

Abstract
Bardet–Biedl syndrome (BBS) is an autosomal recessive ciliopathy characterized by developmental abnormalities and vision loss. To date, mutations in 21 genes have been linked to BBS. The products of eight of these BBS genes form a stable octameric complex termed the BBSome. Mutations in BBS8, a component of the BBSome, cause early vision loss, but the role of BBS8 in supporting vision is not known. To understand the mechanisms by which BBS8 supports rod and cone photoreceptor function, we generated animal models lacking BBS8. The loss of BBS8 protein led to concomitant decrease in the levels of BBSome subunits, BBS2 and BBS5 and increase in the levels of the BBS1 and BBS4 subunits. BBS8 ablation was associated with severe reduction of rod and cone photoreceptor function and progressive degeneration of each photoreceptor subtype. We observed disorganized and shortened photoreceptor outer segments (OS) at post-natal day 10 as the OS elaborates. Interestingly, loss of BBS8 led to changes in the distribution of photoreceptor axonemal proteins and hyper-acetylation of ciliary microtubules. In contrast to properly localized phototransduction machinery, we observed OS accumulation of syntaxin3, a protein normally found in the cytoplasm and the synaptic termini. In conclusion, our studies demonstrate the requirement for BBS8 in early development and elaboration of ciliated photoreceptor OS, explaining the need for BBS8 in normal vision. The findings from our study also imply that early targeting of both rods and cones in BBS8 patients is crucial for successful restoration of vision.
Introduction
Bardet–Biedl syndrome (BBS) is a pleiotropic ciliopathy often accompained by vision loss (1–6). A subset of gene products linked to BBS (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, BBIP1 or BBS18) interact to form an octameric complex termed the BBSome (7,8). In neurons, the BBSome promotes the trafficking of GPCRs to ciliary membranes (7,9). The proposed trafficking of GPCRs by the BBSome is supported by animal models that show mis-localization of Stomatin-like protein 3 (SLP3) in olfactory sensory neurons in the absence of BBS8 and melanin-concentrating hormone receptor 1 (MCHR1) in neurons lacking BBS2 and BBS4 (10,11).
The function of the BBSome in photoreceptor neurons is poorly understood despite multiple mutations in BBS subunits associated with blinding diseases. In highly polarized photoreceptor cells, the major GPCR rhodopsin is present in the outer segment (OS). The outer segment is an elaborated stack of ciliary membranes that house proteins involved in the phototransduction cascade. The outer segment is attached to the photoreceptor cytoplasm (inner segment) by the connecting cilium, a narrow conduit through which proteins synthesized in the cytoplasm move to the OS (12,13). The entire photoreceptor OS is renewed every 10 days as new disks are formed at the base of the OS, while apical disk membranes are phagocytized by the retinal pigment epithelium (RPE) (14). The constant renewal of the OS requires a robust and tightly regulated protein trafficking system and is crucial for the proper function of photoreceptors. By analogy to its role in trafficking GPCRs in neurons, it was thought that rhodopsin, a GPCR, is also transported to the outer segment by a mechanism involving the BBSome. However, the evidence for the involvement of BBSome in trafficking rhodopsin is weak (13). Recent work has suggested an alternative role for the BBSome in photoreceptors, where it acts either as a ciliary gatekeeper or in retrograde protein movement to keep cytoplasmic proteins out of the photoreceptor OS (15).
Bardet–Biedl syndrome protein 8 (BBS8), also known as TTC8, a tetratricopeptide repeat (TPR) protein and a component of the octameric BBSome complex (1,7,8). Bardet–Biedl syndrome patients with mutations in BBS8 were first reported in families of Saudi Arabian and Pakistani lineage. These patients displayed obesity, excess digits, developmental delay and onset of retinitis pigmentosa as early as one year of age (1). Interestingly, two mutations in BBS8, IVS1–2A > G and c.1347G > C (p.Gln449His) cause non-syndromic Retinitis pigmentosa (RP) (16,17). The phenotype of IVS1–2A > G mutation is confined to photoreceptor cells through cell type specific alternative splicing, which causes BBS8 protein ablation specifically in photoreceptors (18). The mechanism of the non-syndromic RP phenotype of c.1347G > C (p.Gln449His) has not been investigated, but possible scenarios include a splicing mechanism similar to the IVS1–2A > G mutation, or a unique role for BBS8 in photoreceptors (16). In this study, we sought to gain insight into the role of BBS8 in photoreceptor neurons. Our studies show an early and crucial role for BBS8 in elaboration of the photoreceptor outer segments, their function and subsequent survival. Interestingly, loss of BBS8 led to changes in the composition of cognate BBS partner subunits that form the octameric BBSome complex, and altered distribution of ciliary markers demonstrating a critical role for BBS8 in development of photoreceptor cilium.
Results
Generation of complete and retina-specific BBS8 knockout animals
To study the role of BBS8 in photoreceptor cells, we recovered a BBS8 global knockout (BBS8-null) mouse model from sperm obtained from UC Davis KOMP repository. This line contains a stop cassette that knocks in a β-galactosidase reporter after exon 2 and terminates the Bbs8 gene transcription (Fig. 1A). While we obtained sufficient number of complete knockout animals for our experiments, the number of knockout pups from heterozygous crosses was always below Mendelian ratios. Additionally, the knockout males were infertile and females were obese (data not shown). To avoid these confounding issues, we generated a retina specific BBS8 knockout (Ret-Bbs8) using a Cre recombinase under Six3 promoter (Fig. 1A). Retinal extracts of BBS8-null and the pan-retina-specific BBS8 knockout (Ret-Bbs8) animals showed no detectable BBS8 protein (Fig. 1B). To demonstrate the specificity of the antibody, we overexpressed BBS8 in Neuro 2a (N2A) cells (N2A +, in Fig. 1) and cell extracts were probed for BBS8 by immunoblotting. In comparison to mock-transfected N2A cells, a robust immunoreactivity corresponding to BBS8 was found in Bbs8 transfected cells, verifying the specificity of the antibody employed in this study (Fig. 1B).
Figure 1.

Validation of animal models used in this study. (A) Schematic of the KOMP Bbs8 null allele and the generation of floxed and conditional Bbs8 alleles. In the KOMP knock-in, exons 3, 4 and 5 of the Bbs8 gene are floxed. In addition, a lacZ reporter and Neomycin resistance cassettes are knocked-in between exons 2 and 3. The reporter cassettes contain poly-adenylation sites that terminate the transcription of the Bbs8 gene. Flp recombination deletes the two cassettes, restores BBS8 protein expression and creates a floxed Bbs8 allele. Subsequent Cre expression from cell or tissue specific promoter excises exons 3, 4 and 5 and creates a conditional knockout allele. (B) Western blot validation of the animal models and the specificity of the BBS8 antibody. Equal amount of retinal extracts (100 µg) from postnatal day 10 (P10) BBS8 null (−/−) and littermate controls (+/+, wild-type and +/−, heterozygous animals) were loaded on SDS-PAGE followed by immunoblotting with an antibody against the C-termini of BBS8 (Left Panel). On the right, retinal extracts from P15 Ret-Bbs8 animals (+/+, Six3-Cre: Bbs8 fl/fl), +/−, [Six3-Cre: Bbs8 fl/+ were loaded. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH)] serves as a loading control. Extracts from cultured N2A cells transfected with plasmid over-expressing Bbs8 (+) or vector control (-) were used to test the specificity of the BBS8 antibody used.
