

Abstract
Fuchs’ endothelial corneal dystrophy (FECD) is the most common repeat expansion disorder. FECD impacts 4% of U.S. population and is the leading indication for corneal transplantation. Most cases are caused by an expanded intronic CUG tract in the TCF4 gene that forms nuclear foci, sequesters splicing factors and impairs splicing. We investigated the sense and antisense RNA landscape at the FECD gene and find that the sense-expanded repeat transcript is the predominant species in patient corneas. In patient tissue, sense foci number were negatively correlated with age and showed no correlation with sex. Each endothelial cell has ∼2 sense foci and each foci is single RNA molecule. We designed antisense oligonucleotides (ASOs) to target the mutant-repetitive RNA and demonstrated potent inhibition of foci in patient-derived cells. Ex vivo treatment of FECD human corneas effectively inhibits foci and reverses pathological changes in splicing. FECD has the potential to be a model for treating many trinucleotide repeat diseases and targeting the TCF4 expansion with ASOs represents a promising therapeutic strategy to prevent and treat FECD.
Introduction
Corneal disorders impacting corneal clarity are a leading cause of vision loss and blindness globally. Fuchs’ endothelial corneal dystrophy (FECD, MIM 136800) is an age-related degenerative disorder of the endothelium that affects 4% of individuals over the age of 40 in the United States and is the leading indication for corneal transplantation (1,2). The corneal endothelium is the inner hexagonal monolayer responsible for maintenance of stromal dehydration and corneal clarity. In FECD, the post-mitotic endothelium undergoes premature senescence and apoptosis (3–9). Descemet’s membrane, the basement membrane of the endothelium, becomes markedly thickened and develops focal excrescences called guttae. These guttae are diagnostic of FECD and are detected by clinical slit-lamp biomicroscopy (10). Confluent central guttae and concomitant loss of endothelial cell density results in corneal edema, scarring and loss of vision.
Recently, the potential for treating FECD has been transformed by the discovery that expansions at the intronic CTG triplet repeat polymorphism of TCF4 (MIM 602272) account for 70% of FECD cases in the United States (11–14). TCF4 expansions of greater than 40 CTG repeats confer significant risk for developing FECD (12). Expanded CUG-repeat RNA transcripts (CUGexp) accumulate as nuclear foci in FECD endothelial tissue that can be visualized by fluorescent in-situ hybridization (FISH) (15). The splicing factor muscleblind-like 1 (MBNL1) co-localizes with CUGexp nuclear foci and its sequestration and functional depletion correlate with mis-splicing of MBNL1-sensitive exons (16). Myotonic dystrophy type 1 (MIM 160900) patients with CTG expansions within the 3′-untranslated region of DMPK (MIM 605377) are also at increased risk for FECD and form CUGexp-MBNL1 foci in corneal endothelium (17).
Currently, the only effective therapy for FECD is corneal transplantation. Surgical outcomes including visual acuity have improved markedly over the last decade as endothelial keratoplasty (EK) techniques have overtaken penetrating keratoplasty for FECD (18). However, modern EK techniques are still associated with significant complications including a 28.8% detachment rate of allograft and 1.7% primary graft failure rate in the immediate post-operative period necessitating additional surgical interventions (19). Reported EK graft failure rates for FECD are 3.8–5% at 5 years in specialized, single-center studies (20,21). Even when the initial surgery is successful, nearly a quarter of patients develop glaucoma after EK that require further medical or surgical treatments (21). Access to tertiary-care corneal transplant surgeons and a suitable corneal donor pool with eye banking support to process tissue limits EK as a treatment option globally.
As a rule, visual disability must be substantial before surgery becomes an acceptable option to patients and many patients suffer loss in quality of vision for years prior to seeking surgical treatment. A less or non-invasive medical treatment option would benefit hundreds of thousands of individuals with early or moderate stage FECD by halting disease pathogenesis.
Over 20 diseases are caused by expansions of nucleotide repeats within genes (22). These expanded repeats can occur within coding regions, introns, or 3′-untranslated regions and the pathogenic mechanisms linking the molecular mutation to disease are diverse. For Huntington’s disease, the disease appears to be caused by the mutant protein, while for Friedreich’s ataxia the disease appears to be caused by repression of wild-type protein expression. Understanding mechanism and the molecular vulnerabilities of the mutant RNA is critical to the design of gene specific drugs capable of treating disease.
FECD has the potential to be a model for studying the pathogenesis and treatment of diseases caused by expanded repeats. Unlike most other repeat diseases, where affected tissue is usually inaccessible and can be examined only by autopsy, the corneal endothelium is only a half-millimeter from the surface of the eye. Both disease progression and treatment outcomes can be followed by simple non-invasive measurements. For preclinical studies such as the one presented here, the ready availability of patient corneal tissue provides an ex vivo model for evaluating molecular pathology and drug action. Some lessons learned from FECD may be broadly applicable to other diseases that affect less accessible locations.
In this report, we qualitatively and quantitatively characterize RNA transcription at the FECD locus as a target for molecular therapy. We show that antisense oligonucleotides (ASOs) can block RNA foci in patient-derived cells. ASOs can enter corneal tissue and correct splicing changes in ex vivo human corneas with FECD. Targeting the TCF4 expansion with oligonucleotides is a promising therapeutic strategy to prevent or treat FECD, the most common human repeat expansion disorder.
Results
Sense and antisense transcription at the TCF4 locus
Because the CTG repeat expansion is outside the protein-encoding region of the TCF4 gene, defining the landscape of RNA synthesis is essential for understanding the molecular basis of FECD. The disease-associated expanded CUG repeat occurs within intron 2 of TCF4 pre-mRNA (Fig. 1A) (23). Many genes, including some with expanded repeats, express transcripts that are in an antisense orientation relative to mRNA and these antisense transcripts have the potential to contribute to disease (24,25). We used quantitative strand-specific PCR (qPCR) to evaluate antisense transcription at the TCF4 locus and detected an antisense transcript containing the expanded CAG repeat in the F35T (1500 CUG repeats) cell line derived from a patient with FECD (Fig. 1B;Supplementary Material, Fig. S1).
Figure 1.

Sense and antisense RNA foci are detected in FECD corneal endothelial cells by fluorescent microscopy. (A, B) Scheme showing TCF4 pre-mRNA and antisense transcripts with the location of the expanded CUG and CAG repeat regions. (C) FISH images of CUGexp RNA foci in two patient-derived FECD endothelial cell lines (F35T, F45) compared with a healthy control endothelial cell line (Zante). A (CAG)6CA RNA probe was used for detecting the sense CUGexp foci. (D) Representative images of sense CUGexp or antisense CAGexp RNA foci in one FECD patient corneal tissue compared with a healthy control endothelial tissue. A (CUG)6CU RNA probe was used for detecting the antisense CAGexp foci.
