

Abstract
Spinal muscular atrophy (SMA) is the leading genetic cause of infant mortality, characterized by progressive degeneration of spinal-cord motor neurons, leading to atrophy of skeletal muscles. However, accumulating evidence indicates that it is a multi-system disorder, particularly in its severe forms. Several studies delineated structural and functional cardiac abnormalities in SMA patients and mouse models, yet the abnormalities have been primarily attributed to autonomic dysfunction. Here, we show in a severe mouse model that its cardiomyocytes undergo G0/G1 cell-cycle arrest and enhanced apoptosis during postnatal development. Microarray and real-time RT-PCR analyses revealed that a set of genes associated with cell cycle and apoptosis were dysregulated in newborn pups. Of particular interest, the Birc5 gene, which encodes Survivin, an essential protein for heart development, was down-regulated even on pre-symptomatic postnatal day 0. Interestingly, cultured cardiomyocytes depleted of SMN recapitulated the gene expression changes including downregulation of Survivin and abnormal cell-cycle progression; and overexpression of Survivin rescued the cell-cycle defect. Finally, increasing SMN in SMA mice with a therapeutic antisense oligonucleotide improved heart pathology and recovered expression of deregulated genes. Collectively, our data demonstrate that the cardiac malfunction of the severe SMA mouse model is mainly a cell-autonomous defect, caused by widespread gene deregulation in heart tissue, particularly of Birc5, resulting in developmental abnormalities through cell-cycle arrest and apoptosis.
Introduction
Spinal muscular atrophy (SMA) is the leading genetic cause of infantile death, characterized by progressive atrophy and weakness of proximal voluntary muscles, due to loss of α-motor neurons in the spinal cord (1). SMA is caused by homozygous loss or mutation of survival of motor neuron 1 (SMN1) (2); this gene encodes an essential multifunctional protein, SMN, whose best-characterized function is assisting the assembly of spliceosomal U snRNPs and other RNPs including the histone RNA processing U7 snRNPs and axonal mRNPs (3–6). In humans, there is a paralogous backup gene, called SMN2, which arose from an intrachromosomal duplication of Chr. 5q13 during evolution (7). SMN2 and SMN1 have a small number of nucleotide differences, the most important of which are a single-nucleotide transition, C6T in exon 7, and to a lesser extent A-44G in intron 6, resulting in predominant skipping of SMN2 exon 7 during pre-mRNA splicing (8,9). The exon-7-skipped protein isoform, SMNΔ7, is dysfunctional and rapidly degraded. As a result, SMN2 generates considerably less functional SMN protein, which is not sufficient to compensate for the lack of SMN1, though it is essential for the survival of SMA patients. SMA is clinically classified into five types, ranging from severe type 0 to mild type IV in terms of age of onset and maximum motor function achieved (9).
Although SMA has been traditionally considered as a neuromuscular disease, increasing evidence indicates that it is a multi-system disorder; various peripheral tissues and cell types have been reported to be affected in mouse models and to a lesser extent in SMA patients, especially in severe SMA forms (10,11). Indeed, we recently showed that restoring normal SMN levels exclusively in peripheral tissues results in long-term phenotypic rescue in both mild and severe mouse models (12).
Structural and functional cardiac abnormalities have been documented in severe SMA patients. For example, some type 0 or type I patients displayed severe bradycardias, atrial arrhythmia, remarkable fluctuation of blood pressure and heart rate, dilated right ventricle, atrial septal defects, and/or ventricular septal defects (13–16). A developmental defect called hypoplastic left heart syndrome was also observed in several type I patients (16–18). Rudnik-Schoneborn et al. revealed that 3/4 SMA patients carrying only one copy of SMN2 suffered congenital cardiac defects, whereas only a small percentage of patients carrying two copies of SMN2 had mild cardiac abnormalities, with the rest being unaffected (16). Similar cardiac defects have also been observed in severe mouse models (19–22). Interestingly, based on studies in mouse models, cardiac structural defects precede motor-neuron loss and can exist independently of motor-unit pathology and muscle paralysis, suggesting an important role of heart anomalies in premature death of these mice (21,23). Cardiovascular defects in patients and mouse models have been primarily attributed to autonomic dysfunction (17,19,23–25). One study suggested that oxidative stress contributes to interstitial fibrosis observed in in the heart of a SMA mouse model (21). However, to date, no thorough study of the heart at the cellular and molecular levels has been reported in the context of SMA.
In this study, we explored the mechanisms of cardiac dysfunction and developmental defects in a severe SMA mouse model. Using tissue staining and flow cytometry analyses, we uncovered a marked proliferation defect of cardiomyocytes and activation of apoptosis. A set of genes involved in the cell cycle and apoptosis were dysregulated in newborn pups. Particularly, the Birc5 gene, which encodes Survivin (also called Birc5), which is essential for heart development, was downregulated as early as postnatal day 0 (P0). Using primary cardiomyocytes we revealed that the widespread dysregulation of gene expression is a cell autonomous effect of SMN deficiency. Furthermore, we showed that over-expression of Survivin in SMN-depleted primary cardiomyocytes promoted cell-cycle progression. Finally, we observed a recovery of heart pathology and expression of deregulated genes, including Birc5, after treatment of these mice with ASO10–29, a therapeutic antisense oligonucleotide that corrects SMN2 splicing. Our data demonstrate that the cardiac malfunction of SMA mice is caused by widespread gene deregulation in the heart tissue, particularly of Birc5, resulting in developmental defects through cell-cycle arrest and enhanced apoptosis.
Results
Heart weight and cell number of severe SMA mice are dramatically reduced
Cardiac defects are common features present in SMA mouse models. To investigate the underlying molecular mechanisms, we utilized the Taiwan model (Smn−/−, SMN22TG/0) which displays severe SMA phenotype including loss of motor neurons in the spinal cord and a mean lifespan of 10 days (26–28). Cardiac pathology of this model has not been thoroughly examined, except for a study that revealed interventricular septum thinning (22). We first compared heart weights at five time points: P0, P2, P4, P6 and P8. As shown in Figure 1A, the hearts of SMA mice were markedly smaller than those of their heterozygous littermates starting from P2, a time when SMA mice are still rescuable and generally considered to be healthy based on their weigh progression and motor performance (27,28). On P4, an early symptomatic stage in disease development, the heart weight was only about half of that of the heterozygotes (9.2 mg versus 20.1 mg, see Fig. 1B). The ratio of heart weight to total body weight is an important parameter in assessing heart conditions, especially because SMA mice are smaller than their heterozygote littermates. We found that the ratio is much lower in SMA mice than their heterozygous littermates, starting from P2 (Fig. 1C). These results indicate that SMN deficiency affects the postnatal growth and development of the heart in this mouse model.
Figure 1.

Heart weight and cardiomyocyte density of SMA mice are dramatically reduced, compared with heterozygotes. (A) Comparison of heart weight between SMA mice and their heterozygous littermates (Het) at five time points (P0, P2, P4, P6, and P8) during postnatal development (n = 8). (B) Representative pictures of hearts obtained from P4 SMA and heterozygous mice. (C) Comparison of the heart weight/body weight ratios from P0 to P8 between SMA and heterozygous mice (n = 8). (D) Hematoxylin and eosin (H&E) staining of sections from hearts of heterozygous (upper) and SMA (lower) mice on P0, P2, P4, and P6. The two enlargements below show irregular nuclei of cardiomyocytes (P4, SMA). Scale bar, 50 μm. (E) Quantitation of the number of cardiomyocytes per high-power field (HPF) (n = 4). **P < 0.01, ***P < 0.001.