Early reduction in rod photoresponse in the absence of BBS8
To assess the photoreceptor function in the absence of BBS8, we measured scotopic (dark-adapted) and photopic (light-adapted) responses by electroretinography (ERG) in Ret-Bbs8−/− animals (Fig. 2). The two primary measures of electroretinography are the amplitudes of the ‘a’ and ‘b’ waves, which reflect photoreceptor activity and inner neuron activity, respectively (19). At P16, an age when retinal development is largely complete, but photoreceptor outer segments (OSs) in wild-type retinas have not yet achieved their full length, scotopic and photopic ERG responses were significantly reduced (Fig. 2A). Scotopic “a”-wave amplitude was reduced by half in Ret-Bbs8−/− animals (218 ± 5 µV) compared with wild-type littermate controls (403 ± 20 µV, n = 6, Student’s t-test, P = 0.0002) (Fig. 2A and C). The scotopic response progressively declined as animals aged (Fig. 2B). In the absence of BBS8, the scotopic light response saturated at lower light intensities (I½ = 0.07 ± 0.01 cd.s m−2 in Ret-Bbs8+/+ vs. 0.03 ± 0.01 cd.s m−2 in Ret-Bbs8−/−) (Fig. 2C). Altogether, our electrophysiology studies show an early reduction of photoresponse and suggest that BBS8 plays a role in the development and function of photoreceptors.
Figure 2.

Reduced photoresponse as measured by electroretinography. (A) Scotopic ERG response from Ret-Bbs8 +/+ (Black) and Ret-Bbs8 −/− (Grey) animals measured at P16 (left panel). Representative trace of a scotopic ERG response measured at -0.8 cd.s−1m2. Photopic ERG response measured under light-adapted conditions at 0.69 log cd.s−1m2 at P16 (right panel). (B) Decline in rod photoreceptor response measured by “a” wave response plotted against the age of the animal at which ERG was recorded. Data are represented as mean ± SEM (n = 6, unpaired two-tailed t-test; *P < 0.05, **P < 0.0009). (C) Stimulus intensity curve of scotopic “a”-wave at P16 of Ret-Bbs8 −/− and littermate controls (n=6). The data were fitted with a hyperbolic function with a half-saturating intensity of 0.07 ± 0.01 cd.s m −2 (Littermate control, n=6) and 0.03 ± 0.01 cd.s m−2 (Ret-Bbs8−/−, n=6), and maximum amplitudes of 403 ± 20 µV (Littermate control, n = 6) and 219 ± 6 µV (Ret-Bbs8−/−, n = 6). Data are represented as mean ± SEM (n=6, unpaired two-tailed t-test; * P ≤ 0.01, **P ≤ 0.0009).
Loss of BBS8 leads to progressive photoreceptor cell death
To determine if the progressive decrease in photoresponse with age was a result of photoreceptor cell death, we stained retinal sections of Ret-Bbs8−/− and wild type littermates with propidium iodide at various ages (Fig. 3). At P10, retinal lamination appeared normal with no changes in the number of photoreceptor nuclei in comparison to littermate controls, suggesting normal retinal development in the absence of BBS8 (Fig. 3A and B; 13–14 nuclear layers, n = 3). At P17, in addition to brightly stained PI nuclei indicative of cell death (arrows), thinning of the photoreceptor outer nuclear layer (ONL) was observed in Ret-Bbs8−/− mice compared with wild type controls (Fig. 3A). By four months of age, retina lacking BBS8 lost the majority of their photoreceptor nuclei (Fig. 3A and B; 12–14 nuclei in littermate vs. 3–4 nuclei, n = 3, P = 0.0001). Quantification of photoreceptor nuclei at various regions in the retina showed uniform thinning in the retina lacking Bbs8 (Fig. 3B). Our results show that the BBS8 is not needed for the early commitment of photoreceptor cells, but is required for the survival of photoreceptors in the mature retina.
Figure 3.
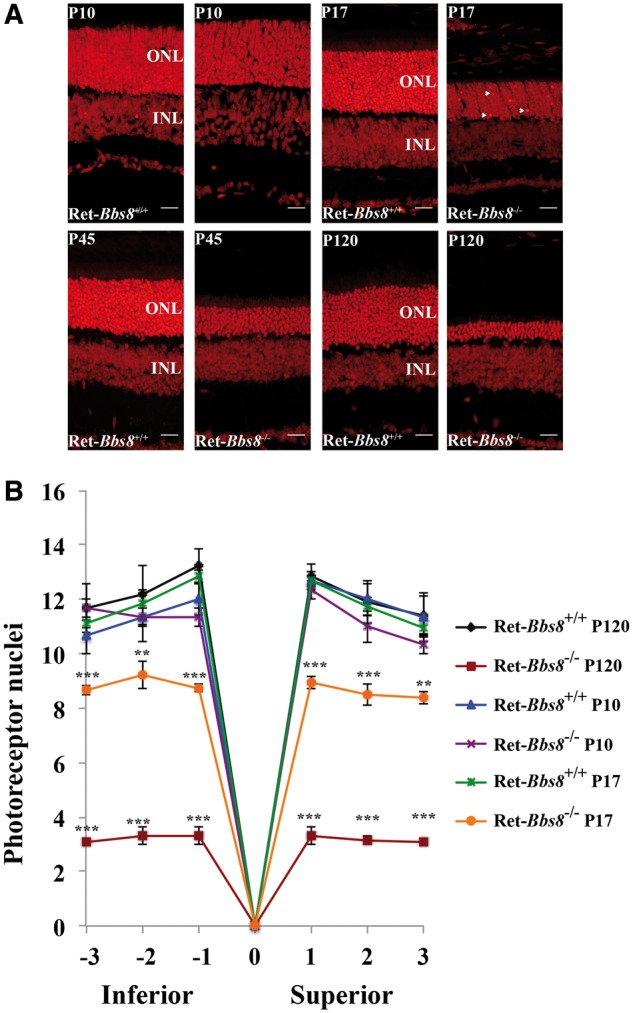
Progressive loss of photoreceptors in the retina lacking BBS8. (A) Retinal sections from animals stained with propidium iodide (PI) at indicated age. At P17, brightly stained nuclei indicative of chromatin condensation and cell death are observed (Arrows). PI staining from littermate controls is shown at the left for each age examined. (B) Quantification of the ONL thickness (number of cell nuclei) at different locations within the retina from the inferior to superior regions between Ret-Bbs8+/+ and Ret-Bbs8−/− animals at P10 and P120; Data are represented as mean ± SEM (n=3, unpaired two-tailed t-test; **P ≤ 0.01, ***P ≤ 0.001).: Outer nuclear layer, INL: Inner nuclear layer.
