

Abstract
Shape variation of human head hair shows striking variation within and between human populations, while its genetic basis is far from being understood. We performed a series of genome-wide association studies (GWASs) and replication studies in a total of 28 964 subjects from 9 cohorts from multiple geographic origins. A meta-analysis of three European GWASs identified 8 novel loci (1p36.23 ERRFI1/SLC45A1, 1p36.22 PEX14, 1p36.13 PADI3, 2p13.3 TGFA, 11p14.1 LGR4, 12q13.13 HOXC13, 17q21.2 KRTAP, and 20q13.33 PTK6), and confirmed 4 previously known ones (1q21.3 TCHH/TCHHL1/LCE3E, 2q35 WNT10A, 4q21.21 FRAS1, and 10p14 LINC00708/GATA3), all showing genome-wide significant association with hair shape (P < 5e-8). All except one (1p36.22 PEX14) were replicated with nominal significance in at least one of the 6 additional cohorts of European, Native American and East Asian origins. Three additional previously known genes (EDAR, OFCC1, and PRSS53) were confirmed at the nominal significance level. A multivariable regression model revealed that 14 SNPs from different genes significantly and independently contribute to hair shape variation, reaching a cross-validated AUC value of 0.66 (95% CI: 0.62–0.70) and an AUC value of 0.64 in an independent validation cohort, providing an improved accuracy compared with a previous model. Prediction outcomes of 2504 individuals from a multiethnic sample were largely consistent with general knowledge on the global distribution of hair shape variation. Our study thus delivers target genes and DNA variants for future functional studies to further evaluate the molecular basis of hair shape in humans.
Introduction
Variation in head hair shape represents a strongly visible and highly heritable trait in humans showing a striking diversity between major continental groups with decreasing prevalence of non-straight hair from Africans via Europeans towards Asians (1). Moreover, within continental groups, hair shape shows a pronounced degree of variation in African and European populations compared with Asian populations (2). The heritability of hair curliness has been estimated up to 95% in Europeans (3). Unveiling the genetic basis of hair shape variation is relevant for understanding the molecular basis of human appearance, is potentially useful in cosmetics, and is expected to contribute towards finding unknown perpetrators of crime from DNA evidence in the emerging field of Forensic DNA Phenotyping (4,5).
Genome-wide association studies (GWASs) have previously identified 8 genes being involved in human variation of head hair shape in different continental groups, including TCHH (Trichohyalin) (6–8), EDAR (ectodysplasin A receptor) (8–10), GATA3 (GATA binding protein 3) (8), PRSS53 (protease, serine 53) (8), WNT10A (Wnt family member 10A) (6,7), FRAS1 (Fraser extracellular matrix complex subunit 1) (6,7), OFCC1 (orofacial cleft 1 candidate 1) (6), and LCE3E (late cornified envelope 3E) (6). The LCE3E is at the same locus with TCHH (about 430 kBp) and was suggested to contribute a TCHH-independent effect on hair curliness in a web-based participant driven GWAS (6). The TCHH gene harbors European specific variants and EDAR harbors East Asian specific variants, indicating independent evolutionary history of hair shape variation in both continental regions. TCHH, EDAR, LINC00708/GATA3, and PRSS53 were found in a recent GWAS in Latin Americans of mixed European and Native American origin (8).
However, these 8 genes explain only a small proportion of the hair shape variation and trait heritability. The previously established predictive capacity of the three most informative DNA variants from three genes (TCHH, WNT10A, and FRAS1) yielded an AUC around 0.62 for straight vs. non-straight hair (11), which emphasizes the need for a more complete list of DNA predictors for potential applications such as in Forensic DNA Phenotyping (4,5).
Aiming to further improve the genetic understanding of shape variation in human head hair, and to find additional DNA predictors for future applications such as in forensics and anthropology, we performed a series of GWASs, replication studies, and prediction studies in a total of 28 964 subjects from 9 cohorts that include Europeans, East Asians, Latin Americans, and admixed individuals from around the world.
Results
This study included a total of 28 964 subjects from nine cohorts (Supplementary Material, Fig. S1 and Table S1). These included five European cohorts: the Queensland Institute of Medical Research study (QIMR, N = 10 607, individuals with North-Western European ancestry from Australia), the Rotterdam Study (RS, N = 2809, North-Western Europeans from the Netherlands), the TwinsUK study (N = 3347, North-Western Europeans from the UK), the Erasmus Rucphen Family study (ERF, N = 977, North-Western Europeans from the Netherlands), and samples from Poland (POL, N = 635, East-Central Europeans from Poland), as well as two multi-origin cohorts: the CANDELA study (N = 6238, Latin Americans of estimated 48% European, 46% Native American and 6% African ancestry), North Americans from the USA (US, N = 743, various origins including Europe, America, Middle East, and Asia), and two East Asian cohorts: the Xinjiang Uyghur Study (UYG, N = 709, Uyghurs of estimated 50% East Asian and 50% European ancestry) and the Taizhou Longitudinal Study (TZL, N = 2899, Han Chinese). The hair shape phenotypes collected in the various cohorts were unified as three broad ordinal levels, straight, wavy, and curly (Supplementary Material, Table S1).
Discovery Meta-analysis of Three GWASs in Europeans
In the discovery stage of the study, we conducted a genome-wide inverse variance, fixed-effect meta-analysis of three hair shape GWASs, which were independently conducted in three European cohorts, i.e. QIMR, RS, and TwinsUK, totalling 16 763 subjects (referred to bellow as META: Discovery). No significant inflation was detected at the genome-wide level (λ = 1.03), and the observed test statistics started to deviate from the expected null after P = 1e-3 as shown in the Q-Q plot (Supplementary Material, Fig. S2), suggesting that the significant findings are unlikely to be false positives caused by residual population sub-stratifications. The META: Discovery identified a total of 706 SNPs at 12 distinct loci/gene regions showing genome-wide significant association with hair shape (Fig. 1, Supplementary Material, Table S2). Among these 12 loci, 8 were novel for involvement in hair shape and 4 had been identified by previous GWASs (Table 1), the latter including the three most significant loci (1q21.3, 4q21.21 and 2q35) (Supplementary Material, Fig. S3) reflecting high consistency with association patterns in the previous studies (6–8). The most significant association was identified for rs17646946 (P = 1.78e-84) at 1q21.3, where the top-associated SNPs (rs17646946, rs11803731, rs12130862, rs4845418) were located within or close to the TCHH and TCHHL1 genes. The associated SNPs in this region spanned ∼800 kbp, including an intergenic SNP rs499697 (P = 2.57e-17) between CRCT1 and LCE3E genes. This SNP is located in a different linkage disequilibrium block and has been previously reported to contribute a TCHH-independent effect on hair curliness in a web-based participant driven GWAS (6). Conditioning on the genotype of rs17646946, rs499697 still showed nominally significant association (P = 1.46e-5) with hair curliness, confirming previous findings. The second significant association was seen for rs506863 (P = 2.16e-15), an intron SNP in FRAS1 at 4q21.21. The third significant association was the SNP rs74333950 (P = 3.98e-15) in WNT10A at 2q35. The associated SNP rs1999874 (P = 3.72e-09) on 10p14 was located between LINC00708 and GATA3. The 8 new loci showing genome-wide significant association with hair shape included 1p36.23 ERRFI1/SLC45A1 (top SNP rs80293268, P = 3.66e-9), 1p36.22 PEX14 (rs6658216, P = 3.02e-9), 1p36.13 PADI3 (rs11203346, P = 4.58e-8), 2p13.3 TGFA (rs12997742, P = 9.28e-9), 11p14.1 LGR4 (rs2219783, P = 3.84e-8), 12q13.13 HOXC13 (rs11170678, P = 1.62e-11), 17q21.2 KRTAP (rs11078976, P = 1.29e-9), and 20q13.33 PTK6 (rs310642, P = 3.74e-10) (Table 1, Fig. 2).
Figure 1.
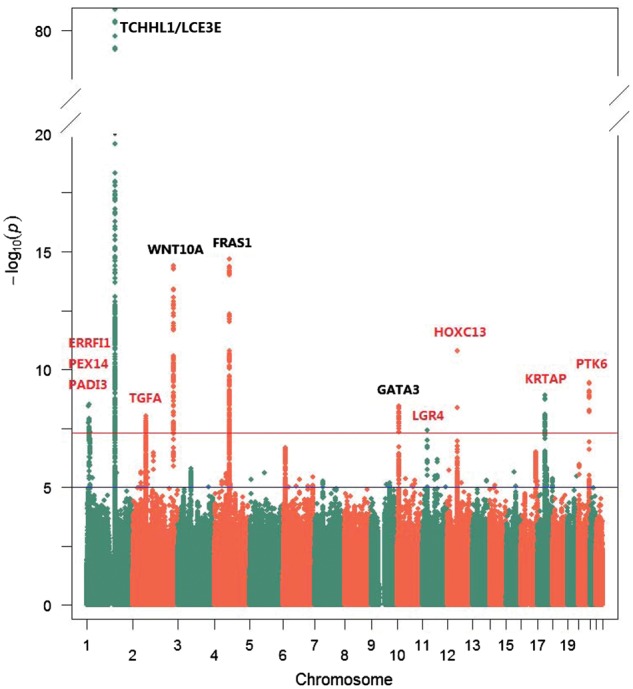
Manhattan plot of meta-analysis of three GWASs for human hair shape in Europeans from QIMR, TwinsUK and RS (totaling 16, 763 subjects). The −log10 P values for association were plotted for each SNP according to its chromosomal position according to assembly GRCh37.p13. Previously known genes with genome-wide significant association in the present study are noted using black text above the figure and genes in the newly identified loci are noted in red. Genome-wide significance threshold (P = 5e-8) is indicated as a red line, while the suggestive significance threshold (P = 1e-5) is indicated as a blue line.
Table 1.