In an attempt to identify what defects at the cellular level might be present in the heart tissue, we performed hematoxylin and eosin (H&E) staining at four time points: P0, P2, P4 and P6. A dramatic reduction (∼25%) of cardiomyocyte numbers was evident starting from P2, and the decrease continued over time (Fig. 1D and E). At the severe symptomatic stage (P6), the cell number decreased by ∼36%. These data indicate that cardiomyocyte proliferation in SMA mice is markedly impaired. We indeed observed enlarged and irregular-shaped nuclei in multiple P4 SMA cardiomyocytes (Fig. 1D).
G0/G1 cell-cycle arrest occurs in cardiomyocytes of SMA mice
To determine if cardiomyocyte proliferation is truly affected during postnatal development of SMA mice, we examined the expression of two cellular protein markers of cell division, at four time points (P0, P2, P4 and P6), using immunofluorescence staining. One marker is Ki67, which is expressed in all active phases of the cell cycle (G1, S, G2 and M phases) but not in resting cells (G0). The other marker is phosphorylated histone H3 (pH3), which is expressed only in mitotic cells. Compared with heterozygous mice, the number of Ki67-positive cardiomyocytes per high-power field (HPF) in SMA mice decreased by ∼50, 72 and 67% on P2, P4 and P6, respectively (Fig. 2A and B), and the percentage of Ki67-positive cardiomyocytes decreased by ∼36, 69 and 47% on P2, P4 and P6, respectively (Fig. 2C). The number of pH3-positive cardiomyocytes per HPF was also markedly decreased from P2 to P6 (about 2-fold lower at all time points, Fig. 2D and E). The percentage of pH3 positive nuclei on P2, P4 or P6, was 34, 50 and 32%, respectively lower than that of heterozygous mice (P < 0.01 for all three time points, Fig. 2F). These data suggest that SMN deficiency results in decreased proliferation of cardiomyocytes in neonatal mice. We did not observe a reduction of Ki67-positive cells on P0 in SMA mice, which is consistent with our observation above that the number of cardiomyocytes were not reduced on P0. It is of interest to note that the number of pH3-positive cells derived from SMA mice on P0 increased ∼58% (P < 0.05).
Figure 2.

Cardiomyocyte proliferation and cell cycle are impaired in SMA mice. Heart sections from SMA (n = 3) and heterozygous (Het, n = 3) mice at four time points (P0, P2, P4, and P6) were stained with anti-Ki67 antibody (A) or anti-pH3 antibody (D) to label mitotic cells; nuclei were counterstained with DAPI. Total number of Ki67+ (B) or pH3+ cardiomyocytes (E) and percentage of Ki67+ (C) or pH3+ (F) cardiomyocytes per high-power field (HPF) was determined from three sections per heart, with three hearts per group (n = 9). **P < 0.01, ***P < 0.001.
To investigate the decreased number of cycling cells in heart tissue of SMN-depleted mice, we isolated cardiomyocytes from neonatal mice aged P0, P2, P4 and P6, and analysed their cell-cycle profile using flow cytometry. As shown in Figure 3, SMA mice had more cardiomyocytes in G0/G1 phases than heterozygotes, starting from P2, with an increase of 14, 10, and 11% on P2, P4 and P6, respectively (P < 0.05), and concomitantly fewer cells in S and G2/M phases. In particular, cells in S phase were reduced by 60–80%. As for P0 mice, no differences in cell number in G0/G1 phases were observed between SMA and heterozygous mice, but the cell number in S phase was significantly higher (Fig. 3A), which is consistent with the pH3 immunofluorescence analysis on P0. Although modest in terms of cell number changes in S/G2/M phases, which is expected for an early postnatal tissue in transition from hyperplasia to hypertrophy, these results confirm that SMN deficiency impairs cardiomyocyte proliferation through G0/G1 cell-cycle arrest.
Figure 3.

Cell cycle of SMA cardiomyocytes was analysed by flow cytometry. (A–D) DNA profiles of cardiomyocytes isolated from SMA mice aged P0, P2, P4, or P6. Heterozygous littermates (Het) were used as controls (n = 3). Cells were stained with propidium iodide. (E–H) Histograms of cell-cycle data from (A–D). *P < 0.05 **P < 0.01.
Cardiomyocytes undergo apoptosis in severe SMA mice
The reduction in cardiomyocyte number could be partially caused by apoptosis. To address if apoptosis is involved, we examined the heart tissue by TUNEL assay. Apoptotic cells in cardiomyocytes were extremely infrequent in mice with either genotype between P0 and P2; no differences between SMA and normal mice were observed (Fig. 4A and B). However, SMA mice had a significant increase in apoptosis incidence (∼3 fold) on P4 and P6. Apoptosis involves characteristic morphological changes of the cell. Therefore, we further examined the ultrastructure of cardiomyocytes sampled from P4 SMA mice using transmission electron microscopy (TEM). As shown in Figure 4C, mitochondria of cardiomyocytes obtained from SMA mice were obviously swollen; some displayed prominent cristae fragmentation and vacuolar degeneration, indicating degenerative changes (Fig. 4C, red arrow), similar to the morphological changes of cardiomyocytes observed in the severe SMNΔ7 mouse model, which has a slightly mild phenotype than the severe Taiwan model (19,29).
Figure 4.
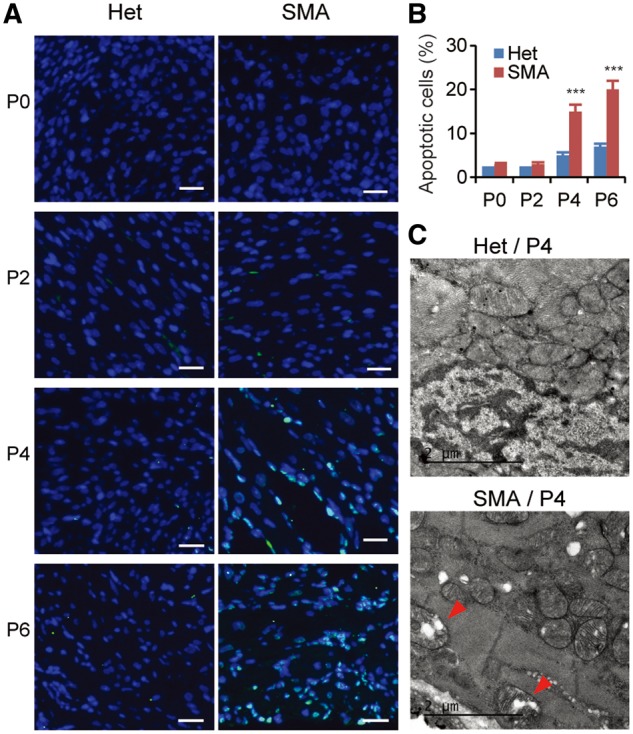
Apoptosis is increased in cardiac tissues of SMA mice. (A) TUNEL staining of cardiac tissues from both heterozygous (Het) and SMA mice aged P0, P2, P4, and P6. Compared with heterozygous mice, the number of TUNEL-positive SMA cardiomyocytes is markedly increased on P4 and P6. Nuclei were counterstained with DAPI. Scale bar, 20 μm. (B) Quantification of apoptotic cardiomyocytes identified by TUNEL staining, as shown in (A) (3 mice per group and 3 counts per mouse). ***P < 0.001. (C) TEM images of heterozygous (upper) and SMA (lower) cardiac tissues. Mitochondria are normal in heterozygous mice, but swollen in SMA cardiomyocytes. The arrowheads point to degenerating mitochondria with cristae deformation.
The TUNEL staining and TEM ultrastructural analyses demonstrate that enhanced apoptosis is present in the tissue and contributes to the heart defects, though it occurs at a relatively late time point, compared with cell-cycle arrest.