Loss of BBS8 leads to altered levels of BBS partner subunits
BBS8 is part of a core octameric BBSome complex (7,8) (Supplementary Material, Fig. S1). Therefore, we were interested in examining if the loss of BBS8 alters the levels of other BBSome subunit proteins (Fig. 4). We measured the protein levels of the seven BBS proteins that make up the core BBSome complex in BBS8-null and wild type littermate animals at P10 and P18 (Fig. 4A and B). BBS5 and BBS2 were reduced by 67% (P < 0.0001) and 20% (P < 0.05) respectively in the absence of BBS8 at P10 (Fig. 4A and C). In contrast, BBS1 expression increased by 75% in BBS8 null animals compared with wild type controls at P10 (Fig. 4A and C;P < 0.0001). In addition, BBS4, a TPR repeat protein similar to BBS8, increased by 86% in BBS8 deficient animals compared with littermate controls (Fig. 4A and C;P < 0.0001) (1,20,21). We observed no significant changes in BBS7 and BBS9 levels. Unfortunately, due to the lack of suitable antibodies to detect BBS18 protein in murine tissues, we were unable to ascertain if BBS18 levels are affected in the absence of BBS8. The change in BBS levels in response to the loss of BBS8 was at the post-transcriptional level; we found no significant alterations in mRNA levels via RT-qPCR (data not shown). Overall, our results show that absence of BBS8 leads to changes in cognate subunits that form the BBSome (Supplementary Material, Fig. S1).
Figure 4.

Alteration of BBSome proteins in the absence of BBS8. (A) Immunoblot showing levels of indicated BBS proteins in retinal extracts from Bbs8 null animals (−/−) prior to photoreceptor degeneration at P10. Littermate controls, both heterozygous and wild-type are shown for comparison. GAPDH serves as a loading control. (B) Similar to Panel A, immunoblot showing the levels of indicated BBS proteins in retinal extracts from P18 animals lacking BBS8. (C) Quantitation of BBS proteins shown in panel A (n = 3) normalized to GAPDH levels at P10. *P < 0.05; *** P < 0.0001 as determined by t-test.
Mislocalization of syntaxin 3 in photoreceptors lacking BBS8
Based on the role of the BBSome in trafficking GPCRs in neurons, one proposed role for this protein complex in photoreceptors was the anterograde trafficking of rhodopsin, the major GPCR in photoreceptor neurons (12). To evaluate the role for BBS8 in protein trafficking to the outer segment, we investigated the localization of rhodopsin in retinal cryosections obtained from retina lacking BBS8 and compared them to their wild-type littermate controls (Fig. 5A and B). At P10, prior to signs of photoreceptor degeneration, the majority of rhodopsin was in the OS in Ret-Bbs8−/− animals, similar to littermate controls (Fig. 5A). We also performed similar staining at P17, where photoreceptor degeneration is pervasive (Fig. 5B). Here, we observed mislocalization of rhodopsin in the inner segments (Fig. 5B). Additionally, we examined localization and expression of other OS-resident proteins in the absence of BBS8. At P17, the OS structural protein peripherin-2/rds (PRPH2) and the effector enzyme of the phototransduction cascade, phosphodiesterase-6 (PDE6) were localized to the OS (Supplementary Material, Fig. S2A). However, in our immunohistochemistry analysis, we noticed shortened OS in the absence of BBS8. Therefore, we verified the levels of PRPH2 and PDE6 by immunoblotting. Indeed, we observe a 63% decrease of PRPH2 in Ret-Bbs8−/− compared with littermate controls (Supplementary Material, Fig. S2B and C; P = 0.0008). Similarly, we observed a 47% reduction PDE6β in Ret-Bbs8−/− at P12 (Supplementary Material, Fig. S2B and C; P = 0.0002). Alternatively, a recent study showed mislocalization of syntaxin 3 (STX3) and other cytoplasmic proteins in the outer segments of retina lacking BBS17 and in the BBS1 M390R animal model (15). Since syntaxin is normally present in the inner segments and synaptic termini of photoreceptor neurons (Fig. 5, Green), this finding suggests a gatekeeper role for the BBSome that prevents cytoplasmic proteins from entering the outer segment (15). Interestingly, we observed robust accumulation of syntaxin 3 in the OS in the absence of BBS8 at both ages tested (P10 and P17) (Fig. 5).
Figure 5.
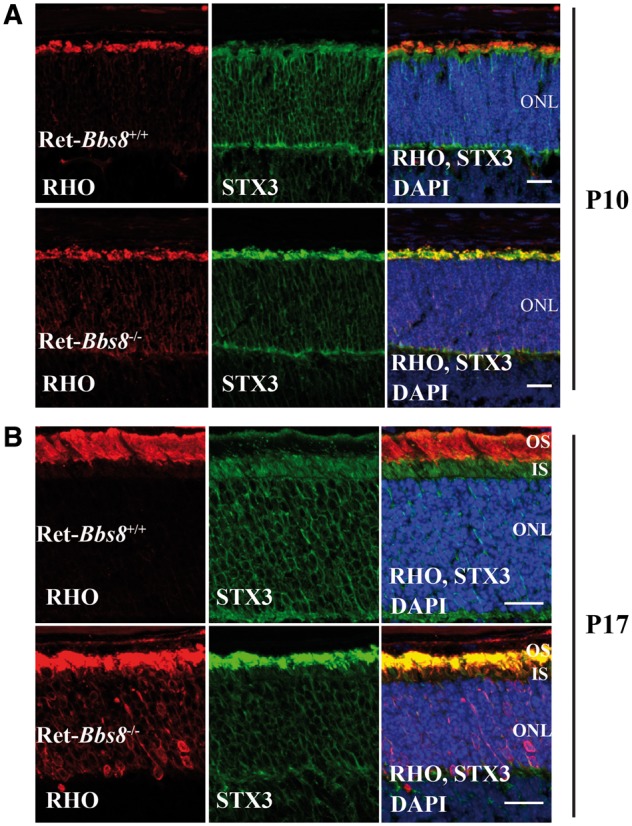
Immunolocalization of rhodopsin and syntaxin 3 in retina lacking BBS8. (A) Retinal cross-sections from Ret-Bbs8+/+ littermate control and from Ret-Bbs8−/− stained against rhodopsin (Rho-red), syntaxin 3 (STX3-green) and DAPI (blue) at post natal day 10 (P10) and (B) P17. Scale bar: 20μm. ONL: outer nuclear layer.
Lack of BBS8 causes aberrant rod outer segment ultrastructure
The reduction in photoresponse coupled with a decrease in peripherin-2/rds and OS-resident protein levels suggests that OS development and/or elaboration may be impaired in the absence of BBS8. Light microscopy of toluidine blue-stained semi-thin sections revealed pyknotic nuclei (Supplementary Material, Fig. S3B, arrow) and shortened inner and outer segments at P18 in Bbs8-null animals. We next examined the photoreceptor ultrastructure both at P10 and at P18 by transmission electron microscopy (TEM). The TEM analysis revealed dysmorphic photoreceptor OS with disoriented disks in animals lacking BBS8 at all ages tested, in comparison to littermate controls (Fig. 6 and Supplementary Material, Fig. S3C–F). Interestingly, we frequently observed extracellular vesicles (EVs) at both P10 and P18 (Fig. 6A and Supplementary Material, Fig. S3G). At P10 the median diameter for EV observed was145 nm (n = 142, pooled from 2 animals), in the absence of BBS8 (Fig. 6B). These vesicles were present at the proximal inner segment at P10 and in greater abundance and broader distribution at P18 (Fig. 6B and Supplementary Material, Fig. S3G and H). These data demonstrate that BBS8 is critical for the morphogenesis of the outer segment.
