

Abstract
Hereditary hemorrhagic telangiectasia (HHT) is a highly debilitating and life-threatening genetic vascular disorder arising from endothelial cell (EC) proliferation and hypervascularization, for which no cure exists. Because HHT is caused by loss-of-function mutations in bone morphogenetic protein 9 (BMP9)-ALK1-Smad1/5/8 signaling, interventions aimed at activating this pathway are of therapeutic value. We interrogated the whole-transcriptome in human umbilical vein ECs (HUVECs) and found that ALK1 signaling inhibition was associated with a specific pro-angiogenic gene expression signature, which included a significant elevation of DLL4 expression. By screening the NIH clinical collections of FDA-approved drugs, we identified tacrolimus (FK-506) as the most potent activator of ALK1 signaling in BMP9-challenged C2C12 reporter cells. In HUVECs, tacrolimus activated Smad1/5/8 and opposed the pro-angiogenic gene expression signature associated with ALK1 loss-of-function, by notably reducing Dll4 expression. In these cells, tacrolimus also inhibited Akt and p38 stimulation by vascular endothelial growth factor, a major driver of angiogenesis. In the BMP9/10-immunodepleted postnatal retina—a mouse model of HHT vascular pathology—tacrolimus activated endothelial Smad1/5/8 and prevented the Dll4 overexpression and hypervascularization associated with this model. Finally, tacrolimus stimulated Smad1/5/8 signaling in C2C12 cells expressing BMP9-unresponsive ALK1 HHT mutants and in HHT patient blood outgrowth ECs. Tacrolimus repurposing has therefore therapeutic potential in HHT.
Introduction
Hereditary hemorrhagic telangiectasia (HHT or Rendu–Osler–Weber syndrome) is an autosomal dominant genetic disease affecting ∼1 in 5000 individuals (1,2). The clinical presentation of HHT includes potentially hemorrhagic vascular anomalies in multiple tissues and organs in the form of arteriovenous malformations (AVMs) and mucocutaneous telangiectasias. The systemic manifestations of HHT make patient management challenging and can lead to highly debilitating and life-threatening hemorrhagic events and secondary cerebral, hepatic, pulmonary and cardiac complications (3,4).
Mutations in the genes ENG (encoding endoglin) or ACVRL1 (activin receptor-like kinase 1, ALK1) are the main cause of HHT and define the two disease subtypes: HHT1 (OMIM #187300) and HHT2 (#600376), respectively (5,6). Mutations in SMAD4 [encoding Smad4; Ref. (7)] and GDF2 [bone morphogenetic protein 9, BMP9; Ref. (8)] cause rare forms of the disease called juvenile polyposis/HHT combined syndrome (OMIM #175050) and HHT-like vascular anomaly syndrome (#615506), respectively. BMP9, ALK1, endoglin and Smad4 functionally interact in the same signaling pathway (9). ALK1 is a BMP type I receptor of the transforming growth factor-β (TGF-β) superfamily, which forms complexes with a BMP type II receptor (e.g. BMPR2) and the co-receptor endoglin. Of note, mutations in BMPR2 cause familial pulmonary arterial hypertension (PAH), a separate clinical entity that is observed in some HHT2 patients (10). ALK1 receptors are abundantly expressed by endothelial cells [ECs; Ref. (11)] and are specifically activated by the circulating ligands BMP9 and BMP10 (12–15). Once activated, ALK1 receptors phosphorylate the signal transducers Smad1, 5 and 8 to trigger the formation of Smad1/5/8–Smad4 complexes. Smad1/5/8–Smad4 complexes then translocate to the nucleus to control specific gene expression programs (16–18).
HHT mutations cause a loss-of-function in the ALK1 signaling pathway. Indeed, HHT-causing mutations in ALK1 or endoglin block Smad1/5/8 signaling by BMP9 (19–22). Studies in mouse and zebrafish models further revealed that ALK1 signaling inactivation leads to robust vascular defects that include vascular hyperproliferation and AVMs (23–27). The exact cellular processes leading to AVM development in HHT, i.e. the formation of direct shunts between arteries and veins, remain poorly understood. Solid evidence suggests, however, that HHT is caused by abnormal reactivation of angiogenesis (28,29) and that inhibition of the pro-angiogenic guidance cue, vascular endothelial growth factor (VEGF), might reduce the pathology in HHT mouse models and HHT patients (28,29), but see also (30,31).
During angiogenesis, ECs engage in specific gene expression programs enabling them to migrate and proliferate to expand a vascular sprout and ultimately form a new vascular bed structured around arteries connecting with veins via the capillaries (32). Angiogenic sprouting is initiated by the activation of VEGF receptor 2 (VEGFR2) by VEGF in a specific subset of ECs. Communication between ECs during angiogenesis is primarily controlled by Dll4/Notch signaling, where VEGFR2 activation triggers Dll4 expression (33). ALK1 receptors interact with Notch signaling during angiogenesis by activating Notch transcriptional targets and by supporting sprouting angiogenesis and EC specification (24–26,34,35), but see also (36). Thus, ALK1 is a key regulator of VEGF/Dll4/Notch signaling and EC functions during angiogenesis and vascular maintenance. It is therefore not surprising that ALK1 loss-of-function in HHT is sufficient to cause significant defects in EC integrity that ultimately lead to vascular pathology.
In this study, by using RNA-Seq analyses in human umbilical vein ECs (HUVECs), we show that ALK1 signaling inhibition was associated with a specific transcriptional signature that significantly increased the gene expression of several key pro-angiogenic markers, such as Dll4. By screening ∼700 FDA-approved drugs, we identified tacrolimus as a potent activator of endothelial Smad1/5/8 signaling. We found that tacrolimus significantly reversed the transcriptional signature associated with ALK1 loss-of-function, by notably opposing the gene expression upregulation of the identified pro-angiogenic markers, including Dll4. In addition, tacrolimus reduced the vascular pathology of an HHT mouse model and potently activated Smad1/5/8 signaling in HHT patient-derived primary ECs. These data show that tacrolimus has the potential to block HHT pathology and restore EC homeostasis by rescuing the signaling and gene expression control defects caused by ALK1 loss-of-function.
Results
ALK1 inhibition controls a pro-angiogenic gene expression response
To gain insights into the global transcriptional deregulations caused by ALK1 signaling inhibition, we profiled gene expression by RNA-Seq in HUVECs treated with ALK1 extracellular domain-derived ligand trap (ALK1-Fc). Incubation with ALK1-Fc for 24 h led to an inhibition of ALK1 signaling, which manifested by a decrease of Smad1/5/8 phosphorylation (pSmad1/5/8) and ID1 (inhibitor of differentiation 1) expression (Fig. 1A, inset). ALK1-Fc treatment significantly changed the expression of 251 genes (n = 6 biological replicates per group, Limma Voom test controlling for sample batches, false discovery rate (FDR) ≤ 0.01; Fig. 1A and B;Supplementary Material, Table S1). Among the up-regulated genes, several had previously been described as pro-angiogenic and/or endothelial tip cell markers (33,37–40), including DLL4, ANGPT2, KDR, CXCR4, PGF and SMOC1 (Fig. 1A and B;Supplementary Material, Table S1). Dll4 is of particular interest because of its central role in angiogenesis (33). We confirmed at the protein level that ALK1 inhibition by ALK1-Fc robustly and significantly increased Dll4 expression by HUVECs (Fig. 2A and B). Importantly, treatment with the physiological activating ALK1 ligand, BMP9, led to the opposite effect, by significantly reducing Dll4 expression (Fig. 2A and B). These whole-transcriptome and protein expression analyses are consistent with the concept that ALK1 inhibition is associated with a pro-angiogenic gene expression program.
Figure 1.