Cell cycle and apoptosis-related genes are dysregulated in SMA mice
To understand the molecular basis of cardiomyocyte defects in the severe SMA mouse model, we performed gene-expression analysis using microarrays on the heart tissue, and identified 205 genes that were down-regulated and 269 genes that were up-regulated on P5 in the heart (Supplementary Material, Table S1). Gene ontology cluster analysis of the down-regulated transcripts revealed significant enrichment for genes involved in regulating the mitotic cell cycle, chromosome condensation, G2 DNA-damage checkpoint, and the cellular response to DNA damage (Fig. 5A).
Figure 5.

Expression of cell cycle-related genes during postnatal cardiac development of SMA mice. Heterozygous littermates (Het) were used as controls. (A) Microarray analysis of genes differentially expressed in the heart of SMA mice (n = 5). Tissue samples were collected on P5. Gene Ontology analysis shows that cell cycle-related genes are primarily affected. (B) Heat map showing log2 fold changes of expression levels for a subset of cell cycle-related genes (SMA over Het), analysed by real-time RT-PCR. Cardiac RNA samples were obtained from P0, P2, P4, and P6 mice (n = 3), respectively; gene-expression levels were normalized to Gapdh. (C–F) Histograms showing six mitosis-related genes that are downregulated in the heart of SMA mice on P0, P2, P4, and/or P6 (n = 4). (G) Western blotting analysis of Survivin, AURKB and SMN levels in hearts of mice aged P0, P2, P4, and P6. Protein levels were normalized to β-Tubulin. (H–J) Quantitation of the protein data from (G) and unpublished data (n = 3). For (C–F) and (H–J), *P< 0.05, **P < 0.01, ***P < 0.001.
Among the 205 downregulated genes, 36 are known regulators of the cell cycle. Real-time RT-PCR analysis confirmed that expression of 18 genes important in cell-cycle regulation was significantly decreased in the heart of SMA mice from P2 to P6 (Fig. 5B). Interestingly, expression of six mitotic genes—Birc5, Aurkb, Cdca8, Incenp, Cenpa, and Ndc80—whose protein products act as key regulators of mitosis via formation of the chromosomal passenger complex (CPC), were markedly decreased already on P2 (Fig. 5C–F). In particular, expression of Birc5, which encodes a protein called Survivin, was already decreased by ∼25% on P0 (Fig. 5C). Western blot analysis showed corresponding changes at the protein level (Fig. 5G–J). Several splicing isoforms of Birc5 have been reported (30); however, we found that all isoforms were equally or similarly down-regulated, indicating that no splicing changes are involved in the gene-expression alteration (Supplementary Material, Fig. S1). These data confirm that SMN deficiency impacts the cell cycle and mitosis in cardiomyocytes during postnatal development.
We also examined the expression of the tumor suppressor Trp53/p53, an SMN-interacting protein (31), and its phosphorylation at Ser18, corresponding to Ser15 of human p53, which can induce both cell-cycle arrest and apoptosis; however, no expression or phosphorylation-level changes were detected (Fig. 6A–D). Interestingly, genes downstream of Trp53, including Cdkn1a/p21, Pmaip1/Noxa, Bbc3/Puma, Fas and Bax, were markedly up-regulated (Fig. 6A–G). Upregulation of these apoptosis-related genes can explain why apoptosis is enhanced in the heart tissue of SMA mice. The late timing (P4) of the upregulation is also consistent with our observations above that decreased cell proliferation occurs prior to apoptosis (Figs 2–4). On the other hand, expression of Bcl-2, a well-characterized member of the Bcl-2 family that negatively modulates apoptosis by several mechanisms, and of Cycs/Cyt c, a determining factor in mitochondrial-mediated apoptosis, were not increased (Fig. 6A).
Figure 6.

Expression of apoptosis-related genes during postnatal cardiac development of SMA mice. Heterozygous littermates (Het) were used as controls. (A) Heat map showing apoptosis-related genes that are upregulated. Heart tissues were collected on P0, P2, P4, and P6 (n = 3). Gene expression was analysed by real-time RT-PCR and presented as log2-transformed fold change. (B) Western blotting analysis of p53, p-p53 (Ser18), PUMA, NOXA and p21 levels. Protein levels were normalized to β-Tubulin. (C–G) Quantitation of the data in (A) plus unpublished data (n = 4). *P< 0.05, **P < 0.01, ***P < 0.001.
Previous studies have shown upregulation of Cdkn1a, Pmaip1, and Fas in the spinal cord of SMA mouse models, and upregulation of Bax in muscle samples from patients (32–37). Taken together, these data suggest that cell death pathways are more or less activated in multiple tissues. Upregulation of Cdkn1a, a major effector of p53’s activities in cell-cycle arrest and apoptosis, has been recently postulated as a critical factor in SMA pathology (33,37). However, its upregulation in the heart initiates at a rather late time point (P4), suggesting an indirect role at least in the cardiac pathology of SMA mice.
Overexpression of Survivin in SMN-depleted cardiomyocytes
Survivin plays a key role in heart development (38,39). To test whether the downregulation of Birc5 indeed affects the cardiac development of SMA mice through cell-cycle arrest, we overexpressed Survivin in SMN-depleted mouse cardiomyocytes. Primary cardiomyocytes were isolated from P2 neonatal Smn+/− heterozygous mice that theoretically express half the amount of SMN as do wild-type mice, and SMN was further depleted by 51% and 72% with two independent siRNAs, respectively (Fig. 7A and B). SMN knockdown induced similar gene expression alterations as observed in the heart of SMA mice (Supplementary Material, Fig. S2), including robust downregulation of Survivin, by 49% and 59% for siRNA-1 and siRNA-2, respectively. We selected siRNA-2 for further flow-cytometry analysis. Depletion of SMN induced cell-cycle arrest, with about 70% cells accumulating in G0/G1 phases after 48 h, a 10% increase in comparison to the control siRNA (Fig. 7E–H). A Survivin cDNA-expressing plasmid, pCGT7-Survivin (0.6 μg), was then transfected into the SMN-depleted cells, which led to approximately a three-fold increase of the total Survivin levels (Fig. 7C and D). Interestingly, after Survivin overexpression, cell-cycle progression was mostly restored, as seen by the proportions of cells in the G0/G1, S and G2/M phases of the cell cycle (Fig. 7G and H). These data indicate that Survivin is a critical regulator of cell division, particularly of cardiomyocytes, consistent with previous studies (38,40).
Figure 7.
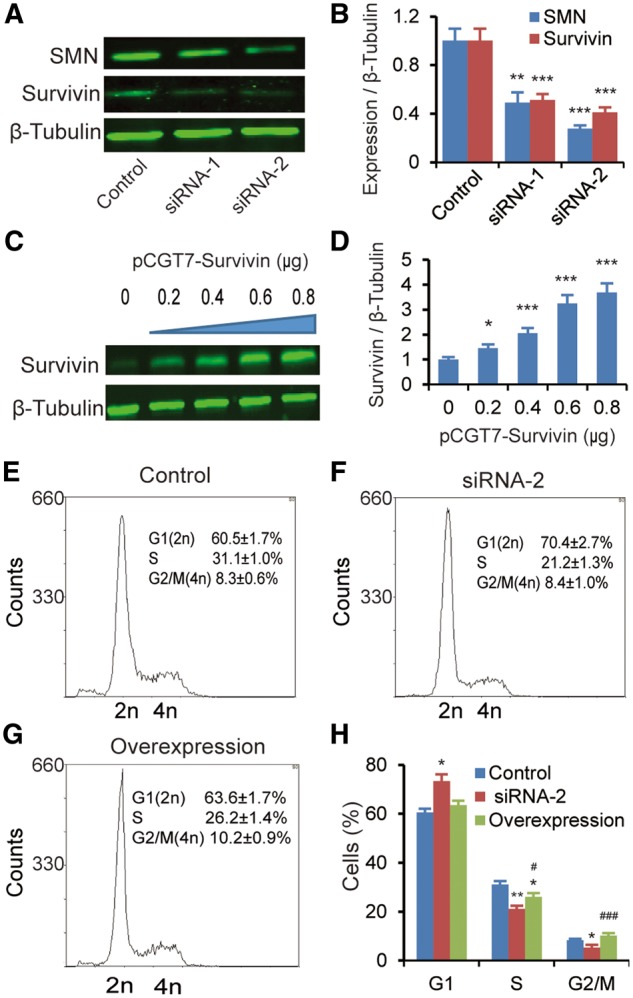
Effect of Survivin overexpression in SMN-depleted neonatal mouse cardiomyocytes. Cardiomyocytes isolated from P2 heterozygous mice were treated with two Smn1-specific siRNAs (siRNA-1 and siRNA-2), respectively, and a scrambled siRNA control. (A) Western blotting analysis showing that Survivin expression was markedly reduced in cardiomyocytes after knockdown of SMN with two independent siRNAs. Tubulin was used as a loading control. (B) Quantitation of SMN and Survivin protein levels after treatment with siRNA-1 or siRNA-2 in cardiomyocytes (n = 3). (C) Western blotting analysis showing proper expression of T7-tagged Survivin after transfection of the cDNA expression plasmid pCGT7-Survivin into SMN-depleted cardiomyocytes. Monoclonal anti-T7 antibody was used to detect T7-Survivin. (D) Quantitation of T7-Survivin levels after transfection of different amounts of pCGT7-Survivin. Tubulin was used as a loading control. (E–G) DNA profiles of cardiomyocytes transfected with control siRNA (E), or siRNA-2 with (G) or without (F) overexpression of Survivin (0.6 μg plasmid). Plasmid pCGT7-Survivin was transfected into cells 24 h post siRNA-2 transfection; 48 h later, cells were harvested for analysis (n = 3). (H) Histogram of cell-cycle data from (E–G). *P < 0.05 **P < 0.01.
Rescue of severe SMA mouse heart pathology with ASO10–29
We next asked if rescue of SMN levels in SMA mice is sufficient to recover expression of Survivin and rescue the heart defects. To this end, we used a MOE-modified antisense oligonucleotide, ASO10–29, that efficiently rescues SMA mouse models when delivered systemically (41). After two subcutaneous injections of ASO10–29 at 90 mg/kg between P0 and P1, the exon 7 inclusion percentage of the SMN2 transgene in the heart, analysed on P6, increased ∼3 fold, from 12 to 35% (Fig. 8A–C). Western blot analysis showed ∼2-fold changes at the protein level (Fig. 8B–D). Treated SMA mice still expressed much less full-length SMN protein than heterozygous mice, reflecting the incomplete inclusion of SMN2 exon 7 in cardiac tissues. After ASO10–29 treatment, both heart weight and its ratio to body weight significantly increased on P6, suggesting that ASO treatment partially rescued heart pathology (Fig. 8E and F), consistent with effects previously published with another SMN2 splice-switching ASO (27). In addition, expression of deregulated genes associated with cell-cycle arrest and apoptosis, including Birc5, Mki67, Aurkb, Cdkn1a, and Pmaip1 was fully or partially restored (Fig. 8G). These data indicate that early treatment with ASO10–29 can rescue both abnormal gene expression and postnatal development of the heart.
Figure 8.
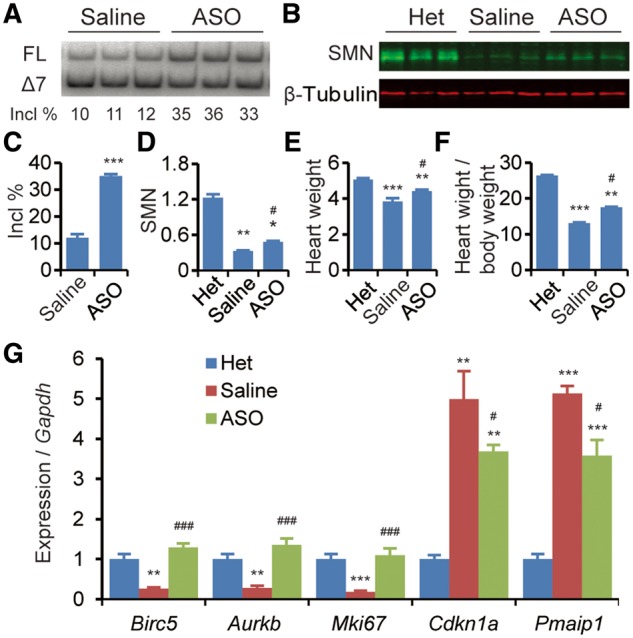
Subcutaneous delivery of ASO10–29 partially corrects the heart pathology of SMA mice. ASO in saline or saline alone was injected into mice at 90 mg/kg, twice between P0 and P1, and cardiac tissues were collected on P6 for various analyses. Heterozygous mice (Het) were used as controls. (A) Analysis of SMN2 exon 7 inclusion by radioactive RT–PCR after ASO treatment. FL, full-length transcript; Δ7, exon 7-skipped transcript; Incl %, 100 × FL/(FL + Δ7). (B) Western blotting analysis of SMN levels, normalized to β-tubulin. (C) Histogram of exon 7 inclusion data after ASO treatment as in (A) (n = 4). (D) Histogram of SMN levels from (B) (n = 3). (E) Heart weight after ASO treatment (n = 10). (F) Ratio of heart weight to body weight (mg/g, n = 10). (G) Quantitation of gene expression (Birc5, Aurkb, Mki67, cdkn1a/p21, Pmaip1/Noxa), normalized to Gapdh (n = 4). For (C–G), *P < 0.05, **P < 0.01, ***P < 0.001 versus Het; #P < 0.05, ###P < 0.001 versus saline.
Discussion
Autonomic imbalance has been considered as the primary cause of cardiac defects observed in severe SMA patients and mouse models. However, whether low levels of SMN may affect cardiomyocytes in a cell-autonomous manner has not previously been addressed. In this study, using tissue staining, we revealed a decrease in cardiomyocyte proliferation and an increase in apoptosis during early postnatal development of the heart in the severe Taiwan mouse model, reflecting their small body size, low heart weight and decreased cell numbers. An undersized heart was previously observed in the SMNΔ7 model (19–21), though the ratio of heart weight to body weight was not significantly changed, which could be attributable to that model being slightly less severe (10 days vs 15 days lifespan). Furthermore, using immunofluorescence and flow cytometry, we demonstrated that impaired cardiomyocyte proliferation was caused by both alteration in cell cycle profiles and interestingly an increase in apoptosis; this was further confirmed by transcriptomic analysis using oligonucleotide microarrays. Our data represent the first report of cell-cycle arrest present in any tissues in the context of SMA, consistent with three earlier transcriptomic studies that reveal sets of genes associated with cell-cycle signaling and apoptosis signaling deregulated in the spinal cord and/or liver in the SMNΔ7 model, severe Taiwan model, or a severe adult SMA phenocopy model generated by an inhibitory ASO that induces SMN2 exon 7 skipping (33,42,43). Defective cell proliferation and apoptosis have been reported in cultured SMN-depleted cells (44,45). In mouse models, reduced cell number and/or decreased proliferation in the hippocampus, retinal and optical nerve, resulting in impaired neurogenesis have been observed (46,47). Fayzullina and Martin first examined tissue cell death events in muscle and spinal cord tissue using multiple methods, including the TUNEL assay, and found that both skeletal muscles and the spinal cord of the severe Taiwan model undergo increased apoptosis, albeit with different times of onset in the two tissues (48). Taken together, these data suggest that alterations in cell cycle and apoptosis may be a global phenomenon present in a broad set of tissues in SMA mice. Of note, alteration in cell cycle profiles occurred at an early time point (P2) and two days before apoptosis was first detectable, suggesting that cell-cycle arrest is the initial defect and therefore may have the primary role in causing cardiac defects during postnatal development.