Figure 6.

Aberrant photoreceptor ultrastructure in Bbs8-null mice. (A) Left: TEM images of photoreceptor OSs in P10 retinas of wild-type (left; scale bar, 500 nm) and Bbs8-null (center panel; scale bar, 500 nm) mice. In contrast to the well-ordered disc stack seen in wild-type controls, knockout animals possessed shortened and dysmorphic OSs, with highly disorganized disc membranes. Retinas from knockout animals also displayed 100-300 nm extracellular vesicles (arrowheads), commonly observed adjacent to the proximal ISs (right panel; scale bar, 500 nm); extracellular vesicles were never observed in the wild-type controls. (B) Distribution of Feret diameter measurements (nm) of extracellular vesicles found in Bbs8−/− animals at P10 (N = 142 from two animals).
Ciliary defects in retina lacking BBS8
BBS proteins are localized to cilia and are associated with ciliogenesis and ciliary protein trafficking in a variety of cell types (9). Given this relationship, we decided to examine the structure of the photoreceptor cilia in retina lacking BBS8. The photoreceptor cilia extending from the basal body can be separated into two distinct compartments, connecting cilium (transition zone) and axoneme. The connecting cilium can be labeled with an antibody to X-linked retinitis pigmentosa GTPase regulator (RPGR) (22,23). The axoneme is marked by Retinitis Pigmentosa protein 1 (RP1) and Male germ cell-associated kinase (MAK) (24–26). Acetylated tubulin stains the connecting cilium and proximal part of the photoreceptor axoneme (26,27). We used these markers to determine if there are any changes in photoreceptor cilia size and organization in retina lacking Bbs8 (Fig. 7). In the absence of BBS8 at P10, we observed an increase in acetylated tubulin labeling (Fig. 7A and Supplementary Material, Fig. S4C; 1.145 μm ± 0.02 in wild type, n = 288; 2.301 μm ± 0.04 in Ret-Bbs8−/−, n = 278; P < 0.0001). Marginal, but statistically significant increase of RGPR stained zones was also observed along the connecting cilium (Fig. 7B and C and Supplementary Material, Fig. S4D; 0.9428 μm ± 0.01, n = 336 in littermate controls, 1.011 μm ± 0.01, n = 350 in Ret-Bbs8−/−; P < 0.0001). In contrast, we observed reduced immnunoreactivity for both axonemal markers RP1 and MAK, suggesting reduced axoneme length. RP1 staining was reduced in Ret-Bbs8−/− animals compared with littermate controls (2.32 μm ± 0.05 in wild type, n = 215; 1.53 ± 0.04 in Ret-Bbs8−/−, n = 190; P < 0.0001) (Fig. 7A and C and Supplementary Material, Fig. S4A). Similarly, MAK staining was reduced in retina lacking BBS8 (1.21 μm ± 0.02, n = 317 in littermate controls, 0.81 μm ± 0.01, n = 310 in Ret-Bbs8−/−;P < 0.0001) (Fig. 7B and C and Supplementary Material, Fig. S4B). The total protein levels of RP1 and MAK in the retina were not significantly altered (Supplementary Material, Fig. S4E and F); however, a trend for increased acetylated tubulin protein levels was observed by immunoblotting (Supplementary Material, Fig. S4E and F). Overall, these data show altered distribution of ciliary markers in the absence of BBS8, which likely reflects changes in the organization of the photoreceptor cilia.
Figure 7.

Increased acetylated tubulin stained ciliary zones in the absence of BBS8 at P10. (A) Immunofluorescence images showing RP1 (Retinitis Pigmentosa-1) and acetylated α-tubulin staining the photoreceptor axoneme and connecting cilia in Ret-Bbs8+/+ and Ret-Bbs8−/− animals, respectively. Scale bar: 20 and 10 μm. (B) Retinal cryosections stained against MAK (male germ cell-associated kinase), Rootletin and RPGR (X-linked retinitis pigmentosa GTPase regulator) in Ret-Bbs8−/− and littermate controls. Scale bar: 10 μm. (C) Violin plots depicting overall distribution of length measurements for RP1, acetylated α-tubulin, MAK and RPGR stained zones in Ret-Bbs8−/− and littermate controls (n = 200–300 cilia pooled from 3 animals). All retinal tissues are derived from P10 animals with littermates used as controls.
BBS8 is crucial for cone photoreceptor viability
We found that the removal of BBS8 in the retina leads to reduction in both rod and cone photoresponses (Fig. 2). This decrease in cone function could be due to a “by-stander” effect (secondary to the loss of rod photoreceptor cells), or it may be due to a direct role of BBS8 in cone photoreceptors. Therefore, to determine if the reduction of cone photoresponse is a cell autonomous effect, we ablated Bbs8 in developing cone photoreceptors using a mouse line that expresses Cre recombinase under the human red-green pigment promoter (HRGP-cre) (Supplementary Material, Fig. S5) (28). We evaluated the specificity of the Cre expression in cone photoreceptors by staining retinal cross-sections from cone-Bbs8−/− and littermate controls against Cre and the cone marker peanut agglutinin (PNA), which stains the cone outer sheath. As expected, Cre expression was specifically found in PNA+ cells (Supplementary Material, Fig. S5B). In this Cre driver line, robust expression of Cre recombinase starts at P10, and recombination as judged by cre reporter expression, is complete by P30 (28). Therefore, we performed photopic ERGs to examine cone function at P30 and observed no significant differences in ERG responses between Cone-Bbs8−/− and their wild type littermate controls (Fig. 8A). Although the cone ERGs were similar at P30, there is a progressive reduction in cone response with age (Supplementary Material, Fig. S6A), and by 4 months this response was completely ablated (Fig. 8A). To investigate if loss of cone response is due to the death of cone photoreceptor cells, we performed immunocytochemistry using various cone markers at P120. GRK1, the kinase expressed in rods and cones (green) in littermate controls was specifically absent in cones from cone-Bbs8−/− animals (Fig. 8B). The loss of cones was confirmed by the reduced staining for PNA, cone arrestin, and cone phosphodiesterase 6 (Supplementary Material, Fig. S7). In addition, we observed uniform reduction in cone density by flat-mount in various regions of the retina (data not shown). We also performed scotopic ERGs at this age and saw no significant changes between experimental and wild-type animals, further corroborating the specificity of the cone-Bbs8 animal model (Supplementary Material, Fig. S6B). Additionally, similar to our Ret-Bbs8 animal model, we observed mislocalization of syntaxin 3 to cone OS at P30 (Fig. 9B). At this stage, the localization of M-opsin was unaltered and was found in the cone OS (Fig. 9A). Altogether, our results show that BBS8 presence in cone photoreceptors is essential for their viability (Fig. 8B and Supplementary Material, Fig. S7).
Figure 8.