Tacrolimus mimics ALK1-mediated gene expression control. (A) RNA-Seq heat map of gene expression changes in HUVECs treated or not (CTRL) for 24 h in complete medium (conditioned for 2 days) with ALK1-Fc (1 μg/ml) or tacrolimus (FK-506, 0.3 μM) (n = 6). Inset, cell extracts were analyzed by western blotting (WB) using antibodies directed against the indicated proteins. (B, C) RNA-Seq MA-plots displaying all differentially expressed transcripts in ALK1-Fc-treated (B) and FK-506-treated (C) HUVECs. Genes with a FDR < 0.01 are shown in red. Genes unchanged by treatment are expected to lie on the horizontal dashed line at log2 fold change = 0. Low expression values reflect experimental measurement noise. Genes discussed in the text were annotated on the plots. (D, E) RNA-Seq scatter plots comparing gene expression changes (log fold change) between ALK1-Fc and FK-506 treatments for transcripts differentially expressed by either treatment (FDR < 0.01, D) and for all transcripts (E). (F) RNA-Seq UpSet plot showing the size of set intersections for transcripts differentially expressed by ALK1-Fc or FK-506 treatments. Bars indicate how many transcripts were differentially expressed in each labelled condition. Lines connect dots to indicate set intersection. Specifically, 572 + 187 transcripts were differentially expressed upon FK-506 treatment, of which 187 were also differentially expressed when HUVECs were treated with ALK1-Fc. 64 genes were differentially expressed following ALK1-Fc treatment, but not in the other condition.
Figure 2.

ALK1 signaling controls Dll4 expression. (A) HUVECs were treated or not (CTRL) for 24 h in complete medium (conditioned for 2 days) with ALK1-Fc (1 μg/ml) or BMP9 (10 ng/ml). Cell extracts were analyzed by WB using antibodies directed against the indicated proteins. (B) Densitometric analyses and quantification of Dll4, pSmad1/5/8, and ID1 relative levels in n = 4 independent experiments, as in (A). Data represent mean ± SEM; **P < 0.01, ***P < 0.001, ****P < 0.0001; one-way ANOVA, Dunnett's multiple comparisons test.
Tacrolimus is a potent Smad1/5/8 signaling activator
We next screened the NIH clinical collections (NCCs) of ∼700 FDA-approved drugs for their potential to activate Smad1/5/8 signaling in reporter C2C12 myoblast cells expressing luciferase under the control of an Id1 promoter response element [C2C12BRA cells; Ref. (41)]. This approach previously identified BMPR2 signaling activators in BMP4-challenged reporter cells (42). Here, we asked whether we could identify drugs that enhance BMP9-specific Smad1/5/8 signaling. To this end, we screened the NCC drugs in C2C12BRA cells maintained in medium depleted of exogenous growth factors and supplemented with a half-maximal effective concentration (EC50) of BMP9. We determined that 0.5 ng/ml was the EC50 of BMP9 in C2C12BRA cells when the cells were challenged for 24 h in depleted medium [0.1% fetal bovine serum (FBS), Supplementary Material, Fig. S1A]. NCC drugs were thus screened using C2C12BRA cells maintained in depleted medium supplemented with 0.5 ng/ml BMP9 for 24 h. The most potent activating drug was tacrolimus (FK-506; fold change = 3.4, Z score = 9.7; Fig. 3A). The effect of tacrolimus in C2C12BRA cells was confirmed using another commercial source of the drug, and the EC50 of tacrolimus was determined to be of 37 nM in depleted medium supplemented with BMP9 at EC50 (Fig. 3B) and of 85 nM in complete culture medium containing 10% FBS (Supplementary Material, Fig. S1B). At the protein level and in a dose-dependent manner, tacrolimus increased ID1 levels in C2C12 cells (Fig. 3C) and HUVECs (Fig. 3D). Importantly, tacrolimus also efficiently elevated pSmad1/5/8 in both C2C12 cells and HUVECs (Fig. 3C and D). Thus, tacrolimus is a potent trigger of Smad1/5/8 signaling in ECs under conditions of specific challenge with BMP9 and in complete medium containing regular serum exogenous trophic factors. These data are in line with the previously reported activating effect of tacrolimus on BMPR2-Smad1/5/8 signaling in pulmonary ECs (42). Thus, tacrolimus is a potent activator of the BMP9-ALK1-Smad1/5/8-ID1 signaling cascade.
Figure 3.

Identification and validation of tacrolimus as a Smad1/5/8 signaling activator. (A) NCC screen using Id1 transactivation luciferase assay in C2C12BRA cells. Drugs were tested in duplicate at 3 μM on cells maintained for 24 h in depleted medium containing 0.1% FBS and 0.5 ng/ml BMP9 (BMP9 EC50). Data (fold change mean ± range) are ranked by mean score and were normalized to DMSO control. (B) Luciferase activity in C2C12BRA cells treated with different concentrations of tacrolimus (FK-506) in depleted medium supplemented with BMP9 at EC50, as in (A). Data represent mean ± SEM (n = 4); ***P < 0.001, ****P < 0.0001; one-way ANOVA, Bonferroni's multiple comparisons test. (C–E) C2C12 cells (C) and HUVECs (D and E) were treated or not (CTRL) for 24 h in complete medium (conditioned for 2 days) with ALK1-Fc (1 μg/ml, D) or tacrolimus at the indicated concentrations (C and D) or 0.3 μM (E). Cell extracts were analyzed by WB using antibodies directed against the indicated proteins. (F) Densitometric analyses and quantification of Dll4, pSmad1/5/8, and ID1 relative levels in n = 3 independent experiments, as in (E). Data represent mean ± SEM; **P < 0.01, ****P < 0.0001; Student’s t-test. (G) Flow cytometry analysis of surface ALK1 expression in HUVECs treated or not (CTRL) for 24 h in complete medium (conditioned for 2 days) with BMP9 (10 ng/ml) or tacrolimus (0.3 μM).
Tacrolimus rescues the gene expression deregulations associated with ALK1 inhibition
At the whole-transcriptome level, we compared the effect of tacrolimus and ALK1-Fc on gene expression in HUVECs. RNA-Seq revealed that Smad1/5/8 signaling activation by tacrolimus (Fig. 1A, inset) significantly altered the expression of 759 genes (FDR ≤ 0.01, Fig. 1A and C;Supplementary Material, Table S1). Strikingly, 187 of these deregulated genes were also found to be deregulated by ALK1-Fc treatment (Fig. 1F), but in the opposite direction (Fig. 1A–C). Indeed, a significant inverse correlation in fold change (log2) was found for genes either differentially expressed by ALK1-Fc treatment (versus control) or tacrolimus treatment (versus control) (FDR < 0.01 either condition, Fig. 1D). The same comparison between all genes did not show a similar correlation (Fig. 1E), validating the specificity of the observed effect. When restricting this intersection to the 187 genes differentially expressed in both conditions (changed by both ALK1-Fc and tacrolimus treatments), we identified DLL4, ANGPT2, KDR, CXCR4, PGF and SMOC1 (Fig. 1A–C and F). In agreement with the effect of BMP9 treatment on Dll4 expression (Fig. 2), Smad1/5/8 signaling activation by tacrolimus robustly and significantly reduced Dll4 protein expression by HUVECs (Fig. 3E and F).
To provide further evidence that tacrolimus activates Smad1/5/8 signaling and to increase our understanding of the mechanism by which this signaling is activated, we asked whether tacrolimus affects ALK1 trafficking, as TGF-β receptors are internalized upon ligand binding during signal transduction (43). Flow cytometry analyses using an antibody directed against the extracellular region of ALK1 revealed an almost complete loss of the endogenous levels of ALK1 at the cell surface upon BMP9 treatment in HUVECs (Fig. 3G), suggesting that ALK1, like TGF-β receptors, might internalize upon receptor activation. Interestingly, tacrolimus, at a concentration activating Smad1/5/8, also caused a decrease of surface ALK1 (Fig. 3G), indicating that tacrolimus mimics several important aspects of BMP9 activation and that it acts upstream in the signaling cascade at the level of the receptor.
Tacrolimus inhibits VEGF-mediated Akt and p38 activation
Increased Dll4 expression is a critical transcriptional response to VEGF/VEGFR2 signaling in ECs during angiogenesis (44). We sought to determine whether the repressing effect of ALK1 activation on Dll4 expression could be owing to inhibition of VEGFR2 signal transduction. At least three major kinase pathways are activated in ECs upon VEGFR2 signaling: Akt/PKB, p38 MAPK and ERK1/2. Compelling evidence has highlighted the key role of the PI3K/Akt axis in Dll4 transcriptional activation by VEGF (45,46). We found that pretreatment with BMP9 for 24 h had no effect on Akt, p38 or ERK1/2 phosphorylation upon VEGF stimulation in HUVECs, whereas BMP9 efficiently activated ALK1 signaling by increasing pSmad1/5/8 and ID1 levels under these conditions (Fig. 4A and B). Surprisingly, tacrolimus treatment of HUVECs for 24 h prior to VEGF stimulation resulted in a complete inhibition of Akt phosphorylation (both at Thr-308 and Ser-473), and in a partial but significant reduction in p38 phosphorylation (Fig. 4A and B). The effect of tacrolimus was specific towards Akt and p38 because the drug did not affect VEGF-mediated ERK1/2 phosphorylation (Fig. 4A and B). Together, these data show that ALK1 signaling can control Dll4 expression independent of VEGF signaling. Therefore, the potent repressing effect of tacrolimus on Dll4 expression is likely to be owing to a dual and independent control of the Smad1/5/8 and VEGF pathways (see model in Fig. 4C).
Figure 4.

Tacrolimus inhibits VEGF-mediated Akt and p38 activation. (A) HUVECs treated or not (CTRL) for 24 h (in 0.05% FBS medium) with BMP9 (10 ng/ml) or tacrolimus (FK-506, 0.3 μM), were stimulated for 5 min with VEGF (25 ng/ml). Cell extracts were analyzed by WB using antibodies directed against the indicated proteins. (B) Densitometric analyses and quantification of the relative levels of the indicated proteins in n = 3 independent experiments, as in (A). Data represent mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; one-way ANOVA, Dunnett's and Sidak’s multiple comparisons tests. (C) Schematic illustration of the proposed effect of tacrolimus on ALK1 and VEGFR2 signaling pathways.
Tacrolimus improves vascular pathology in the BMP9/10-immunoblocked retina
Recently, we described a new HHT mouse model, which has the advantage of being practical, non-invasive and robust, as well as particularly suitable and reliable for the analysis of large cohorts (47). In this model, we showed that transmammary delivery of BMP9 and BMP10 blocking antibodies to nursing mouse pups led to the inhibition of ALK1 signaling and development of an HHT-like pathology in their postnatal retinal vasculature. This pathology included the presence of robust hypervascularization and AVM development (47). Using this model, we assessed the in vivo potential of tacrolimus to modulate Smad1/5/8 signaling and HHT vascular pathology. Pathology in the newborn retina was triggered by one intraperitoneal (i.p.) injection of anti-BMP9/10 antibodies in lactating dams on postnatal day 3 (P3), as reported before (47). In parallel, pups were treated with tacrolimus (0.5 mg/kg/d, i.p.) from P3 until they were euthanized on P6 to analyze their retinal vasculature. Tacrolimus slightly but significantly decreased both the number of AVMs and their diameter (Fig. 5A and B). More strikingly, tacrolimus significantly reduced vein dilation (Fig. 5C–I) and the density of the vascular plexus by preventing the appearance of the hyperbranched phenotype caused by BMP9/10 immunoblocking (Fig. 5J–L). We quantified the surface area occupied by the retinal vasculature at the capillary plexus and found that tacrolimus treatment significantly reduced the abnormal vascular density caused by BMP9/10 immunoblocking (Fig. 5M).
Figure 5.

Tacrolimus improves vascular pathology in the BMP9/10-immunoblocked retina. (A and B) AVM number (A) and AVM diameter (B) in the retinal vasculature of P6 pups treated via the transmammary route with BMP9 and BMP10 blocking antibodies [see Methods and Ref. (47)], and treated with tacrolimus (FK-506, 0.5 mg/kg/d) or vehicle (DMSO). Data represent mean ± SEM per retina (n = 10–20 pups per group); *P < 0.05; Student’s t-test (A) and Mann Whitney U test (B). (C–H) and (J–L) Representative images of retinas stained with fluorescent isolectin B4 from pups injected or not (DMSO) with tacrolimus (FK-506, 0.5 mg/kg/d), and treated via the transmammary route with control IgG2a/b (C, F and J) or BMP9/10 blocking antibodies (D, E, G, H, K and L). Higher magnifications in (F–H and J–L) show retinal vein diameter (F–H, see yellow arrowheads) and retinal vasculature fields (plexus area) between an artery (a) and a vein (v) (J–L). Arrows in (D, G and K) denote AVMs. (I, M) Scatter plots measuring retinal vein diameter (I) and retinal vascular plexus density (M) in pups treated as in (C–E). Data represent mean ± SEM (n = 6–8); *P<0.05, **P<0.01, ****P<0.0001; one-way ANOVA, Tukey’s multiple comparisons test. (N–S) Histochemistry analysis of the vascular front of P6 retinas treated as in (C–H) and stained with fluorescent isolectin B4 (N–P, green) and anti-Dll4 antibody (N–S, red). (T) Retinal ECs isolated with anti-CD31 microbeads from pups treated as in (A) were analyzed for Id1 mRNA levels by RT-qPCR. The results are expressed as relative levels of the control condition (n = 3 determinations). (U) Quantification of Dll4 levels in 3 experiments as in (R and S). Data in (T and U) represent mean ± SEM (n = 3–5); *P < 0.05; Student’s t-test (T) and Mann–Whitney U test (U). Scale bars, 500 μm (C–E), 100 μm (J–L), 30 μm (N–S).
In agreement with the in vitro data using ALK1-Fc in HUVECs (Fig. 2), we further found that ALK1 signaling interference via BMP9/10 immunoblocking in mouse pups caused a pronounced increase in Dll4 expression in vivo in their retinal ECs at the developing vascular front (Fig. 5N, O, Q and R). Importantly, in retinal ECs, tacrolimus treatment not only increased Smad1/5/8 signaling by elevating Id1 gene expression (Fig. 5T), but also prevented the Dll4 overexpression caused by BMP9/10 immunoblocking (Fig. 5N–S and U). In summary, tacrolimus activated Smad1/5/8 signaling and reduced Dll4 overexpression in vivo in ECs, and reduced HHT pathology in a mouse model of ALK1 loss-of-function.
Tacrolimus activates Smad1/5/8 in cells expressing ALK1 HHT mutants
The in vivo data obtained in the BMP9/10-immunoblocked retina show that tacrolimus activates endothelial Smad1/5/8 signaling even in the presence of strong ALK1 blockade. In cell cultures, we further found that, although ALK1-Fc treatment reduced the effect of tacrolimus on Smad1/5/8 signaling, it still allowed the drug to strongly increase luciferase activity in C2C12BRA cells (Supplementary Material, Fig. S1B) and pSmad1/5/8 and ID1 levels in HUVECs (Fig. 3D). These results strongly support the concept that tacrolimus is able to bypass a partial, or even complete, ALK1 loss-of-function caused by mutations in HHT patients. To address this possibility, we investigated the effect of tacrolimus on Smad1/5/8 activation in C2C12 cells transfected with two inactive HHT ALK1 mutants, R411P and T372fsX (20,48,49). We found that over-expression of R411P-ALK1 (Fig. 6A) or T372fsX-ALK1 (Fig. 6B) resulted in a nearly complete inhibition of Smad1/5/8 activation by BMP9, when compared with cells overexpressing comparable levels of wild-type (WT) ALK1, in which BMP9, as expected, triggered a robust increase of pSmad1/5/8 (Fig. 6A and B). In contrast, tacrolimus treatment efficiently activated Smad1/5/8 in cells expressing either WT ALK1 or the ALK1 HHT mutants (Fig. 6A and B). To extend our observations and demonstrate efficacy using a cell system more directly relevant to the pathophysiology of human HHT, we generated blood outgrowth ECs (BOECs) from circulating endothelial progenitors in peripheral blood of an HHT patient genetically confirmed to carry the ALK1 T372fsX truncation (Fig. 6C). Strikingly, in these cells, tacrolimus robustly increased pSmad1/5/8 and ID1 levels, showing that tacrolimus promoted Smad1/5/8 signaling in HHT patient ECs (Fig. 6D).
Figure 6.
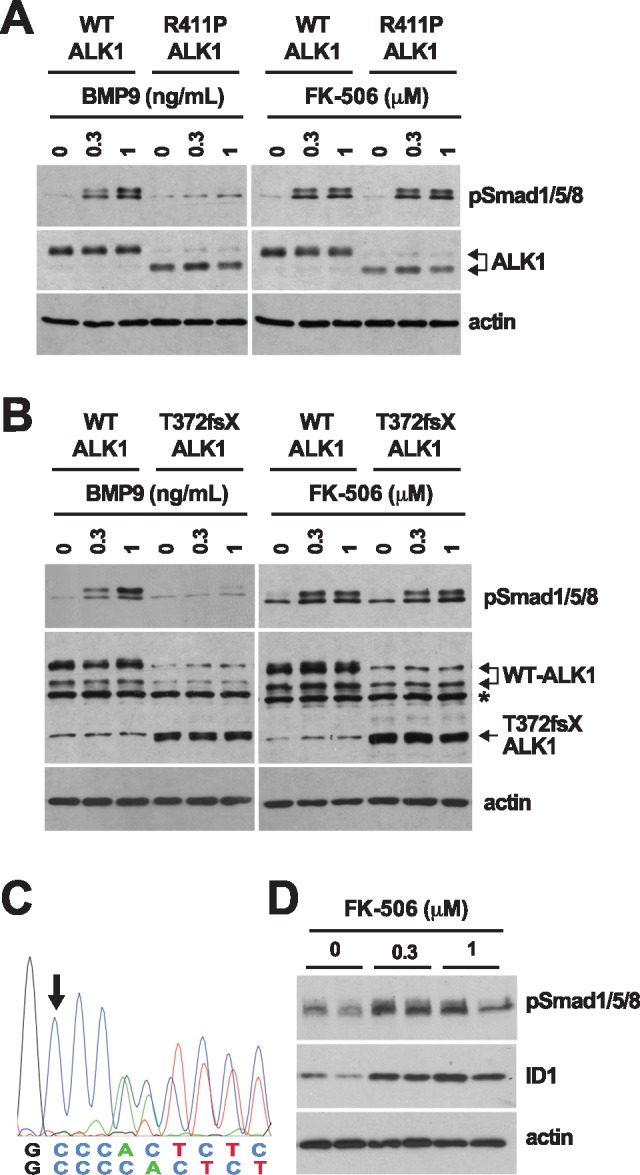
Tacrolimus activates Smad1/5/8 in cells expressing ALK1 HHT mutants. (A, B) C2C12 cells transfected with human WT ALK1 or ALK1 HHT mutants R411P (A) and T372fsX (B) were serum-starved for 3 h (0.1% FBS medium) and then treated for 3 h with the indicated concentrations of BMP9 or tacrolimus (FK-506). Please note that two immunoreactive bands were detected for WT ALK1, and that the R411P mutation increased the abundance of the low molecular weight band. Asterisk in (B) denotes a nonspecific band. (C) Partial genomic DNA sequences of the HHT patient ACVRL1 gene. Arrow indicates the c.1112dup insertion (T372fsX mutation) in the mutated allele. (D) HHT BOECs were treated with the indicated concentrations of tacrolimus for 24 h in depleted medium (0.1% FBS medium). Cell extracts in (A, B and D) were analyzed by WB using antibodies directed against the indicated proteins.
Discussion
Whole-transcriptome analyses in primary ECs revealed that tacrolimus is a potent ALK1 signaling mimetic at the transcriptional level. Specifically, we report that tacrolimus treatment almost diametrically opposed the transcriptional deregulation caused by ALK1 inhibition by significantly changing in the reverse direction, the expression of 187 genes among the 251 found to be deregulated in the ALK1 loss-of-function transcriptional response (Fig. 1). Notably, ALK1 loss-of-function transcriptional response was associated with expression elevations of several key pro-angiogenic regulators, such as DLL4, ANGPT2, KDR, CXCR4, PGF and SMOC1. Previous studies have demonstrated that BMP9/10-ALK1 signaling is required for the maintenance of vascular quiescence (50–52). Disruption of this quiescence mechanism is believed to facilitate the reactivation of angiogenesis and cause the appearance of abnormal vessels in HHT. In adult inducible Alk1 and Eng knockout mice, for instance, wound-induced AVMs were reported to originate from angiogenesis-dependent vascular elongation (53,54). In addition, extensive and concordant evidence from our laboratory (47) and others (26,50,55–59) has demonstrated using different models that the expression of ANGPT2, KDR and CXCR4 is negatively controlled by ALK1 signaling. The control of DLL4 expression by ALK1 signaling has also been reported in an alk1 mutant zebrafish model (60) and in BMP9/10-treated pulmonary arterial ECs (24). In line with these studies, we found clear evidence that, at steady-state, ALK1 inhibition in HUVECs increased Dll4 expression at the transcript and protein levels. We further show that Smad1/5/8 signaling activation with tacrolimus or BMP9 had the opposite effect by repressing Dll4 expression (Figs 2 and3). Together, these results further highlight the central role of ALK1 in the transcriptional regulation of angiogenesis and thus, fully support the concept that ALK1 loss-of-function and HHT pathogenesis are associated with deregulated activation of angiogenesis.
Because VEGF signaling is critical for Dll4 expression during angiogenesis, we asked whether tacrolimus impacts VEGF signaling activation. Our data in primary ECs showed that tacrolimus is a potent inhibitor of VEGF-mediated activation of Akt and p38 (Fig. 4). These results are in line with previous work showing that tacrolimus could interfere with VEGF-mediated EC tube formation in vitro (61). Because BMP9 treatment failed to recapitulate the repressing effect of tacrolimus on VEGF-mediated Akt/p38 activation, we concluded that the drug controlled VEGF signaling independent from its effect on ALK1 (Fig. 4C). We therefore postulate that tacrolimus lowered Dll4 expression via a dual control of ALK1 and VEGF signaling, two pathways critically, but independently, involved in Dll4 expression regulation. Although these data strengthen the concept that ALK1 cross talks with Notch signaling during angiogenesis, it is worth mentioning that studies performed in zebrafish have demonstrated that ALK1 can also regulate vascular quiescence and AVM development via a mechanism independent of Notch (60,62). It will therefore be important going forward to determine the exact contributions of the different Dll4/Notch signaling pathways in AVM development in ALK1 loss-of-function models.
A recent study has demonstrated that BMP9/ALK1 blockade was associated with PI3K/Akt overactivation and that PI3K inhibition prevented HHT vascular pathology in mouse models (63). In this context, our data suggest that tacrolimus treatment might also be beneficial by mitigating the aberrant Akt activation observed in HHT models. It is interesting to note that the authors of this study (63) also showed that, although ALK1 inhibition enhanced VEGF-mediated angiogenesis, its effect on Akt overactivation was, at least in part, not dependent on VEGF signaling. It appears therefore that ALK1 might control Akt signaling in ECs via several independent pathways. Future studies will be needed to assess the specific contributions of these different Akt pathways in the aberrant angiogenic process of HHT pathology and to determine whether tacrolimus affects their signaling responses differently.
An important question relates to the mechanism by which tacrolimus activates Smad1/5/8 signaling. FK-506-binding protein-12 (FKBP12) is known to interact with TGF-β type I family receptors within their receptor glycine-serine-rich phosphorylation domain to mute receptor activation in the absence of ligand (64,65). More recently, compelling data showed that tacrolimus can activate BMPR2 and Smad1/5/8 signaling in pulmonary EC by blocking the inhibitory binding of FKBP12 to ALK1, but also to ALK2 and ALK3 (42), two other BMP type I receptors that are widely expressed (9). In this study, we confirmed that tacrolimus intervened upstream at the level of the receptor by activating Smad1/5/8 and by promoting a decrease of surface ALK1 in primary ECs, demonstrating that upon tacrolimus treatment, ALK1 responded to receptor activation through internalization.
Our data further demonstrated that tacrolimus was capable of potently activating Smad1/5/8 in cells overexpressing R411P- and T372fsX-ALK1, two well-described ALK1 HHT mutations that rendered the cells unresponsive to BMP9 (Fig. 6). These results are important because it was proposed that HHT pathology might arise from complete ALK1 loss-of-function (66). A drug with therapeutic potential is thus expected to activate Smad1/5/8 signaling in the presence of non-functional ALK1 receptors. In line with these results using transfected cells, we found that tacrolimus also activated Smad1/5/8 in BOECs isolated from an HHT patient carrying the ALK1 T372fsX truncation. These results not only show that tacrolimus activates Smad1/5/8 signaling in the presence of defective ALK1 receptor and in HHT patient-derived ECs, but also are consistent with the proposed model that tacrolimus can control Smad1/5/8 signaling in ECs by bypassing ALK1 loss-of-function through the parallel activation of ALK2 and ALK3, for instance (42).
Treatment of newborn mice with BMP9/10 blocking antibodies leads to the development of a specific pathology in the developing retinal vasculature, which includes abnormal hypervascularization (24,25), but also AVMs when ALK1 signaling is inhibited at a vascular developmental stage that allows a normal blood flow (47,50–52). Recently, we reported that antibody delivery and BMP9/10 immunoneutralization in mouse pups could be achieved via the transmammary route by injecting lactating females. We further showed that this model is more practical because it is non-invasive, robust, and particularly suitable and reliable for the analysis of large cohorts (47). Tacrolimus administration in this model elicited a robust reduction in retinal hypervascularization and a small but significant decrease in AVM numbers and diameter (Fig. 5). Strikingly, tacrolimus also potently increased Id1 levels in retinal ECs, while reducing the elevation of Dll4 levels observed in the BMP9/10-immunoblocked retinas. Immunohistochemistry analyses suggested that the Dll4 elevation triggered by BMP9/10 immunoblocking was restricted to the ECs located at the developing vascular front. Knowing that Dll4 is a tip cell marker (33), it is tempting to speculate that tacrolimus, by activating Smad1/5/8 signaling in this model, has anti-angiogenic properties that limits a pro-tip cell phenotype required for the hypervascularization observed in HHT models. Further investigation will be required to test this hypothesis and delineate the exact effect of tacrolimus on EC specification deregulations in HHT.
Finally, our data concur with previous studies that provide indirect evidence supporting the potential beneficial effect of tacrolimus in HHT patients. Indeed, a Canadian case study reported the regression of angiodysplasia and reduction of mucosal hemorrhage in a probable HHT patient who underwent liver transplantation following high-output cardiac failure and hepatic AVM development (67). The authors proposed that the anti-rejection regimen, which included tacrolimus, contributed to the observed beneficial effects in this HHT patient. In addition, the potential clinical benefit of tacrolimus in patients with end-stage PAH has been reported (68).
In conclusion, we show in this study that tacrolimus is a potent activator of Smad1/5/8 signaling in cell and mouse models of ALK1 loss-of-function, including in HHT patient-derived primary ECs. Consequently, tacrolimus treatment corrected the signaling and gene expression defects caused by endothelial ALK1 inhibition and significantly improved HHT vascular pathology in mice. We propose that tacrolimus has therapeutic potential in HHT.
Materials and Methods
Cell cultures and transfections
HUVECs were isolated from anonymous umbilical veins and subcultured in 5% FBS-containing EC growth medium (Sciencell), as described before (47,69). C2C12 cells were obtained from ATCC. C2C12 cells expressing pNeo-(BRE)2-Luc reporter plasmid (C2C12BRA) were kindly provided by Dr Daniel Rifkin (41). C2C12 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS, penicillin and streptomycin. Cells were transiently transfected with pcDNA3 plasmids encoding C-terminally HA-tagged WT ALK1 or ALK1 HHT mutants T372fsX (also annotated G371fsX391) and R411P [kindly provided by Dr. Sabine Bailly (20)], using Lipofectamine 2000 reagent (Invitrogen) and as per the manufacturer’s instructions.
Human BOECs isolation and characterization
BOECs were isolated from blood draws obtained from an HHT2 patient carrying the ALK1 T372fsX truncation [ACVRL1 c.1112dup mutation, first reported in Ref. (49)]. Cells were isolated as described before (70). Flow cytometry was performed to characterize the generated BOECs (data not shown), using the surface markers: VEGFR2 and CD31 (endothelial), and CD45 (hematopoietic, negative control), following a procedure described in (70). BOEC genomic DNA was isolated using DNeasy blood and tissue kit (Qiagen), as per the manufacturer’s recommendations. ACVRL1 exon 8 partial sequence was amplified by PCR using forward primer ACTCACAGGGCAGCGATTAC and reverse primer CCAAAGGCCCAGATGTCAGT. The generated 142 pb PCR product was then sequenced using reverse primer AAAGGCCCAGATGTCAGTCC.
RNA extraction and sequencing
HUVECs were rinsed with PBS and processed for RNA extraction using the RNeasy mini kit (Qiagen), according to the manufacturer’s instructions. Total RNA quality was verified using Thermo Scientific NanoDrop and Agilent Bioanalyzer. RNA was processed for RNA-Seq at the Genomics Resources Core Facility, Weill Cornell Medical College, New York, NY, USA. Briefly, cDNA conversion and library preparation were performed using the TrueSeq v2 Illumina library preparation kit, following the manufacturers’ recommended protocols. Samples were multiplexed 6 per lane and sequenced on an Illumina HiSeq 4000 instrument.
RNA-Seq data analysis
Reads were uploaded to the GobyWeb system (71) and aligned to the 1000 genome human reference sequence (72) with the STAR aligner (73). Ensembl annotations for transcripts were automatically obtained from Biomart and Ensembl using GobyWeb. Annotations were used to determine read counts using the Goby alignment-to-annotation-counts mode (74), integrated in GobyWeb in the differential expression analysis with EdgeR plugin. Counts were downloaded from GobyWeb as a tab delimited file and analyzed with MetaR (75). Statistical analyses were conducted with Limma Voom (76), as integrated in MetaR 1.7.2, using the rocker-metar docker image version 1.6.0.1. P-values were adjusted for multiple testing using the FDR method (77). Heat maps were constructed with MetaR, using the pheatmap R package. Gene annotations were determined with Ensembl/Biomart, using the biomart micro-language in MetaR (75). MetaR analysis scripts are presented in Supplementary Material, Figures S2–S4. UpSet plot were generated with MetaR and the UpSet R package (78).
Drug screening and luciferase assays
BMP9 EC50 was determined in C2C12BRA cells following treatments with different concentrations of recombinant BMP9 for 24 h in depleted medium containing 0.1% FBS. FDA-approved drug libraries (NIH clinical collections, NCCs) were screened at 3 μM on C2C12BRA cells maintained for 24 h in depleted medium containing 0.1% FBS and 0.5 ng/ml BMP9 (BMP9 EC50). Cells were solubilized in Luciferase Cell Culture Lysis 5× Reagent (E1531, Promega) and processed for luciferase measurements using Luciferase Assay System (E1501, Promega), following the manufacturer’s instructions and using Victor3 1420 Multilabel counter luminometer (PerkinElmer).
Mice
Timed-pregnant C57BL/6 J mice (3- to 4-month-old, The Jackson Laboratory) were used in this study.
Transmammary-delivered immunoblocking of BMP9 and BMP10, tacrolimus treatments, and retinal vasculature analyses in mice
Antibody injections and retinal whole-mount histochemistry were performed in mice as previously described (47). Briefly, lactating dams were injected i.p. once on P3 with mouse monoclonal isotype control antibodies (15 mg/kg, IgG2b, MAB004; 15 mg/kg, IgG2a, MAB003; R&D Systems) or mouse monoclonal anti-BMP9 and anti-BMP10 antibodies (15 mg/kg, IgG2b, MAB3209; 15 mg/kg, IgG2a, MAB2926; R&D Systems, respectively). On P3, P4, and P5, pups were injected i.p. with tacrolimus (FK-506, 0.5 mg/kg) or vehicle (1% DMSO saline). Pups were euthanized on P6 by CO2 asphyxiation and were enucleated. Eyes were fixed in 4% paraformaldehyde for 20 min on ice and retinas were isolated and analyzed by histochemistry, as before (47), see also Supplementary Material, Methods. Images for the analysis of the vascular network density were acquired using a laser confocal microscopy Olympus FV300. Quantifications were performed using ImageJ. Using a 20× lens, images (two to five fields per retina) were acquired at the vascular plexus (between an artery and a vein). Quantification was done by using the measure particles tool, working with 8-bit images, adjusting the threshold, and measuring the area occupied by the vasculature in a region of interest of 200× 200 μm2.
Retinal EC isolation and RT-qPCR
ECs were isolated from neonatal retinal tissue using anti-CD31 microbeads (Miltenyi Biotec GmbH), as per the manufacturer’s instructions. One-step RT-qPCR was performed on pellets of 40–60 000 cells per sample using Cells-to-Ct™ 1-Step TaqMan® Kit following the manufacturer’s protocol (Ambion, Thermo Fisher Scientific, Inc.). PCR was performed using TaqMan assays on ABI7900HT (Applied Biosystems, Life Technologies). Id1 expression levels were normalized to the reference gene Polr2a. Relative changes in gene expression were determined by the ΔΔCt method (79) and using control values normalized to 1.0.
Statistics
Figure legends indicate the test used for each experiment to determine whether the differences between the experimental and control groups were statistically significant. A P value <0.05 was considered to be statistically significant. The analyses were performed using GraphPad Prism 7.
Study approvals
Study subject (HHT2 patient carrying the ALK1 T372fsX mutation) provided voluntary and written informed consent using a form approved by the Feinstein Institute for Medical Research Institutional Review Board (IRB). Study subject BOECs were isolated and cultured using a protocol approved by the Institute’s IRB. All animal procedures were performed in accordance with protocols approved by the Feinstein Institute for Medical Research Institutional Animal Care and Use Committee, and conformed to the NIH Guide for the Care and Use of Laboratory Animals and ARRIVE guidelines (80), see Supplementary Material, Methods.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank Dr Daniel Rifkin (NYU School of Medicine, New York, NY) for providing us with C2C12BRA cells and Dr Sabine Bailly (CEA Grenoble, France) for the ALK1 plasmids. We are grateful to Drs LaQueta Hudson and Kevin J. Tracey (The Feinstein Institute for Medical Research) for assistance with the NIH clinical collections. We thank Dr Florencia Palacios (The Feinstein Institute for Medical Research) for assistance with flow cytometry.
Conflict of Interest statement. None declared.
Funding
Feinstein Institute for Medical Research fund (to P.M.).
References
- 1. Guttmacher A.E., Marchuk D.A., White R.I. (1995) Hereditary hemorrhagic telangiectasia. N. Engl. J. Med., 333, 918–924. [DOI] [PubMed] [Google Scholar]
- 2. Richards-Yutz J., Grant K., Chao E.C., Walther S.E., Ganguly A. (2010) Update on molecular diagnosis of hereditary hemorrhagic telangiectasia. Hum. Genet., 128, 61–77. [DOI] [PubMed] [Google Scholar]
- 3. Dupuis-Girod S., Bailly S., Plauchu H. (2010) Hereditary hemorrhagic telangiectasia: from molecular biology to patient care. J. Thromb. Haemost., 8, 1447–1456. [DOI] [PubMed] [Google Scholar]
- 4. Shovlin C.L. (2010) Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev., 24, 203–219. [DOI] [PubMed] [Google Scholar]
- 5. McAllister K.A., Grogg K.M., Johnson D.W., Gallione C.J., Baldwin M.A., Jackson C.E., Helmbold E.A., Markel D.S., McKinnon W.C., Murrel J.. et al. (1994) Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet., 8, 345–351. [DOI] [PubMed] [Google Scholar]
- 6. Johnson D.W., Berg J.N., Baldwin M.A., Gallione C.J., Marondel I., Yoon S.J., Stenzel T.T., Speer M., Pericak-Vance M.A., Diamond A.. et al. (1996) Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet, 13, 189–195. [DOI] [PubMed] [Google Scholar]
- 7. Gallione C.J., Repetto G.M., Legius E., Rustgi A.K., Schelley S.L., Tejpar S., Mitchell G., Drouin E., Westermann C.J., Marchuk D.A. (2004) A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet, 363, 852–859. [DOI] [PubMed] [Google Scholar]
- 8. Wooderchak-Donahue W.L., McDonald J., O’Fallon B., Upton P.D., Li W., Roman B.L., Young S., Plant P., Fülöp G.T., Langa C.. et al. (2013) BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet., 93, 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goumans M.J., Zwijsen A., Ten Dijke P., Bailly S. (2017) Bone morphogenetic proteins in vascular homeostasis and disease. Cold Spring Harb. Perspect. Biol., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Newman J.H., Trembath R.C., Morse J.A., Grunig E., Loyd J.E., Adnot S., Coccolo F., Ventura C., Phillips J.A., Knowles J.A.. et al. (2004) Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J. Am. Coll. Cardiol., 43, 33S–39S. [DOI] [PubMed] [Google Scholar]
- 11. Seki T., Yun J., Oh S.P. (2003) Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ. Res., 93, 682–689. [DOI] [PubMed] [Google Scholar]
- 12. David L., Mallet C., Mazerbourg S., Feige J.J., Bailly S. (2007) Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood, 109, 1953–1961. [DOI] [PubMed] [Google Scholar]
- 13. Scharpfenecker M., van Dinther M., Liu Z., van Bezooijen R.L., Zhao Q., Pukac L., Löwik C.W., ten Dijke P. (2007) BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci., 120, 964–972. [DOI] [PubMed] [Google Scholar]
- 14. David L., Mallet C., Keramidas M., Lamandé N., Gasc J.M., Dupuis-Girod S., Plauchu H., Feige J.J., Bailly S. (2008) Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res., 102, 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown M.A., Zhao Q., Baker K.A., Naik C., Chen C., Pukac L., Singh M., Tsareva T., Parice Y., Mahoney A.. et al. (2005) Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J. Biol. Chem., 280, 25111–25118. [DOI] [PubMed] [Google Scholar]
- 16. Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol., 13, 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cai J., Pardali E., Sánchez-Duffhues G., ten Dijke P. (2012) BMP signaling in vascular diseases. FEBS Lett., 586, 1993–2002. [DOI] [PubMed] [Google Scholar]
- 18. Akhurst R.J., Hata A. (2012) Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov., 11, 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alaa el Din F., Patri S., Thoreau V., Rodriguez-Ballesteros M., Hamade E., Bailly S., Gilbert-Dussardier B., Abou Merhi R., Kitzis A, Buratti E. (2015) Functional and splicing defect analysis of 23 ACVRL1 mutations in a cohort of patients affected by Hereditary Hemorrhagic Telangiectasia. PLoS One, 10, e0132111.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ricard N., Bidart M., Mallet C., Lesca G., Giraud S., Prudent R., Feige J.J., Bailly S. (2010) Functional analysis of the BMP9 response of ALK1 mutants from HHT2 patients: a diagnostic tool for novel ACVRL1 mutations. Blood, 116, 1604–1612. [DOI] [PubMed] [Google Scholar]
- 21. Mallet C., Lamribet K., Giraud S., Dupuis-Girod S., Feige J.J., Bailly S., Tillet E. (2015) Functional analysis of endoglin mutations from hereditary hemorrhagic telangiectasia type 1 patients reveals different mechanisms for endoglin loss of function. Hum. Mol. Genet., 24, 1142–1154. [DOI] [PubMed] [Google Scholar]
- 22. Ruiz-Llorente L., Gallardo-Vara E., Rossi E., Smadja D.M., Botella L.M., Bernabeu C. (2017) Endoglin and alk1 as therapeutic targets for hereditary hemorrhagic telangiectasia. Expert Opin. Ther. Targets, 21, 933–947. [DOI] [PubMed] [Google Scholar]
- 23. Tual-Chalot S., Oh S.P., Arthur H.M. (2015) Mouse models of hereditary hemorrhagic telangiectasia: recent advances and future challenges. Front Genet., 6, 25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ricard N., Ciais D., Levet S., Subileau M., Mallet C., Zimmers T.A., Lee S.J., Bidart M., Feige J.J., Bailly S. (2012) BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood, 119, 6162–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen H., Brady Ridgway J., Sai T., Lai J., Warming S., Chen H., Roose-Girma M., Zhang G., Shou W., Yan M. (2013) Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc. Natl. Acad. Sci. U.S.A., 110, 11887–11892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Larrivée B., Prahst C., Gordon E., del Toro R., Mathivet T., Duarte A., Simons M., Eichmann A. (2012) ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell, 22, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Niessen K., Zhang G., Ridgway J.B., Chen H., Yan M. (2010) ALK1 signaling regulates early postnatal lymphatic vessel development. Blood, 115, 1654–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ardelean D.S., Letarte M. (2015) Anti-angiogenic therapeutic strategies in hereditary hemorrhagic telangiectasia. Front Genet., 6, 35.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thalgott J., Dos-Santos-Luis D., Lebrin F. (2015) Pericytes as targets in hereditary hemorrhagic telangiectasia. Front Genet., 6, 37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dupuis-Girod S., Ambrun A., Decullier E., Fargeton A.-E., Roux A., Bréant V., Colombet B., Rivière S., Cartier C., Lacombe P.. et al. (2016) Effect of bevacizumab nasal spray on epistaxis duration in hereditary hemorrhagic telangectasia. A randomized clinical trial. JAMA, 316, 934–942. [DOI] [PubMed] [Google Scholar]
- 31. Whitehead K.J., Sautter N.B., McWilliams J.P., Chakinala M.M., Merlo C.A., Johnson M.H., James M., Everett E.M., Clancy M.S., Faughnan M.E.. et al. (2016) Effect of topical intranasal therapy on epistaxis frequency in patients with hereditary hemorrhagic telangiectasia. A randomized clinical trial. JAMA, 316, 943–951. [DOI] [PubMed] [Google Scholar]
- 32. Potente M., Gerhardt H., Carmeliet P. (2011) Basic and therapeutic aspects of angiogenesis. Cell, 146, 873–887. [DOI] [PubMed] [Google Scholar]
- 33. Phng L.K., Gerhardt H. (2009) Angiogenesis: a team effort coordinated by notch. Dev. Cell, 16, 196–208. [DOI] [PubMed] [Google Scholar]
- 34. Aspalter I.M., Gordon E., Dubrac A., Ragab A., Narloch J., Vizán P., Geudens I., Collins R.T., Franco C.A., Abrahams C.L.. et al. (2015) Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting downstream of Notch. Nat. Commun., 6, 7264.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moya I.M., Umans L., Maas E., Pereira P.N., Beets K., Francis A., Sents W., Robertson E.J., Mummery C.L., Huylebroeck D.. et al. (2012) Stalk cell phenotype depends on integration of Notch and Smad1/5 signaling cascades. Dev. Cell, 22, 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Meeteren L.A., Thorikay M., Bergqvist S., Pardali E., Stampino C.G., Hu-Lowe D., Goumans M.J., ten Dijke P. (2012) Anti-human activin receptor-like kinase 1 (ALK1) antibody attenuates bone morphogenetic protein 9 (BMP9)-induced ALK1 signaling and interferes with endothelial cell sprouting. J. Biol. Chem., 287, 18551–18561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. del Toro R., Prahst C., Mathivet T., Siegfried G., Kaminker J.S., Larrivee B., Breant C., Duarte A., Takakura N., Fukamizu A.. et al. (2010) Identification and functional analysis of endothelial tip cell-enriched genes. Blood, 116, 4025–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu C., Hasan S.S., Schmidt I., Rocha S.F., Pitulescu M.E., Bussmann J., Meyen D., Raz E., Adams R.H., Siekmann A.F. (2014) Arteries are formed by vein-derived endothelial tip cells. Nat. Commun., 5, 5758.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Strasser G.A., Kaminker J.S., Tessier-Lavigne M. (2010) Microarray analysis of retinal endothelial tip cells identifies CXCR4 as a mediator of tip cell morphology and branching. Blood, 115, 5102–5110. [DOI] [PubMed] [Google Scholar]
- 40. Awwad K., Hu J., Shi L., Mangels N., Abdel Malik R., Zippel N., Fisslthaler B., Eble J.A., Pfeilschifter J., Popp R.. et al. (2015) Role of secreted modular calcium-binding protein 1 (SMOC1) in transforming growth factor beta signalling and angiogenesis. Cardiovasc. Res., 106, 284–294. [DOI] [PubMed] [Google Scholar]
- 41. Zilberberg L., ten Dijke P., Sakai L.Y., Rifkin D.B. (2007) A rapid and sensitive bioassay to measure bone morphogenetic protein activity. BMC Cell Biol., 8, 41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spiekerkoetter E., Tian X., Cai J., Hopper R.K., Sudheendra D., Li C.G., El-Bizri N., Sawada H., Haghighat R., Chan R.. et al. (2013) FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Invest., 123, 3600–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kang J.S., Liu C., Derynck R. (2009) New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol., 19, 385–394. [DOI] [PubMed] [Google Scholar]
- 44. Carmeliet P., Jain R.K. (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature, 473, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wei Y., Gong J., Thimmulappa R.K., Kosmider B., Biswal S., Duh E.J. (2013) Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc. Natl. Acad. Sci. U.S.A., 110, E3910–E3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dharaneeswaran H., Abid M.R., Yuan L., Dupuis D., Beeler D., Spokes K.C., Janes L., Sciuto T., Kang P.M., Jaminet S.C.. et al. (2014) FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ. Res., 115, 238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ruiz S., Zhao H., Chandakkar P., Chatterjee P.K., Papoin J., Blanc L., Metz C.N., Campagne F., Marambaud P. (2016) A mouse model of hereditary hemorrhagic telangiectasia generated by transmammary-delivered immunoblocking of BMP9 and BMP10. Sci. Rep., 6, 37366.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lesca G., Plauchu H., Coulet F., Lefebvre S., Plessis G., Odent S., Rivière S., Leheup B., Goizet C., Carette M.F.. et al. (2004) Molecular screening of ALK1/ACVRL1 and ENG genes in hereditary hemorrhagic telangiectasia in France. Hum. Mutat., 23, 289–299. [DOI] [PubMed] [Google Scholar]
- 49. Abdalla S.A., Cymerman U., Johnson R.M., Deber C.M., Letarte M. (2003) Disease-associated mutations in conserved residues of ALK-1 kinase domain. Eur. J. Hum. Genet., 11, 279–287. [DOI] [PubMed] [Google Scholar]
- 50. Corti P., Young S., Chen C.Y., Patrick M.J., Rochon E.R., Pekkan K., Roman B.L. (2011) Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development, 138, 1573–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Laux D.W., Young S., Donovan J.P., Mansfield C.J., Upton P.D., Roman B.L. (2013) Circulating Bmp10 acts through endothelial Alk1 to mediate flow-dependent arterial quiescence. Development, 140, 3403–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baeyens N., Larrivée B., Ola R., Hayward-Piatkowskyi B., Dubrac A., Huang B., Ross T.D., Coon B.G., Min E., Tsarfati M.. et al. (2016) Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol., 214, 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Park S.O., Wankhede M., Lee Y.J., Choi E.J., Fliess N., Choe S.W., Oh S.H., Walter G., Raizada M.K., Sorg B.S.. et al. (2009) Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Invest., 119, 3487–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garrido-Martin E.M., Nguyen H.L., Cunningham T.A., Choe S.W., Jiang Z., Arthur H.M., Lee Y.J., Oh S.P. (2014) Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models—brief report. Arterioscler. Thromb. Vasc. Biol., 34, 2232–2236. [DOI] [PubMed] [Google Scholar]
- 55. Oh S.P., Seki T., Goss K.A., Imamura T., Yi Y., Donahoe P.K., Li L., Miyazono K., ten Dijke P., Kim S.. et al. (2000) Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. U.S.A., 97, 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ardelean D.S., Jerkic M., Yin M., Peter M., Ngan B., Kerbel R.S., Foster F.S., Letarte M. (2014) Endoglin and activin receptor-like kinase 1 heterozygous mice have a distinct pulmonary and hepatic angiogenic profile and response to anti-VEGF treatment. Angiogenesis, 17, 129–146. [DOI] [PubMed] [Google Scholar]
- 57. Milton I., Ouyang D., Allen C.J., Yanasak N.E., Gossage J.R., Alleyne C.H., Seki T. (2012) Age-dependent lethality in novel transgenic mouse models of central nervous system arteriovenous malformations. Stroke, 43, 1432–1435. [DOI] [PubMed] [Google Scholar]
- 58. Kim J.H., Peacock M.R., George S.C., Hughes C.C. (2012) BMP9 induces EphrinB2 expression in endothelial cells through an Alk1-BMPRII/ActRII-ID1/ID3-dependent pathway: implications for hereditary hemorrhagic telangiectasia type II. Angiogenesis, 15, 497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Young K., Conley B., Romero D., Tweedie E., O'Neill C., Pinz I., Brogan L., Lindner V., Liaw L., Vary C.P. (2012) BMP9 regulates endoglin-dependent chemokine responses in endothelial cells. Blood, 120, 4263–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rochon E.R., Wright D.S., Schubert M.M., Roman B.L. (2015) Context-specific interactions between Notch and ALK1 cannot explain ALK1-associated arteriovenous malformations. Cardiovasc. Res., 107, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Siamakpour-Reihani S., Caster J., Bandhu Nepal D., Courtwright A., Hilliard E., Usary J., Ketelsen D., Darr D., Shen X.J., Patterson C.. et al. (2011) The role of calcineurin/NFAT in SFRP2 induced angiogenesis—a rationale for breast cancer treatment with the calcineurin inhibitor tacrolimus. PLoS One, 6, e20412.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rochon E.R., Menon P.G., Roman B.L. (2016) Alk1 controls arterial endothelial cell migration in lumenized vessels. Development, 143, 2593–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ola R., Dubrac A., Han J., Zhang F., Fang J.S., Larrivee B., Lee M., Urarte A.A., Kraehling J.R., Genet G.. et al. (2016) PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat. Commun., 7, 13650.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen Y.G., Liu F., Massague J. (1997) Mechanism of TGFbeta receptor inhibition by FKBP12. Embo j, 16, 3866–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huse M., Chen Y.G., Massague J., Kuriyan J. (1999) Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell, 96, 425–436. [DOI] [PubMed] [Google Scholar]
- 66. McDonald J., Wooderchak-Donahue W., VanSant Webb C., Whitehead K., Stevenson D.A., Bayrak-Toydemir P. (2015) Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front. Genet., 6, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Skaro A.I., Marotta P.J., McAlister V.C. (2006) Regression of cutaneous and gastrointestinal telangiectasia with sirolimus and aspirin in a patient with hereditary hemorrhagic telangiectasia. Ann. Intern. Med., 144, 226–227. [DOI] [PubMed] [Google Scholar]
- 68. Spiekerkoetter E., Sung Y.K., Sudheendra D., Bill M., Aldred M.A., van de Veerdonk M.C., Vonk Noordegraaf A., Long-Boyle J., Dash R., Yang P.C.. et al. (2015) Low-dose FK506 (Tacrolimus) in end-stage pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med., 192, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chatterjee P.K., Al-Abed Y., Sherry B., Metz C.N. (2009) Cholinergic agonists regulate JAK2/STAT3 signaling to suppress endothelial cell activation. Am. J. Physiol. Cell Physiol., 297, C1294–C1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ormiston M.L., Toshner M.R., Kiskin F.N., Huang C.J., Groves E., Morrell N.W., Rana A.A. (2015) Generation and culture of blood outgrowth endothelial cells from human peripheral blood. J. Vis. Exp., 106, e53384.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dorff K.C., Chambwe N., Zeno Z., Simi M., Shaknovich R., Campagne F., Provart N.J. (2013) GobyWeb: simplified management and analysis of gene expression and DNA methylation sequencing data. PLoS One, 8, e69666.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Durbin R.M., Altshuler D.L., Durbin R.M., Abecasis G.R., Bentley D.R., Chakravarti A., Clark A.G., Collins F.S., De La Vega F.M., Donnelly P.. et al. (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Campagne F., Dorff K.C., Chambwe N., Robinson J.T., Mesirov J.P., Lisacek F. (2013) Compression of structured high-throughput sequencing data. PLoS One, 8, e79871.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Campagne F., Digan W.E.R., Simi M. (2015) MetaR: simple, high-level languages for data analysis with the R ecosystem. bioRxiv, 030254. [Google Scholar]
- 76. Law C.W., Chen Y., Shi W., Smyth G.K. (2014) voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol., 15, R29.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.), 57, 289–300. [Google Scholar]
- 78. Conway J.R., Lex A., Gehlenborg N. (2017) UpSetR: an R package for the visualization of intersecting sets and their properties. bioRxiv, 120600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Livak K.J., Schmittgen T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 80. Kilkenny C., Browne W.J., Cuthill I.C., Emerson M., Altman D.G. (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol, 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.