Widespread defects in gene expression including pre-mRNA splicing have been observed in SMA mouse models from a number of studies using microarrays or RNA-Seq (32,33,36,42,43,49–52). However, the key genes act downstream of SMN in affected tissues such as the spinal cord and heart remain elusive. Here, we identified Birc5 (Survivin) as a key gene in SMA cardiac pathogenesis. Our finding is not surprising considering the established roles of Survivin in regulating cell-cycle progression and organogenesis. As the smallest member of the inhibitor-of-apoptosis family, Survivin has a key role in mitotic cell division. It represents a central node in multiple networks in cell-division including forming the chromosomal passenger complex (CPC) with the products of five other regulatory genes (Aurkb, Cdca8, Incenp, Cenpa, and Ndc80) (53–55). The CPC regulates chromosomal and cytoskeletal events to warrant proper completion of mitosis and avoid unequal segregation of chromosomal material into daughter cells. Due to its essential role in cell-cycle regulation, Survivin is critical during organ development, as well as in regeneration, and its ablation leads to abnormal cell division (including abnormal oriented cell division that affects morphogenesis) in various organs, such as kidney, heart, brain, gut, and liver (38,56–59). Most importantly, Survivin determines the total cardiomyocyte number and ploidy, and is indispensable for postnatal cardiac development (38). The prominent features of lack of Survivin are cell-number reduction, enlarged and abnormally-shaped nuclei, and polyploidization (60). We did not observe obvious polyploid myocytes in our study, which may reflect incomplete Survivin downregulation in the SMA mice as opposed to the complete knockout of Survivin in the studies reporting polyploid myocytes (38). However, the decrease in Survivin expression observed in SMA mice may be sufficient to result in alteration in cell proliferation.
The importance of Survivin in cardiac pathogenesis of SMA mice is strongly supported by our in vitro study using primary cardiomyocytes obtained from P2 heterozygous neonates. Further depletion of SMN with specific siRNAs recapitulated gene expression alteration including a decrease in expression of Survivin, and caused cell-cycle arrest, indicating that Survivin is downstream of SMN, and that the cardiac defects are caused by a tissue-autonomous mechanism. Overexpression of Survivin alone could markedly restore cell-cycle progression indicating a critical role of Survivin deficiency in the cell arrest of SMA cardiomyocytes. It also suggests that dysregulation of other genes could be tolerated to a certain extent provided that the expression of Survivin is restored. Therefore, we conclude that Survivin is one of the major factors in causing cardiac failure in SMA. In agreement with this notion, we observed that treatment with a therapeutic ASO not only ameliorates the heart conditions of SMA mice, but also restores expression of Birc5.
Survivin has also been implicated in apoptosis. However, its role and mechanisms in inhibiting this pathway remain controversial (40,61). Loss of Survivin in several tissues, such as the brain, gut, and kidney, induces apoptosis (56–58), whereas in other tissues, such as the liver, it does not affect apoptosis (59), suggesting that its anti-apoptotic role is tissue-dependent. Levkau et al. reported that apoptosis was unaffected in Survivin-deficient mouse cardiomyocytes (38). However, a more recent study by Lee et al. showed that overexpression of Survivin reduced apoptosis and caspase activity in a rat model of cardiomyopathy, and improved heart function (62). It is likely that Survivin provides a heightened cell-survival threshold in the heart, as previously suggested (40). In this case, the increased apoptosis in the heart of SMA mice could be caused by both down-regulation of Birc5/Survivin and upregulation of pro-apoptotic genes, including Pmaip1, Bbc3, and Cdkn1a.
Cardiac fibrosis is a consequence of excessive deposition of extracellular matrix in the cardiac muscle; it causes tissue stiffness and cardiac arrhythmias, due to electrophysiological disturbances, and predisposes patients with heart failure to sudden cardiac death (63,64). Shababi et al. reported that SMNΔ7 mice have interstitial fibrosis in the heart, which initially develops on P2 and intensifies on P5 and P9 (21). High levels of fibrosis were correlated with cardiac defects, such as a slower heart rate, suggesting that cardiac fibrosis is an important contributing factor in cardiac pathology of SMA mice. Shababi et al. further found high levels of reactive oxygen species present in the neonatal heart tissue, and thus proposed that oxidative stress, a known pro-fibrotic factor, is the underlying mechanism of cardiac fibrosis (21). Though we did not examine cardiac fibrosis and oxidative stress in the Taiwan model, we expect that lack of SMN likewise induces cardiac fibrosis given the level of pathology observed on H&E stains. Interestingly, however, Levkau et al. reported that cardiac-specific depletion of Survivin alone drastically increases interstitial fibrosis in the mouse heart (38). Moreover, Lee et al. reported that overexpression of Survivin in a rat model with doxorubicin-induced cardiomyopathy strongly reduces interstitial fibrosis in the left ventricle (62). These data suggest that Survivin is a key regulator of cardiac fibrosis. Considering our finding that Survivin expression was downregulated on P0, we propose that both Survivin dysregulation and oxidative stress contribute to the interstitial fibrosis in the heart of SMA mice.
In the Levkau et al. study, cardiac-specific depletion of Survivin in mice had no effect on the heart weight, and median survival was 28 weeks (38). The much milder phenotype of these mice in comparison to the Taiwan model suggests that the cardiac dysfunction in the latter model involves additional factors, such as other dysregulated signaling pathways in the heart, and/or negative cues released from other affected tissues.
Recently, Jangi et al. observed a marked increase in intron-retention events in SMN-depleted cells, which they proposed to lead to DNA damage through aberrant R-loop formation and subsequent activation of p53 signaling (37). These observations provide a potential mechanistic explanation for our findings, as p53 suppresses expression of Survivin, while promoting expression of p21, Noxa, and Puma (65–68). However, we did not detect increases in total p53 or phopho-p53 (Ser18) levels. One reason could be the presence of an undetected or unknown modification of p53 that activates p53 signaling. Alternatively, the cardiac defects could involve a p53-independent pathway, considering that several genes regulate transcription of Survivin and of the pro-apoptotic genes whose expression is altered in SMA mice. Nonetheless, based on the gene expression analysis in cultured cardiomyocytes depleted of SMN by siRNA treatment, we can rule out cell-non-autonomous mechanisms causing dysregulation of most of the cell-cycle- and apoptosis-related genes, including robust downregulation of Birc5 in the mouse model. Further work to determine the signaling pathway that links SMN to Survivin will shed light on how limiting SMN affects heart development, and enable the design of appropriate medical interventions.
Materials and Methods
Animals
All mice were handled in accordance with the guidelines and protocols approved by the Care and Use of Animals Committee of Soochow University (China) and Cold Spring Harbor Laboratory’s Institutional Animal Care and Use Committee (USA). All experiments except for the microarray analysis were performed with female mice to avoid confounding effects caused by gender differences. The severe Taiwan SMA mouse model (Smn−/−, SMN22TG/0) in FVB background was generated by crosses as described (27). For tissue sample collection, mice were sacrificed by CO2 asphyxiation; tissues were immediately rinsed with saline, weighed, and snap-frozen in liquid nitrogen for storage at −80 °C.
Histology
Dissected mouse hearts were fixed in 4% (v/v) formaldehyde at 4 °C overnight, washed in phosphate buffered saline (PBS) and embedded in paraffin blocks. Five-µm-thick paraffin sections were cut for either hematoxylin and eosin (H&E) or immunofluorescence staining. For immunofluorescence, following antigen retrieval with 10 mM sodium citrate buffer (pH 6.0) in boiling water for 15 min, paraffin sections were permeabilized with 0.3% (v/v) Triton-X100 for 10 min and then blocked with 10% (v/v) goat serum for 30 min. Primary antibodies including polyclonal anti-Ki67 (Bioss) and polyclonal anti-phospho-histone H3 (Ser10) (Cell Signaling Technology) were incubated overnight at 4 °C. Sections were subsequently washed with PBS and incubated with the corresponding anti-rabbit secondary antibodies conjugated to Alexa Fluor 488 (Lianke). Nuclei were counter-stained with DAPI (Invitrogen). For TUNEL staining to detect apoptotic nuclei, paraffin sections were processed using the In Situ Cell Death Detection Kit (TMR Red, Roche Applied Sciences), according to the manufacturer’s instructions (48).
Transmission electron microscope imaging
Hearts collected immediately after euthanasia were fixed in 2.5% (v/v) glutaraldehyde for a minimum of 24 h and post-fixed in 2% (m/v) osmium tetroxide in 0.1 M cacodylate buffer. Tissues were rapidly dehydrated in increasing concentrations of ethanol (30–100%), infiltrated with 100% acetone and embedded in low-viscosity polymerized epoxy resin. Sections were stained on grids with 1% uranyl acetate and lead citrate. Electron micrographs were taken with HT7700 transmission electron microscope (Hitachi).
Flow cytometry
Neonatal mouse cardiomyocytes were dissociated with 0.25% trypsin-EDTA and collagenase and spun down (200 g for 10 min) to obtain a cell pellet. The cell pellet was resuspended in cold PBS to obtain a single-cell suspension. Cells were fixed in ice-cold 70% ethanol at 4 °C overnight and spun down (200 g for 10 min). The cell pellet was then resuspended in propidium iodide staining solution (5 ng/μl propidium iodide in PBS with RNase A) and incubated at 37 °C for 30 min, followed by immediate flow-cytometry analysis on a Beckman Coulter FC500. The distribution of cells in different cell-cycle phases was assessed by Flowjo software.
Quantitative and radioactive RT-PCR
Trizol (Invitrogen) was used to extract total RNA from mouse heart tissues after pulverizing them in liquid N2 with mortar and pestle. One microgram of total RNA was reverse-transcribed with ImProm-II Reverse Transcriptase (Vazyme). Quantitative real-time RT-PCR was performed with an Applied Biosystems StepOnePlus instrument and an Express One-Step SuperScript Kit, according to the manufacturer’s instructions. Primer pairs for all genes are listed in Supplementary Material, Table S2. For radioactive RT–PCR, the human-specific primer pair E4–33to55-F and E8–15to36-R was used for amplifying human SMN2 transcripts in RNA samples from mouse tissues, as previously described (69); all PCR products were labeled with a-32P-dCTP and analysed by 6% native PAGE, followed by phosphorimage analysis. The extent of exon 7 inclusion was calculated as described (70), and the signal intensity of each cDNA band was normalized according to its G + C content.
Western blotting
Twenty milligrams of mouse heart was pulverized in liquid N2 and homogenized in 0.4 ml of protein sample buffer containing 2% (w/v) SDS, 10% (v/v) glycerol, 50 mM Tris-HCl (pH 6.8), and 0.1 M DTT. Protein samples were separated by 12% SDS-PAGE and electro-blotted onto nitrocellulose membranes. The membranes were blocked for 2 h with 5% (w/v) non-fat milk in Tris-buffered saline containing 0.05% Tween-20 (TBST), and then incubated overnight at 4 °C with the following primary antibodies: monoclonal anti-SMN (BD Biosciences, 1: 500), monoclonal anti-p53 (Santa Cruz), polyclonal anti-phospho-p53 (Ser18) (Abcam), monoclonal anti-p21 (Abcam), monoclonal anti-PMAIP1/NOXA (Abcam), polyclonal anti-PUMA (Abcam), polyclonal anti-Survivin (Abcam), polyclonal anti-AURKB (Proteintech), or polyclonal anti-β-tubulin (Lianke). After washing with TBST three times, the membrane was incubated with secondary IRDye 700CW-conjugated goat anti-mouse or anti-rabbit antibody. Protein signals were detected with an Odyssey instrument (LI-COR Biosciences).
Cell culture, transfection and siRNA knockdown
Primary cardiomyocytes were isolated from minced heart tissues from P2 neonatal heterozygous mice by enzymatic digestion using trypsin, collagenase Type II and DNase I (Sigma), according to the manufacturer’s recommended protocol. Isolated cardiomyocytes were further treated with 5-Bromo-2-deoxyUridine (BrdU) (Sigma) for 48 h to prevent proliferation of non-cardiomyocyte cells. Cells were cultured in DMEM/M199 (4:1) supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 units/ml) (Gibco) (71). The siRNAs to knock down mouse Smn were siRNA-1 (sense: 5’-CAGUUUCAAUGGUCAUAAATT-3’) and siRNA-2 (sense: 5’-GAGGCAAUUACCAGUUUCATT-3’) with a scramble siRNA control (sense: 5’- UUCUCCGAACGUGUCACGUTT -3’). Cells were seeded in 6-well plates at 70% confluency, 1 × 105 per well, and 50 nM of each siRNA was transfected into cells in each well, using Lipofectamine LTX (Invitrogen). Six hours post-transfection, cells were added with cardiomyocyte medium and further incubated for 24 h before harvesting. For the Survivin overexpression study, 0.6 μg of the expression plasmid pCGT7-Survivin was transfected into cardiomyocytes in each well of 6-well plates with Lipofectamine LTX, and cells were incubated for 48 h before harvesting for flow-cytometry analysis.
Microarray
Oligonucleotide microarray analysis on RNA samples extracted from the hearts of SMA mice (Smn−/−, SMN22TG/0) with heterozygotes (Smn+/−, SMN22TG/0) as controls was performed using the Illumina MouseWG-6 v2.0 Expression BeadChip and GenomeStudio software. Statistically significant gene-expression changes were determined for each sample as the log2 (SMA/control) ratio. Genes that showed a log2 ratio higher than 3 were selected.
Antisense oligonucleotide treatment
MOE-modified ASO10–29 (5′-ATTCACTTTCATAATGCTGG-3′) with phosphorothioate backbone and all 5-methylcytosines (27,41). ASO10–29 in saline was injected subcutaneously at 90 mg/kg twice between P0 and P1. Tissue samples were collected on P6.
Statistical analysis
Differences between sets of data were analysed by two-tailed Student’s t test using SPSS16.0 statistical software (IBM); a value of P < 0.05 was considered statistically significant. In all of the figures, data are expressed as mean ± SEM.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
National Natural Science Foundation of China (grants 81271423, 81471298, 81530035 to Y.H.) and Priority Academic Program Development of Jiangsu Higher Education Institutions. NIH grant GM42699 to A.R.K. China Scholarship Council grant 201606920049 to L.S.
Supplementary Material
References
- 1. Crawford T.O., Pardo C.A. (1996) The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis., 3, 97–110. [DOI] [PubMed] [Google Scholar]
- 2. Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 80, 155–165. [DOI] [PubMed] [Google Scholar]
- 3. Donlin-Asp P.G., Fallini C., Campos J., Chou C.C., Merritt M.E., Phan H.C., Bassell G.J., Rossoll W. (2017) The Survival of Motor Neuron Protein Acts as a Molecular Chaperone for mRNP Assembly. Cell Rep., 18, 1660–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meister G., Buhler D., Pillai R., Lottspeich F., Fischer U. (2001) A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat. Cell Biol., 3, 945–949. [DOI] [PubMed] [Google Scholar]
- 5. Pellizzoni L., Yong J., Dreyfuss G. (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science, 298, 1775–1779. 10.1126/science.1074962 [DOI] [PubMed] [Google Scholar]
- 6. Tisdale S., Lotti F., Saieva L., Van Meerbeke J.P., Crawford T.O., Sumner C.J., Mentis G.Z., Pellizzoni L. (2013) SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3'-end formation of histone mRNAs. Cell Rep., 5, 1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rochette C.F., Gilbert N., Simard L.R. (2001) SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet., 108, 255–266. [DOI] [PubMed] [Google Scholar]
- 8. Lorson C.L., Hahnen E., Androphy E.J., Wirth B. (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA, 96, 6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu X., Wang S.H., Sun J., Krainer A.R., Hua Y., Prior T.W. (2017) A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum. Mol. Genet., 26, 2768–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamilton G., Gillingwater T.H. (2013) Spinal muscular atrophy: going beyond the motor neuron. Trends Mol. Med., 19, 40–50. [DOI] [PubMed] [Google Scholar]
- 11. Simone C., Ramirez A., Bucchia M., Rinchetti P., Rideout H., Papadimitriou D., Re D.B., Corti S. (2016) Is spinal muscular atrophy a disease of the motor neurons only: pathogenesis and therapeutic implications?. Cell. Mol. Life Sci., 73, 1003–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hua Y., Liu Y.H., Sahashi K., Rigo F., Bennett C.F., Krainer A.R. (2015) Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev., 29, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elkohen M., Vaksmann G., Elkohen M.R., Farncart C., Foucher C., Rey C. (1996) [Cardiac involvement in Kugelberg-Welander disease. A prospective study of 8 cases]. Arch. Mal. Coeur. Vaiss., 89, 611–617. [PubMed] [Google Scholar]
- 14. El-Matary W., Kotagiri S., Cameron D., Peart I. (2004) Spinal muscle atrophy type 1 (Werdnig-Hoffman disease) with complex cardiac malformation. Eur. J. Pediatr., 163, 331–332. [DOI] [PubMed] [Google Scholar]
- 15. Bach J.R. (2007) Medical considerations of long-term survival of Werdnig-Hoffmann disease. Am. J. Phys. Med. Rehabil., 86, 349–355. [DOI] [PubMed] [Google Scholar]
- 16. Rudnik-Schoneborn S., Heller R., Berg C., Betzler C., Grimm T., Eggermann T., Eggermann K., Wirth R., Wirth B., Zerres K. (2008) Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet., 45, 635–638. [DOI] [PubMed] [Google Scholar]
- 17. Finsterer J., Stollberger C. (1999) Cardiac involvement in Werdnig-Hoffmann's spinal muscular atrophy. Cardiology, 92, 178–182. 10.1159/000006968 [DOI] [PubMed] [Google Scholar]
- 18. Menke L.A., Poll-The B.T., Clur S.A., Bilardo C.M., van der Wal A.C., Lemmink H.H., Cobben J.M. (2008) Congenital heart defects in spinal muscular atrophy type I: a clinical report of two siblings and a review of the literature. Am. J. Med. Genet. A, 146A, 740–744. [DOI] [PubMed] [Google Scholar]
- 19. Bevan A.K., Hutchinson K.R., Foust K.D., Braun L., McGovern V.L., Schmelzer L., Ward J.G., Petruska J.C., Lucchesi P.A., Burghes A.H.. et al. (2010) Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet., 19, 3895–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heier C.R., Satta R., Lutz C., DiDonato C.J. (2010) Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum. Mol. Genet., 19, 3906–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shababi M., Habibi J., Yang H.T., Vale S.M., Sewell W.A., Lorson C.L. (2010) Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum. Mol. Genet., 19, 4059–4071. [DOI] [PubMed] [Google Scholar]
- 22. Schreml J., Riessland M., Paterno M., Garbes L., Roßbach K., Ackermann B., Krämer J., Somers E., Parson S.H., Heller R.. et al. (2013) Severe SMA mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ-26481585. Eur. J. Hum. Genet., 21, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gogliotti R.G., Quinlan K.A., Barlow C.B., Heier C.R., Heckman C.J., Didonato C.J. (2012) Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J. Neurosci., 32, 3818–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arai H., Tanabe Y., Hachiya Y., Otsuka E., Kumada S., Furushima W., Kohyama J., Yamashita S., Takanashi J., Kohno Y. (2005) Finger cold-induced vasodilatation, sympathetic skin response, and R-R interval variation in patients with progressive spinal muscular atrophy. J. Child Neurol., 20, 871–875. [DOI] [PubMed] [Google Scholar]
- 25. Hachiya Y., Arai H., Hayashi M., Kumada S., Furushima W., Ohtsuka E., Ito Y., Uchiyama A., Kurata K. (2005) Autonomic dysfunction in cases of spinal muscular atrophy type 1 with long survival. Brain Dev., 27, 574–578. [DOI] [PubMed] [Google Scholar]
- 26. Gogliotti R.G., Hammond S.M., Lutz C., Didonato C.J. (2010) Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem. Biophys. Res. Commun., 391, 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hua Y., Sahashi K., Rigo F., Hung G., Horev G., Bennett C.F., Krainer A.R. (2011) Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature, 478, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Riessland M., Ackermann B., Forster A., Jakubik M., Hauke J., Garbes L., Fritzsche I., Mende Y., Blumcke I., Hahnen E.. et al. (2010) SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum. Mol. Genet., 19, 1492–1506. [DOI] [PubMed] [Google Scholar]
- 29. Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. (2005) SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet., 14, 845–857. [DOI] [PubMed] [Google Scholar]
- 30. Conway E.M., Pollefeyt S., Cornelissen J., DeBaere I., Steiner-Mosonyi M., Ong K., Baens M., Collen D., Schuh A.C. (2000) Three differentially expressed survivin cDNA variants encode proteins with distinct antiapoptotic functions. Blood, 95, 1435–1442. [PubMed] [Google Scholar]
- 31. Young P.J., Day P.M., Zhou J., Androphy E.J., Morris G.E., Lorson C.L. (2002) A direct interaction between the survival motor neuron protein and p53 and its relationship to spinal muscular atrophy. J. Biol. Chem., 277, 2852–2859. [DOI] [PubMed] [Google Scholar]
- 32. Murray L.M., Beauvais A., Gibeault S., Courtney N.L., Kothary R. (2015) Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/-) mouse model of spinal muscular atrophy. Acta Neuropathol. Commun., 3, 55.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Staropoli J.F., Li H., Chun S.J., Allaire N., Cullen P., Thai A., Fleet C.M., Hua Y., Bennett C.F., Krainer A.R.. et al. (2015) Rescue of gene-expression changes in an induced mouse model of spinal muscular atrophy by an antisense oligonucleotide that promotes inclusion of SMN2 exon 7. Genomics, 105, 220–228. 10.1016/j.ygeno.2015.01.007 [DOI] [PubMed] [Google Scholar]
- 34. Tews D.S., Goebel H.H. (1997) Apoptosis-related proteins in skeletal muscle fibers of spinal muscular atrophy. J. Neuropathol. Exp. Neurol., 56, 150–156. [DOI] [PubMed] [Google Scholar]