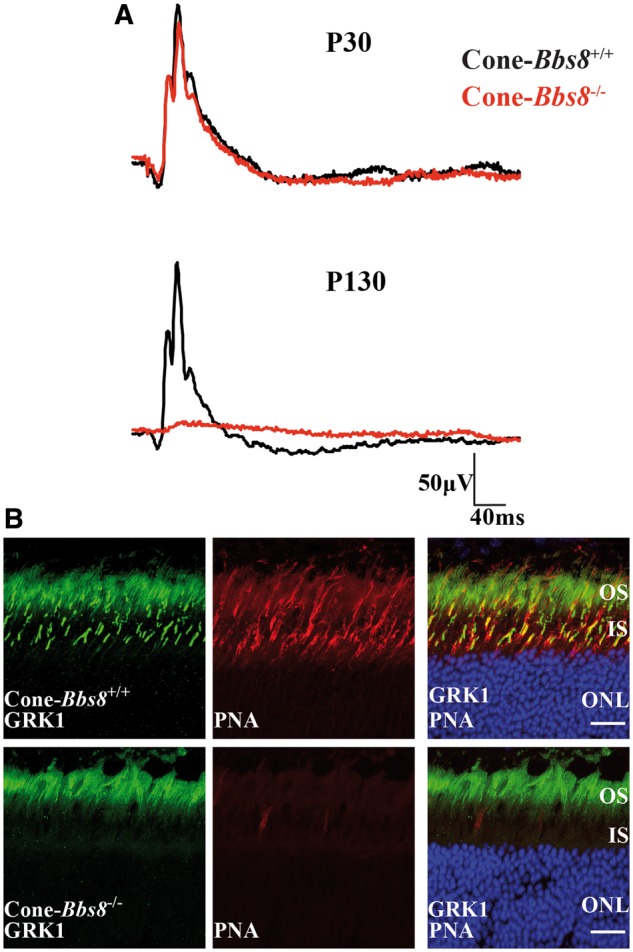
Degeneration of cone photoreceptors missing BBS8. (A) Representative trace of photopic ERG responses measured under light adapted conditions at 0.69 cd.s−1m2 in Cone-Bbs8−/− and littermate controls at P30 and P130. (B) Retinal cryosections stained against G-protein-coupled Receptor Kinase 1 (GRK1; green) and Peanut agglutinin (PNA) in Ret-Bbs8−/− and wild type littermate controls at P130. GRK1 stains both rods and cones while PNA marks the extracellular matrix surrounding the cones. Scale bar: 20 μm.
Figure 9.
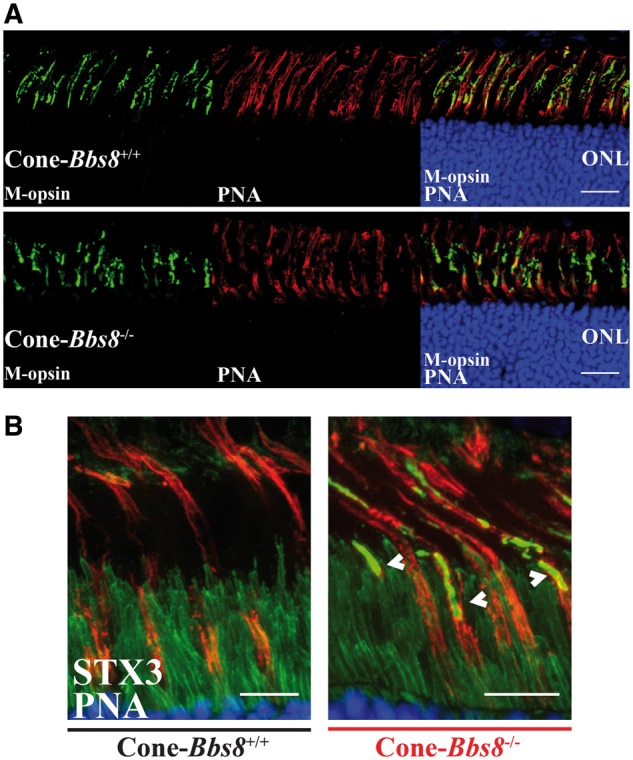
Mislocalization of Syntaxin 3 in cone photoreceptors lacking BBS8. (A) Immunolocalization of M-opsin and peanut agglutinin (PNA) in retinal cross-sections of Cone-Bbs8−/− animals compared with littermate controls at P30. Scale bar: 20μm. (B) Retinal cross-sections showing mislocalized synatxin 3 (white arrows; STX3 in green), co-stained with peanut agglutinin (PNA, red), in cones lacking BBS8 (Cone-Bbs8−/−) compared with wild –type littermate controls at P30. Scale bar: 10μm.
BBS8 is critical for maintaining cone photoreceptor OS structure
We observed defects in rod OS development in our Ret-Bbs8−/− animals, and immunocytochemistry hinted at potential defects in cone OS in the Cone-Bbs8−/− animals. Therefore, we examined the structure of the cone outer segment by TEM in perfusion-fixed Cone-Bbs8−/− animals and wild-type controls at P30. We chose this age because photopic responses were comparable between Cone-Bbs8−/− and wild-type controls. Ultrastructural analysis revealed less tightly packed, elongated, and mis-oriented OS disk membranes of Cone-Bbs8−/− animals (Fig. 10). In conclusion, despite no significant changes in cone photoresponse at this age, absence of BBS8 leads to aberrant cone OS morphology.
Figure 10.

Defective cone OS ultrastructure in Cone-Bbs8−/− retinas. (A) Two representative images of cone OS each from wild-type and Cone-Bbs8−/− animals are shown. The vast majority of cones in knockout animals possessed abnormal OSs; disc stacks were less dense, of larger diameters, and were misaligned. In contrast, cones in the wild-type control possessed tightly stacked, properly aligned, and normal diameter discs. Scale bars: 500 nm.
Discussion
Mutations in BBS8 cause ciliopathies affecting multiple organs and invariably lead to loss of vision in early childhood (1). The importance of BBS8 in photoreceptor function is further exemplified by mutations in BBS8 that lead to non-syndromic retinitis pigmentosa, yet the role for BBS8 in photoreceptors is not known (16–18). In this study, we sought to investigate the role of BBS8 in the development and function of photoreceptor cells. Our studies using multiple murine models, including a complete knockout, retina and cone- specific removal of BBS8 lead to the following novel findings. We show that BBS8 is required for the development and maintenance of the photoreceptor cilia and outer segments in both rods and cones. Although mutations in BBS proteins are known to affect rod cells, this is the first study to demonstrate that BBSome function is also essential for cone photoreceptor structure, function, and viability. We observed dynamic changes in BBSome subunits that included increased levels of BBS1 and BBS4 in the absence of BBS8. Our studies indicate that altered photoreceptor OS development in the absence of BBS8 is likely due to defective ciliary structure indicated by sizable changes in acetylated tubulin staining zones or as a result of altered ciliary trafficking.