SNPs associated with hair shape variation in the discovery meta-analysis of three European cohorts and in meta-analysis of all 9 cohorts of European and non-European origins
|
META: Discovery |
META: Non-Asian |
META: ALL |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
N = 16 763 |
N = 25 356 |
N = 28 964 |
|||||||||||
| SNP | Gene | CHR | MBp | EA | fEA | beta | P-value | fEA | beta | P-value | fEA | beta | P-value |
| rs80293268 | ERRFI1 | 1p36.23 | 8.21 | C | 0.04 | −0.17 | 3.66E-09 | 0.03 | −0.18 | 5.85E-14 | 0.03 | −0.18 | 5.85E-14 |
| rs6658216 | PEX14 | 1p36.22 | 10.56 | C | 0.34 | −0.06 | 3.02E-09 | 0.38 | −0.02 | 1.58E-03 | 0.38 | −0.01 | 8.31E-03 |
| rs11203346 | PADI3 | 1p36.13 | 17.60 | G | 0.17 | −0.07 | 4.58E-08 | 0.15 | −0.07 | 9.24E-17 | 0.15 | −0.07 | 9.24E-17 |
| rs17646946 | TCHHL1 | 1q21.3 | 152.06 | A | 0.19 | −0.22 | 1.78E-84 | 0.17 | −0.21 | 5.82E-134 | 0.17 | −0.21 | 5.82E-134 |
| rs12997742 | TGFA | 2p13.3 | 70.79 | C | 0.36 | −0.05 | 9.28E-09 | 0.35 | −0.04 | 3.34E-08 | 0.36 | −0.03 | 7.35E-07 |
| rs74333950 | WNT10A | 2q35 | 219.75 | G | 0.15 | 0.10 | 3.98E-15 | 0.15 | 0.09 | 3.53E-26 | 0.16 | 0.06 | 9.47E-18 |
| rs506863 | FRAS1 | 4q21.21 | 79.26 | C | 0.34 | 0.08 | 2.16E-15 | 0.35 | 0.05 | 6.67E-14 | 0.41 | 0.04 | 2.73E-10 |
| rs1999874 | GATA3 | 10p14 | 8.35 | A | 0.38 | 0.06 | 3.72E-09 | 0.36 | 0.06 | 3.55E-18 | 0.34 | 0.04 | 6.53E-15 |
| rs2219783 | LGR4 | 11p14.1 | 27.41 | G | 0.10 | 0.09 | 3.84E-08 | 0.09 | 0.06 | 2.32E-06 | 0.09 | 0.02 | 4.13E-03 |
| rs11170678 | HOXC13 | 12q13.13 | 54.15 | G | 0.26 | −0.07 | 1.62E-11 | 0.22 | −0.06 | 1.98E-13 | 0.22 | −0.06 | 1.98E-13 |
| rs11078976 | KRTAP | 17q21.2 | 39.19 | T | 0.83 | 0.12 | 1.29E-09 | 0.81 | 0.05 | 6.65E-08 | 0.80 | 0.03 | 1.12E-04 |
| rs310642 | PTK6 | 20q13.33 | 62.16 | C | 0.05 | 0.13 | 3.74E-10 | 0.05 | 0.10 | 3.30E-10 | 0.05 | 0.06 | 3.42E-14 |
META: Discovery, meta-analysis of 3 European cohorts (QIMR, TwinsUK and RS);
META: Non-Asian, meta-analysis of 5 European cohorts (QIMR, TwinsUK, RS, ERF, POL) and 2 European admixed cohorts from America (CANDELA and US);
META: ALL, meta-analysis of all 9 cohorts used in this study including QIMR, TwinsUK, RS, ERF, POL, CANDELA, US, UYG, and TZL;
SNP, the top-associated SNP per locus in META: Discovery, bold indicate new loci for hair curliness;
EA, fEA, the effect allele and the frequency of effect allele; P-values < 5e-8 are in bold.
Figure 2.


Regional Manhattan plot for eight novel loci associated with hair shape variation in the meta-analysis of three European GWASs (QIMR, TwinsUK, RS). (A) 1p36.23 - ERRFI1/SLC45A1; (B) 1p36.22 - PEX14; (C) 1p36.13 - PADI3; (D) 2p13.3 – TGFA; (E) 11p14.1 - LGR4; (F) 12q13.13 - HOXC13; (G) 17q21.2 – KRTAP; (H) 20q13.33 - PTK6. The index SNP in each region (Table 1) is shown as a purple diamond. At the top of the figure are shown the association P-values on a -log10 P scale (y-axis) for all genotyped and imputed SNPs according to their physical positions (x-axis) using human genome sequence build 37. Genes in the region and LD heatmap (r2) patterns according to 1000 genomes EUR data set are aligned bellow. Plots for identified regions not shown here are presented in Supplementary Material, Figure S3.
Out of the 706 SNPs significantly associated with hair shape in the META: Discovery, 5 SNPs in TCHH and one in WNT10A had existing entries in the GWAS Catalog noted for common traits and hair shape and one SNP (rs636291) at PEX14 was noted for prostate cancer (Supplementary Material, Table S3). An enrichment analysis of 172 genes harbored by the 706 significant SNPs highlighted a total of 25 biological process terms under the FDR < 0.05 level (Supplementary Material, Table S4), among which ‘keratinization’, ‘keratinocyte differentiation’, ‘epidermis development’, ‘epidermal cell differentiation’, and ‘skin development’ were the most significantly enriched terms (FDR < 1e-16), which is highly consistent with established knowledge about the important role of keratin in hair shape formation (12). The potential association of the most associated polymorphisms and expression of genes located nearest to them was investigated. As expected, the majority of these genes had eQTL effects in several tissues. The most obvious expression were observed in both sun exposed and non-sun exposed skin tissue for the TCHHL1, WNT10A, GATA3, HOXC13, KRTAP and PTK6 (Supplementary Material, Fig. S4).
Next, we explored the allele frequency distribution for the 12 top-associated SNPs in 2504 individuals from the 1000-Genomes Project consisting of 26 worldwide populations covering all major geographic regions except Oceania (Supplementary Material, Fig. S5). The frequency patterns for all 12 SNPs demonstrated various degrees of differences between major continental groups (Supplementary Material, Table S5). Remarkable for SNPs initially identified in Europeans, all but one (rs80293268 at 1p36.23 ERRFI1/SLC45A1) showed considerable variation in Africans, Asians and Native Americans. For some SNPs, such rs2219783 (11p14.1 LGR4), rs310642 (20q13.33 PTK6), and rs12997742 (2p13.3 TGFA), the allele associated with non-straight hair showed a higher frequency in Africans compared with Europeans, Asians, and Americans. For other SNPs, such as rs11203346 (1p36.13 PADI3) and rs17646946 (1q21.3 TCHH/TCHH1), the non-straight allele was nearly fixed in East Asians, and thus more frequent than in Europeans, Americans, and Africans. Two SNPs, rs11170678 (12q13.13 HOXC13) and rs80293268 at 1p36.23 ERRFI1/SLC45A1, were only polymorphic in Europeans and (less so) in Latin Americans from CANDELA likely due to their European admixture.
Replication and Meta-analysis in Additional Europeans, Native Americans and East Asians
For the 8 newly identified loci we conducted a replication study focusing on the top-associated SNP per locus in two additional European cohorts (ERF, POL, N = 1612) and two additional multi-ethnic cohorts with European as the major origin (CANDELA, US, N = 6981), and performed a meta-analysis in the 7 cohorts containing individuals of European ancestry (QIMR, RS, TwinsUK, ERF, POL, CANDELA, and US), referred to below as META: Non-Asian, N = 25 356 (Table 1, Fig. 3, Supplementary Material, Table S2). This analysis revealed sufficient evidence of replication for 5 out of the 8 novel loci involved in head hair shape. The 1p36.23 locus (ERRFI1/SLC45A1 rs80293268) showed consistent replication in the four cohorts used for replication testing (ERF, POL, CANDELA, and US) at nominal significance (P < 0.05), and greatly enhanced genome-wide significance in META: Non-Asian (from 3.66e-9 to P = 5.85e-14). The 1p36.13 locus (PADI3 rs11203346) was nominally significant in ERF (P = 3.40e-5) and CANDELA (P = 7.35e-3) and showed greatly enhanced significance in META: Non-Asian (from P = 4.58e-8 to P = 9.24e-17). The 12q13.13 locus (HOXC13 rs11170678) was replicated in ERF (P = 1.6e-3) and showed enhanced significance in META: Non-Asian (from P = 1.62e-11 to P = 1.98e-13). Note rs80293268, rs11203346, and rs11170678 were all nearly monomorphic in East Asians in the 1000-Genome Project reference panel. The 20q13.33 locus (PTK6 rs310642) was significant in POL (P = 6.23e-3) and showed enhanced significance in META: Non-Asian (from P = 3.74e-10 to P = 3.30e-10). The 2p13.3 locus (TGFA rs12997742) was significant in CANDELA (P = 0.019) and showed slightly reduced but still genome-wide significant association in META: Non-Asian (from P = 9.28e-9 to P = 3.34e-8) (Fig. 3). Two other loci were nominally significant in at least one replication cohort but reduced the significance of the META: Non-Asian, i.e. 1p14.1 (LGR4 rs2219783, P = 0.04 in ERF, P = 0.02 in US, meta from 3.84e-8 to 2.32e-6); and 17q21.2 (KRTAP rs11078976, P = 0.04 in CANDELA, meta from 1.29e-9 to 6.65e-8). For all nominally significantly associated SNPs in the replication analysis, their allelic effects were consistent with the European discovery meta-analysis. The only locus that did not show any replication was 1p36.22 (PEX14 rs6658216) where the P-value dropped considerably from 3.02e-9 to 1.58e-3 in the META: Non-Asian. Note the straight-associated C allele showed a reversed frequency between European (f = 0.31) and African (f = 0.84) populations. Four out of the 12 loci from META: Discovery have been previously reported for association with hair shape and all these 4 loci demonstrated genome-wide significant association in the META: Non-Asian, i.e. TCHH (P = 5.82e-134), WNT10A (P = 3.53e-26), LINC00708/GATA3 (P = 3.55e-18), FRAS1 (P = 6.67e-14) (Supplementary Material, Fig. S6).
Figure 3.

Effect sizes for the derived allele at index SNPs (Table 1) in eight novel genomic regions associated with hair shape variation in 9 cohorts and a meta-analysis of all 9 cohorts (META). (A) 1p36.23—rs80293268; (B) 1p36.22 – rs6658216; (C) 1p36.13—rs11203346; (D) 2p13.3—rs12997742; (E) 11p14.1 – rs2219783; (F) 12q13.13—rs11170678; (G) 17q21.2—rs11078976; (H) 20q13.33—rs310642. Orange color indicates European cohorts, green color indicates admixed European cohorts, and purple color indicated East Asian. META: Discovery, meta-analysis of 3 European cohorts (QIMR, TwinsUK and RS), META: Non-Asian, meta-analysis of European cohorts (QIMR, TwinsUK, RS, ERF, and POL) and admixed European cohorts (CANDELA and US), and META: ALL indicates meta-analysis of all 9 cohorts (QIMR, TwinsUK, RS, ERF, POL, CANDELA, US, UYG, and TZL). Blue boxes represent linear regression coefficients (x-axis) estimated in each cohort. Red boxes represent effect sizes estimated in the combined meta-analysis. Box sizes are proportional to sample size. Horizontal bars indicate a 95% confidence interval of width equal to 1.96 standard errors. The right y-axis indicates P values in each cohort on –log10 scale. Similar plots for regions previously associated with hair shape are shown in Supplementary Material, Figure S6.