- 35. Wu C.Y., Whye D., Glazewski L., Choe L., Kerr D., Lee K.H., Mason R.W., Wang W. (2011) Proteomic assessment of a cell model of spinal muscular atrophy. BMC Neurosci., 12, 25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Z., Pinto A.M., Wan L., Wang W., Berg M.G., Oliva I., Singh L.N., Dengler C., Wei Z., Dreyfuss G. (2013) Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA, 110, 19348–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jangi M., Fleet C., Cullen P., Gupta S.V., Mekhoubad S., Chiao E., Allaire N., Bennett C.F., Rigo F., Krainer A.R.. et al. (2017) SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. USA, 114, E2347–E2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levkau B., Schafers M., Wohlschlaeger J., von Wnuck Lipinski K., Keul P., Hermann S., Kawaguchi N., Kirchhof P., Fabritz L., Stypmann J.. et al. (2008) Survivin determines cardiac function by controlling total cardiomyocyte number. Circulation, 117, 1583–1593. [DOI] [PubMed] [Google Scholar]
- 39. Schrickel J.W., Lickfett L., Lewalter T., Tiemann K., Nickenig G., Baba H., Heusch G., Schulz R., Levkau B. (2012) Cardiomyocyte-specific deletion of survivin causes global cardiac conduction defects. Basic Res. Cardiol., 107, 299.. [DOI] [PubMed] [Google Scholar]
- 40. Altieri D.C. (2008) Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer, 8, 61–70. [DOI] [PubMed] [Google Scholar]
- 41. Bogdanik L.P., Osborne M.A., Davis C., Martin W.P., Austin A., Rigo F., Bennett C.F., Lutz C.M. (2015) Systemic, postsymptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy. Proc. Natl. Acad. Sci. USA, 112, E5863–E5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baumer D., Lee S., Nicholson G., Davies J.L., Parkinson N.J., Murray L.M., Gillingwater T.H., Ansorge O., Davies K.E., Talbot K. (2009) Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet., 5, e1000773.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Doktor T.K., Hua Y., Andersen H.S., Broner S., Liu Y.H., Wieckowska A., Dembic M., Bruun G.H., Krainer A.R., Andresen B.S. (2017) RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res., 45, 395–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li D.K., Tisdale S., Espinoza-Derout J., Saieva L., Lotti F., Pellizzoni L. (2013) A cell system for phenotypic screening of modifiers of SMN2 gene expression and function. PLoS One, 8, e71965.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ilangovan R., Marshall W.L., Hua Y., Zhou J. (2003) Inhibition of apoptosis by Z-VAD-fmk in SMN-depleted S2 cells. J. Biol. Chem., 278, 30993–30999. [DOI] [PubMed] [Google Scholar]
- 46. Wishart T.M., Huang J.P., Murray L.M., Lamont D.J., Mutsaers C.A., Ross J., Geldsetzer P., Ansorge O., Talbot K., Parson S.H.. et al. (2010) SMN deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy. Hum. Mol. Genet., 19, 4216–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu H., Beauvais A., Baker A.N., Tsilfidis C., Kothary R. (2011) Smn deficiency causes neuritogenesis and neurogenesis defects in the retinal neurons of a mouse model of spinal muscular atrophy. Dev. Neurobiol., 71, 153–169. [DOI] [PubMed] [Google Scholar]
- 48. Fayzullina S., Martin L.J. (2014) Skeletal muscle DNA damage precedes spinal motor neuron DNA damage in a mouse model of Spinal Muscular Atrophy (SMA). PLoS One, 9, e93329.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boulisfane N., Choleza M., Rage F., Neel H., Soret J., Bordonne R. (2011) Impaired minor tri-snRNP assembly generates differential splicing defects of U12-type introns in lymphoblasts derived from a type I SMA patient. Hum. Mol. Genet., 20, 641–648. [DOI] [PubMed] [Google Scholar]
- 50. Lotti F., Imlach W.L., Saieva L., Beck E.S., Hao L. T., Li D.K., Jiao W., Mentis G.Z., Beattie C.E., McCabe B.D.. et al. (2012) An SMN-dependent U12 splicing event essential for motor circuit function. Cell, 151, 440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murray L.M., Lee S., Baumer D., Parson S.H., Talbot K., Gillingwater T.H. (2010) Pre-symptomatic development of lower motor neuron connectivity in a mouse model of severe spinal muscular atrophy. Hum. Mol. Genet., 19, 420–433. [DOI] [PubMed] [Google Scholar]
- 52. Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M., Dreyfuss G. (2008) SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell, 133, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li F., Ambrosini G., Chu E.Y., Plescia J., Tognin S., Marchisio P.C., Altieri D.C. (1998) Control of apoptosis and mitotic spindle checkpoint by survivin. Nature, 396, 580–584. [DOI] [PubMed] [Google Scholar]
- 54. Vader G., Medema R.H., Lens S.M. (2006) The chromosomal passenger complex: guiding Aurora-B through mitosis. J. Cell Biol., 173, 833–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Altieri D.C. (2008) New wirings in the survivin networks. Oncogene, 27, 6276–6284. 10.1038/onc.2008.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martini E., Wittkopf N., Gunther C., Leppkes M., Okada H., Watson A.J., Podstawa E., Backert I., Amann K., Neurath M.F.. et al. (2016) Loss of survivin in intestinal epithelial progenitor cells leads to mitotic catastrophe and breakdown of gut immune homeostasis. Cell Rep., 14, 1062–1073. [DOI] [PubMed] [Google Scholar]
- 57. Jiang Y., de Bruin A., Caldas H., Fangusaro J., Hayes J., Conway E.M., Robinson M.L., Altura R.A. (2005) Essential role for survivin in early brain development. J. Neurosci., 25, 6962–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aboualaiwi W.A., Muntean B.S., Ratnam S., Joe B., Liu L., Booth R.L., Rodriguez I., Herbert B.S., Bacallao R.L., Fruttiger M.. et al. (2014) Survivin-induced abnormal ploidy contributes to cystic kidney and aneurysm formation. Circulation, 129, 660–672. 10.1161/CIRCULATIONAHA.113.005746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hagemann S., Wohlschlaeger J., Bertram S., Levkau B., Musacchio A., Conway E.M., Moellmann D., Kneiseler G., Pless-Petig G., Lorenz K.. et al. (2013) Loss of Survivin influences liver regeneration and is associated with impaired Aurora B function. Cell Death Differ., 20, 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uren A.G., Wong L., Pakusch M., Fowler K.J., Burrows F.J., Vaux D.L., Choo K.H. (2000) Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr. Biol., 10, 1319–1328. [DOI] [PubMed] [Google Scholar]
- 61. Zumbragel F.K., Machtens D.A., Curth U., Luder C.G., Reubold T.F., Eschenburg S. (2017) Survivin does not influence the anti-apoptotic action of XIAP on caspase-9. Biochem. Biophys. Res. Commun., 482, 530–535. [DOI] [PubMed] [Google Scholar]
- 62. Lee P.J., Rudenko D., Kuliszewski M.A., Liao C., Kabir M.G., Connelly K.A., Leong-Poi H. (2014) Survivin gene therapy attenuates left ventricular systolic dysfunction in doxorubicin cardiomyopathy by reducing apoptosis and fibrosis. Cardiovasc. Res., 101, 423–433. [DOI] [PubMed] [Google Scholar]
- 63. Gourdie R.G., Dimmeler S., Kohl P. (2016) Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat. Rev. Drug Discov., 15, 620–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Talman V., Ruskoaho H. (2016) Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res., 365, 563–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vousden K.H. (2000) p53: death star. Cell, 103, 691–694. 10.1016/S0092-8674(00)00171-9 [DOI] [PubMed] [Google Scholar]
- 66. Hoffman W.H., Biade S., Zilfou J.T., Chen J., Murphy M. (2002) Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem., 277, 3247–3257. [DOI] [PubMed] [Google Scholar]
- 67. Armstrong J.L., Veal G.J., Redfern C.P., Lovat P.E. (2007) Role of Noxa in p53-independent fenretinide-induced apoptosis of neuroectodermal tumours. Apoptosis, 12, 613–622. [DOI] [PubMed] [Google Scholar]
- 68. Nickson P., Toth A., Erhardt P. (2007) PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress. Cardiovasc. Res., 73, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hua Y., Vickers T.A., Okunola H.L., Bennett C.F., Krainer A.R. (2008) Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet., 82, 834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hua Y., Vickers T.A., Baker B.F., Bennett C.F., Krainer A.R. (2007) Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol., 5, e73.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Louch W.E., Sheehan K.A., Wolska B.M. (2011) Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell. Cardiol., 51, 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.