Alterations in cognate BBS subunits
Our study sought to understand the effects of BBS8 on the levels of cognate partner subunits that are part of the core octameric BBSome complex. The core of the BBSome is formed by BBS2/7/9 proteins, followed by the addition of BBS1/5/8, and the final incorporation of BBS4 (8). In BBS8 animal models, the expression of the core BBSome proteins is preserved in the retina with a minor reduction in BBS2 levels. BBS4, a tetratricopeptide repeat (TPR) protein similar to BBS8, was highly upregulated in the absence of BBS8 prior to any significant photoreceptor degeneration (1,20,21). Interestingly BBS1, a protein that interacts with the small GTPase ARL6 (BBS3), an interaction necessary for the targeting of the BBSome to the membrane, was also up-regulated in the absence of BBS8 (20). Lastly, BBS5, a protein that is present in the photoreceptor axoneme and is absent in the connecting cilia was severely down-regulated. BBS5 interacts with arrestin and is hypothesized to alter its light-dependent translocation (29). Interestingly, previous reports indicate absence of BBS4 leads to impaired light-dependent translocation of transducin and arrestin in photoreceptor neurons (30). In BBS8 knockouts, we did not find any changes in arrestin or transducin translocation in response to light stimulus (data not shown). This could be due to the increased expression of BBS4 in BBS8 models, leading to proper arrestin and transducin translocation, or that arrestin and transducin can be transported by an alternative mechanism. All changes observed in BBS levels in the absence of BBS8 were post-transcriptional, as message levels were unaltered (data not shown). It is not clear if the observed phenotype is exclusively due to loss of BBS8 or the effect of altered BBSome subunits but further experiments are needed to address the mechanism behind the loss of OS development. One interesting question stemming from these results is if the changes in BBSome subunits after Bbs8 depletion reflect a dynamic nature of this protein complex, which may exist in different states depending on subunit availability, presence of interacting molecules, or external signals.
Normal rhodopsin trafficking in the absence of BBS8
Studies from multiple murine models lacking individual BBS subunits suggest a role for the BBSome in rhodopsin trafficking to the outer segments (30,31). This is in agreement with results from primary cilia showing the need for the BBSome in GPCR trafficking (10). Alternatively, the BBSome is thought to be involved in retrograde trafficking of cargo from the OS to the IS and/or a role as a ciliary ‘gatekeeper’, as evidenced by mislocalization of non-OS proteins in the OS in BBS17 (Lztfl1) mutant mice (15). In our BBS8 models, we did not observe significant defects in anterograde trafficking of OS-resident cargoes, including rhodopsin. The vast majority of rhodopsin at P10, prior to photoreceptor degeneration, was present in the OS, similar to the littermate controls. However, we observed slight mislocalization at P17, which we believe is likely due to the indirect effect of dysmorphic outer segments in BBS8 knockouts (Supplementary Material, Figs S2 and S3). Our studies indicate that BBS8 is not needed for rhodopsin trafficking but we cannot rule out the possibility that rhodopsin is able to reach the OS through a partially formed BBSome which may be compensating for the lack of BBS8 in our animal models. Our assays to investigate the formation of the BBSome complex failed because of the early morphological defects observed and the low levels of BBSome subunits in retina at P10. Importantly, animal models lacking BBS8 in rods or cones showed mislocalization of syntaxin in the OS, which agrees with current thinking that the BBSome is either involved in retrograde trafficking of proteins or acts as a ciliary gate in photoreceptors (15). However, further studies in photoreceptors are needed to confirm either of these hypotheses.
BBS8 is crucial for OS morphogenesis
BBS8 animal models generated in this study show the need for BBS8 in the initial elaboration of the photoreceptor outer segments. The changes in photoreceptor ultrastructure correlated with reduced rod response in complete and retina-specific knockouts where BBS8 was conditionally removed in the developing embryonic retina.
Although mutations in BBS are linked to retinitis pigmentosa, which implies a primary impact on rod cells, a recent report documented predominant cone dysfunction in BBS patients (32). We investigated the potential importance of BBS8 for murine cones using a conditional knockout model, which ablates BBS8 from cones between P8 and 10. While we did not detect any abnormalities in cone function at P30 (by ERG), cone outer segments were dysmorphic and showed abnormal ultrastructure, with discs stacked vertically rather than horizontally. Unlike rod photoreceptors, where significant number of functioning cells survived at four months in the absence of BBS8, cone degeneration was far more profound, and function was lost in parallel. The reason for cone degeneration is likely linked to the morphological changes in cone OS. Overall, the findings from our studies show a critical need for BBS8 in cone survival and function.
We also observed an accumulation of electron dense extracellular vesicles (EVs), in the extracellular space adjacent to the photoreceptor inner segments in BBS8 null retina. The accumulation of ectosomes, a class of extra cellular vesicles that are derived from ciliary plasma membranes, were also observed in the absence of BBS and IFT proteins in primary cilium in cultured kidney cells and mouse embryonic fibroblasts (33). Intriguingly, mutation of another ciliopathy protein, IFT88, has also been shown to generate EVs of similar appearance and distribution (34). That study documented the presence of rhodopsin in the EVs, and noted that numerous other mouse models defective for OS assembly also accumulate EVs. A more recent investigation identified subretinal EVs in the retinal degeneration slow (rds) null mouse as ectosomes (35). Because that study observed that ectosome shedding was stimulated by peripherin-2/rds loss, and we detected a major reduction (65%) in peripherin-2/rds levels in the absence of BBS8 at P10, it is conceivable that peripherin-2/rds loss in the BBS8 knockout likewise stimulates EV accumulation. It is also possible that a more general disruption of OS disk membrane morphogenesis contributed to this phenotype.
Hyper-acetylation of ciliary microtubules in the absence of BBS8
Our ultrastructural analyses in BBS8 knockout animals show connecting cilium that are of comparable length to the littermate controls (Supplementary Material, Fig. S3). This is in agreement with RPGR staining, a maker for the connecting cilium (22,23). However, the acetylated tubulin staining on photoreceptor cilia was longer in photoreceptors lacking BBS8 (Fig. 7). In photoreceptors, acetylated tubulin marks connecting cilium and the proximal part of the axoneme (27). RP1 exclusively stains the entire axoneme (24,25). In the absence of BBS8, it appears that the increase in acetylated tubulin staining comes at the loss in axonemal staining by RP1 (Supplementary Material, Fig. S4A and C). A corresponding reduction in MAK, another axonemal marker confirms this finding (26). The observed changes in MAK or RP1 staining of the axoneme could also be due to altered ciliary trafficking. Acetylation of tubulin marks long-lived tubulin molecules and recent studies show the importance of this tubulin post-translational modification in increased mechanical stabilization and protection against breakage (36,37). The observed increase in acetylated tubulin staining could be due to the established interaction between BBS proteins and α-Tubulin N-acetyltransferase-1 (αTAT1), the major α-tubulin acetyl transferase that acetylates tubulin in the cilium or alterations in the BBSome subunit, BBS18, a known regulator of tubulin acetylation (9,38). Interestingly, hyperacetylation of tubulin in the absence of BBS8 was accompanied by an increase in tubulin glutamylation, a C-terminal post-translational modification that marks stable microtubules (Supplementary Material, Fig. S8). Hyperglutamylation is a known cause of photoreceptor neuronal degeneration in pcd (Purkinje cell degeneration) and in TTLL3 (Tubulin Tyrosine ligase-like) mouse models. In these models, increased glutamylation leads to a compensatory decrease in tubulin glycation (39–41). Currently, it is unclear how these changes in photoreceptor cilia relate to defects in outer segment formation and photoreceptor cell death. Further studies are underway to investigate the mechanism behind the altered tubulin posttranslational modifications and impaired outer segment formation.