Finally, we conducted a replication analysis in two additional East Asian cohorts (UYG, TZL, N = 3608) and a meta-analysis of the 9 European and non-European cohorts used in this study (referred to below as META: All, N = 28 964, Table 1, Fig. 3, Supplementary Material, Table S2). Compared with the results of META: Non-Asian, the PTK6 rs310642 was the only locus that showed an enhanced significance of association (from P = 3.30e-10 to P = 3.42e-14) and this was the only SNP with nominally significant association in TZL (East Asian). None of the 8 novel SNPs were significant in UYG (East Asian-European admixed) (Fig. 3) despite its previously estimated 50% European genomic admixture (9). The non-enhanced association in the META: ALL compared with META: Non-Asian is likely explained by differences in allele frequency and allelic heterogeneity between East Asian and Non-Asian cohorts.
Examination of Previously Suggested Candidates
To compare our META: Discovery results with previous GWAS findings, we selected 8 SNPs in 7 genomic regions TCHH/TCHHL1/LCE3E, EDAR, WNT10A, FRAS1, OFCC1, LINC00708/GATA3, and PRSS53 which have been previously reported showing genome-wide significant association with hair shape in Europeans, Latin Americans, or East Asians (6–9). The SNPs in TCHH, WNT10A, and FRAS1 showed genome-wide significant, the SNP in OFCC1 showed boarder line genome-wide significant, and the SNPs in EDAR, GATA3, and PRSS53 showed nominally significant association with hair curliness in our META: Discovery (Supplementary Material, Table S6). Interestingly, the association at PRSS53 and GATA3 was initially identified in Latin Americans (8), and EDAR is known to have a predominant effect in East Asians (9). As expected, the straight hair associated EDAR rs3827760 G allele known to have a high frequency in East Asians (9) had a low frequency in our European samples (MAF=0.02, P = 0.008), a high frequency in the East Asian-European mixed Uyghurs from UYG (MAF=0.46, P = 2.7e-14), and a pronounced frequency in Han Chinese from TZL (MAF=0.95, P = 2.19e-18).
In a recently published review paper on the biology and genetics of curly hair (13), novel data on South Africans of African origin (N = 2417) were presented, including a GWAS on 25% highest and lowest curl subjects that did not reveal any genome-wide significant (P < 5e-8) results. Nevertheless, these authors emphasized on three genes, KRT74, TCHH and CUTC as being involved in curly hair of Africans based on suggestive association and other evidence. Of these, only SNPs in TCHH showed genome-wide significant results in our European META: Discovery GWAS and this gene was known before to be involved in European hair shape (7). The noted the SNP rs3912631 from KRT74 was not significant in our data but about 200kbp to another SNP rs12311316 in KRT82 which showed borderline genome-wide significant association in META: Discovery (P = 1.13e-7, Supplementary Material, Fig. S7). This region on 12q13.13 contains a cluster of KRT genes (KRT82, KRT83, KRT84, KRT85, KRT86, KRT75, KRT6A, KRT6B, KRT6C, etc). The noted variants in CUTC did not pass quality control in our data, likely due to low frequency in European populations (e.g. rs4919394, MAF = 0 in HapMap CEU).
Multivariable, Prediction and Interaction Analyses
To build a prediction model, a step-wise multiple logistic regression was conducted in 6068 unrelated QIMR subjects to examine the independent effects of the 12 strongest-associated SNPs from the current study and additionally 4 SNPs from previous studies (EDAR rs3827760, OFCC1 rs1556547, PRSS53 rs11150606, LCE3E rs499697). This analysis revealed 14 SNPs significantly and independently contribute to hair shape variation (Table 2) within this European sample. As expected, given that the QIMR participants were included in the META: Discovery all SNPs identified with genome-wide significant association in the META: Discovery demonstrated nominally significant and independent effect (Table 2). In addition, OFCC1 rs1556547, which was reported by a previous GWAS (6), also demonstrated a significant effect in this analysis (P = 1.85e-7, Table 2). Interestingly, female subjects were significantly more likely to have non-straight rather than straight hair (OR = 1.43, P = 8.71e-9, Table 2) based on the self-reported phenotypes used. This is largely consistent with the observation in RS, where the hair curl phenotyping was based on a photo numeric approach, suggesting a real effect of sex influencing true hair curl and not just socially driven preferences. Using the QIMR data, the model fitting using ten-fold cross validation was estimated at AUC = 0.66 and Nagelkerke r2 =0.10 (Table 2, Fig. 4). Applying this model to the independent 977 ERF samples showed a similar accuracy of AUC = 0.64 (Supplementary Material, Table S7, Fig. 4). The prediction results were practically informative for about 7.1% of QIMR and 3.9% of ERF subjects, that is, individuals with predicted probabilities of non-straight < 0.2 or > 0.8 (7.1% < 0.2 in QIMR and 3.9% < 0.2 in ERF, 0% > 0.8 in both cohorts Fig. 4).
Table 2.
Multiple logistic regression for SNPs associated with hair shape variation in 6068 Europeans from QIMR
| Marker | Gene | CHR | EA | OR | Lower95 | Upper95 | P-value | R2 | AUC |
|---|---|---|---|---|---|---|---|---|---|
| rs17646946 | TCHHL1 | 1q21.3 | A | 0.50 | 0.45 | 0.56 | 2.35E-38 | 0.04 | 0.58 |
| rs80293268 | ERRFI1/SLC45A1 | 1p36.23 | C | 0.40 | 0.30 | 0.53 | 3.33E-10 | 0.05 | 0.60 |
| Sex (female) | 1.43 | 1.27 | 1.62 | 8.71E-09 | 0.05 | 0.61 | |||
| rs506863 | FRAS1 | 4q21.21 | C | 1.27 | 1.17 | 1.38 | 6.06E-09 | 0.06 | 0.62 |
| rs1556547 | OFCC1 | 6p24.3 | G | 0.81 | 0.75 | 0.88 | 1.85E-07 | 0.07 | 0.63 |
| rs74333950 | WNT10A | 2q35 | G | 1.35 | 1.20 | 1.51 | 2.68E-07 | 0.07 | 0.63 |
| rs310642 | PTK6 | 20q13.33 | C | 1.56 | 1.32 | 1.86 | 2.76E-07 | 0.08 | 0.64 |
| rs11170678 | HOXC13 | 12q13.13 | G | 0.78 | 0.70 | 0.86 | 6.99E-07 | 0.08 | 0.64 |
| rs11203346 | PADI3 | 1p36.13 | G | 0.79 | 0.71 | 0.88 | 1.53E-05 | 0.09 | 0.65 |
| rs11078976 | KRTAP | 17q21.2 | C | 0.75 | 0.66 | 0.86 | 2.73E-05 | 0.09 | 0.65 |
| rs2847344* | PEX14 | 1p36.22 | G | 0.86 | 0.79 | 0.93 | 3.14E-04 | 0.09 | 0.65 |
| rs1999874 | LINC00708/GATA3 | 10p14 | A | 1.15 | 1.07 | 1.25 | 3.91E-04 | 0.10 | 0.65 |
| rs2219783 | LGR4 | 11p14.1 | G | 1.26 | 1.11 | 1.44 | 5.05E-04 | 0.10 | 0.65 |
| rs12997742 | TGFA | 2p13.3 | C | 0.88 | 0.81 | 0.95 | 1.13E-03 | 0.10 | 0.66 |
| rs499697 | LCE3E | 1q21.3 | G | 1.13 | 1.04 | 1.22 | 5.54E-03 | 0.10 | 0.66 |
The markers were ordered according to P-values in the multiple logistic regression and hair curliness is dichotomized as non-straight (1) vs. straight (0); Marker, initial analysis includes sex, age, and 16 SNPs associated with hair curliness, 12 from the current study (see Table 1) and 4 from previous studies (EDAR rs3827760, OFCC1 rs1556547, PRSS53 rs11150606, LCE3E rs499697, also see Supplementary Material, Table S6), non-significant SNPs in the final model are not presented; *rs2847344 is used as a replacement for rs6658216, these 2 SNPs are in nearly complete LD (r2 > 0.9); R2, Accumulative Nagelkerke pseudo R2 while the current marker is included; AUC, accumulative Area Under the ROC Curve value while the current marker is included.
Figure 4.

Hair shape prediction accuracy in unrelated QIMR subjects (N = 6068) and ERF (N = 977). (A) Accuracy of hair straight vs. non-straight hair prediction using DNA variants, sex and age in unrelated Europeans from QIMR based on 10-fold cross validation and the accuracy of applying model to the testing set of QIMR and to all ERF subjects. Prediction performance measured by AUC for the model based on binomial logistic regression (Y-axis) was plotted against the number of markers included in the model (X-axis). Sex and age were always included and fixed at the position 1 and 2 in all analyses. (B) Frequency (left y-axis) of predicted probability (x-axis) for non-straight hair and percentage (right y-axis) of non-straight hair in each probability bin in Europeans from QIMR by self-cross-validation. (C) Frequency (left y-axis) of predicted probability (x-axis) for non-straight hair and percentage (right y-axis) of non-straight hair in each probability bin in Europeans from ERF by applying model of QIMR to ERF. (D) The distribution of predicted non-straight hair probabilities for 2504 subjects from the 1000-Genomes Project panel; samples are grouped according to the 5 continents they originate from: AFR—Sub-Saharan Africans, AMR—Native Americans, EAS—East Asians, EUR—Europeans, and SAS - South Asians. (E) The distribution of predicted non-straight hair probabilities for 2504 subjects from 19 worldwide countries from the 1000-Genomes Project panel; Except 3 populations, 26 populations were grouped to 19 countries including 2248 subjects according to the official population description from the 1000 Genomes Project.
We then included the EDAR rs3827760 in the prediction model, estimated its effect in East Asians from TZL, and applied this enhanced model to predict hair shape in the 2504 worldwide subjects from the 1000-Genomes Project panel. On the continental level, this analysis demonstrated an increasing degree of predicted non-straight hair probabilities for Africans and South Asians, relative to Europeans and Americans, while it was considerably lower for East Asians (Fig. 4). However, the degree of within-continental variance showed a different pattern, i.e. East-Asia (Var = 0.0014), Africa (Var = 0.015), South-Asia (Var =0.017), Europe (Var = 0.026), and America (Var = 0.046). Although no phenotype information is available for the 1000-Genomes Project samples, hair shape phenotypes can be roughly assumed from regional knowledge and was consistent with the prediction outcome. For instance, the predicted very low non-straight hair probabilities in East Asians, i.e. in China, Japan, and Vietnam (Fig. 4E) are in line with general knowledge that East Asians belonged to the straightest hair groups worldwide and are consistent with the observation in our Chinese samples (92% straight hair in TZL, Supplementary Material, Table S1). On the other side, Sub-Saharan Africans i.e. people from Gambia, Kenya, Nigeria and Sierra Leone, for which our prediction analysis reveals the highest probabilities for non-straight hair on the level of continental groups as well as populations (Fig. 4), belong to most non-straight hair groups worldwide. This pattern of the predicted non-straight hair is largely consistent with the known hair curliness distribution around the world (2). Interestingly, East Asians in our prediction analysis displayed the smallest degree of variation in the non-straight hair probabilities (Fig. 4) and the majority (71%) had small probability estimates (P < 0.2) for non-straight hair, which is likely explained by the strong effect of the East Asian-specific straight-associated EDAR rs3827760 G allele. However, there were also a large number of outliers (29%) with non-straight probabilities > 0.2, which is likely explained by a combined effect of previously known and our newly identified SNP predictors.
A pair-wise SNP-SNP interaction analysis between all 3632 SNPs that had a P-value < 1e-4 in the META: Discovery did not identify significant inter-locus interaction after strict Bonferroni correction for multiple testing (Supplementary Material, Fig. S8). SNP-sex and SNP-age interaction analyses did not reveal any significant results. Dominant and recessive effect analysis for the 13 SNPs showing significant association in the multivariable analysis in Table 2 did not detect any obvious effects (Supplementary Material, Table S8). To investigate a potential link between genetic susceptibility to hair loss and hair morphology (14) we looked up the 12 hair shape associated SNPs from the present study in our previous GWAS of male pattern baldness in 2725 German and Dutch males (15). Of those, rs17646946 in TCHHL1, which showed the strongest hair shape association in our study, showed nominally significant association with male pattern baldness (allelic OR for A allele =1.23, P = 0.002, Supplementary Material, Table S9).
Discussion
A series of GWASs, meta analyses, and replication analyses on head hair shape in multiple sizable European, Latin American, North American, East Asian and admixed cohorts highlighted 8 novel loci and confirmed 8 previously known loci. Fourteen SNPs in 14 genes independently contributed to hair shape variation in Europeans according to a multivariable analysis. A statistical prediction model provided an improved accuracy in a European dataset compared with a previous model (11) and the distribution of the predicted shape variation in world-wide subjects is largely consistent with the known global distribution of hair shape variation. Given the large study size, the high rate of successful replication, the high rate of confirmation for previously known genes, and the high consistency between our prediction results and general phenotype knowledge, we believe that most of our new findings are true positives. In addition, our GO analysis highlighted 25 biological pathways showing significant enrichment, all of which represent well-known hair morphogenesis pathways, which further supports the reliability of our findings.
Given current knowledge, 6 of the 8 loci showing significant association with hair morphology harbor plausible candidate genes likely to be involved in hair morphogenesis, i.e. 1p36.13 PADI3, 1p36.23 ERRFI1, 2p13.3 TGFA, 11p14.1 LGR4, 12q13.13 HOXC13, 12q13.13 KRT and 17q21.2 KRTAP. At chromosome 1p36.13 the top-associated SNP rs11203346 is an intron variant of PADI3. The allele effect was on the same direction for all 7 full or partially European cohorts, significantly replicated in CANDELA and ERF, the significance was greatly enhanced in the META: Non-Asian; this SNP is nearly monomorphic in TZL (Han Chinese) as well as in East Asians from the 1000-Genomes Project. This locus consists of a cluster of five paralogous PAD (peptidyl arginine deiminase) genes, i.e. PADI1, PADI2, PADI3, PADI4, and PADI6. PADI3 encodes type III peptidyl arginine deiminase, which modulates hair structural proteins, such as filagrin in the hair follicle and trichohyalin in the inner root sheath, during hair follicle formation by converting arginine residues to citrullines. Five isoforms are known that are present in distinct tissue locations. In the epidermis, like skin, only PAD1, 2, and 3 are expressed. PAD3 is the enzyme that deiminates trichohyalin in the medulla and the inner root sheath Henle layer (16). The enzymatic properties of PAD1 and 3 and their co-localization with filaggrin within the fibrous matrix of deep corneocytes suggest that they are responsible for the deimination of this protein. These two isoforms participate in, and possibly control, the production of the natural moisturizing factor, and play a major role in the stratum corneum homeostasis (17). Multiple missense mutations within PADI3 in homozygote or compound heterozygote states caused spun glass hair syndrome and Padi3 knockout mice expressed whisker and hair anomalies (18). Suggestive association of PADI3 gene (rs11585118) with hair morphology was previously shown in Eriksson et al., but it did not reach genome-wide significance (6).
At 1p36.23 the top-associated SNP rs80293268 is intergenic of ERRFI1 and SLC45A1, with several uncharacterized or pseudo genes in between. The SNP is in the same LD block with ERRFI1 and in lower LD with SLC45A1. The allele effect was consistent in all replication cohorts and nominally significant in most replication cohorts, and the association signal was greatly enhanced in the META: Non-Asian. The SNP is nearly monomorphic in TZL and in the East Asians from the 1000 Genomes Project. ERRFI1 (ERBB receptor feedback inhibitor 1), also known as RALT or MIG6, encodes an inhibitor of epidermal growth factor receptor, which plays a key role in epidermal homeostasis, hair follicle development, inhibitor-associated skin toxicities, and cell migration and invasion in carcinogenesis (19,20). Ballaro et al., previously reported that overexpression of RALT in the mouse skin suppresses epidermal growth factor receptor signalling and causes a waved-like phenotype, characterized by wavy coat and curly whiskers (21). SLC45A1, as the member of Solute Carrier 45A Family, encoded protein which belongs to the glycoside-pentoside-hexuronide cation symporter transporter family, plays a role in glucose uptake, and have been implicated in the regulation of glucose homoeostasis in the brain (22). There is little evidence supporting a potential link between SLC45A1 and hair morphology.
At 2p13.3 the top-associated SNP rs12997742 is about 5 kBp upstream of TGFA. The association signal was replicated at nominal significance in CANDELA and remained at genome-wide significance level in the META: Non-Asian. TGFA encodes transforming growth factor alpha, which is a ligand for the epidermal growth factor receptor. Disrupting the mouse TGFA by homologous recombination in embryonic stem cells showed that homozygous mutant mice displayed pronounced waviness of the whiskers and fur, accompanied by abnormal curvature, disorientation, and misalignment of the hair follicles (23). Since then TGFA mutant mice have been used as a model to study hair abnormalities (24,25).
At 11p14.1 the top-associated SNP rs2219783 is an intron variant of LGR4 and in the same LD block with CCDC34. The slightly reduced significance in the META: Non-Asian is driven by the Polish sample, i.e. little or no effect was observed. LGR4 (leucine rich repeat containing G protein-coupled receptor 4) encodes a G-protein coupled receptor that binds R-spondins and activates the Wnt signaling pathway, which is long known for its key role in controlling hair growth and structure. Lgr4-deficient mice showed partial impairment in hair follicle development with reduced expression of Edar, Lef1, and Shh (26) and premature hair cell differentiation in the embryonic cochlea (27).
At 12q13.13, the top associated SNP rs11170678 is an intron variant of an uncharacterized LOC105378250 and in the same LD block with two known genes, CALCOCO1 (Calcium-binding and coiled-coil domain-containing protein) and HOXC13 (homeobox C13). The allele effect was consistent in all replication cohorts and the association signal was replicated in ERF as well as greatly enhanced in the META: Non-Asian, and the SNP is nearly monomorphic in Han Chinese from TZL and East Asians from the 1000-Genomes Project. There is lack of existing evidence supporting a role of CALCOCO1 in hair morphogenesis while the product of HOXC13 may play a role in the development of hair, nail, and filiform papilla. A whole-exome sequencing study in a consanguineous Chinese family has identified a homozygous nonsense mutation in HOXC13 being responsible for pure hair, i.e. loss of scalp hair, beard, eyebrows, eyelashes, axillary hair, and pubic hair, and a reduced level of HOXC13 expression led to nearly absent protein staining in hair follicles (28).
At 17q21.2 the top-associated SNP rs11078976 is intergenic near several KRTAP and KRT genes that are in high LD. The allele effect was on the same direction in all cohorts except the two Chinese cohorts, where the SNP had no effect, explaining the reduced association signal in the META: All. KRT genes encode keratins that are heteropolymeric structural proteins which form the intermediate filament and KRTAP encodes a cluster of keratin associated proteins. Studies have shown that KRT and KRTAP are major structural proteins of the hair fibre and sheath (29,30), and their content is important for fleece quality (29) and Cashmere goat hair morphology (31). Ageing processes influence expression of KRT and KRTAP in human hair follicles (30) and dysregulation of KRTAP genes can cause hair disorders (32). Mutations in various KRT genes have been linked to monogenic forms of hair abnormalities. For example, a missense variant in the coil1A domain of the keratin 25 gene has been recently reported to cause dominant curly hair in horse (33). Mutations in the KRT14 gene have been recently found to cause autosomal dominant Naegeli-Franceschetti-Jadassohn syndrome, in which hair changes represent a key symptom (34). A heterozygous mutation in exon 7 of the KRT86 gene was found to cause Monilethrix (35). Interestingly, a novel locus at 12q13.13 where a cluster of KRT genes is located (i.e. KRT82, KRT83, KRT84, KRT85, KRT86, KRT75, KRT6A, KRT6B, KRT6C, KRT74) showed border-line genome-wide significant association in our META: Discovery. Further, a previous GWAS in South African populations showed a borderline genome-wide significant association for SNP rs3912631 in KRT74, which is about 200kbp to rs12311316 in KRT82 (13). Note that this locus and the HOXC13 locus, which we identified in our GWAS meta-analysis, although falling into the same cytoband 12q13.13, are considered as two different loci because (i) KRT rs12311316 is 1.5 MBp upstream of HOXC13; (ii) the associated SNPs at the KRT genes were in different LD blocks with the ones at HOXC13; and (iii) the association of rs12311316 remained at the same level of significance (P = 5.49e-8) after additionally adjusting for the HOXC13 SNP rs11170678.
The remaining two loci (PEX14 and PTK6) are currently less documented for their potential involvement in hair morphology. PEX14 at 1p36.22 encodes peroxisomal biogenesis factor 14, an essential component of the peroxisomal import machinery (36). This locus is the only locus which did not show any replication in all replication cohorts. At 20q13.33 the top associated SNP rs310642 is an intronic variant of PTK6; this is the only locus that showed considerably enhanced association when additional East Asian cohorts were included in the META: ALL. PTK6 encodes protein tyrosine kinase 6, a cytoplasmic nonreceptor protein kinase which may function as an intracellular signal transducer in epithelial tissues. Among several other known genes at 20q13.33, the expression level of KCNQ2 (encoding potassium voltage-gated channel subfamily Q member 2) has been related to down-hair sensitivity in mice (37).