We used FISH to examine the presence of sense or antisense transcript foci within two different corneal endothelial cell lines derived from patients with FECD. For both F35T (1500 CUG repeats) and F45 (71 CUG repeats) cell lines, sense foci were detected (Fig. 1C). No obvious antisense foci were detected. We also used FISH to examine foci in corneal endothelial tissue from a FECD patient with the TCF4 repeat expansion and observed both sense and antisense foci (Fig. 1D). No foci were detected in endothelial cells of control donor cornea endothelial tissue lacking the expansion.
Analysis of sense and antisense foci in patient tissue
After establishing that both sense and antisense foci were detectable in FECD corneal endothelial tissue, we initiated a quantitative analysis of corneal surgery samples from 20 different FECD patients to assess the correlation of RNA expression with disease, age, expanded allele length and sex. Tissue samples were used for FISH to evaluate the percentage of endothelial cells with at least one detectable focus and the number of foci per 100 cells.
Both sense and antisense foci were detected in all 20 FECD tissues with the TCF4 triplet repeat expansion (Table 1). Sense foci were found in 62–95% of cells while the antisense foci were found in only 3–27% of the cells. Foci are not numerous, with 1–2.7 sense foci and .07–0.6 antisense foci per cell. No foci were observed in a healthy control tissue or a FECD tissue without expansion. Genetic heterogeneity of FECD (17) would account for the lack of foci in endothelial tissue explants from patients without repeat expansions resulting in the same phenotype.
Table 1.
Summary table of patient corneal endothelial tissue samples
| ID | Age | Sex | Ethnicity | CTG repeat | % of total cells with sense foci | Number of sense foci per 100 cells | % of total cells with antisense foci | Number of antisense foci per 100 cells |
|---|---|---|---|---|---|---|---|---|
| Control | ||||||||
| 2014-1670 | 45 | M | Black | 23, 35 | 0.0 | 0.0 | 0.0 | 0.0 |
| CA052 | 75 | F | White | 12, 17 | 0.0 | 0.0 | 0.0 | 0.0 |
| FECD | ||||||||
| CA056 | 59 | F | White | 24, 150 | 92.0 | 211 | 6.0 | 7.2 |
| VVM573 | 50 | F | White | 12, 130 | 91.2 | 217 | 7.7 | 9.0 |
| VVM672 | 55 | F | White | 20, 130 | 93.0 | 171 | 2.8 | 5.0 |
| CA105 | 58 | M | White | 12, 130 | 82.8 | 195 | 7.1 | 8.0 |
| CA120 | 58 | F | White | 16, 130 | 84.9 | 168 | 5.8 | 7.0 |
| AK009 | 61 | F | White | 18, 130 | 79.7 | 171 | 8.3 | 13.0 |
| CA033 | 62 | M | White | 71, 130 | 95.0 | 277 | 10.0 | 12.0 |
| CA063 | 64 | F | White | 12, 74 | 92.3 | 210 | 6.2 | 7.0 |
| CA045 | 66 | F | White | 15, 120 | 91.7 | 210 | 8.0 | 9.0 |
| CA026 | 67 | M | White | 12, 67 | 85.0 | 160 | 5.0 | 6.0 |
| CA047 | 67 | M | White | 12, 91 | 69.0 | 129 | 41.0 | 62.0 |
| CA054 | 68 | M | White | 12, 79 | 90.0 | 272 | 6.0 | 6.7 |
| VVM513 | 70 | M | White | 12, 88 | 93.7 | 212 | 6.1 | 7.0 |
| CA062 | 71 | M | White | 17, 87 | 89.0 | 173 | 17.0 | 20.0 |
| CA027 | 71 | F | White | 12, 74 | 86.3 | 143 | 27.0 | 13.0 |
| CA050 | 73 | F | White | 17, 74 | 90.0 | 191 | 13.0 | 16.0 |
| CA048 | 74 | F | White | 12, 54 | 62.0 | 127 | 12.6 | 20.0 |
| CA049 | 75 | F | White | 17, 1000 | 81.0 | 144 | 15.0 | 17.0 |
| CA053 | 78 | F | White | 14, 84 | 70.0 | 134 | 4.0 | 4.0 |
| VVM289 | 86 | F | White | 12, 87 | 61.9 | 96 | 3.8 | 4.0 |
Sample 2014-1670 is a healthy control. CA052 is a FECD tissue without CTG expansion (negative control). Twenty FECD samples are with CTG expansions. Sense foci were found in 84.0% of cells [standard deviation (SD) = 10.4] compared to the antisense foci found in 10.6% of the cells (SD = 9.1) (). Foci are not numerous, with 1.8 sense foci (SD = 4.7) and 0.13 antisense foci per cell (SD = 0.13) (). n = 300–500 cells evaluated per patient sample.
We observed a significant negative correlation between age and both the percentage of cells with sense foci ( and the number of sense foci per cell ( (Fig. 2A). Tissues were harvested from patients with late stage disease and the correlation of decreased foci with age may be due to earlier disappearance of cells with relatively large numbers of foci. There was a trend towards a positive correlation between the number of sense foci and the length of the triplet repeat allele, but it did not reach the statistical significance (Fig. 2B). Although women are two to four times more likely to be affected by FECD (26,27), there was no significant correlation between sex of subjects and sense foci number (Fig. 2C). There was no significant correlation between foci number for the antisense transcript and age or sex (Supplementary Material, Fig. S2).
Figure 2.

Effect of age, repeat length, and sex on foci. (A) Graphs show significant negative correlation between % of cells with sense RNA foci or number of sense foci per 100 cells with age of subject. (B) There is a trend toward a positive correlation between sense foci and triplet repeat allele length. (C) Graphs show no correlation between sense foci and patient gender.
Copy number of mutant RNA molecules in patient cells
Defining the number of disease-causing molecules is fundamental to understanding of FECD pathology and successful drug development. Using qPCR (28), we measured a copy number of less than 3 TCF4 intronic transcripts per cell in patient tissue samples, control tissue, as well as in F35T cells (Fig. 3; Supplementary Material, Figs S3 and S4). The patient-derived cells used in this study were heterozygous for mutant TCF4 and our qPCR methods detect both the mutant and wild-type TCF4 intron 2 transcripts. Therefore, the number of mutant transcripts is a fraction of the total measured RNA molecules.
Figure 3.

One-to-one correspondence between sense foci and mutant TCF4 RNA molecules in FECD. (A) Analysis of sense foci in F35T cells. (B) Sense foci distribution in F35T cell line. Majority of F35T cells have one or two sense foci in nuclei. (C) Copy number of GAPDH, TCF4 intron 2, TCF4 exon 2/3, TCF4 exon 18 transcripts measured with qPCR in F35T cells. F35T cells have less than three TCF4 intron 2 molecules. (D) Copy number of TCF4 transcripts in healthy control corneal endothelial tissues (n = 3). (E) Copy number of TCF4 transcripts in FECD corneal endothelial tissues (n = 3). TCF4 intron 2 molecules are rare RNA species in endothelial tissue. One-to-one correspondence between intronic RNA transcripts per cell and sense foci per cell suggests that each focus is a single mutant TCF4 RNA molecule.
Using FISH, we determined that most cells have only one or two sense foci (Fig. 3A and B;Table 1). Thus, there is an approximately a one-to-one correspondence between mutant intronic RNA transcripts per cell and sense foci per cell, suggesting each focus is a single mutant TCF4 RNA molecule. Our laboratory recently reported that c9orf72 disease-related foci are also each composed of one mutant expanded repeat RNA (28). Similarly, Wansink and colleagues have reported that the mutant RNA expanded CUG transcript associated with myotonic dystrophy is also low abundance (29). These data cumulatively suggest that small numbers of RNA molecules have a big impact on repeat expansion disease pathogenesis making them ideal therapeutic targets.
Antisense oligonucleotides inhibit foci in vitro
ASOs have several advantages as starting points for FECD drug discovery. ASOs are synthetic nucleic acids that can recognize complementary RNA sequences inside cells and sequence-specifically modulate gene expression in vivo (30). Several ASOs have shown encouraging results in late stage clinical trials, including two recent FDA approvals to alleviate spinal muscular atrophy (31) and muscular dystrophy (32).
We chose to test the ability of anti-CUG ASOs to block foci and serve as a starting point for therapeutic development. ASOs targeting the DMPK CUG repeat RNA can reverse cellular phenotypes of myotonic dystrophy (33). Our laboratory and others have shown that synthetic nucleic acids can target expanded CAG repeats within genes causing Huntington’s disease, Machado Joseph Disease and dentatorubral-pallidoluysian atrophy (34–36).
FECD sense foci are much more prevalent than antisense foci, making the expanded repeat within the sense transcript the logical target of a molecular therapy. We synthesized locked nucleic acid (LNA) ASOs complementary to the CUG repeat in all three registers relative to the target sequence (Table 2). LNA substitutions were spread through the ASO, creating designs that would not be expected to recruit RNase H to cause degradation of target RNA. LNA is an advantageous chemical modification for drug development because it contains a conformationally restricted ribose that enables high-affinity binding to cellular RNA targets (37,38).
Table 2.
List of CUG repeat-targeting LNA-ASOs
| Name | Sequence 5′-3′ | Mass (observed/ calculated) | Tm (°C) | |
|---|---|---|---|---|
| LNA 1 | CAGCAGCAGCAGCAGCAGC | 6273.56/6273.05 | 78.1 | 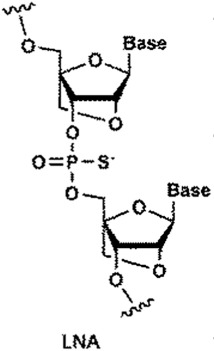 |
| LNA 2 | AGCAGCAGCAGCAGCAGCA | 6325.52/6325.08 | 77.4 | |
| LNA 3 | GCAGCAGCAGCAGCAGCAG | 6313.56/6313.07 | 77.9 | |
| LNA 4 | AGCAGCAGCAGCAGCA | 5317.50/5317.27 | 74.1 |
LNA base: bold; DNA base: normal caps; all oligomers are fully phosphorothioate backbones. Tm was detected by equal amount (1 µm each) of LNAs with complementary DNAs.
All three 19-mers (LNA 1–3) were potent inhibitors of CUG foci in F35T or F45 endothelial cell lines (Fig. 4A and B). The 16-mer LNA 4 is less effective. LNAs 1, 2 and 3 had higher affinities for complementary targets, as measured by melting temperature, than LNA 4 (Table 2), probably explaining their greater efficacy. We were unable to assess the impact of the ASO-LNAs complementary to the sense strand on the antisense foci because the lack of any detectable antisense foci in the FECD cell lines utilized in this study. We also tested a 21-mer ASO that was entirely substituted with 2′O-methyl RNA and found that it was less effective (Supplementary Material, Fig. S5). Transfection of LNAs did not reduce levels of TCF4 RNA (Supplementary Material, Fig. S6).
Figure 4.

LNA-ASOs inhibit CUGexp foci in F35T (CUG 22/1500) and F45 (CUG 16/71) endothelial cell lines. (A) Effect of LNAs on inhibition of CUGexp RNA foci in F35T cells. CM: non-complementary negative control duplex RNA; LC: LNA analog of CM. (B) Effect of LNAs on inhibition of CUGexp foci in F45 cells. Error bars represent SEM. *P < 0.05; ** P < 0.01; *** P < 0.001 compared with control LC. At least one hundred cells were analyzed for each experiment.
Free ex vivo uptake and activity of ASOs in FECD human corneas
To evaluate whether delivery to corneal tissue was feasible, we first tested ‘gymnotic’ or ‘naked’ (transfection reagent free) uptake (39) of a fluorophore-labeled oligonucleotide into the endothelium of ex vivo human corneas (Supplementary Material, Fig. S7). Uptake of ASO was observed within both the cytoplasm and nuclei.
After establishing that ex vivo uptake was possible, we were fortunate to be able to extend our investigation by accessing human corneas deemed unsuitable for transplantation because of findings of FECD and subsequent confirmation of the mutation by genotyping. We treated the corneas of human FECD-patient donors using a non-complementary control (LNA LC) or ASO LNA 1 (Fig. 5A;Supplementary Material, Fig. S8). After dissection, the Descemet’s membrane with endothelium was analyzed by FISH. We found that LNA 1, but not the non-complementary LNA LC, blocked foci formation (Fig. 5B and C).
Figure 5.

An LNA-ASO reduces CUGexp foci and reverses mis-splicing in ex vivo human FECD corneas. (A) Scheme outlining the experiment in ex vivo human corneas. Pairs of corneas from FECD-patient donors and control donors were obtained from eye bank and were treated with ASOs. Corneas from right eyes were used to assess CUGexp foci. Corneas from corresponding left eyes were used to assess splicing events. (FECD Tissue 1 from 54-year-old FECD-patient donor, 17/110 CTG repeat; FECD Tissue 2 from 51-year-old FECD-patient donor, 13/130 CTG repeat). (B) FISH images of FECD corneal endothelial tissue 1 treated with control LNA-LC and LNA 1. (C) Effect of LNA 1 on inhibition of CUGexp foci in two FECD corneas. About 200 cells or more were analyzed for each treatment. (D) RT-PCR gel images showing effect of LNA 1 on the splicing of INF2, MBNL1 and ADD3 in FECD corneal endothelial tissues. Flanking RT-PCR primers were used to assess exon inclusion or exon exclusion. (E) Averaged quantitative bar graphs of splicing events shown in (D). ImageJ was used to compare density of bands on gel. Error bars represent SEM.
The expanded repeat within TCF4 has been shown to affect splicing of MBNL1-sensitive exons in FECD corneal tissue (16). Binding of the LNAs would block the mutant repeat, preventing the RNA from interacting with MBNL1 or other factors that influence splicing. To determine whether LNA 1 was affecting a cellular phenotype, we examined splicing of INF2, MBNL1 and ADD3, all genes previously identified as being influenced by mutant TCF4. We observed alteration of splicing in all three genes upon administration of LNA 1, partially restoring alternative splicing to that observed in normal corneal tissues (Fig. 5D and E).
Discussion
ASOs as treatment for FECD
FECD is a leading cause of vision loss and by far the most common disease caused by mutant expanded trinucleotide repeats. We introduce ASOs as a potential molecular therapy to correct a genetic disorder in human corneas. ASOs targeting the mutant TCF4 repeat effectively reduced CUGexp foci formation and pathologic splicing in ex vivo human FECD corneas.
Our laboratory and others have reported that a greater than 30-fold increased risk for FECD is correlated with an expanded CUG repeat within the TCF4 gene (11–13). There are at least three possible explanations linking the mutation to disease: 1). The expansion with the chromosomal DNA affects gene expression directly, either at the TCF4 locus or at nearby genes; 2). the expansion within the RNA affects gene expression or gene splicing or 3). the expansion within the RNA acts as a template for synthesis of RAN peptides, a phenomenon that has been noted for other expanded repeat disease genes (40).
Our study was not intended to definitively distinguish between these options but does provide some insights into potential roles for mutant RNA. There are an average of 1–2 foci or mutant RNA molecules per cell suggesting that, if the mutant RNA is involved directly, small numbers of RNA molecules must have an outsized impact on disease.
Evidence suggesting that these small numbers can impact disease includes our previous report that FECD is observed in myotonic dystrophy (DM1) patients that have a CUG expansion within the DMPK gene (17). DM1 cells are also characterized by 1–2 foci per cell (29) and the association of FECD with CUG expanded repeats in two different genes implicates the mutant RNA as causative rather than the host gene. The repeat DM1 CUG RNA transcripts within the DMPK gene have been demonstrated to serve as templates for repeat-associated non-ATG (RAN) translation resulting in toxic peptides (40). We did not examine the expression of RAN peptides in this study and such experiments are warranted to understand how repeat CUG RNA transcripts result in dysfunction of the corneal endothelium.
In DM1, experimental evidence suggests that the mutant expanded repeat CUG RNA transcripts sequester the splicing factor MBNL1 and contribute to cellular dysfunction by triggering mis-splicing (41) and splice defects have previously been noted in FECD pathogenesis (16). We provide experimental data suggesting that ASOs targeting the sense RNA transcript reverses foci formation and pathologic splicing. The ability of ASOs to target the mutant expanded RNA and at least partially correct a splicing defect is further evidence supporting the hypothesis that small numbers of mutant RNA can contribute to disease.
ASOs are experiencing a wave of success in the clinic and the eye may be a particularly advantageous organ for targeting with synthetic oligonucleotides. Vitravene, the first ASO drug approved by the FDA, was administered by intraocular (intravitreal) injection for cytomegalovirus retinitis (42). Intracameral injection would place an ASO into the anterior chamber of the eye in direct contact with the corneal endothelium. The small volume of the eye will greatly reduce the cost of treatment relative to systemic administration. Other trials with ASOs have shown long duration of effects (31), suggesting that infrequent dosing may be possible, further lowering cost and increasing patient convenience.
The affected tissue in FECD is only a half-millimeter from the surface of the eye and topical administration by eye drops may be possible. Topical administration of ASO eye drops have already been found to be effective, safe and well tolerated in phase II FDA trials of aganirsen for viral keratitis-induced corneal neovascularization (43). Because systemic administration is unnecessary, the likelihood of systemic off-target effects that might cause toxicity is low.
Early detection of FECD in patients with the TCF4 trinucleotide repeat expansion is possible by genetic testing and routine examination of the cornea. In parallel with further testing of compounds that modulate mutant repeat RNA, it will be essential to rigorously explore the molecular natural history of FECD to guide clinical trials. Our group has documented incomplete penetrance of the FECD trait in the setting of the triplet repeat expansion in the TCF4 gene (12). Additional studies are necessary to develop a FECD risk propensity scoring system based on parameters such as TCF4 triplet repeat allele length, sex, age, ethnicity, family history and corneal examination findings to identify individuals who would benefit from preventative molecular therapies (14).
Any drug treatments for FECD would likely need to start early in the disease process and continue for an extended period. Identification of biomarkers will provide critical guides for convenient and timely monitoring of disease progression so that the length of clinical trials can be minimized. Treatments will therefore need to be minimally invasive and convenient for patients. A safe, non-toxic and convenient preventative therapy may be indicated even in subjects where physical manifestation of early stage FECD is not yet apparent but would certainly be warranted after early stage disease is detected but before significant loss of vision occurs.
Conclusions
Our data provides a rationale of the use of ASOs targeting the small number of mutant TCF4 repeat RNA molecules within cells as a therapeutic strategy to prevent and treat vision loss caused by FECD. Reversal of the molecular phenotype in an ex vivo human FECD cornea model may serve as relevant pre-clinical development data to justify use of a lead ASO therapeutic compound in human trials once dosing and delivery protocols are optimized in animal and ex vivo human corneal models (44,45). An oligonucleotide treatment for FECD would advance clinical practice by realizing the promise of precision medicine for a common human disease. Beyond FECD, the ability to introduce oligonucleotides into corneal tissue suggests the potential to control gene expression associated with many other disorders, such as the group of TGFBI-associated corneal dystrophies and broaden the options for using ASOs in the eye.
Materials and Methods
Subjects
Subjects underwent a complete eye examination including slit lamp biomicroscopy by a cornea fellowship-trained ophthalmologist. Subjects underwent EK for FECD severity Krachmer grade 5 (≥5 mm central confluent guttae without stromal edema) or 6 (≥5 mm central confluent guttae with stromal edema) assessed by slit lamp microscopy (46). Surgically explanted endothelium-Descemet’s membrane monolayers were fixed in a 4% phosphate-buffered formaldehyde, equilibrated in a 30% sucrose solution for cytoprotection, and frozen in Tissue-Tek Optimal Cutting Tissue (OCT) compound (Sakura, Torrance, CA) for FISH studies (15). Genomic DNA was extracted from peripheral blood leukocytes of each study subject using Autogen Flexigene (Qiagen, Valencia, CA).
Corneal endothelial samples from post-mortem donor corneas preserved in Optisol GS corneal storage media (Bausch & Lomb, Rochester, NY) were obtained from the eye bank of Transplant Services at UT Southwestern. Certified eye bank technicians screened the donor corneal endothelium with slit lamp biomicroscopy and Cellchek EB-10 specular microscopy (Konan Medical). Donor corneal tissue with FECD was identified by the presence of confluent central guttae. Endothelium-Descemet’s membrane monolayers from donor corneas were micro-dissected and stored as previously described (15). DNA from the remaining corneal tissue of each sample was extracted with TRIzol reagent (ThermoScientific).
TCF4 CTG18.1 polymorphism genotyping
Genomic DNA from subjects’ peripheral leukocytes or corneal tissue was used for genotyping. The CTG18.1 trinucleotide repeat polymorphism in the TCF4 gene was genotyped using a combination of short tandem repeat (STR) and triplet repeat primed polymerase chain reaction (TP-PCR) assays as we have previously described (12). For the STR assay, a pair of primers flanking the CTG18.1 locus was utilized for PCR amplification with one primer labeled with FAM on 5′ end. The TP-PCR assay was performed using the 5′ FAM-labeled primer specific for the repeat locus paired with repeat sequence targeted primers for PCR amplification. PCR amplicons were loaded on an ABI 3730XL DNA analyzer (Applied Biosystems, Foster City, CA) and the results analyzed with ABI GeneMapper 4.0 (Applied Biosystems). Large triplet repeat expansions were sized by southern blot analysis using digoxigenin-labeled probes (14).
Detection of TCF4 repeat transcript
TCF4 expression was analyzed by qPCR on a 7500 real-time PCR system (Applied Biosystems) using iTaq SYBR Green Supermix (Bio-rad). Data was normalized relative to levels of HPRT1 mRNA. Primers specific for TCF4 exon18 are as follows: F 5′-TGACGATGAGGACCTGACAC-3′; R 5′-GTCTGGGGCTTGTCACTCTT-3′. Primers for TCF4 intron2: F 5′-GAGAGAGGGAGTGAAAGAGAGA-3′; R 5′-GGCAATGTCCATTTCCATCT-3′.
To detect antisense transcript, cDNA was generated from 0.25 μg of total RNA using the SuperScript III system (Invitrogen) with strand-specific reverse primers attaching a linker sequence LK. Next, cDNA was amplified by PCR with strand-specific forward and LK primers (Supplementary Material, Fig. S1). The PCR products were cloned and Sanger sequenced to verify their specificity.
Human corneal endothelial cell culture
The ‘Zante’ human corneal endothelial cell line derived from a healthy control subject expressing TCF4 transcript with approximately 16–19 CUG repeats was a generous gift of Dr Danielle Robertson (UT Southwestern) (47). The F35T corneal endothelial cell line derived from FECD patient expressing TCF4 transcript with approximately 1500 CUG repeats was a generous gift of Dr Albert Jun (Johns Hopkins). The F45 corneal endothelial cell line was a primary culture from a donor cornea from FECD patient expressing TCF4 transcript with approximately 71 CUG repeats. The dissected Descemet’s membrane monolayer was incubated with 2 mg/ml collagenase A (Roche) in culture media at 37°C for 4 h to dissociate the cells, spun down at 800 g for 5 min, and plated on culture dish pre-coated with fibronectin (FNC) (Athena Environmental Sciences). Cells were grown in modified Eagle’s minimal essential media (OptiMEM) (ThermoFisher) supplemented with 8% fetal bovine serum, 5 ng/mL human epidermal growth factor (ThermoFisher), 20 ng/mL nerve growth factor (Fisher Scientific), 100 µg/mL bovine pituitary extract (ThermoFisher), 20 µg/mL ascorbic acid (Sigma-Aldrich), 200 mg/L calcium chloride (Sigma-Aldrich), 0.08% chondroitin sulfate (Sigma-Aldrich), 50 µg/mL gentamicin (ThermoFisher) and antibiotic/antimycotic solution (diluted 1/100) (Sigma-Aldrich) (48). Cultures were incubated at 37°C in 5% CO2 and passed when confluent.
Transcript copy number measurement by qPCR
Transcript copy number measurement was performed as we described previously (28). Briefly, standard RNA was made by in vitro transcription with corresponding purified PCR fragment containing SP6 promoter sequence. After checking RNA with Bioanalyzer (Agilent), a serial dilution of purified standard RNA ranging from 10 to 105 copies was used to construct standard curve for qPCR efficiency following reverse transcription. The qPCR was performed on a CFX Connect Real-Time System (Bio-Rad). Ct values in each dilution were measured in triplicate. To determine transcript copy number per cell, total RNA was extracted by TRIzol (ThermoScientific) for F35T cell line or by NucleoSpin RNA XS kit (Fisher Scientific) for endothelial tissue samples and used to do reverse transcription. Cell number of F35T was counted by two methods: hemocytometer and Z1 Particle Counter (Beckman Coulter). Cell number of control endothelial tissue was calculated as π × (d/2)2 × cell density (d is the diameter of trephine used for dissection; endothelial cell density measured by specular microscopy was provided by eye bank). Endothelial cell density of FECD tissue was estimated as 500 cells/mm2. For each transcript, a serial dilution of cDNA was used. Ct values in each dilution were measured in triplicate. Copy number per cell was calculated by using the standard curve for qPCR efficiency of each transcript. For cells, data were analyzed from two batches of F35T cells. For tissues, data were analyzed from three control tissues and three FECD tissues. Results are showed as the mean ± SD. Primers used to make PCR product are: GAPDH E1F1 SP6 (5′-ATTTAGGTGACACTATAGAACTCTGCTCCTCCTGTTCGAC-3′), GAPDH E5R1 (5′-TTGATTTTGGAGGGATCTCG-3′), TCF4 E2F1 SP6 (5′-ATTTAGGTGACACTATAGAATGCTAAAATGCATCACCAACA-3′), TCF4 E3R1 (5′-GAGCCAGTAAAATGTCCACTTG-3′), TCF4 intron2dwF1 SP6 (5′-ATTTAGGTGACACTATAGAATGTTGCACTTTCTCCATTCG-3′), TCF4 intron2dwR1 (5′-CAGGAGAGCAATTTGAAGCA-3′), TCF4 E18F1 SP6 (5′-ATTTAGGTGACACTATAGAATGACGATGAGGACCTGACAC-3′) and TCF4 E18R1 (5′-AGGATCAGGAGCTTGGTCTG-3′). Primers and probes used for qPCR are: Human GAPD (GAPDH) Endogenous Control (Thermo Fisher 4310884E), TCF4 E2F2 (5′-TAGGGACGGACAAAGAGCTG-3′), TCF4 E3R2 (5′-CCACTTGCCAAAGAAGTTGG-3′), TCF4 E2/3 probe (5′-TTCACCTCCTGTGAGCAGTG-3′), TCF4 intron2dwF2 (5′-TTCTCCATTCGTTCCTTTGC-3′), TCF4 intron2dwR2 (5′-CACTCCCTCTCTCTCCAGCA-3′), TCF4 intron2dw probe (5′-GCTCTGACTCAGGGAAGGTG-3′), TCF4 E18F2 (5′-ACGATGAGGACCTGACACCA-3′), TCF4 E18R2 (5′-GGGCTTGTCACTCTTGAGGT-3′) and TCF4 E18 probe (5′-AGAAGGCAGAGCGTGAGAAG-3′).
Synthesis and transfection of oligonucleotides
LNA phosphoramidites were synthesized from the 3′-hydroxyl precursors (Rasayan) and assembled into oligonucleotides as described previously (49). ASOs were deprotected using concentrated aqueous ammonia for 16 h at 55°C and were characterized by LCMS (Table 2). LNA ASOs were transfected into cells with Lipofectamine RNAiMAX (Life Technologies) as described previously (34). Cells were plated at a density of 300 000 per well of a 6-well plate and transfected at the same time using an oligonucleotide concentration of 25 nM. After 72 h, the cells were transfected again as mentioned above and harvested 4 days later to assess the effects for reduction of CUGexp foci using RNA FISH/immunofluorescence.
Fluorescence in-situ hybridization
Cornea endothelial cells post oligonucleotide transfection were harvested by trypsin and replated on glass slides. Cells were fixed with 4% formaldehyde in 1X phosphate-buffered saline (PBS) and permeabilized in 70% ethanol at 4°C overnight. After removing the permabilization solution, cells were washed with buffer (10% formamide in 2X saline sodium citrate buffer [SSC]) for 5 min, and then incubated with prehybridization buffer (40% formamide in 2XSSC) at 45°C for 20 min. (CAG)6CA-5′ Texas red-labeled 2-Omethyl RNA 20-mers probe in hybridization buffer (100 mg/mL dextran sulfate and 40% formamide in 2XSSC) was added. The slides were placed in a humidified chamber and incubated in the dark at 37°C overnight. On the next day, cells were washed twice with wash buffer at 37°C for 15 min, and then stained with mounting media with DAPI (H-1500; Vector Labs).
Cells were imaged at 60× magnification using a Widefield Deltavision microscope. Images were processed by blind deconvolution with AutoQuant X3. Visualization of RNA foci were made using ImageJ. For quantification, at least 20 pictures were taken from randomly chosen microscopic fields, containing 100–300 cells for each treatment. Counting of foci was performed by different investigators.
FISH of corneal endothelial tissue monolayers were performed using previously described methods (15). Twenty random archived EK samples from patients with the CTG18.1 expansion were studied. Genotyping of CTG18.1 locus was performed after the time of EK using genomic DNA from subject leukocytes. Each endothelial tissue sample was bisected with one half used to detect sense foci using (CAG)6CA RNA probe and other half used to detect antisense foci using (CUG)6CU RNA probe (Integrated DNA Technologies).
Ex vivo oligonucleotide treatment of human corneas
Post-mortem human donor corneas were obtained from UT Transplant Services. The donor corneas were incubated in Optisol GS corneal storage media with a single stranded anti-sense oligonucleotide LC-Cy5 (final concentration 10 μm) at 37°C. The Descemet’s membrane-endothelium monolayers were dissected as reported previously (15) either after 1 day or 7 days of treatment, stained with DAPI (Vector Labs), and imaged at 60× magnification using a Widefield Deltavision microscope.
FECD donor corneas with confluent corneal endothelial guttae on specular microscopy and the TCF4 expansion and control donor corneas were bisected and incubated in Optisol GS corneal storage media with control LNA (LC) or LNA1 (final concentration 10 μm) at 37°C. After treatment for 7 days, RNA FISH analysis was performed on the dissected Descemet’s membrane-endothelium monolayers from the right eye from each donor to assess CUGexp sense foci as described above. After treatment for 9 days, the total RNA was extracted from the dissected Descemet’s membrane-endothelium monolayers using TRIzol (ThermoScientific) per manufacturer’s instructions. Alternative splicing of MBNL1-sensitive exons were assessed by RT-PCR as described previously (16) using endothelial tissue from corresponding left eyes of donors.
Statistics
Correlation between four foci parameters (percentage of total cells with sense foci, number of sense foci per 100 cells, percentage of total cells with antisense foci and number of antisense foci per 100 cells) and age, sex and CTG repeat number were examined on the 20 FECD subjects by linear regression models.
Study approval
The study was approved by the institutional review board of the University of Texas Southwestern Medical Center and conducted in adherence with the tenets of the Declaration of Helsinki. Written informed consent was obtained from participants prior to inclusion in the study.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors thank the patients for their participation in this study. We appreciate the efforts of Dr H. Dwight Cavanagh and Donna Drurry at Transplant Services at UT Southwestern. We thank the collaborating corneal specialists Drs Wayne R. Bowman, James P. McCulley, Steven Verity, Brad Bowman, Walter Beebe and Aaleya Koreishi. We thank Drs Danielle Robertson and Albert Jun for generously sharing cell lines. We acknowledge Chelsea Burroughs for artistic assistance with figures.
Conflict of Interest statement: Authors are inventors on a planned patent related to findings reported in the study.
Funding
This study was supported by grants R01EY022161 (V.V.M.), P30EY020799 (V.V.M.), and R35GM1118103 (D.R.C.) from the National Institutes of Health, Bethesda, MD, an unrestricted department grant from Research to Prevent Blindness, New York (V.V.M.), the Robert A Welch Foundation (D.R.C.), and UMass Medical School (startup funding to JKW). V.V.M. is the Paul T. Stoffel/Centex Professor in Clinical Care. D.R.C. is the Rusty Kelley Professor of Biomedical Science. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding to pay the Open Access publication charges for this article was provided by the Department of Ophthalmology.
References
- 1. Lorenzetti D.W., Uotila M.H., Parikh N., Kaufman H.E. (1967) Central cornea guttata. Incidence in the general population. American Journal of Ophthalmology, 64, 1155–1158. [PubMed] [Google Scholar]
- 2. 2015 Eye Bank Association of America (2016). Washington, DC. [Google Scholar]
- 3. Chi H.H., Teng C.C., Katzin H.M. (1958) Histopathology of primary endothelial-epithelial dystrophy of the cornea. American Journal of Ophthalmology, 45, 518–535. [DOI] [PubMed] [Google Scholar]
- 4. Laing R.A., Leibowitz H.M., Oak S.S., Chang R., Berrospi A.R., Theodore J. (1981) Endothelial mosaic in Fuchs’ dystrophy. A qualitative evaluation with the specular microscope. Archives of Ophthalmology, 99, 80–83. [DOI] [PubMed] [Google Scholar]
- 5. Bigar F. (1982) Specular microscopy of the corneal endothelium. Optical solutions and clinical results. Developments in Ophthalmology, 6, 1–94. [PubMed] [Google Scholar]
- 6. Borderie V.M., Baudrimont M., Vallee A., Ereau T.L., Gray F., Laroche L. (2000) Corneal endothelial cell apoptosis in patients with Fuchs’ dystrophy. Investigative Ophthalmology & Visual Science, 41, 2501–2505. [PubMed] [Google Scholar]
- 7. Li Q.J., Ashraf M.F., Shen D.F., Green W.R., Stark W.J., Chan C.C., O’Brien T.P. (2001) The role of apoptosis in the pathogenesis of Fuchs endothelial dystrophy of the cornea. Archives of Ophthalmology, 119, 1597–1604. [DOI] [PubMed] [Google Scholar]
- 8. Matthaei M., Zhu A.Y., Kallay L., Eberhart C.G., Cursiefen C., Jun A.S. (2014) Transcript profile of cellular senescence-related genes in Fuchs endothelial corneal dystrophy. Experimental Eye Research, 129, 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jurkunas U.V., Bitar M.S., Funaki T., Azizi B. (2010) Evidence of oxidative stress in the pathogenesis of Fuchs endothelial corneal dystrophy. The American Journal of Pathology, 177, 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hogan M.J., Wood I., Fine M. (1974) Fuchs’ endothelial dystrophy of the cornea. 29th Sanford Gifford Memorial lecture. American Journal of Ophthalmology, 78, 363–383. [DOI] [PubMed] [Google Scholar]
- 11. Wieben E.D., Aleff R.A., Tosakulwong N., Butz M.L., Highsmith W.E., Edwards A.O., Baratz K.H., Lewin A. (2012) A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One, 7, e49083.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mootha V.V., Gong X., Ku H.C., Xing C. (2014) Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs’ endothelial corneal dystrophy. Investigative Ophthalmology & Visual Science, 55, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xing C., Gong X., Hussain I., Khor C.C., Tan D.T., Aung T., Mehta J.S., Vithana E.N., Mootha V.V. (2014) Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs’ corneal dystrophy in Chinese implies common causal variant. Investigative Ophthalmology & Visual Science, 55, 7073–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soliman A.Z., Xing C., Radwan S.H., Gong X., Mootha V.V. (2015) Correlation of severity of Fuchs endothelial corneal dystrophy with triplet repeat expansion in TCF4. JAMA Ophthalmology, 133, 1386–1391. [DOI] [PubMed] [Google Scholar]
- 15. Mootha V.V., Hussain I., Cunnusamy K., Graham E., Gong X., Neelam S., Xing C., Kittler R., Petroll W.M. (2015) TCF4 triplet repeat expansion and nuclear RNA foci in Fuchs’ endothelial corneal dystrophy. Investigative Ophthalmology & Visual Science, 56, 2003–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Du J., Aleff R.A., Soragni E., Kalari K., Nie J., Tang X., Davila J., Kocher J.-P., Patel S.V., Gottesfeld J.M.. et al. (2015) RNA toxicity and missplicing in the common eye disease Fuchs endothelial corneal dystrophy. The Journal of Biological Chemistry, 290, 5979–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mootha V.V., Hansen B., Rong Z., Mammen P.P., Zhou Z., Xing C., Gong X. (2017) Fuchs’ endothelial corneal dystrophy and RNA foci in patients with myotonic dystrophy. Investigative Ophthalmology & Visual Science, 58, 4579–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee W.B., Jacobs D.S., Musch D.C., Kaufman S.C., Reinhart W.J., Shtein R.M. (2009) Descemet’s stripping endothelial keratoplasty: safety and outcomes: a report by the American Academy of Ophthalmology. Ophthalmology, 116, 1818–1830. [DOI] [PubMed] [Google Scholar]
- 19. Deng S.X., Lee W.B., Hammersmith K.M., Kuo A.N., Li J.Y., Shen J.F., Weikert M.P., Shtein R.M. (2017) Descemet membrane endothelial keratoplasty: safety and outcomes: a report by the American Academy of Ophthalmology. Ophthalmology, doi:10.1016/j.ophtha.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 20. Price M.O., Fairchild K.M., Price D.A., Price F.W. Jr (2011) Descemet’s stripping endothelial keratoplasty five-year graft survival and endothelial cell loss. Ophthalmology, 118, 725–729. [DOI] [PubMed] [Google Scholar]
- 21. Ang M., Soh Y., Htoon H.M., Mehta J.S., Tan D. (2016) Five-year graft survival comparing Descemet stripping automated endothelial keratoplasty and penetrating keratoplasty. Ophthalmology, 123, 1646–1652. [DOI] [PubMed] [Google Scholar]
- 22. Rohilla K.J., Gagnon K.T. (2017) RNA biology of disease-associated microsatellite repeat expansions. Acta Neuropathologica Communications, 5, 63.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Breschel T.S., McInnis M.G., Margolis R.L., Sirugo G., Corneliussen B., Simpson S.G., McMahon F.J., MacKinnon D.F., Xu J.F., Pleasant N.. et al. (1997) A novel, heritable, expanding CTG repeat in an intron of the SEF2-1 gene on chromosome 18q21.1. Human Molecular Genetics, 6, 1855–1863. [DOI] [PubMed] [Google Scholar]
- 24. Gendron T.F., Bieniek K.F., Zhang Y.J., Jansen-West K., Ash P.E., Caulfield T., Daughrity L., Dunmore J.H., Castanedes-Casey M., Chew J.. et al. (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathologica, 126, 829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zu T., Liu Y., Banez-Coronel M., Reid T., Pletnikova O., Lewis J., Miller T.M., Harms M.B., Falchook A.E., Subramony S.H.. et al. (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proceedings of the National Academy of Sciences of the United States of America, 110, E4968–E4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Afshari N.A., Pittard A.B., Siddiqui A., Klintworth G.K. (2006) Clinical study of Fuchs corneal endothelial dystrophy leading to penetrating keratoplasty: a 30-year experience. Archives of Ophthalmology, 124, 777–780. [DOI] [PubMed] [Google Scholar]
- 27. Musch D.C., Niziol L.M., Stein J.D., Kamyar R.M., Sugar A. (2011) Prevalence of corneal dystrophies in the United States: estimates from claims data. Investigative Ophthalmology & Visual Science, 52, 6959–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu J., Hu J., Ludlow A.T., Pham J.T., Shay J.W., Rothstein J.D., Corey D.R. (2017) c9orf72 disease-related foci are each composed of one mutant expanded repeat RNA. Cell Chemical Biology, 24, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gudde A., van Heeringen S.J., de Oude A.I., van Kessel I.D.G., Estabrook J., Wang E.T., Wieringa B., Wansink D.G. (2017) Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biol, 14, 1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khvorova A., Watts J.K. (2017) The chemical evolution of oligonucleotide therapies of clinical utility. Nature Biotechnology, 35, 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Corey D.R. (2017) Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nature Neuroscience, 20, 497–499. [DOI] [PubMed] [Google Scholar]
- 32. Aartsma-Rus A., Krieg A.M. (2017) FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Therapeutics, 27, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wojtkowiak-Szlachcic A., Taylor K., Stepniak-Konieczna E., Sznajder L.J., Mykowska A., Sroka J., Thornton C.A., Sobczak K. (2015) Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Research, 43, 3318–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu J., Matsui M., Gagnon K.T., Schwartz J.C., Gabillet S., Arar K., Wu J., Bezprozvanny I., Corey D.R. (2009) Allele-specific silencing of mutant Huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nature Biotechnology, 27, 478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu J., Liu J., Corey D.R. (2010) Allele-selective inhibition of Huntingtin expression by switching to an miRNA-like RNAi mechanism. Chemistry & Biology, 17, 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu J., Liu J., Narayanannair K.J., Lackey J.G., Kuchimanchi S., Rajeev K.G., Manoharan M., Swayze E.E., Lima W.F., Prakash T.P.. et al. (2014) Allele-selective inhibition of mutant atrophin-1 expression by duplex and single-stranded RNAs. Biochemistry, 53, 4510–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Braasch D.A., Corey D.R. (2001) Locked nucleic acid (LNA): fine-tuning the recognition of DNA and RNA. Chemistry & Biology, 8, 1–7. [DOI] [PubMed] [Google Scholar]
- 38. Watts J.K. (2013) Locked nucleic acid: tighter is different. Chemical Communications (Cambridge), 49, 5618–5620. [DOI] [PubMed] [Google Scholar]
- 39. Stein C.A., Hansen J.B., Lai J., Wu S.J., Voskresenskiy A., H⊘g A., Worm J., Hedtjärn M., Souleimanian N., Miller P.. et al. (2010) Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Research, 38, e3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zu T., Gibbens B., Doty N.S., Gomes-Pereira M., Huguet A., Stone M.D., Margolis J., Peterson M., Markowski T.W., Ingram M.A.. et al. (2011) Non-ATG-initiated translation directed by microsatellite expansions. Proceedings of the National Academy of Sciences of the United States of America, 108, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang H., Mankodi A., Swanson M.S., Moxley R.T., Thornton C.A. (2004) Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Human Molecular Genetics, 13, 3079–3088. [DOI] [PubMed] [Google Scholar]
- 42. Geary R.S., Henry S.P., Grillone L.R. (2002) Fomivirsen: clinical pharmacology and potential drug interactions. Clinical Pharmacokinectics, 41, 255–260. [DOI] [PubMed] [Google Scholar]
- 43. Cursiefen C., Viaud E., Bock F., Geudelin B., Ferry A., Kadlecova P., Levy M., Al Mahmood S., Colin S., Thorin E.. et al. (2014) Aganirsen antisense oligonucleotide eye drops inhibit keratitis-induced corneal neovascularization and reduce need for transplantation: the I-CAN study. Ophthalmology, 121, 1683–1692. [DOI] [PubMed] [Google Scholar]
- 44. Johnson D.H., Tschumper R.C. (1987) Human trabecular meshwork organ culture. A new method. Investigative Ophthalmology & Visual Science, 28, 945–953. [PubMed] [Google Scholar]
- 45. Mao W., Tovar-Vidales T., Yorio T., Wordinger R.J., Clark A.F. (2011) Perfusion-cultured bovine anterior segments as an ex vivo model for studying glucocorticoid-induced ocular hypertension and glaucoma. Investigative Ophthalmology & Visual Science, 52, 8068–8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krachmer J.H., Purcell J.J. Jr, Young C.W., Bucher K.D. (1978) Corneal endothelial dystrophy. A study of 64 families. Archives of Ophthalmology, 96, 2036–2039. [DOI] [PubMed] [Google Scholar]
- 47. Sheerin A.N., Smith S.K., Jennert-Burston K., Brook A.J., Allen M.C., Ibrahim B., Jones D., Wallis C., Engelmann K., Rhys-Williams W.. et al. (2012) Characterization of cellular senescence mechanisms in human corneal endothelial cells. Aging Cell, 11, 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Toyono T., Usui T., Villarreal G. Jr, Kallay L., Matthaei M., Vianna L.M., Zhu A.Y., Kuroda M., Amano S., Jun A.S. (2016) MicroRNA-29b overexpression decreases extracellular matrix mRNA and protein production in human corneal endothelial cells. Cornea, 35, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pendergraff H.M., Krishnamurthy P.M., Debacker A.J., Moazami M.P., Sharma V.K., Niitsoo L., Yu Y., Tan Y.N., Haitchi H.M., Watts J.K. (2017) Locked nucleic acid gapmers and conjugates potently silence ADAM33, an asthma-associated metalloprotease with nuclear-localized mRNA. Molecular Therapy. Nucleic Acids, 8, 158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.