Materials and Methods
Animals, genotyping and maintenance
BBS8 knockout mouse line (Ttc8 tm1a(KOMP)Wtsi) was recovered using sperm obtained from KOMP (https://www.komp.org/geneinfo.php? geneid=85321) (Project id: CSD28015, Knock out first). Along with LoxP sites flanking exon 4 to 6 of the BBS8 gene, the KOMP targeting construct knocks-in a cassette containing a splice acceptor with LacZ reporter, polyadenylation signal and neomycin selection at Bbs8 locus between exons 3 and 4 that terminates the transcript. The LacZ/neomycin cassette is flanked by FRT sites and can be excised with FLP recombinase to restore the expression of the BBS8 gene. The animals were backcrossed with C57BL/6J (Jackson laboratory) to eliminate the rd8 allele (42). Heterozygous mice were intercrossed, and the progeny were genotyped using genomic DNA prepared from ear punches by PCR. The following primers [5’ GGA TCA CAG TCA AGA GTA GAG TC 3’ and (5’ CAC ACG TGT TTC TCC TTA GAG GC 3’] were used to verify the presence of the Neomycin gene. (5’ ATC ACG ACG CGC TGT ATC 3’ and 5’ ACA TCG GGC AAA TAA TAT CG 3’) primers were used to assess the presence of the LacZ reporter gene.
To produce the conditional knockouts, we first removed the termination cassette (LacZ/neomycin) by crossing BBS8 animals heterozygous for KOMP cassette with transgenic mice expressing FLP recombinase (Jax stock #012930). This cross resulted in a floxed Bbs8 allele (Bbs8 +/fl). The homozygous floxed animals were subsequently crossed with animals expressing Cre recombinase under the retina and forebrain specific Six3 promoter and bred to produce Six3-Cre: Bbs8 fl/fl animals (43). A similar strategy was used to produce cone specific knockouts using Cre recombinase expressed under human red green pigment (HRGP) promoter (28). Oligonucleotides used in PCR to detect Cre recombinase were 5’ CCT GGA AAA TGC TTC TGT CCG 3’ and 5’ CAG GGT GTT ATA AGC AAT CCC 3’. Primers used to detect floxed alleles were 5’ GGT GAC CAG AAG CAA GCA CAT A 3’ and 5’ GGC TGC TGT CTG GTT GAG TAA T 3’. Institutional Animal Care and Use Committee of the West Virginia University approved all experimental procedures involving animals conducted in this study.
Electroretinography
Electroretinographies (ERGs) were carried out on the UTAS Visual Diagnostic System with Big-Shot Ganzfeld with UBA-4200 amplifier and interface, and EMWIN 9.0.0 software (LKC Technologies, Gaithersburg, MD, USA). Prior to testing, mice were dark-adapted for 24 h. Animals were anesthetized [2.0% isoflurane with 2.5 l/min oxygen flow rate] for 10 min and eyes were topically dilated with a 1: 1 mixture of tropicamide: phenylephrine hydrochloride. For ERG testing, mice were positioned on a heated platform with continuous flow of isoflurane through a nose cone [1.5% isoflurane with 2.5 l/pm) oxygen flow rate]. A reference electrode was inserted subcutaneously in the scalp. ERG responses were recorded from both eyes with silver wire electrodes placed on top of each cornea, with contact being made with hypromellose solution (2% hypromellose in PBS) (Gonioscopic Prism Solution, Wilson Ophthalmic, Mustang, OK, USA). Scotopic (Rod-dominant) responses were obtained in the dark with flashes of LED white light at increasing flash intensities. For photopic response, animals were light adapted with rod-saturating white background light (30 cd · m–2) for 10 min and cone responses were subsequently recorded.
Antibodies
The antibodies used in this study are described in Supplementary Material, Table S1.
Western blot
Mice were euthanized by CO2 inhalation followed by cervical dislocation and eyes were enucleated. For immunoblots, flash-frozen retinae dissected from enucleated eyes were sonicated in phosphate buffered saline [PBS: 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4·7H2O, 1.4 mM KH2PO4, with protease inhibitor cocktail (Roche)]. Protein concentrations were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc.). Equal amounts of samples (100 μg total protein per well) were separated in a polyacrylamide SDS–PAGE gel and transferred onto polyvinylidene difluoride (PVDF) membranes (Immunobilon-FL, Millipore, Billerica). The membranes were then blocked with blocking buffer (Rockland Inc.) for 30 min at room temperature and further incubated with primary antibodies overnight at 4 °C. Following incubation, membranes were washed in PBST (PBS with 0.1% Tween-20) three times for 5 min each at room temperature and incubated in secondary antibody, goat anti-rabbit Alexa 680 (or 800), rabbit anti-goat Alexa 680 or goat anti-mouse Alexa 680 (Invitrogen) for 45 min at room temperature. After washes with PBST, membranes were scanned using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA).
Immunocytochemistry
For immunofluorescence, enucleated eyes were submerged in 4% paraformaldehyde fixative for 5 min prior to removal of the cornea and lens. Eyecups were fixed for an additional h, then washed in PBS three times for 5 min each, and incubated in 20% sucrose in PBS overnight at 4 °C. Eyes were then incubated in 1: 1 mixture of 20% sucrose in PBS: OCT (Cryo Optimal Cutting Temperature Compound, Sakura) for 1 h and flash-frozen in OCT. The cryosectioning was performed using Leica CM1850 Cryostat, and retinal sections of 16 μm and/or 10 μm (for ciliary staining) thickness were mounted on Superfrost Plus slides (Fisher Scientific). Retinal sections were mounted on slides, washed with PBST and incubated for 1 h in blocking buffer at room temperature (PBS with 5% Goat Sera, 0.5% TritonX-100, 0.05% Sodium Azide). After blocking, retinal sections were incubated with primary antibodies at the dilutions described in Supplementary Material, Table S1 at 4 °C overnight. Afterwards, retinal sections were washed two times for 10 min with PBST and once for 5 min with PBS before incubation with secondary antibody at a 1: 1000 dilution [DAPI nuclear stain 405, anti-Rabbit 488 (or 568), anti-mouse 488 (or 568)] for 1 h. For ciliary staining, enucleated eyes were fixed in 4% paraformaldehyde in PBS for 30 s and flash-frozen in OCT. Sectioning and staining were performed as stated above. Slides were mounted with ProLong Gold (Life Technologies) and cover slipped. Confocal imaging was performed at the WVU Microscope Imaging Facility with a Zeiss LSM 510 laser scanning confocal on a LSM Axioimager upright microscope using excitation wavelengths of 405, 488, 543 and 647 nm.
Ultrastructural analysis
Enucleated eyes were fixed (2% paraformaldehyde, 2.5% glutaraldehyde, 0.1 M cacodylate buffer, pH 7.5) for 30 min prior to dissection and removal of the cornea and lens, followed by 48 h fixation at room temperature. Dissection, embedding and transmission electron microscopy was done according to previously established methods (44–46). For analyses of cone outer segment morphology, prior to fixation of enucleated eyes, mice underwent transcardial fixation perfusion with 2% paraformaldehyde, 2.5% glutaraldehyde, 0.1 M cacodylate buffer, pH 7.5.