Our META: Discovery also confirmed four previously known loci (1q21.3 TCHH/TCHHL1/LCE3E, 2q35 WNT10A, 4q21.21 FRAS1, and 10p14 LINC00708/GATA3) showing genome-wide significant association with hair shape. LCE3E is about 430 kBp upstream of TCHHL1 and thus considered as the same locus, and our data support a possible independent residual effect of LCE3E as reported in an earlier study (6). The TCHH/TCHHL1/LCE3E gene variants showed the largest effect with hair shape in our European cohorts; the association is greatly enhanced in the META: All at hundreds order of magnitude higher level than any other loci; and its predictive value is top-ranked in all genes studied. These findings are highly consistent with two previous GWAS of hair morphology (7,8). Therefore, this gene represents the major gene of hair shape in European and Latin American populations. Its effect may also be extrapolated to Sub-Saharan African and South Asian populations as suggested by our prediction analysis of the 1000-Genomes Project subjects, while no effect is seen in East Asians in our and previous data (9). Its effect is secondary in East Asian-European admixed Uyghurs after the East Asian-specific effect of EDAR as shown previously (9), which we used as replication set in our study. The association between gene variants at WNT10A, FRAS1, and LINC00708/GATA3 and hair morphology has been described in a previous GWAS in over 6000 Latin Americans (8), which we use as replication sample in our study. These gene variants also showed genome-wide significant association with hair shape in the META: Discovery so that it may be assumed that the effect seen in Latin Americans derives from the partial European admixture. Interestingly, by looking up our previously published data (15), we found a nominally significant association of rs17646946 in TCHH with male pattern baldness, indicating a potential link between genetic susceptibility to human hair loss and hair morphology.
The META: Discovery also confirmed the other three previously known loci at nominal significance, i.e. EDAR, OFCC1, and PRSS53. In respect of EDAR, this is explained by the sole use of Europeans. A recent GWAS in 2899 Han Chinese and 709 Uyghurs showed that EDAR was the only predominant gene affecting hair morphology in East Asians (9). These samples are included in the current study as replication cohorts. In the META: Discovery, the EDAR variant rs3827760 showed nominal significant association with hair curliness in RS (not available in the QIMR and the TwinsUK due to low frequency), and as expected, the straight hair associated G allele had a low frequency in our European samples and increasingly higher frequencies in the Uyghurs and the Han Chinese. This indicates that the effect of EDAR persists in Europeans although its frequency is low. The association between PRSS53 variants and hair curliness is also reported by the GWAS of Latin Americans (top-associated SNP rs11150606) (8). In our META: Discovery, the noted SNP also showed nominally significant association with hair shape variation. The involvement of OFCC1 in hair morphology was discovered by a previous web-based, participant-driven GWAS in Europeans (top-associated SNP rs1556547) (6). The noted SNP showed a border-line genome-wide significant association in the META: Discovery and a genome-wide significant association in a multivariable analysis, confirming the previous finding.
Basmanav et al., recently reported that mutations in PADI3, TGM3 (transglutaminase 3) and TCHH cause spun glass hair syndrome, characterized by silvery, blond, or straw-colored scalp hair that is dry, frizzy, and wiry (18). A nonsynonymous homozygous mutation in PADI3 (c.1372C > A; p.Pro458Thr) was found to be likely causative for congenital anonychia and uncombable sparse hair (38). PADI3 and TCHH have been described above while the most significant SNP among 923 SNPs within 100kb up- and down-stream of TGM3 on chromosome 20p13 (rs11696560, crude P = 0.004) did not survive the Bonferroni correction (adjusted P > 0.05).
Our data suggest that the genetic architecture of hair morphology in East Asians is substantially different than the rest of the world. Except TCHHL1 rs17646946, none of the 12 SNPs showed significant association in UYG (Asian-European admixed Uyghurs) and the PTK6 rs310642 was the only locus with nominally significant association in TZL (East Asian Han Chinese). This likely is explained by the fact that the three cohorts used in the discovery meta-analysis of GWAS were all of European origin, and these newly identified SNPs have only subtle effects in East Asians, if any. However, a unique pattern of allelic distribution was observed in the East Asia populations of the 1000-Genomes Project panel as the non-straight hair associated alleles from 8 out of the 12 genome-wide associated SNPs from our European discovery GWAS showed a high degree of frequency in the East Asian populations. This strongly indicates that genetic background of hair curliness in East Asian populations is substantially different to the rest of the world. For example, the straight hair associated A allele of rs17646946 in TCHHL1 is the most frequent allele in Europeans (∼24.3%) and less frequent in Americans (∼11.2%), Africans (∼11.0%), South Asians (∼10.0%), but it is nearly absent in East Asian (<0.2%) in the 1000-Genomes Project panel, which is also similar to the frequency distribution observed in our multiple ethnic cohorts, i.e. no observation in Chinese samples. These data indicate that either the A-allele has little or no effect in East Asian populations or its effect is nearly completely masked by East Asian specific gene variants, for instance from the EDAR gene. However, we were surprised to see that 10 of the 12 SNPs we identified with genome-wide significant association in our European GWAS discovery analysis did show considerable variation in Africans. This preliminary finding may indicate a link between the genetics underlying hair morphology in Europeans and Africans. Clearly, more work to unveil hair shape variation in Asians but also in Africans is needed in the future.
A recent hair curliness prediction model-based logistic regression using three SNPs from three genes i.e. rs11803731 (TCHH), rs7349332 (WNT10A) and rs1268789 (FRAS1) as predictors in 528 Polish Europeans explained 8.2% of the trait variance (Nagelkerke r2) with an estimated AUC of 0.62 (11). Our prediction model using 14 SNPs in 14 genes explained 10% of the trait variance (Nagelkerke r2) with a self-cross-validated AUC value of 0.66 in QIMR and an AUC value of 0.64 as validated in an independent European cohort (ERF). Therefore, our study represents an improvement, albeit not large, in the predictability of hair shape from genotype data, providing further promises for future applications in forensics and anthropology. Although the AUC value has not reached those previously obtained for eye and hair colour categories, which range from 0.74 to 0.95 depending on the eye/hair color category (39), our model provided informative prediction for a fraction of tested European subjects (∼7%), i.e. the subjects predicted with large or small probabilities of non-straight hair. Furthermore, our prediction model may be generalizable to non-Europeans except Asians as the distribution of the predicted probabilities in the African and American subjects from the 1000-Genomes Project panel is consistent with known global distribution of hair curliness (2) while its predictive capacity in East Asians appears limited. In any case, the generalizability of our findings needs to be evaluated in additional sizable Non-European cohorts.
In this study, we considered three broad curliness levels to capture the major dimension of the variation in hair curl, but likely missed some specific hair shape features such as frizzy hair. Future studies with detailed phenotypic information are needed to find genes that are specific for these additional hair features commonly found in Africans, as well as New Guinean and Australian Aborigines. Finally, although several genes have been suggested as promising functional candidates in the associated loci, there may well exist alternative interpretations, e.g. long-range functional connections via chromatin-loop formation (40,41). Therefore, future functional evaluations of the causal variants and genes are warranted.
In conclusion, with the 8 novel loci identified here and the 8 previously known loci confirmed here, we have substantially improved the human genetic knowledge of head hair shape variation in Europeans and beyond. We have increased the accuracy of predicting hair shape phenotypes from DNA genotypes over a previous model, which is relevant for forensics and cosmetics. Moreover, with newly reported hair shape genes and DNA variants we provide targets for future functional studies to further unveil the molecular basis of this externally visible trait expressing variation in people from around the world.
Materials and Methods
Queensland Institute of Medical Research study
After phenotype and genotype quality control, data were available for 10 607 participants. The data available for this cohort were collected over a number of research studies. All participants, and where appropriate their parent or guardian, gave informed consent, and all studies were approved by the Queensland Institute of Medical Research (QIMR) Berghofer Human Research Ethics Committee. Hair curliness was collected via questionnaires using 3 levels (Straight, Wavy, and Curly). Participants were genotyped on the Illumina Human610-Quad and Core + Exome SNP chips. These samples were genotyped in the context of a larger genome-wide association project that resulted in the genotyping of 28 028 individuals using the Illumina 317, 370, 610, 660, Core + Exome, PsychChip, Omni2.5 and OmniExpress SNP chips which included data from twins, their siblings and their parents. Genotype data were screened for genotyping quality (GenCall < 0.7), SNP and individual call rates (< 0.95), HWE failure (P < 1e-6) and MAF (< 0.01). As these samples were genotyped in the context of a larger project, the data were integrated with the larger QIMR genotype project and the data were checked for pedigree, sex and Mendelian errors and for non-European ancestry. As the QIMR genotyping project included data from the multiple chip sets, to avoid introducing bias to the imputed data individuals genotyped on the Human Hap Illumina chips (the 317, 370, 610, 660K chips) were imputed separately from those genotyped on the Omni chips (the Core + Exome, PsychChip, Omni2.5 and OmniExpress chips). Individuals were imputed to the Haplotype Reference Consortium (HRC.1.1) using a set of SNPs common to the first generation genotyping platforms (N ∼ 278 000). Imputation was performed on the Michigan Imputation Server using the SHAPEIT/minimac Pipeline.
Rotterdam study
The Rotterdam study (RS) is a population based cohort study of 14 926 participants aged 45 years and older, living in the same suburb of Rotterdam, the Netherlands (42). The Rotterdam Study has been approved by the medical ethics committee according to the Population Study Act Rotterdam Study and written informed consent was obtained from all participants. The present study includes 2809 participants of Dutch European ancestry, for whom high-resolution digital photographs of frontal and the left side of the face were taken. Using the portrait and side photos, 2 independent graders rated hair curliness in a 6-point scale levels according to a previous study (6): straight, slightly wavy, wavy, big curls, small curls and frizzy hair. The graders discussed the grades in advance and practiced 50 photos together. Exclusion criteria included baldness, hair length shorter than 2 cm (except for frizzy hair), hair not visible on the photograph and perm. We evaluated inter-rater agreement between two graders using weighted Cohen’s Kappa coefficient. Scoring concordance between the two graders was reasonably high (kappa = 0.70). For consistency with other cohorts, we combined the 6 levels into 3 levels, i.e. straight (straight and slightly wavy), wavy (wavy), and curly (big curls, small curls) in the subsequent genetic analysis. Genotyping was carried out using the Infinium II HumanHap 550K Genotyping BeadChip version 3 (Illumina, San Diego, California USA). Collection and purification of DNA have been described previously (43). All SNPs were imputed using MACH software (www.sph.umich.edu/csg/abecasis/MaCH/) based on the 1000-Genomes Project reference populaton information (44). Genotype and individual quality controls have been described in detail previously (45). After all quality controls, the current study included a total of 8 397 532 autosomal SNPs (MAF > 0.01, imputation R2 > 0.3, SNP call rate > 0.97, HWE > 1e-4) and 2809 individuals (individual call rate > 0.95, pair-wise IBD coefficient < 0.25, excluding x-mismatches and outliers from MDS analysis).
TwinsUK study
The TwinsUK study included 3347 phenotyped participants (all female and all of Caucasian ancestry) within the TwinsUK adult twin registry based at St. Thomas’ Hospital in London. Twins largely volunteered unaware of the hair form research interests at the time of enrollment and gave fully informed consent under a protocol reviewed by the St. Thomas’ Hospital Local Research Ethics Committee. Hair curliness was collected via questionnaires using 6 levels as described above (straight, slightly wavy, wavy, big curls, small curls and frizzy hair). Genotyping of the TwinsUK cohort was done with a combination of Illumina HumanHap300 and HumanHap610Q chips. Intensity data for each of the arrays were pooled separately and genotypes were called with the Illuminus32 calling algorithm, thresholding on a maximum posterior probability of 0.95 as previously described (46). Imputation was performed using the IMPUTE 2.0 software package using haplotype information from the 1000 Genomes Project (Phase 1, integrated variant set across 1092 individuals, v2, March 2012). After all quality controls, the current study included a total of 9.35 million autosomal SNPs (MAF > 0.01, imputation R2 > 0.3, SNP call rate > 0.95, HWE > 1e-6) and 3347 individuals.
Erasmus Rucphen family study
Erasmus Rucphen family (ERF) is a cohort derived from a region in the southwest of the Netherlands (47). The population was established in the middle of the 18th century by a limited number of founders, has experienced minimal immigration and emigration, and has exponentially increased in size in the last century. The ERF study was instituted in this population to determine the genes underlying quantitative trait variation in humans (47). Interviews at the time of blood sampling were performed by medical practitioners. The Medical Ethics Committee of the Erasmus University Medical Center approved the ERF study protocol (approval #MEC 213.575/2002/114) and all participants, or their legal representatives, provided written informed consent. In the present study, we collected 3-level hair curliness data (Straight, Wavy, and Curly) for 977 subjects using questionnaires.
Genotyping in ERF was performed using Illumina 318/370 K, Affymetrix 250 K, and Illumina 6 K micro-arrays. All SNPs were imputed using MACH software (www.sph.umich.edu/csg/abecasis/MaCH/) based on the 1000-Genomes reference population information (44). Individuals were excluded for excess autosomal heterozygosity, mismatches between called and phenotypic gender, and if there were outliers identified by an IBS clustering analysis. The exclusion criteria for SNPs were Hardy–Weinberg equilibrium (HWE) P < 1e-6 or SNP call rate < 98%.
Polish (POL) study
Samples involved 635 unrelated individuals containing 307 females and 328 males (>18 years old) from Poland. Hair morphology of the participants has been assessed by a physician specializing in dermatology using a simple three-point scale that categorized hair as straight, wavy or curly. Hair types corresponded to the previously proposed Loussouarn hair morphology classification system (2). Categories I and II define straight hair, type III reflects wavy hair, types IV-V refer to curly hair typical for people of European ancestry. Written informed consent was obtained from all the samples donors, and the study protocol was approved by the Commission on Bioethics of the Regional Board of Medical Doctors in Krakow (48 KBL/OIL/2008).
Samples were genotyped with next-generation sequencing using Ion AmpliSeqTM Technology and Ion PGMTM system (Thermo Fisher Scientific). 35 SNPs under the replication study were genotyped within 96-SNPs panel with primers designed using Ion AmpliSeq Designer and Thermo Fisher Scientific support. DNA (1–5 ng) was amplified in 10 μl of PCR reaction and one primer pool. DNA libraries were prepared using Ion AmpliSeq Library Kits, quantified with Agilent High Sensitivity DNA Kit (Agilent Technologies) or Qubit dsDNA High-Sensitivity Assay Kit (Thermo Fisher Scientific) and normalized to 100 pM. Thirty-two of 100 pM DNA library samples were combined in equal ratios and subjected to template preparation with Ion PGM HiQ OT2 Kit and Ion OneTouch 2 System (Thermo Fisher Scientific). Templating was performed according to manufacturer’s directions with the exception of 100 pM DNA library volume that was increased from 2 to 5μl. Sequencing was conducted with Ion PGM Hi-Q Sequencing Kit using Ion 318 Chip v2, 200 bp read chemistry and 520 flows per run. Raw data were analysed using Torrent Server v5.0.5 and DNA variants were typed using variantCaller v5.0.4.0 (Thermo Fisher Scientific).
CANDELA study
CANDELA samples (48) consist of 6630 volunteers recruited in five Latin American countries (Brazil, Colombia, Chile, México and Perú). Ethics approval was obtained from: the Universidad Nacional Autónoma de México (México), the Universidad de Antioquia (Colombia), the Universidad Peruana Cayetano Heredia (Perú), the Universidad de Tarapacá (Chile), the Universidade Federal do Rio Grande do Sul (Brazil) and the University College London (UK). All participants provided written informed consent. These individuals were genotyped on Illumina’s Omni Express BeadChip. After applying quality control filters 669, 462 SNPs and 6357 individuals were retained for further analyses (2922 males, 3435 females). Average admixture proportions for this sample were estimated as: 48% European, 46% Native American and 6% African, but with substantial inter-individual variation. In these individuals we performed a categorical assessment (in men and women) of scalp hair shape (curliness), recorded by physical examination of the volunteers. Hair curliness was scored as 1-straight, 2-wavy, 3-curly or 4-frizzy. Individuals with frizzy hair were excluded from the final hair GWAS, as it was a rare phenotype (2.4%). After all genomic and phenotypic quality controls this study included 6238 individuals. The genetic PCs were obtained from the LD-pruned dataset of 93 328 SNPs using PLINK 1.9. These PCs were selected by inspecting the proportion of variance explained and checking scatter and scree plots. The final imputed dataset used in the GWAS analyses included genotypes for 9 143 600 SNPs using the 1000 Genomes Phase I reference panel. Association analysis on the imputed dataset were performed using the best-guess imputed genotypes in PLINK 1.9 using linear regression with an additive genetic model incorporating age, sex and 5 genetic PCs as covariates.
US study
US sample consists of 743 unrelated volunteers (498 females, 245 males) recruited in the USA, who were of European (98%), and non-European ancestry (2% South American/South Asian/Middle Eastern). In these individuals we collected 3-level hair curliness data (Straight, Wavy, and Curly) on what hair type they had in their 20’s, as well as country of birth, parents’ country of birth, age and sex using self-reported questionnaires. Genotyping was performed by massive parallel sequencing, using an in house custom library designed assay that consisted of candidate hair structure variants, on a MiSeq FGx (in RUO mode) desktop sequencer using reagent kit v2, 300 cycle – 2 ×150 bp output (Illumina). Raw data were separated into individual samples based on the ligated adapter tags using the MiSeq™ reporter software. Sequences within these FASTQ files were aligned to a custom reference sequence using a Burrows-Wheeler alignment (BWA) algorithm (49) and the maximum entropy method (mem) algorithm. The sequence alignment/map (SAM) file generated was converted using SAMtools (50) into a BAM file that was analysed by the Genome Analysis Toolkit (GATK) (51) to target specific SNP/variant positions and their coverage in these aligned sequences. The requested variant sites were then reported to a .vcf file for final genotype calls. All participants from this study were collected with approval from the Institutional review board of Indiana University IRB00000222; Protocol number 1409306349.
Chinese Taizhou longitudinal study and Xinjiang Uyghur study
The Han Chinese samples were collected in Taizhou, Jiangsu Province in 2014, as part of the Taizhou Longitudinal Study (52). In total, 2899 individuals (including 1038 males and 1861 females, with an age range of 31–87) were enrolled. The Institutional Research Board at Fudan University approved the Han Chinese study protocols. The Xinjiang Uyghur (UYG) samples were collected at Xinjiang Medical University in 2013–2014. In total, 709 individuals (including 276 males and 433 females, with an age range of 17–25) were enrolled. The research was conducted with the official approval from the Ethics Committee of the Shanghai Institutes for Biological Sciences, Shanghai, China. All participants had provided written consent. In both Taizhou Longitudinal (TZL) and UYG, hair curliness was rated on a three-point scale (straight, wavy, and curly) by investigators. All samples were genotyped using the Illumina HumanOmniZhongHua-8 chips, which interrogates 894 517 SNPs. Individuals with more than 5% missing data, related individuals, and the ones that failed the X-chromosome sex concordance check or had ethnic information incompatible with their genetic information were excluded. SNPs with more than 2% missing data, with a minor allele frequency smaller than 1%, and the ones that failed the Hardy–Weinberg deviation test (P < 1e-5) were also excluded. After applying these filters, we obtained a dataset of 2899 samples with 776 213 SNPs for the Han Chinese, and 709 samples with 810, 648 SNPs for the Uyghurs. The chip genotype data were firstly phased using SHAPEIT (53). IMPUTE2 (54) was then used to impute genotypes at ungenotyped SNPs using the 1000 Genomes Phase 3 data as reference. Finally, for the Uyghur sample, a total of 6 414 304 imputed SNPs passed quality control and were combined with 810, 648 genotyped SNPs for further analyses. For the Han Chinese sample, a total of 6 343 243 imputed SNPs passed quality control and were combined with 776 213 genotyped SNPs for association analysis.
Statistical analyses
Three GWASs in three cohorts of European origin (QIMR, TwinsUK and RS, totalling 16 763 subjects) were independently carried out. The GWAS in the RS was conducted in PLINK v1.9 (55) using linear regression (considering 3 curliness levels as a continuous trait) adjusted for age, sex and 4 genetic principal components, assuming an additive allele effect. The GWAS in QIMR was conducted using Rare Metal Worker using linear regression (considering curliness levels as a continuous trait) adjusted for age, sex, data source (self vs research nurse report) and wave of data collection assuming an additive allele effect. The GWAS in TwinsUK was conducted using in GEMMA (56) using linear regression (considering curliness levels as a continuous trait) adjusted for age and sex assuming an additive allele effect. All GWASs outputs were meta-analysed using inverse variance fixed-effect meta-analysis using the METAL software (57). P-values equal or smaller than 5e-8 in the meta-analysis was considered as genome-wide significant. The inflation factor was close to 1.0 (λ = 1.03) and not further considered. GWAS results were visualized using Manhattan plots and Q-Q plots. Regional LD analysis was conducted using HaploView and regional Manhattan plots were produced using LocusZoom. Allele frequency distribution in 2504 subjects from the 1000 Genomes Project was visualized using MapViewer.
All SNPs with genome-wide significant association in the discovery meta-analysis were selected for replication with a focus on the top-associated SNP per region. The replication was carried out separately in ERF, POL, CANDELA, US, UYG, and TZL (totaling 12 201 samples), using linear regression adjusted for age and sex, assuming an additive allele effect. We then conducted an inverse variance fixed effect meta-analysis in all 7 Non-Asian cohorts as well as in all 9 cohorts.
To further study the involved functions and regulation relationships of the genome-wide significantly associated SNP markers (P < 5e-8), 172 genes located within 100kb up- and down-stream of the SNPs were selected to perform biological process enrichment analysis based on the Gene Ontology (GO) database. The enrichment analysis was performed using the MATLAB R2015a (The MathWorks, Inc., Natick, MA, USA).
Fine-tuned individual-level data analyses were conducted in 6068 unrelated QIMR participants (mean age 40 year) of European ancestry. A multiple logistic regression analysis was conducted to access the independent effect of 16 SNPs. The model considers hair curliness as a binary phenotype (straight vs. non-straight), 12 top-associated SNPs (one per region) from current study, and 4 SNPs from previous association studies (EDAR rs3827760, OFCC1 rs1556547, PRSS53 rs11150606, LCE3E rs499697), together with sex and age as explanatory factors. The prediction analysis was conducted considering 14 SNPs showed significant association with hair morphology in multivariable analysis, sex and age as predictors using binary logistic regression model in QIMR. Markers were ordered according to their contribution to model. We then applied the model developed in the QIMR cohort to participants from the ERF cohort. Prediction accuracy was estimated using the area under the receiver operating characteristic (ROC) curves, or AUC. AUC is the integral of ROC curves and ranges from 0.5 representing total lack of prediction (no better than flipping a coin) to 1.0 representing perfect prediction. Sensitivities and specificities were calculated using confusion matrices considering the predicted probability > t as the predicted shape type, where t optimized the sum of sensitivity and specificity. Finally, we applied our prediction model to the 2504 worldwide subjects from the 1000-Genomes Project panel (58). A SNP–SNP interaction analysis was conducted for 3632 SNPs with P-values < 1e-4 from the discovery meta-analysis of GWASs. Linear regression analysis was used for interaction testing, y ∼ sex + age + SNP1 + SNP2 + SNP1*SNP2, where SNP1*SNP2 was considered as the interaction term at the multiplicative scale. The analysis was conducted in a pair-wise manner for all selected SNPs and the resultant P-values were adjusted using the Bonferroni correction (crude P = 3.8e-9 corresponds to adjusted P = 0.05). SNP-sex and SNP-age interaction analysis was conducted in QIMR using linear model by adding the interaction term at multiplicative scale. All statistical analyses and result visualization were conducted in R version 3.2.3 unless otherwise specified.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors thank all sample donors for their contribution to this project. MK additionally thanks Maren von Köckritz-Blickwede for discussions on PAD knockout mice.
The generation and management of GWAS genotype data for the Rotterdam Study (RS I, RS II, RS III) was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Rotterdam, The Netherlands.
QIMR gratefully acknowledges the participants and their families. We would like to thank Marlene Grace and Ann Eldridge for data collection and Scott Gordon for data management.
For CANDELA study, the following institutions kindly provided facilities for the assessment of volunteers: Escuela Nacional de Antropología e Historia and Universidad Nacional Autónoma de México (México); Pontificia Universidad Católica del Perú, Universidad de Lima and Universidad Nacional Mayor de San Marcos (Perú); Universidade Federal do Rio Grande do Sul (Brazil); 13° Companhia de Comunicações Mecanizada do Exército Brasileiro (Brazil).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest statement. None declared.
Funding
European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 285487 (EUROFORGEN-NoE).
Rotterdam Study: Erasmus MC University Medical Center, Rotterdam, Unilever and funds from the Netherlands Genomics Initiative/Netherlands Organization of Scientific Research (NWO) within the framework of the Netherlands Consortium of Healthy Ageing (NCHA). The generation and management of GWAS genotype data for the Rotterdam Study (RS I, RS II, RS III) was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Rotterdam, The Netherlands. The GWAS datasets are supported by the Netherlands Organisation of Scientific Research NWO Investments (nr. 175.010.2005.011, 911–03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014–93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project nr. 050–060-810. Author FL is supported by the Erasmus University Rotterdam (EUR) fellowship and a Chinese recruiting program ‘The National Thousand Young Talents Award’. The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam.
QIMR study: Australian National Health and Medical Research Council (NHMRC) grants 241944, 339462, 389927, 389875, 389891, 389892, 389938, 442915, 442981, 496739, 552485, 552498 and Australian Research Council grants A7960034, A79906588, A79801419, DP0770096, DP0212016, DP0343921 for building and maintaining the adolescent twin family resource through which samples were collected. SEM is supported by an NHMRC fellowship APP1103623.
TwinsUK study: Wellcome Trust, Medical Research Council, European Union (FP7/2007–2013), the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London.
ERF Study: Joint grant from the Netherlands Organization for Scientific Research (NWO, 91203014), the Center of Medical Systems Biology (CMSB), Hersenstichting Nederland, Internationale Stichting Alzheimer Onderzoek (ISAO), Alzheimer Association project number 04516, Hersenstichting Nederland project number 12F04(2).76, and the Interuniversity Attraction Poles (IUAP) program. As a part of EUROSPAN (European Special Populations Research Network) ERF was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006–01947) and also received funding from the European Community's Seventh Framework Programme (FP7/2007–2013)/grant agreement HEALTH-F4–2007-201413 by the European Commission under the programme ‘Quality of Life and Management of the Living Resources’ of 5th Framework Programme (no. QLG2-CT-2002–01254). High-throughput analysis of the ERF data was supported by joint grant from Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043).
CANDELA study: Leverhulme Trust (F/07 134/DF to ARL), BBSRC (BB/I021213/1 to ARL); Universidad de Antioquia, Colombia (CPT-1602, Estrategia para sostenibilidad 2016–2018 grupo GENMOL to GB).
TZL and UYG studies: National Natural Science Foundation of China (NSFC) (grant numbers 91631307 to SW, 30890034 and 31271338 to LJ, 91731303 and 31771388 to SX), the Science and Technology Commission of Shanghai Municipality (grant number 16JC1400504 to LJ and SW), the Chinese Academy of Sciences (grant number XDB13041000 to SW and SX), the National Basic Research Program (2015FY111700 to LJ and SW), the National Program for Top-notch Young Innovative Talents of The ‘Wanren Jihua’ Project to SX, the National Thousand Young Talents Award to SW, Max Planck-CAS Paul Gerson Unna Independent Research Group Leadership Award to SW.
POL study: National Science Centre in Poland SONATA 8 no 2014/15/D/NZ8/00282. Foundation for Polish Science within the programme START 2017 to EP.
US study: US National Institute of Justice (NIJ) Grant 2014-DN-BX-K031 and the US Department of Defense (DOD) DURIP-66843LSRIP-2015.
Funding to pay the Open Access publication charges for this article was provided by the National Thousand Young Talents Award to FL.
References
- 1. Loussouarn G., Lozano I., Panhard S., Collaudin C., El Rawadi C., Genain G. (2016) Diversity in human hair growth, diameter, colour and shape. An in vivo study on young adults from 24 different ethnic groups observed in the five continents. Eur. J. Dermatol., 26, 144–154. [DOI] [PubMed] [Google Scholar]
- 2. Loussouarn G., Garcel A.L., Lozano I., Collaudin C., Porter C., Panhard S., Saint-Leger D., de La Mettrie R. (2007) Worldwide diversity of hair curliness: a new method of assessment. Int. J. Dermatol., 46 Suppl 1, 2–6. [DOI] [PubMed] [Google Scholar]
- 3. Medland S.E., Zhu G., Martin N.G. (2009) Estimating the heritability of hair curliness in twins of European ancestry. Twin Res. Hum. Genet., 12, 514–518. [DOI] [PubMed] [Google Scholar]
- 4. Kayser M., de Knijff P. (2011) Improving human forensics through advances in genetics, genomics and molecular biology. Nat. Rev. Genet., 12, 179–192. [DOI] [PubMed] [Google Scholar]
- 5. Kayser M. (2015) Forensic DNA Phenotyping: Predicting human appearance from crime scene material for investigative purposes. Forensic Sci. Int. Genet., 18, 33–48. [DOI] [PubMed] [Google Scholar]
- 6. Eriksson N., Macpherson J.M., Tung J.Y., Hon L.S., Naughton B., Saxonov S., Avey L., Wojcicki A., Pe'er I., Mountain J., Gibson G. (2010) Web-based, participant-driven studies yield novel genetic associations for common traits. PLoS Genet., 6, e1000993.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Medland S.E., Nyholt D.R., Painter J.N., McEvoy B.P., McRae A.F., Zhu G., Gordon S.D., Ferreira M.A.R., Wright M.J., Henders A.K.. et al. (2009) Common variants in the trichohyalin gene are associated with straight hair in Europeans. Am. J. Hum. Genet., 85, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adhikari K., Fontanil T., Cal S., Mendoza-Revilla J., Fuentes-Guajardo M., Chacon-Duque J.C., Al-Saadi F., Johansson J.A., Quinto-Sanchez M., Acuna-Alonzo V.. et al. (2016) A genome-wide association scan in admixed Latin Americans identifies loci influencing facial and scalp hair features. Nat. Commun., 7, 10815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu S.J., Tan J.Z., Yang Y.J., Peng Q.Q., Zhang M.F., Li J.X., Lu D.S., Liu Y., Lou H.Y., Feng Q.D.. et al. (2016) Genome-wide scans reveal variants at EDAR predominantly affecting hair straightness in Han Chinese and Uyghur populations. Hum. Genet., 135, 1279–1286. [DOI] [PubMed] [Google Scholar]
- 10. Fujimoto A., Kimura R., Ohashi J., Omi K., Yuliwulandari R., Batubara L., Mustofa M.S., Samakkarn U., Settheetham-Ishida W., Ishida T.. et al. (2008) A scan for genetic determinants of human hair morphology: EDAR is associated with Asian hair thickness. Hum. Mol. Genet., 17, 835–843. [DOI] [PubMed] [Google Scholar]
- 11. Pospiech E., Karlowska-Pik J., Marcinska M., Abidi S., Andersen J.D., Berge M.V.D., Carracedo A., Eduardoff M., Freire-Aradas A., Morling N.. et al. (2015) Evaluation of the predictive capacity of DNA variants associated with straight hair in Europeans. Forensic Sci. Int. Genet., 19, 280–288. [DOI] [PubMed] [Google Scholar]
- 12. Thibaut S., Barbarat P., Leroy F., Bernard B.A. (2007) Human hair keratin network and curvature. Int. J. Dermatol., 46 Suppl 1, 7–10. [DOI] [PubMed] [Google Scholar]
- 13. Westgate G.E., Ginger R.S., Green M.R. (2017) The biology and genetics of curly hair. Exp. Dermatol., 26, 483–490. [DOI] [PubMed] [Google Scholar]
- 14. Heilmann S., Kiefer A.K., Fricker N., Drichel D., Hillmer A.M., Herold C., Tung J.Y., Eriksson N., Redler S., Betz R.C.. et al. (2013) Androgenetic alopecia: identification of four genetic risk loci and evidence for the contribution of WNT signaling to its etiology. J. Invest. Dermatol., 133, 1489–1496. [DOI] [PubMed] [Google Scholar]
- 15. Liu F., Hamer M.A., Heilmann S., Herold C., Moebus S., Hofman A., Uitterlinden A.G., Nothen M.M., van Duijn C.M., Nijsten T.E.. et al. (2016) Prediction of male-pattern baldness from genotypes. Eur. J. Hum. Genet., 24, 895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nachat R., Mechin M.C., Charveron M., Serre G., Constans J., Simon M. (2005) Peptidylarginine deiminase isoforms are differentially expressed in the anagen hair follicles and other human skin appendages. J. Invest. Dermatol., 125, 34–41. [DOI] [PubMed] [Google Scholar]
- 17. Mechin M.C., Enji M., Nachat R., Chavanas S., Charveron M., Ishida-Yamamoto A., Serre G., Takahara H., Simon M. (2005) The peptidylarginine deiminases expressed in human epidermis differ in their substrate specificities and subcellular locations. Cell. Mol. Life. Sci., 62, 1984–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ü Basmanav F.B., Cau L., Tafazzoli A., Mechin M.C., Wolf S., Romano M.T., Valentin F., Wiegmann H., Huchenq A., Kandil R.. et al. (2016) Mutations in three genes encoding proteins involved in hair shaft formation cause uncombable hair syndrome. Am. J. Hum. Genet., 99, 1292–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nanba D., Toki F., Barrandon Y., Higashiyama S. (2013) Recent advances in the epidermal growth factor receptor/ligand system biology on skin homeostasis and keratinocyte stem cell regulation. J. Dermatol. Sci., 72, 81–86. [DOI] [PubMed] [Google Scholar]
- 20. Vu H.L., Rosenbaum S., Capparelli C., Purwin T.J., Davies M.A., Berger A.C., Aplin A.E. (2016) MIG6 Is MEK Regulated and Affects EGF-Induced Migration in Mutant NRAS Melanoma. J. Invest. Dermatol., 136, 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ballaro C., Ceccarelli S., Tiveron C., Tatangelo L., Salvatore A.M., Segatto O., Alema S. (2005) Targeted expression of RALT in mouse skin inhibits epidermal growth factor receptor signalling and generates a Waved-like phenotype. EMBO Rep., 6, 755–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bartolke R., Heinisch J.J., Wieczorek H., Vitavska O. (2014) Proton-associated sucrose transport of mammalian solute carrier family 45: an analysis in Saccharomyces cerevisiae. Biochem. J., 464, 193–201. [DOI] [PubMed] [Google Scholar]
- 23. Luetteke N.C., Qiu T.H., Peiffer R.L., Oliver P., Smithies O., Lee D.C. (1993) TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell, 73, 263–278. [DOI] [PubMed] [Google Scholar]
- 24. Johnson K.R., Lane P.W., Cook S.A., Harris B.S., Ward-Bailey P.F., Bronson R.T., Lyons B.L., Shultz L.D., Davisson M.T. (2003) Curly bare (cub), a new mouse mutation on chromosome 11 causing skin and hair abnormalities, and a modifier gene (mcub) on chromosome 5. Genomics, 81, 6–14. [DOI] [PubMed] [Google Scholar]
- 25. Lee D., Cross S.H., Strunk K.E., Morgan J.E., Bailey C.L., Jackson I.J., Threadgill D.W. (2004) Wa5 is a novel ENU-induced antimorphic allele of the epidermal growth factor receptor. Mamm. Genome, 15, 525–536. [DOI] [PubMed] [Google Scholar]
- 26. Mohri Y., Kato S., Umezawa A., Okuyama R., Nishimori K. (2008) Impaired hair placode formation with reduced expression of hair follicle-related genes in mice lacking Lgr4. Dev. Dyn., 237, 2235–2242. [DOI] [PubMed] [Google Scholar]
- 27. Zak M., van Oort T., Hendriksen F.G., Garcia M.I., Vassart G., Grolman W. (2016) LGR4 and LGR5 Regulate Hair Cell Differentiation in the Sensory Epithelium of the Developing Mouse Cochlea. Front. Cell. Neurosci., 10, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin Z., Chen Q., Shi L., Lee M., Giehl K.A., Tang Z., Wang H., Zhang J., Yin J., Wu L.. et al. (2012) Loss-of-function mutations in HOXC13 cause pure hair and nail ectodermal dysplasia. Am. J. Hum. Genet., 91, 906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Langbein L., Schweizer J. (2005) Keratins of the human hair follicle. Int. Rev. Cytol., 243, 1–78. [DOI] [PubMed] [Google Scholar]
- 30. Giesen M., Gruedl S., Holtkoetter O., Fuhrmann G., Koerner A., Petersohn D. (2011) Ageing processes influence keratin and KAP expression in human hair follicles. Exp. Dermatol., 20, 759–761. [DOI] [PubMed] [Google Scholar]
- 31. Gao Y., Wang X., Yan H., Zeng J., Ma S., Niu Y., Zhou G., Jiang Y., Chen Y., Zhang M. (2016) Comparative Transcriptome Analysis of Fetal Skin Reveals Key Genes Related to Hair Follicle Morphogenesis in Cashmere Goats. PLoS One, 11, e0151118.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Callea M., Willoughby C.E., Nieminen P., Di Stazio M., Bellacchio E., Giglio S., Sani I., Vinciguerra A., Maglione M., Tadini G., Clarich G. (2015) Identification of a novel frameshift mutation in the EDAR gene causing autosomal dominant hypohidrotic ectodermal dysplasia. J. Eur. Acad. Dermatol. Venereol., 29, 1032–1034. [DOI] [PubMed] [Google Scholar]
- 33. Morgenthaler C., Diribarne M., Capitan A., Legendre R., Saintilan R., Gilles M., Esquerré D., Juras R., Khanshour A., Schibler L., Cothran G. (2017) A missense variant in the coil1A domain of the keratin 25 gene is associated with the dominant curly hair coat trait (Crd) in horse. Genet. Sel. Evol., 49, 85.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shah B.J., Jagati A.K., Gupta N.P., Dhamale S.S. (2015) Naegeli-Franceschetti-Jadassohn syndrome: A rare case. Indian Dermatol. Online J., 6, 403–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu J., Lin Y., Xu W., Li Z., Fan W. (2011) A mutation in the type II hair keratin KRT86 gene in a Han family with monilethrix. J. Biomed. Res., 25, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fuchs L., Bintein T., Laget P. (1976) [Development of tectum-striatum and striatum-tectum connections in the chick]. C. R. Seances Soc. Biol. Fil., 170, 553–557. [PubMed] [Google Scholar]
- 37. Schutze S., Orozco I.J., Jentsch T.J. (2016) KCNQ potassium channels modulate sensitivity of skin down-hair (D-hair) mechanoreceptors. J. Biol. Chem., 291, 5566–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsu C.-K., Romano M.T., Nanda A., Rashidghamat E., Lee J.Y.W., Huang H.-Y., Songsantiphap C., Lee J.Y.-Y., Al-Ajmi H., Betz R.C.. et al. (2017) Congenital anonychia and uncombable hair syndrome: coinheritance of homozygous mutations in RSPO4 and PADI3. J. Invest. Dermatol., 137, 1176–1179. [DOI] [PubMed] [Google Scholar]
- 39. Walsh S., Chaitanya L., Clarisse L., Wirken L., Draus-Barini J., Kovatsi L., Maeda H., Ishikawa T., Sijen T., de Knijff P.. et al. (2014) Developmental validation of the HIrisPlex system: DNA-based eye and hair colour prediction for forensic and anthropological usage. Forensic Sci. Int. Genet., 9, 150–161. [DOI] [PubMed] [Google Scholar]
- 40. Visser M., Kayser M., Palstra R.J. (2012) HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res., 22, 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smemo S., Tena J.J., Kim K.H., Gamazon E.R., Sakabe N.J., Gomez-Marin C., Aneas I., Credidio F.L., Sobreira D.R., Wasserman N.F.. et al. (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature, 507, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hofman A., Darwish Murad S., van Duijn C.M., Franco O.H., Goedegebure A., Ikram M.A., Klaver C.C., Nijsten T.E., Peeters R.P., Stricker B.H.. et al. (2013) The Rotterdam Study: 2014 objectives and design update. Eur. J. Epidemiol., 28, 889–926. [DOI] [PubMed] [Google Scholar]
- 43. Kayser M., Liu F., Janssens A.C.J.W., Rivadeneira F., Lao O., van Duijn K., Vermeulen M., Arp P., Jhamai M.M., van IJcken W.F.J.. et al. (2008) Three genome-wide association studies and a linkage analysis identify HERC2 as a human iris color gene. Am. J. Hum. Genet., 82, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Genomes Project C., Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. (2012) An integrated map of genetic variation from 1, 092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lango Allen H., Estrada K., Lettre G., Berndt S.I., Weedon M.N., Rivadeneira F., Willer C.J., Jackson A.U., Vedantam S., Raychaudhuri S.. et al. (2010) Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature, 467, 832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Small K.S., Hedman Å.K., Grundberg E., Nica A.C., Thorleifsson G., Kong A., Thorsteindottir U., Shin S.-Y., Richards H.B., Soranzo N.. et al. (2011) Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet., 43, 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pardo L.M., MacKay I., Oostra B., van Duijn C.M., Aulchenko Y.S. (2005) The effect of genetic drift in a young genetically isolated population. Ann. Hum. Genet., 69, 288–295. [DOI] [PubMed] [Google Scholar]
- 48. Ruiz-Linares A., Adhikari K., Acuna-Alonzo V., Quinto-Sanchez M., Jaramillo C., Arias W., Fuentes M., Pizarro M., Everardo P., de Avila F.. et al. (2014) Admixture in Latin America: geographic structure, phenotypic diversity and self-perception of ancestry based on 7, 342 individuals. PLoS Genet., 10, e1004572.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li H., Durbin R. (2010) Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics, 26, 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing S. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M.. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang X., Lu M., Qian J., Yang Y., Li S., Lu D., Yu S., Meng W., Ye W., Jin L. (2009) Rationales, design and recruitment of the Taizhou longitudinal study. BMC Public Health, 9, 223.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. O'Connell J., Gurdasani D., Delaneau O., Pirastu N., Ulivi S., Cocca M., Traglia M., Huang J., Huffman J.E., Rudan I.. et al. (2014) A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet., 10, e1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Howie B.N., Donnelly P., Marchini J. (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet., 5, e1000529.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J.. et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou X., Stephens M. (2012) Genome-wide efficient mixed-model analysis for association studies. Nat. Genet., 44, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Willer C.J., Li Y., Abecasis G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H.. et al. (2015) An integrated map of structural variation in 2, 504 human genomes. Nature, 526, 75–81. 10.1038/nature15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.