 29. Guo J.U., Su Y., Shin J.H., Shin J., Li H., Xie B., Zhong C., Hu S., Le T., Fan G.. et al. (2014) Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci., 17, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ladd-Acosta C., Hansen K.D., Briem E., Fallin M.D., Kaufmann W.E., Feinberg A.P. (2014) Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry, 19, 862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jones P.A. (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet., 13, 484–492. [DOI] [PubMed] [Google Scholar]
- 32. Houtmeyers R., Souopgui J., Tejpar S., Arkell R. (2013) The ZIC gene family encodes multi-functional proteins essential for patterning and morphogenesis. Cell. Mol. Life Sci., 70, 3791–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brown S.A., Warburton D., Brown L.Y., Yu C.Y., Roeder E.R., Stengel-Rutkowski S., Hennekam R.C., Muenke M. (1998) Holoprosencephaly due to mutations in ZIC2, a homologue of drosophila odd-paired. Nat. Genet., 20, 180–183. [DOI] [PubMed] [Google Scholar]
- 34. Aruga J., Inoue T., Hoshino J., Mikoshiba K. (2002) Zic2 controls cerebellar development in cooperation with Zic1. J. Neurosci., 22, 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frank C.L., Liu F., Wijayatunge R., Song L., Biegler M.T., Yang M.G., Vockley C.M., Safi A., Gersbach C.A., Crawford G.E.. et al. (2015) Regulation of chromatin accessibility and Zic binding at enhancers in the developing cerebellum. Nat. Neurosci., 18, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ciani L., Salinas P.C. (2005) WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat. Rev. Neurosci., 6, 351–362. [DOI] [PubMed] [Google Scholar]
- 37. Rosso S.B., Inestrosa N.C. (2013) WNT signaling in neuronal maturation and synaptogenesis. Front. Cell. Neurosci., 7, 103.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhong X., Desilva T., Lin L., Bodine P., Bhat R.A., Presman E., Pocas J., Stahl M., Kriz R. (2007) Regulation of secreted Frizzled-related protein-1 by heparin. J. Biol. Chem., 282, 20523–20533. [DOI] [PubMed] [Google Scholar]
- 39. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A.. et al. (2015) Tissue-based map of the human proteome. Science (New York, N., 347, 1260419 Proteomics. Y.), [DOI] [PubMed] [Google Scholar]
- 40. Konno D., Iwashita M., Satoh Y., Momiyama A., Abe T., Kiyonari H., Matsuzaki F., Alsina B. (2012) The mammalian DM domain transcription factor Dmrta2 is required for early embryonic development of the cerebral cortex. PLoS One, 7, e46577.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Machon O., Masek J., Machonova O., Krauss S., Kozmik Z. (2015) Meis2 is essential for cranial and cardiac neural crest development. BMC Dev. Biol., 15, 40–015. 0093-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tian L., Wu X., Lin Y., Liu Z., Xiong F., Han Z., Zhou Y., Zeng Q., Wang Y., Deng J., Chen H. (2009) Characterization and potential function of a novel pre-implantation embryo-specific RING finger protein: TRIML1. Mol. Reprod. Dev., 76, 656–664. [DOI] [PubMed] [Google Scholar]
- 43. Baskerville S., Bartel D.P. (2005) Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA (New York, N.11, 241–247. Y.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nowak J.S., Choudhury N.R., de Lima Alves F., Rappsilber J., Michlewski G. (2014) Lin28a regulates neuronal differentiation and controls miR-9 production. Nat. Commun., 5, 3687.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Laneve P., Gioia U., Andriotto A., Moretti F., Bozzoni I., Caffarelli E. (2010) A minicircuitry involving REST and CREB controls miR-9-2 expression during human neuronal differentiation. Nucleic Acids Res., 38, 6895–6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yasunaga S., Grati M., Cohen-Salmon M., El-Amraoui A., Mustapha M., Salem N., El-Zir E., Loiselet J., Petit C. (1999) A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness . Nat. Genet, 21, 363–369., [DOI] [PubMed] [Google Scholar]
- 47. Yasunaga S., Grati M., Chardenoux S., Smith T.N., Friedman T.B., Lalwani A.K., Wilcox E.R., Petit C. (2000) OTOF encodes multiple long and short isoforms: genetic evidence that the long ones underlie recessive deafness DFNB9. Am. J. Hum. Genet., 67, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Du P., Zhang X., Huang C.C., Jafari N., Kibbe W.A., Hou L., Lin S.M. (2010) Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics, 11, 587–2105. 2111-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Price M.E., Cotton A.M., Lam L.L., Farre P., Emberly E., Brown C.J., Robinson W.P., Kobor M.S. (2013) Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin, 6, 4–8935. 8936-8934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li L.C., Dahiya R. (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics, 18, 1427–1431. 10.1093/bioinformatics/18.11.1427 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.