Statistics
All data are presented as mean ± standard error margin. ERG data were analysed by unpaired, two-tailed t test. For cilia measurements, 80–100 cilia were measured for each animal (n = 3) and data were visualized with the ggplot2 package in R version 3.3.2. Image and densitometry analysis were performed using ImageJ 1.50i along with the Bio-Formats plugin (NIH).
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors thank Zachary Wright, Caity Haring, Jessica Clemente and Dr. Eric Tucker (WVU) for help with data collection.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health Grants R01 EY025536 (to V.R. and P.S.), R21 EY027707 (to V.R.), West Virginia Lions and Lions Club International Foundation.
References
- 1. Ansley S.J., Badano J.L., Blacque O.E., Hill J., Hoskins B.E., Leitch C.C., Kim J.C., Ross A.J., Eichers E.R., Teslovich T.M.. et al. (2003) Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature, 425, 628–633. 10.1038/nature02030 [DOI] [PubMed] [Google Scholar]
- 2. Forsythe E., Beales P.L. (2013) Bardet-Biedl syndrome. Eur J Hum Genet, 21, 8–13. 10.1038/ejhg.2012.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Novas R., Cardenas-Rodriguez M., Irigoin F., Badano J.L. (2015) Bardet-Biedl syndrome: Is it only cilia dysfunction? FEBS Lett, 589, 3479–3491. [DOI] [PubMed] [Google Scholar]
- 4. Zaghloul N.A., Katsanis N. (2009) Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest, 119, 428–437. 10.1172/JCI37041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heon E., Kim G., Qin S., Garrison J.E., Tavares E., Vincent A., Nuangchamnong N., Scott C.A., Slusarski D.C., Sheffield V.C. (2016) Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum Mol Genet, 25, 2283–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lindstrand A., Frangakis S., Carvalho C.M., Richardson E.B., McFadden K.A., Willer J.R., Pehlivan D., Liu P., Pediaditakis I.L., Sabo A.. et al. (2016) Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am J Hum Genet, 99, 318–336. 10.1016/j.ajhg.2015.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nachury M.V., Loktev A.V., Zhang Q., Westlake C.J., Peranen J., Merdes A., Slusarski D.C., Scheller R.H., Bazan J.F., Sheffield V.C.. et al. (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell, 129, 1201–1213. 10.1016/j.cell.2007.03.053 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Q., Yu D., Seo S., Stone E.M., Sheffield V.C. (2012) Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable bardet-biedl syndrome protein complex, the BBSome. J Biol Chem, 287, 20625–20635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Loktev A.V., Zhang Q., Beck J.S., Searby C.C., Scheetz T.E., Bazan J.F., Slusarski D.C., Sheffield V.C., Jackson P.K., Nachury M.V. (2008) A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell, 15, 854–865. [DOI] [PubMed] [Google Scholar]
- 10. Berbari N.F., Lewis J.S., Bishop G.A., Askwith C.C., Mykytyn K. (2008) Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci U S A, 105, 4242–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tadenev A.L., Kulaga H.M., May-Simera H.L., Kelley M.W., Katsanis N., Reed R.R. (2011) Loss of Bardet-Biedl syndrome protein-8 (BBS8) perturbs olfactory function, protein localization, and axon targeting. Proc Natl Acad Sci U S A, 108, 10320–10325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pearring J.N., Salinas R.Y., Baker S.A., Arshavsky V.Y. (2013) Protein sorting, targeting and trafficking in photoreceptor cells. Prog Retin Eye Res, 36, 24–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wensel T.G., Zhang Z., Anastassov I.A., Gilliam J.C., He F., Schmid M.F., Robichaux M.A. (2016) Structural and molecular bases of rod photoreceptor morphogenesis and disease. Prog Retin Eye Res, 55, 32–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Young R.W. (1967) The renewal of photoreceptor cell outer segments. J Cell Biol, 33, 61–72. 10.1083/jcb.33.1.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Datta P., Allamargot C., Hudson J.S., Andersen E.K., Bhattarai S., Drack A.V., Sheffield V.C., Seo S. (2015) Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in Bardet-Biedl syndrome. Proc Natl Acad Sci U S A, 112, E4400–E4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goyal S., Jager M., Robinson P.N., Vanita V. (2015) Confirmation of TTC8 as a disease gene for nonsyndromic autosomal recessive retinitis pigmentosa (RP51). Clin Genet, in press. [DOI] [PubMed] [Google Scholar]
- 17. Riazuddin S.A., Iqbal M., Wang Y., Masuda T., Chen Y., Bowne S., Sullivan L.S., Waseem N.H., Bhattacharya S., Daiger S.P.. et al. (2010) A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am J Hum Genet, 86, 805–812. 10.1016/j.ajhg.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murphy D., Singh R., Kolandaivelu S., Ramamurthy V., Stoilov P. (2015) Alternative Splicing Shapes the Phenotype of a Mutation in BBS8 To Cause Nonsyndromic Retinitis Pigmentosa. Mol Cell Biol, 35, 1860–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robson J.G., Frishman L.J. (1998) Dissecting the dark-adapted electroretinogram. Doc Ophthalmol, 95, 187–215. 10.1023/A:1001891904176 [DOI] [PubMed] [Google Scholar]
- 20. Jin H., White S.R., Shida T., Schulz S., Aguiar M., Gygi S.P., Bazan J.F., Nachury M.V. (2010) The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell, 141, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim J.C., Badano J.L., Sibold S., Esmail M.A., Hill J., Hoskins B.E., Leitch C.C., Venner K., Ansley S.J., Ross A.J.. et al. (2004) The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet, 36, 462–470. 10.1038/ng1352 [DOI] [PubMed] [Google Scholar]
- 22. Hong D.H., Pawlyk B., Sokolov M., Strissel K.J., Yang J., Tulloch B., Wright A.F., Arshavsky V.Y., Li T. (2003) RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Vis Sci, 44, 2413–2421. [DOI] [PubMed] [Google Scholar]
- 23. Zhao Y., Hong D.H., Pawlyk B., Yue G., Adamian M., Grynberg M., Godzik A., Li T. (2003) The retinitis pigmentosa GTPase regulator (RPGR)- interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci U S A, 100, 3965–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Q., Lyubarsky A., Skalet J.H., Pugh E.N. Jr., Pierce E.A. (2003) RP1 is required for the correct stacking of outer segment discs. Invest Ophthalmol Vis Sci, 44, 4171–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Q., Zuo J., Pierce E.A. (2004) The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J Neurosci, 24, 6427–6436. 10.1523/JNEUROSCI.1335-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Omori Y., Chaya T., Katoh K., Kajimura N., Sato S., Muraoka K., Ueno S., Koyasu T., Kondo M., Furukawa T. (2010) Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc Natl Acad Sci U S A, 107, 22671–22676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arikawa K., Williams D.S. (1993) Acetylated alpha-tubulin in the connecting cilium of developing rat photoreceptors. Invest Ophthalmol Vis Sci, 34, 2145–2149. [PubMed] [Google Scholar]
- 28. Le Y.Z., Ash J.D., Al-Ubaidi M.R., Chen Y., Ma J.X., Anderson R.E. (2004) Targeted expression of Cre recombinase to cone photoreceptors in transgenic mice. