

Abstract
Spinal muscular atrophy with respiratory distress type 1 (SMARD1) is an autosomal recessive motor neuron disease causing distal limb muscle atrophy that progresses proximally and is accompanied by diaphragmatic paralysis. Neuromuscular junction (NMJ) alterations have been reported in muscles of SMARD1 model mice, known as nmd mice, with varying degrees of severity, suggesting that different muscles are specifically and selectively resistant or susceptible to denervation. To evaluate the extent of NMJ pathology in a broad range of muscles, a panel of axial and appendicular muscles were isolated and immunostained from nmd mice. These analyses revealed that selective distal appendage muscles were highly vulnerable to denervation. Susceptibility to pathology was not limited to NMJ alterations, but included defects in myelination within those neurons innervating susceptible muscles. Interestingly, end plate fragmentation was present within all muscles independent of the extent of NMJ alterations, suggesting that end plate fragmentation is an early hallmark of SMARD1 pathogenesis. Expressing the full-length IGHMBP2 cDNA using an adeno-associated virus (AAV9) significantly decreased all aspects of muscle and nerve disease pathology. These results shed new light onto the pathogenesis of SMARD1 by identifying specific motor units that are resistant and susceptible to neurodegeneration in an important model of SMARD1.
Introduction
Spinal muscular atrophy with respiratory distress type 1 (SMARD1, OMIM 604320) is a devastating motor neuron disorder that manifests during infancy and leads to premature death, typically within 2 years of age (1,2). SMARD1 presents with a broad spectrum of clinical features but is primarily associated with severe muscle atrophy that initiates within distal limb muscles and progresses proximally (1,3–5). Although distal limb muscle atrophy is a major symptom, the most critical complication is respiratory failure due to diaphragmatic paralysis, typically manifesting within the first few months of life (1,3,5,6). In the majority of cases, children with SMARD1 require permanent artificial mechanical ventilation (1,2,4–6). Selective loss of alpha motor neurons in the ventral horns of the spinal cord and subsequent neuromuscular junction (NMJ) denervation is the main cause of muscle atrophy (7,8). The molecular basis of SMARD1 arises from loss-of-function mutations in the immunoglobulin-μ-binding protein 2 (IGHMBP2) gene located in chromosome 11q13.3 (3,9–11). To date, no effective SMARD1 therapeutic treatments exist.
IGHMBP2 is a ubiquitously expressed protein that contains a RNA/DNA helicase domain, an ATP binding motif, a zinc finger domain, and nucleic acid binding domain with homology to the UPF1-like superfamily 1 helicases (9,12–14). Most intragenic mutations identified in SMARD1 patients reside within these conserved domains (1,2,15,16). Biochemical studies of IGHMBP2 have demonstrated transcription (17) and pre-mRNA processing activity; however, the most well-described function is in translation where it interacts with factors including tRNA Tyr, Reptin, Potin, TFIIIC220, and ABT1 (17). A genetic suppressor of the nmd phenotype was shown to encode several tRNA Tyr genes (17), suggesting that IGHMBP2’s role in translation was directly related to disease development. The specific role that IGHMBP2 plays in translation remains to be clarified, and the mechanisms by which loss of function leads to selective loss of alpha motor neurons is not understood (18–20).
The murine Ighmbp2 gene is highly homologous to the human IGHMBP2 gene. A mouse model for SMARD1, called nmd for neuromuscular degeneration, was identified as a spontaneous mutation within Ighmbp2 (21,22). This mutation generates a splice site alteration within intron 4, resulting in aberrant splicing of Ighmbp2 mRNA and an approximately 75%–80% reduction in functional Ighmbp2 protein (21,22). The first disease phenotype associated with nmd mice, namely hindlimb muscle weakness, occurs approximately at postnatal day (PND) 21 with severe paralysis by 5 weeks and survival up to 14 weeks (18,21,23). Unlike spinal muscular atrophy (SMA) mice, nmd mice show significant motor neuron loss prior to disease symptoms with approximately a 40% reduction by PND10, followed by a plateau and then progressive motor neuron degeneration until death (8,18). No motor end plate degeneration is detected prior to motor neuron degeneration and compound motor action potential values were significantly decreased while motor nerve conduction velocity values were normal in nmd mice (8,18). Early loss of motor neurons as well as loss of large caliber axons in distal peripheral nerves, which correlate with NMJ denervation, are features shared by the nmd mouse model and SMARD1 patients (5,8,18,24,25). Unlike SMARD1 patients, nmd mice do not demonstrate severe respiratory distress until the end stage of the disease and appear to die as a result of dilated cardiomyopathy not typically present in SMARD1 patients (1,23). While no SMARD1-specific therapeutics exist, gene replacement via an AAV9-IGHMBP2 vector significantly reduced disease severity in the nmd mouse in multiple preclinical studies (19,20).
Loss of alpha motor neurons is a common feature of motor neuron diseases such as amyotrophic lateral sclerosis (ALS) (26,27), spinal muscular atrophy (SMA) (28–30), and spinal and bulbar muscular atrophy (SBMA) (31). Moreover, systemic analyses of different SMA mouse models has revealed selective vulnerability within specific motor units, suggesting that a subset of motor neurons are more vulnerable to denervation (27,29,32,33). To uncover the molecular mechanisms underlying differential susceptibility, transcriptomic analyses of differentially vulnerable motor neurons was performed in ALS, SBMA, and SMA disease contexts, revealing a discrete number of potential disease-modifying targets common to each motor neuron disease (34). Interestingly, several studies on nmd mice report different levels of NMJ denervation in diaphragm (20), gastrocnemius (8,19,20), and quadriceps (20) muscles, suggesting that selective vulnerability of motor neurons is also a feature of SMARD1. Finding specific selective resistant and vulnerable motor units in SMARD1 will allow cross-disease comparisons to identify disease-modifying targets for therapeutic treatment.
In this report, NMJ pathology was systematically evaluated in clinically relevant muscles from the neck, trunk, and appendages to determine whether selective vulnerability to denervation existed within the nmd mouse. Since SMARD1 patients exhibit axonal loss and myelination pathologies (6,24), axonal pathology was also examined to determine if myelination alterations were present in a similar pattern in nmd mice. We also determined whether NMJ and myelin pathologies within these vulnerable populations could be alleviated by gene replacement therapy using the AAV9-IGHMBP2 vector. Our results revealed that NMJ denervation in nmd mice is increased in muscles of the distal limbs compared to trunk and neck muscles. Consistent with the NMJ pathology, axonal myelination alterations were also identified within nerves of highly vulnerable muscles, but not in nerves of resistant muscles. Furthermore, NMJ end plate fragmentation, which has not been previously described in nmd mice, was present in all muscles independent of denervation vulnerability. Finally, presymptomatic administration of AAV9-IGHMBP2 prevents NMJ pathology independent of resistant/susceptible status. Collectively, these results provide insight into the neurodegenerative phenotype of the nmd mouse and SMARD1 pathology and demonstrate that even the severe axonal and NMJ pathologies detected in the vulnerable populations can be reversed by early administration of AAV9-IGHMBP2.
Results
Distal appendicular muscles are highly vulnerable to NMJ denervation in nmd mice
To investigate whether particular motor units were resistant or vulnerable to disease development in nmd mice, muscles were isolated from clinically relevant anatomical regions of the body. Muscles involved in head movement [Longissimus capitis (LC) and splenius capitis (SC)], ear movement [Levator auris longus (caudal), cLAL], and mastication (masseter) were selected and categorized as neck muscles. Muscles important for respiration [Diaphragm, transversus abdominis (TVA), serratus posterior inferior (SPI]) and trunk movement [Latissimus dorsi (LD)] were categorized as trunk muscles. Muscles involved in limb movement [biceps, triceps, gastrocnemius, tibialis anterior (TA), extensor digitorum longus (EDL), lumbricals, and flexor digitorum brevis (FDB) 2, 3, and 4] were categorized as distal appendicular muscles. Immunohistochemical labeling using antibodies against neurofilament-heavy (NF-H) and synaptophysin was to visualize the axon and the presynaptic axon termini, while fluorochrome-tagged alpha-bungarotoxin was used to visualize the postsynaptic end plates.
For these analyses, muscles from 8-week-old nmd and control mice were harvested and analyzed in a blinded manner (Fig. 1). At this stage, all nmd animals are symptomatic and display the characteristic hindlimb paralysis phenotype. NMJ defects were quantified within each anatomical area based on: 1) denervation, 2) partial denervation, or 3) fully innervated. NMJ innervation was determined according to the overlap between the axon terminal and the NMJ end plate. Complete overlap between terminal and end plate was considered full innervation, while incomplete overlap was considered partial denervation, and absence of axon terminal over an end plate was considered full denervation. In nmd mice, a subset of skeletal muscles were identified that were resistant to denervation, including the cLAL and the diaphragm; in contrast, muscles including the gastrocnemius and the FDB muscles displayed significant denervation (Figs 1 and 2). Of the resistant muscles, the cLAL (ear movement) and masseter (jaw) muscles exhibited no detectable denervation at a symptomatic stage of disease development (Figs 1A and 2). However, within the LC and SC (neck movement) muscles, there was a significant decrease in fully innervated NMJs as only 40%–60% of all end plates were fully innervated (Fig. 1A). These results suggest that within muscles of the anterior region, there are resistant muscles (cLAL and masseter) and susceptible muscles (LC and SC) to denervation. A similar pattern was observed in the trunk muscles (LD) and those involved in respiration (diaphragm, TVA, SPI). This group of muscles also comprised resistant and vulnerable muscles. Denervation was not substantial within diaphragm and SPI muscles, whereas a significant reduction in fully innervated NMJs was observed in the LD and TVA muscles as only ∼40% and ∼60% of NMJs, respectively, were fully innervated (Figs 1B and 2). Moreover, no difference in denervation levels was observed between the diaphragm, SPI, cLAL, and masseter. Interestingly, the diaphragm, TVA, and SPI muscles, all associated with respiration, demonstrated varying degrees of denervation.
Figure 1.

Vulnerability to NMJ denervation is more pronounced in distal appendicular muscles and can be prevented by AAV9-IGHMBP2 treatment. Quantification of NMJ pathology showing percentages of fully innervated, partially innervated, and fully denervated end plates from 8-week-old wild type, nmd, and nmd + AAV9-IGHMBP2 treated mice. (A) Quantification of NMJ pathology in resistant (cLAL and masseter) and slightly vulnerable (LC and SC) muscles from the neck. (B) Quantification of NMJ pathology in resistant (diaphragm and SPI) and vulnerable (LD and TVA) muscles from the trunk. (C) Quantification of NMJ pathology in vulnerable (gastrocnemius, TA, FDB-2, FDB-3, and FDB-4) and resistant (EDL and lumbricals) muscles from distal appendages. Quantification of NMJ pathology also shows that AAV9-IGHMBP2 treatment of nmd mice significantly reduces NMJ pathology in all muscle groups analyzed (A, B, and C). Data were analyzed by one-way ANOVA followed by a Bonferroni post hoc test for multiple comparisons. Data expressed as mean ± SEM. *P < 0.05, **P < 0.001, and ***P < 0.0001. n.s., not significant. n = 3 animals per treatment.
Figure 2.

Increased vulnerability to NMJ denervation in distal appendicular muscles and prevention with AAV9-IGHMBP2 treatment. Neck (anterior), trunk (middle), and distal appendicular muscles (posterior) were immunostained to label axon (NF-H), axon terminal (SV2), and end plate (AChRs). (Top panel) Images of representative muscle (cLAL) from the neck showing resistance to NMJ denervation in nmd (middle) and nmd+ AAV9-IGHMBP2 treated (right) compared to an unaffected/wild-type control (left) (scale bars = 100 µm). (Middle panel) Images of a representative resistant muscle (diaphragm) from the trunk area in nmd (middle) and nmd+ AAV9-IGHMBP2 treated (right) compared to an unaffected control (left) (scale bars = 30 µm). (Bottom two panels) Representative muscles (gastrocnemius and FDB-2) from distal appendicular area with high vulnerability to NMJ denervation in nmd (middle) compared to control (left) (scale bars = 30 µm). AAV9-IGHMPB2 treatment successfully prevents NMJ denervation in highly vulnerable muscles (bottom right). Maximum projection confocal microscope images taken at 20× magnification.
The greatest discrepancy between vulnerable and resistant populations was observed within the NMJ integrity of distal appendicular muscles. Of this group of muscles, the gastrocnemius muscle was most severely affected as <5% of the total NMJs were fully innervated (Figs 1C and 2). Similarly, the TA, biceps, and triceps (Fig. 1C and Supplementary Material, Fig. S1) displayed a significant decrease in the number of fully innervated end plates, ranging from ∼35% to ∼60%. In contrast, while the EDL and lumbrical muscles had a decrease in fully innervated end plates (∼65% fully innervated), there was no significant change in denervation as these muscles were at least partially innervated (Fig. 1C). FDB-2, 3, and 4 muscles, a collection of muscles within the foot, all showed significant denervation as each muscle had only <10%, ∼20% and ∼18% of fully innervated NMJs, respectively (Figs. 1C and 2). Collectively, these results demonstrate that subpopulations of resistant and susceptible neurons are present within nmd mice that exhibit significant disease phenotypes.
Distal appendicular muscles are the most vulnerable to NMJ denervation
NMJs within the gastrocnemius and the foot muscles FDB-2, 3, and 4 appeared to be most susceptible to partial and full denervation. To determine whether there was a correlation between the anatomical region and/or function, the percentage of fully innervated NMJs was compared across all muscles groups between wild-type and nmd mice (Fig. 3). Out of the 16 muscles analyzed, the gastrocnemius muscle of nmd mice was the most vulnerable, as very few detectable fully innervated NMJs were present (<1.0%) (Fig. 3). The foot muscles FDB-2 (∼3%), FDB-3 (∼21%), and FDB-4 (∼18%) were minimally innervated, whereas the lumbricals showed decreased innervation with significant variability (Fig. 3). Forelimb (biceps and triceps) and hindlimb (TA) muscles all demonstrated significant denervation, although less than the gastrocnemius (Fig. 3). Interestingly, adjacent muscles in the hindlimb, the gastrocnemius, EDL, and TA showed significant variability in the number of fully innervated NMJs (<1% to ∼65%) (Fig. 3). At the 8-week time point, each of the muscles was examined, and the diaphragm, which becomes paralyzed in SMARD1 patients, showed a high degree of resistance to NMJ denervation in nmd mice with nearly 80% fully innervated NMJs (Fig. 3). Similar results were observed within the SPI muscle that showed ∼80% fully innervated NMJs. In contrast, the TVA muscle, a muscle also used for respiration, showed significant decrease in full innervation as only ∼57% of its NMJs were fully innervated. These results indicate that, although a wide range of muscles from different anatomical regions show vulnerability to NMJ denervation, the distal appendage muscles are the most vulnerable to denervation.
Figure 3.

Differential vulnerability to denervation predominantly affects neurons of distal muscles. Quantification of immunostained muscles and comparison of percent available fully innervated end plates across all muscles analyzed in 8-week wild-type and nmd mice. Individual comparisons were analyzed by Student’s t-test. Data are expressed as mean ± SEM. *P < 0.05. n = 3 animals per treatment.
End plate fragmentation occurs in NMJ of nmd muscles independent of denervation status
End plate fragmentation is a key pathological feature in mouse models of Duchenne muscular dystrophy (DMD) and in aging deteriorating muscle fibers (35,36). To determine if end plate fragmentation was also observed in muscles affected with SMARD1, muscles from nmd mice were examined at 8 weeks of age (Fig. 4). In the masseter and diaphragm muscles, which showed low levels of NMJ denervation, end plate fragmentation was observed within ∼8% to 20% of total end plates; the difference did not reach statistical significance (Fig. 4A). Interestingly, the LAL muscle, which did not show significant NMJ denervation, demonstrated significant end plate fragmentation (∼50%) (Fig. 4). Representative double-labeled images revealed fragmented end plates (Fig. 4B, white arrows) in LAL muscle of untreated nmd mice that were not found in wild-type controls.
Figure 4.

End plate fragmentation is present in nmd muscles regardless of resistance to NMJ denervation. (A) Quantification of percent normal and fragmented end plates in muscles from 8-week wild-type, nmd, and nmd+AAV9-IGHMBP2 treated mice. End plate fragmentation in various selected muscles with differential vulnerability to NMJ denervation encompassing neck, trunk, and distal appendages. AAV9-IGHMPB2 treatment of nmd mice prevents end plate fragmentation in all muscles analyzed. (B) Representative images of immunostained muscle showing presence of end plate fragmentation (arrows) in a muscle resistant to NMJ denervation (cLAL) and prevention of end plate fragmentation by AAV9-IGHMBP2 treatment. Data were analyzed by one-way ANOVA followed by a Bonferroni post hoc test for multiple comparisons. Data are expressed as mean ± SEM. **P < 0.001; ***P < 0.0001. n.s., not significant. n = 3 animals per treatment.
Muscles from the neck, trunk, and distal appendicular regions with insignificant denervation (SPI and EDL and LC) all demonstrated substantial end plate fragmentation (Supplementary Material, Fig. S2). The TA, gastrocnemius, and FDB muscles with significant denervation also showed significant end plate fragmentation (62%, 95%, and (0%–97%, respectively) (Fig. 4B and Supplementary Material, Fig. S2). Further analysis of the rest of the muscles analyzed from each of the three anatomical regions (neck, trunk, and appendages) displayed a significant percent of fragmented end plates (Supplementary Material, Fig. S2). Remarkably, the presence and abundance of fragmented end plates had no correlation with the percent decrease in fully innervated NMJs.
Vulnerability to pathology within innervating nerves of vulnerable muscles
In peripheral nerves, a single Schwann cell innervates a segment of an axon, referred to as the internode. During development, a correlation between internode length and fiber diameter is established, allowing the nerves to optimally transmit impulses (37–39). Alterations to this correlation, by demyelination or remyelination, disrupt optimal impulse speed and lead to pathology (40,41). To investigate if vulnerability extends to the nerves of affected muscles, myelin structure/integrity was examined in nerves that innervate highly vulnerable or resistant muscles: the gastrocnemius and the masseter, respectively. Individual fibers were teased from the tibial nerve, which innervates the gastrocnemius muscle, and the distance between the nodes of Ranvier, or the internode length, and fiber diameter were measured from wild-type and nmd mice (Fig. 5A and B). As expected, there existed a correlation between fiber diameter and internode length in both wild-type and nmd-teased fibers (Fig. 6A). Linear regressions of fiber diameter relative to internode length were calculated and found to be significantly different (P < 0.0001), with r2 of 0.204 and 0.01239 for nmd and wild type mice, respectively. Interestingly, these comparisons suggested a shift in the overall fiber diameter and internode length in nmd tibial fibers toward smaller values. Additionally, a significant decrease (P = 0.0001) in mean fiber diameter of tibial nerve fibers was observed when compared to wild type (6.19 ± 0.07 μm versus 7.89 ± 0.09 μm, respectively) (Fig. 6B). Mean internode length was also significantly reduced (P < 0.0001) in nmd tibial nerve fibers compared to controls (554 ± 5.6 μm versus 707 ± 6.0 µm, respectively) (Fig. 6C). Reduced internode length and fiber diameter are typical alterations that result from demyelination/remyelination events (40,41). Since changes in fiber diameter and internode length were present, the frequency of axons showing myelin defects indicative of demyelination were quantified (Fig. 5B). Strikingly, the tibial nerve had a significantly higher number of randomly selected fibers showing segmental demyelination, myelin fragmentation, or complete demyelination in nmd mice as compared to controls (∼37% versus ∼0.6%, respectively, P = 0.0135) (Fig. 6D).
Figure 5.

Tibial nerve fibers show demyelination in nmd mice. (A) (Left panel) Representative sample of wild-type tibial nerve fibers indicating location of Schwann cell nucleus (star) and nodes of Ranvier (arrows). (Right panel) High magnification images showing wild-type axons with normal myelin, node of Ranvier (arrows), and Schwann cell nucleus (star). (B) (Left panel) Representative sample of nmd tibial nerve teased fibers showing a bundle of axons completely demyelinated (lightning bolt), axons with myelin fragmentation (triangle) and axons with normal myelin and nodes of Ranvier (arrows). Note that in demyelinated axons and fragmented myelin axons, no nodes of Ranvier can be identified. (Left panel) High magnification images of axons with fragmented myelin.
Figure 6.

Pathology in tibial nerve from nmd mice. (A) Scatter plots showing the correlation between internode length and fiber diameter of nmd and wild type controls. r2 values of regression correlations showed a significant shift towards smaller fiber diameter with shorter internode lengths in the population of nmd fibers compared with wild-type controls (P < 0.0001). (B) Mean fiber diameter comparisons revealed a significant (P = 0.0001) decrease in nmd fiber diameter (6.19 ± 0.07 µm) compared to wild-type fiber diameter (7.89 ± 0.09 µm). (C) Mean internode length comparisons revealed a significant (P < 0.0001) decrease in nmd (554.8 ± 5.69 µm) compared to wild-type (707.6 ± 6.1 µm) internode length. (D) Quantification of abundance of fibers with myelin alterations (myelin fragmentation, segmental demyelination, complete demyelination) revealed a significant increase (0 = 0.0135) in percent of total fibers teased with myelin defects in nmd (37%) compared to wild-type controls (0.6%). Individual comparisons were analyzed by Student’s t-test. Data expressed as mean ± SEM. *P < 0.05. n = 3 animals per treatment.
To investigate if similar nerve pathology was present in nerves of resistant muscles, fiber morphology was analyzed in the anterior mandibular division of the trigeminal nerve, which innervates the masseter (resistant) muscle (Supplementary Material, Fig. S3). In contrast to tibial nerve fibers, no differences were observed in the linear correlation between fiber diameter and internode length of nmd and wild-type trigeminal nerve fibers (r2 = 0.00095 versus 0.2343, respectively) (P = 0.2343) (Fig. 7A). No differences in mean fiber diameter (5.68 ± 0.04 μm and 5.59 ± 0.05 μm) (Fig. 7B) or internode length (396 ± 4.4 μm and 404 ± 4.3 μm) (Fig. 7C) between nmd and wild-type controls, respectively, were observed. In all axons examined, no signs of myelin alterations were found in trigeminal nerve fibers of nmd or wild-type controls (Fig. 7D).
Figure 7.

No pathology was found in nerve innervating the masseter muscle. (A) Scatter plot showing correlations of internode length versus fiber diameter correlations in nmd and wild-type controls. No differences were found in r2 regression correlation values between wild-type and nmd mice. (B) Mean fiber diameter comparisons between wild-type and nmd mice showed no statistical differences (P = 0.095). (C) Comparisons of mean internode length also revealed no differences (P = 0.123) between wild-type and nmd mice. (D) Quantification of abundance of fibers with myelin defects revealed no difference (P = 0.528) between wild-type and nmd mice. Individual comparisons were analyzed by Student’s t-test. Data are expressed as mean ± SEM. n = 3 animals per treatment.
These data demonstrate that vulnerability to pathology is not restricted to just muscle or neuronal tissue but also extends to the myelinating Schwann cells of nerves that innervate vulnerable muscles. These data bring to light a new and important target for the investigation of disease pathology in the nmd mouse model. Therefore, therapeutics designed to prevent/restore nmd phenotype must address the complete motor unit: neuron myelinating Schwann cells and muscle.
AAV9-IGHMBP2 treatment prevents widespread NMJ pathology development
To determine whether the nmd-associated pathology was preventable and/or reversible, a potent gene therapy vector, AAV9-IGHMBP2, was delivered to neonatal nmd mice. We and others have demonstrated that neonatal delivery of AAV9-IGHMPB2 significantly extends survival and reduces disease severity in nmd mice (19,20). Neonatal delivery (PND 2) of AAV9-IGHMBP2 significantly improved the pathology of vulnerable muscles by preventing the development of NMJ defects (Figs 1 and 2 and Supplementary Material, Fig. S1). As anticipated, AAV9-IGHMBP2 treatment did not further enhance or diminish NMJ structural integrity in resistant muscles (cLAL, masseter, diaphragm) (Figs 1 and 2 and Supplementary Material, Fig. S1). Independent of the degree of denervation caused by SMARD1 development, early administration (PND 2) of AAV9-IGHMBP2 reduced NMJ alterations in nmd mice. The percent of end plates displaying full innervation within the LC, LD, SC, and TVA was significantly increased at 8 weeks of age (Figs 1 and 2). Even the most susceptible muscles, the gastrocnemius, TA, biceps, triceps, and FDBs, demonstrated significant changes in innervation (Fig. 1 and Supplementary Material, Fig. S1). Moreover, neonatal delivery of AAV9-IGHMBP2 prevents mild and severe denervation across a broad range of disease-relevant muscles (Fig. 1).
End plate fragmentation was significantly improved to normal across all muscles analyzed following AAV9-IGHMBP2 treatment (Fig. 4 and Supplementary Material, Fig. S2), although as noted previously, end plate fragmentation did not correlate with denervation susceptibility. LAL NMJs, which were resistant to denervation but susceptible to end plate fragmentation, were fully restored and were indistinguishable from wild-type samples (Fig. 4). Interestingly, AAV9-IGHMBP2 nearly eliminated end plate fragmentation within NMJs of the even most susceptible muscles (Fig. 4 and Supplementary Material, Fig. S2). These results show that early administration of AAV9-IGHMBP2 successfully prevents development of NMJ pathology in nmd mice.
Discussion
While initially identified as a spontaneous mutation resulting in a neurodegenerative phenotype, the nmd mouse has been an important tool in the molecular and cellular understanding of SMARD1. Similar to SMARD1 patients, muscle weakness and atrophy attributed to NMJ denervation in the nmd mice initiates in distal limb muscles like the gastrocnemius and quadriceps muscles as early as P16 (8,18) (or 1 month of age in humans) and progressively deteriorates with age (5,24,42). Here, we provide further evidence that shows a differential degree of vulnerability to NMJ pathology in a panel of clinically relevant muscles. Distal limb muscles, like the gastrocnemius and FDBs muscles, were found to be the most vulnerable to denervation as they showed near, or over, 90% NMJ denervation by 8 weeks of age. These data are consistent with the previous reports investigating the gastrocnemius muscle in mice (8,18–20) and humans (5,24,42). A major disparity between the nmd model and human SMARD1 pathogenesis is the absence of early diaphragmatic paralysis in the mice, which in humans has been attributed to loss of end plate innervation (24). Reports on nmd diaphragm pathology have been variable with some reporting about 50% NMJ denervation by 4 weeks of age (20) or no loss of NMJ denervation at 5 weeks (8) or even at 14 weeks (18). This lack of NMJ denervation in the diaphragm has been in part attributed to axonal sprouting and increased motor unit size (8). Our current data show further evidence that no major NMJ denervation is found in the diaphragm of the nmd mice at 8 weeks of age, which has previously been attributed by motor axon sprouting and reinnervation of vacated end plates (8). However, in the current study, we found an abundance of fragmented end plates in all muscles examined, including the diaphragm at 8 weeks of age, indicating end plate deterioration (35,36). While the presence or absence of end plate fragmentation did not correlate with resistant or susceptible NMJ phenotypes, the most severely denervated muscles (gastrocnemius and FDBs) were composed of almost exclusively of NMJs with a fragmented end plate structure. These data suggest that severe fragmentation of the end plate in muscle fibers might hinder the ability of sprouting axons to reinnervate vacated end plates, thus resulting in severe loss of innervation in muscles with high levels of fragmented end plates. The presence of fragmented end plates in all muscles analyzed regardless of denervation degree supports the idea that IGHMBP2 has independent effects in different terminally differentiated tissues (23,43) and highlights the need for system-wide therapeutics. Whether IGHMBP2 plays a direct role in preventing end plate fragmentation or whether it is due to downstream events remains to be determined.
SMARD1 shares common pathology features with other motor neuron diseases, like selective motor neuron loss that leads to muscle atrophy despite having a distinct genetic cause (19,20,26,27,29,32). Comparative analysis of NMJ pathology identified specific groups of motor units with selective vulnerability in SMA, ALS, and SBMA, and subsequent transcriptomic analysis revealed genes that were differentially regulated in vulnerable versus resistant tissues (44–46). In this study, we provide detailed analysis of the nmd pathogenesis and demonstrate that not all functionally relevant muscles develop NMJ pathology to the same degree. We identified two muscle groups, gastrocnemius and FDBs, which show the highest vulnerability to denervation in nmd mice. In addition, we demonstrate that vulnerability to pathology also affected the structural integrity of the nerve fibers innervating selectively vulnerable muscle groups. Thus, these findings identify specific motor units that could be used for further analyses aiming at investigating the potential mechanisms by which selective vulnerability occurs in the nmd mouse.
Axonal demyelination is a significant pathological component in demyelinating diseases such as multiple sclerosis and Charcot–Marie–Tooth, which leads to axonal nerve dysfunction by directly impairing electrophysiological properties of the axon (47–51). Initial reports have also indicated that myelin alterations exist in SMARD1 (4,24). Similarly, reports on ALS have demonstrated development of myelin structure and stability in ALS patients (52,53). Moreover, defects in the peripheral nervous system that are intrinsic to the myelinating Schwann cells have been reported in multiple models of SMA (54,55). These data indicate that myelinating Schwann cells play an important role in disease severity in these neuromuscular diseases. Our current analysis suggests that in the nmd mouse, Schwann cells might also be susceptible various levels of disease vulnerability. Utilizing Schwann cells as disease-modifying targets in the nmd phenotype offers a new avenue for therapeutic investigation since Schwann cells, which unlike motor neurons, can be useful due to their regenerative properties. Further investigation into the contributions of Schwann cells to the pathology in SMARD1 will be required to assess their potential for therapeutic treatments of the disease.
Our analysis revealed that fragmentation of the end plate is a prominent feature of nmd pathology that has not been reported or examined in previously (8,19,20). Given that end plate fragmentation was found in all muscles analyzed regardless of denervation and that this alteration was successfully prevented by AAV9-IGHMBP2 treatment suggests that IGHMBP2 plays an important role within muscle fibers for end plate maintenance. End plate fragmentation is an alteration commonly found in mouse models of DMD, in which it has been demonstrated to impair NMJ function and prevent proper muscle contraction leading to pathology (35,56,57). Moreover, end plate fragmentation has also been observed in SMA (58), SBMA (59), and in ALS patients (60). These observations indicate that fragmentation of the end plate is a common alteration seen in multiple neuromuscular diseases, which suggests that this alteration might be critical across multiple disease settings and highlights the importance for future study. Investigation into the mechanisms leading to end plate fragmentation in nmd could reveal pathways that are common across multiple neuromuscular diseases, which in turn can help identify potential therapeutic targets for disease treatment. Consistent with this notion, recently a discrete subset of differentially expressed genes was identified between vulnerable and resistant muscles (34). Across several neurodegenerative diseases, such as SMA, ALS, and SBMA, these genes were aberrantly expressed; however, reintroduction of one of the significantly reduced genes—SNCA1—partially rescues the NMJ pathology and significantly extended survival. This type of analysis in nmd could lead to the identification of modifying genes as well as druggable pathways and genes that reduce disease severity.
Materials and Methods
Animal procedures and injection
All animal procedures were carried out in accordance with procedures approved by NIH and MU animal Care and Use Committee. The nmd animals were genotyped at P1 according to the genotyping procedure outlined by the distributor Jackson Laboratories (Jackson Laboratories, Bar Harbor, ME). Mice underwent intracerebroventricular (19,61,62) injection on P2 and P3 with 1.25 × 1011 or 2.5 × 1011 viral genomes. At 8 weeks of age, the mice were anesthetized with 2.5% isoflurane and then perfused transcardially with cold 0.1M PBS (pH = 7.4) followed by 4% paraformaldehyde in PBS. Mice were post-fixed for 48 h at 4 °C in 4% PFA. After 48 h of post-fixing, animals were placed in 0.1M PBS and stored at 4 °C until used for muscle or nerve dissection.
Immunohistochemistry of NMJs
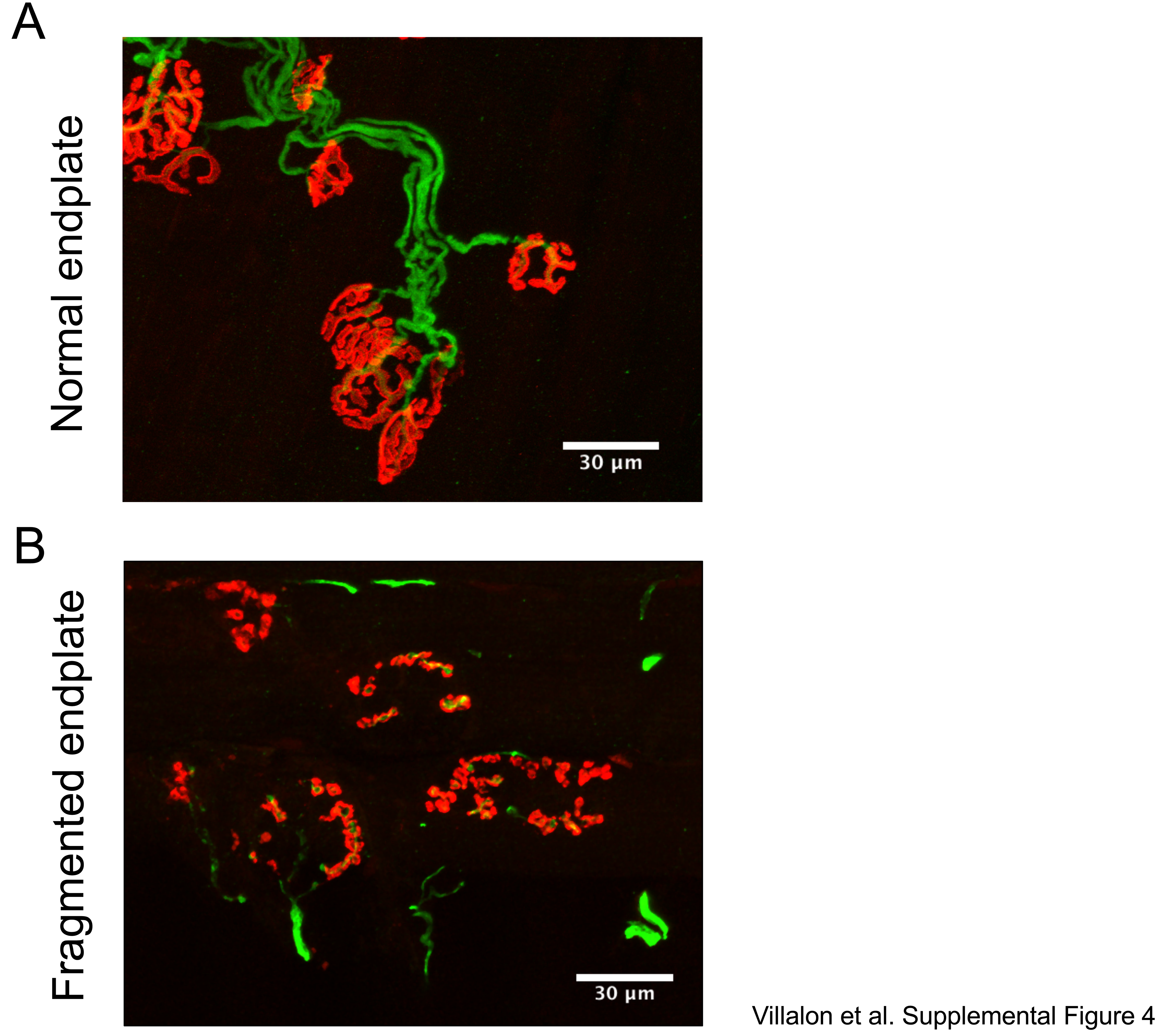
Eight-week-old wild-type, nmd, and nmd+AAV9-IGHMBP2 treaded male mice were used. Muscles were dissected out from previously fixed mice. Whole-mount preparations were done for several muscles from different areas of the body. Neck muscles included LC, SC, cLAL, and masseter muscles. Trunk muscles included SPI, LD, TVA, and Diaphragm. Appendage muscles included biceps, triceps, gastrocnemius, TA, EDL, Lumbricals, FDB-2 FDB-3, and FDB-4. Thick bulkier muscles, like gastrocnemius, masseter, and TA, were teased into bundles of 10–15 muscle fibers to promote penetration of antibodies. Thin ribbon-like muscles were used intact as antibody penetration was not an issue. Muscles were stained using specific antibodies, including anti-NF-H (1:2000; catalog AB5539, Chemicon, EMD Millipore, Burlington, MA), anti-synaptophysin (1: 200, catalog YE269, Life Technologies). Acetylcholine receptors were labeled with Alexa Fluor 594–conjugated α-bungarotoxin (1: 200; Life Technologies, Carlsbad, CA). Representative muscle images for were taken using a laser scanning confocal microscope (×40 objective; Leica TCS SP8, Leica Microsystems Inc.). NMJ analysis was performed on at least three randomly selected fields of view per muscle per mouse (×20 objective; Leica DM5500 B, Leica Microsystems Inc.). Images were analyzed using freely available Fiji Software (NIH). End plates missing overlapping nerve terminal staining were considered completely denervated. End plates with partial overlap were considered partially denervated, and end plates with complete overlap were considered fully innervated. End plate morphology was assessed for normal (pretzel shape) or fragmented structure (Supplementary Material, Fig. S4A and B, correspondingly). End plates showing pretzel-like structure on the entire end plate area were classified as normal end plates (Supplementary Material, Fig. S4A). In contrast, end plates showing full or partial fragmentation (Supplementary Material, Fig. S4B) were classified as fragmented end plates.
Teased fiber analysis
Tibial nerve and the anterior mandibular division of the trigeminal nerve were dissected out from previously fixed animals. Nerves were rinsed in 1× PBS for 5 min and transferred to a saturated solution of Sudan black in 70% ethanol and allowed to incubate for 1 h at room temperature. The samples were then rinsed twice in 70% ethanol for 5 min and transferred to a 10% glycerol solution. Fifty to 100 randomly selected axon bundles were separated from each nerve and transferred to a microscope slide containing a thin layer of 20% glycerol:BSA (1:1). Individual fibers were teased apart from the bundle and laid out on the glass slide. Fibers were then allowed to dry overnight on a 50 °C hot plate. Digital photos of the fibers were taken using a Zeiss Axiovert 200M inverted microscope and analyzed using Fiji software (NIH). Internode length and respective axon diameter were measured from at least 200 internodes per sample and used to calculate the average internode length, fiber diameter. The frequency of fibers with segmental demyelination or myelin fragmentation were counted and represented as percentage of the total fibers teased per genotype.
Statistics
Statistical analyses were performed using GraphPad Prism version 6.0 (GraphPad Software Inc.) and Microsoft Excel version 15.19.1 (Microsoft Corp.) NMJ denervation and end plate structure data were analyzed by one-way ANOVA with a Bonferroni posthoc test for multiple comparisons. Internode, fiber diameter, and demyelination data were analyzed by Student’s t-test. P-value < 0.5 was considered statistically significant.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors would like to thank, Siri O’Day, and Madeline Simon for technical assistance and Monique Lorson for critical reviews of the manuscript. They also appreciate the services rendered by MU Molecular Cytology Core and Electron Microscopy Core.
Conflict of Interest statement. None declared.
Funding
This work was supported by a MU Research Board Grant (C.L.L.); MU College of Veterinary Medicine Faculty Research Grant (M.S.); The Sims' Fund to Cure SMARD (M.S.); NIH/NINDS grant (R21NS093175 to C.L.L. and M.S.). The authors would also like to thank SMA Europe for previous funding support leading up to this project.
References
- 1. Eckart M., Guenther U.P., Idkowiak J., Varon R., Grolle B., Boffi P., Van Maldergem L., Hubner C., Schuelke M., von Au K. (2012) The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics, 129, e148–e156. [DOI] [PubMed] [Google Scholar]
- 2. Grohmann K., Schuelke M., Diers A., Hoffmann K., Lucke B., Adams C., Bertini E., Leonhardt-Horti H., Muntoni F., Ouvrier R.. et al. (2001) Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet., 29, 75–77. [DOI] [PubMed] [Google Scholar]
- 3. Grohmann K., Wienker T.F., Saar K., Rudnik-Schoneborn S., Stoltenburg-Didinger G., Rossi R., Novelli G., Nurnberg G., Pfeufer A., Wirth B.. et al. (1999) Diaphragmatic spinal muscular atrophy with respiratory distress is heterogeneous, and one form is linked to chromosome 11q13-q21. Am. J. Hum. Genet., 65, 1459–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Porro F., Rinchetti P., Magri F., Riboldi G., Nizzardo M., Simone C., Zanetta C., Faravelli I., Corti S. (2014) The wide spectrum of clinical phenotypes of spinal muscular atrophy with respiratory distress type 1: a systematic review. J. Neurol. Sci., 346, 35–42. [DOI] [PubMed] [Google Scholar]
- 5. Grohmann K., Varon R., Stolz P., Schuelke M., Janetzki C., Bertini E., Bushby K., Muntoni F., Ouvrier R., Van Maldergem L.. et al. (2003) Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann. Neurol., 54, 719–724. [DOI] [PubMed] [Google Scholar]
- 6. Pitt M., Houlden H., Jacobs J., Mok Q., Harding B., Reilly M., Surtees R. (2003) Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain, 126, 2682–2692. [DOI] [PubMed] [Google Scholar]
- 7. Wong V.C., Chung B.H., Li S., Goh W., Lee S.L. (2006) Mutation of gene in spinal muscular atrophy respiratory distress type I. Pediatr. Neurol., 34, 474–477. [DOI] [PubMed] [Google Scholar]
- 8. Krieger F., Elflein N., Ruiz R., Guerra J., Serrano A.L., Asan E., Tabares L., Jablonka S. (2013) Fast motor axon loss in SMARD1 does not correspond to morphological and functional alterations of the NMJ. Neurobiol. Dis., 54, 169–182. [DOI] [PubMed] [Google Scholar]
- 9. Guenther U.P., Handoko L., Laggerbauer B., Jablonka S., Chari A., Alzheimer M., Ohmer J., Plottner O., Gehring N., Sickmann A.. et al. (2009) IGHMBP2 is a ribosome-associated helicase inactive in the neuromuscular disorder distal SMA type 1 (DSMA1). Hum. Mol. Genet., 18, 1288–1300. [DOI] [PubMed] [Google Scholar]
- 10. Viollet L., Barois A., Rebeiz J.G., Rifai Z., Burlet P., Zarhrate M., Vial E., Dessainte M., Estournet B., Kleinknecht B.. et al. (2002) Mapping of autosomal recessive chronic distal spinal muscular atrophy to chromosome 11q13. Ann. Neurol., 51, 585–592. [DOI] [PubMed] [Google Scholar]
- 11. Fukita Y., Mizuta T.R., Shirozu M., Ozawa K., Shimizu A., Honjo T. (1993) The human S mu bp-2, a DNA-binding protein specific to the single-stranded guanine-rich sequence related to the immunoglobulin mu chain switch region. J. Biol. Chem., 268, 17463–17470. [PubMed] [Google Scholar]
- 12. Jankowsky A., Guenther U.P., Jankowsky E. (2011) The RNA helicase database. Nucleic Acids Res., 39, D338–D341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen N.N., Kerr D., Chang C.F., Honjo T., Khalili K. (1997) Evidence for regulation of transcription and replication of the human neurotropic virus JCV genome by the human S(mu)bp-2 protein in glial cells. Gene, 185, 55–62. [DOI] [PubMed] [Google Scholar]
- 14. Miao M., Chan S.L., Fletcher G.L., Hew C.L. (2000) The rat ortholog of the presumptive flounder antifreeze enhancer-binding protein is a helicase domain-containing protein. Eur. J. Biochem., 267, 7237–7246. [DOI] [PubMed] [Google Scholar]
- 15. Guenther U.P., Handoko L., Varon R., Stephani U., Tsao C.Y., Mendell J.R., Lutzkendorf S., Hubner C., von Au K., Jablonka S.. et al. (2009) Clinical variability in distal spinal muscular atrophy type 1 (DSMA1): determination of steady-state IGHMBP2 protein levels in five patients with infantile and juvenile disease. J. Mol. Med. (Berl.), 87, 31–41. [DOI] [PubMed] [Google Scholar]
- 16. Maystadt I., Zarhrate M., Landrieu P., Boespflug-Tanguy O., Sukno S., Collignon P., Melki J., Verellen-Dumoulin C., Munnich A., Viollet L. (2004) Allelic heterogeneity of SMARD1 at the IGHMBP2 locus. Hum. Mutat., 23, 525–526. [DOI] [PubMed] [Google Scholar]
- 17. de Planell-Saguer M., Schroeder D.G., Rodicio M.C., Cox G.A., Mourelatos Z. (2009) Biochemical and genetic evidence for a role of IGHMBP2 in the translational machinery. Hum. Mol. Genet., 18, 2115–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grohmann K., Rossoll W., Kobsar I., Holtmann B., Jablonka S., Wessig C., Stoltenburg-Didinger G., Fischer U., Hubner C., Martini R.. et al. (2004) Characterization of Ighmbp2 in motor neurons and implications for the pathomechanism in a mouse model of human spinal muscular atrophy with respiratory distress type 1 (SMARD1). Hum. Mol. Genet., 13, 2031–2042. [DOI] [PubMed] [Google Scholar]
- 19. Shababi M., Feng Z., Villalon E., Sibigtroth C.M., Osman E.Y., Miller M.R., Williams-Simon P.A., Lombardi A., Sass T.H., Atkinson A.K.. et al. (2016) Rescue of a mouse model of spinal muscular atrophy with respiratory distress type 1 by AAV9-IGHMBP2 is dose dependent. Mol. Ther., 24, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nizzardo M., Simone C., Rizzo F., Salani S., Dametti S., Rinchetti P., Del Bo R., Foust K., Kaspar B.K., Bresolin N.. et al. (2015) Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model. Sci. Adv., 1, e1500078.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cook S.A., Johnson K.R., Bronson R.T., Davisson M.T. (1995) Neuromuscular degeneration (nmd): a mutation on mouse chromosome 19 that causes motor neuron degeneration. Mamm. Genome, 6, 187–191. [DOI] [PubMed] [Google Scholar]
- 22. Cox G.A., Mahaffey C.L., Frankel W.N. (1998) Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron, 21, 1327–1337. [DOI] [PubMed] [Google Scholar]
- 23. Maddatu T.P., Garvey S.M., Schroeder D.G., Hampton T.G., Cox G.A. (2004) Transgenic rescue of neurogenic atrophy in the nmd mouse reveals a role for Ighmbp2 in dilated cardiomyopathy. Hum. Mol. Genet., 13, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Diers A., Kaczinski M., Grohmann K., Hubner C., Stoltenburg-Didinger G. (2005) The ultrastructure of peripheral nerve, motor end-plate and skeletal muscle in patients suffering from spinal muscular atrophy with respiratory distress type 1 (SMARD1). Acta Neuropathologica, 110, 289–297. [DOI] [PubMed] [Google Scholar]
- 25. Luan X., Huang X., Liu X., Zhou H., Chen S., Cao L. (2016) Infantile spinal muscular atrophy with respiratory distress type I presenting without respiratory involvement: Novel mutations and review of the literature. Brain Dev., 38, 685–689. [DOI] [PubMed] [Google Scholar]
- 26. Nijssen J., Comley L.H., Hedlund E. (2017) Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathologica, 133, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Comley L.H., Nijssen J., Frost-Nylen J., Hedlund E. (2016) Cross-disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J. Comp. Neur., 524, 1424–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piepers S., van der Pol W.-L., Brugman F., Wokke J.H.J., van den Berg L.H., Deymeer F., Serdaroglu P., Parman Y., Poda M. (2009) Natural history of SMA IIIb: muscle strength decreases in a predictable sequence and magnitude. Neurology, 72, 2057–2058. author reply 2058. [DOI] [PubMed] [Google Scholar]
- 29. Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K., Gillingwater T.H. (2008) Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet., 17, 949–962. [DOI] [PubMed] [Google Scholar]
- 30. Murray L.M., Lee S., Baumer D., Parson S.H., Talbot K., Gillingwater T.H. (2010) Pre-symptomatic development of lower motor neuron connectivity in a mouse model of severe spinal muscular atrophy. Hum. Mol. Genet., 19, 420–433. [DOI] [PubMed] [Google Scholar]
- 31. Grunseich C., Rinaldi C., Fischbeck K.H. (2014) Spinal and bulbar muscular atrophy: pathogenesis and clinical management. Oral Dis., 20, 6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ling K.K., Gibbs R.M., Feng Z., Ko C.P. (2012) Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet., 21, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murray L.M., Beauvais A., Bhanot K., Kothary R. (2013) Defects in neuromuscular junction remodelling in the Smn(2B/-) mouse model of spinal muscular atrophy. Neuro. Dis., 49, 57–67. [DOI] [PubMed] [Google Scholar]
- 34. Kline R.A., Kaifer K.A., Osman E.Y., Carella F., Tiberi A., Ross J., Pennetta G., Lorson C.L., Murray L.M., Cox G.A. (2017) Comparison of independent screens on differentially vulnerable motor neurons reveals alpha-synuclein as a common modifier in motor neuron diseases. PLoS Genet., 13, e1006680.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pratt S.J., Valencia A.P., Le G.K., Shah S.B., Lovering R.M. (2015) Pre- and postsynaptic changes in the neuromuscular junction in dystrophic mice. Front. Physiol., 6, 252.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Willadt S., Nash M., Slater C.R. (2016) Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci. Rep., 6, 24849.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Waxman S.G. (1980) Determinants of conduction velocity in myelinated nerve fibers. Muscle Nerve, 3, 141–150. [DOI] [PubMed] [Google Scholar]
- 38. Brill M.H., Waxman S.G., Moore J.W., Joyner R.W. (1977) Conduction velocity and spike configuration in myelinated fibers: computed dependence on internode distance. J. Neurol. Neurosurg. Psychiatry, 40, 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hursh J.B. (1939) Conduction velocity and diameter of nerve fibers. Am. J. Phys., 127, 131–139. [Google Scholar]
- 40. Vizoso A.D., Young J.Z. (1948) Internode length and fibre diameter in developing and regenerating nerves. J. Anat., 82, 191–195. [PMC free article] [PubMed] [Google Scholar]
- 41. Saporta M.A., Katona I., Lewis R.A., Masse S., Shy M.E., Li J. (2009) Shortened internodal length of dermal myelinated nerve fibres in Charcot-Marie-Tooth disease type 1A. Brain, 132, 3263–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jedrzejowska M., Madej-Pilarczyk A., Fidzianska A., Mierzewska H., Pronicka E., Obersztyn E., Gos M., Pronicki M., Kmiec T., Migdal M. (2014) Severe phenotypes of SMARD1 associated with novel mutations of the IGHMBP2 gene and nuclear degeneration of muscle and Schwann cells. Eur. J. Paediatr. Neurol., 18, 183–192. [DOI] [PubMed] [Google Scholar]
- 43. Maddatu T.P., Garvey S.M., Schroeder D.G., Zhang W., Kim S.Y., Nicholson A.I., Davis C.J., Cox G.A. (2005) Dilated cardiomyopathy in the nmd mouse: transgenic rescue and QTLs that improve cardiac function and survival. Hum. Mol. Genet., 14, 3179–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaplan A., Spiller K.J., Towne C., Kanning K.C., Choe G.T., Geber A., Akay T., Aebischer P., Henderson C.E. (2014) Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron, 81, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hedlund E., Karlsson M., Osborn T., Ludwig W., Isacson O. (2010) Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain, 133, 2313–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim S.H., Chung J.M. (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain, 50, 355–363. [DOI] [PubMed] [Google Scholar]
- 47. Drac H., Kabzińska D., Moszyńska I., Strugalska-Cynowska H., Hausmanowa-Petrusewicz I., Kochański A. (2011) Dysmyelinating and demyelinating Charcot–Marie–Tooth disease associated with two myelin protein zero gene mutations. J. Appl. Genet., 52, 177–183. [DOI] [PubMed] [Google Scholar]
- 48. Sander S., Nicholson G.A., Ouvrier R.A., McLeod J.G., Pollard J.D. (1998) Charcot-Marie-Tooth disease: histopathological features of the peripheral myelin protein (PMP22) duplication (CMT1A) and connexin32 mutations (CMTX1). Muscle Nerve, 21, 217–225. [DOI] [PubMed] [Google Scholar]
- 49. Shy M.E., Patzko A. (2011) Axonal Charcot-Marie-Tooth disease. Curr. Opin. Neurol., 24, 475–483. [DOI] [PubMed] [Google Scholar]
- 50. Waxman S.G., Brill M.H. (1978) Conduction through demyelinated plaques in multiple sclerosis: computer simulations of facilitation by short internodes. J. Neurol. Neurosurg. Psychiatry, 41, 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Waxman S.G. (2006) Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nature Rev. Neurosci, 7, 932–941. [DOI] [PubMed] [Google Scholar]
- 52. Sawa N., Kataoka H., Sugie K., Kawahara M., Horikawa H., Kusunoki S., Ueno S. (2012) Clinical analysis and outcomes of amyotrophic lateral sclerosis with demyelinating polyneuropathy. Amyotroph. Lateral Scler., 13, 125–131. [DOI] [PubMed] [Google Scholar]
- 53. Nishijima H., Tomiyama M., Suzuki C., Kon T., Funamizu Y., Ueno T., Haga R., Miki Y., Arai A., Kimura T.. et al. (2012) Amyotrophic lateral sclerosis with demyelinating neuropathy. Intern. Med., 51, 1917–1921. [DOI] [PubMed] [Google Scholar]
- 54. Hunter G., Aghamaleky Sarvestany A., Roche S.L., Symes R.C., Gillingwater T.H. (2014) SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum. Mol. Genet., 23, 2235–2250. [DOI] [PubMed] [Google Scholar]
- 55. Hunter G., Powis R.A., Jones R.A., Groen E.J., Shorrock H.K., Lane F.M., Zheng Y., Sherman D.L., Brophy P.J., Gillingwater T.H. (2016) Restoration of SMN in Schwann cells reverses myelination defects and improves neuromuscular function in spinal muscular atrophy. Hum. Mol. Genet., 25, 2853–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pratt S.J., Shah S.B., Ward C.W., Kerr J.P., Stains J.P., Lovering R.M. (2015) Recovery of altered neuromuscular junction morphology and muscle function in mdx mice after injury. Cell. Mol. Life Sci., 72, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pratt S.J., Shah S.B., Ward C.W., Inacio M.P., Stains J.P., Lovering R.M. (2013) Effects of in vivo injury on the neuromuscular junction in healthy and dystrophic muscles. J. Phys., 591, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Valsecchi V., Boido M., De Amicis E., Piras A., Vercelli A. (2015) Expression of muscle-specific MiRNA 206 in the progression of disease in a murine SMA model. PloS One, 10, e0128560.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Poort J.E., Rheuben M.B., Breedlove S.M., Jordan C.L. (2016) Neuromuscular junctions are pathological but not denervated in two mouse models of spinal bulbar muscular atrophy. Hum. Mol. Genet., 25, 3768–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bruneteau G., Bauche S., Gonzalez de Aguilar J.L., Brochier G., Mandjee N., Tanguy M.L., Hussain G., Behin A., Khiami F., Sariali E.. et al. (2015) Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol., 2, 362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Glascock J.J., Shababi M., Wetz M.J., Krogman M.M., Lorson C.L. (2012) Direct central nervous system delivery provides enhanced protection following vector mediated gene replacement in a severe model of spinal muscular atrophy. Biochem. Biophys. Res. Commun, 417, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shababi M., Glascock J., Lorson C.L. (2011) Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum. Gene Ther., 22, 135–144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.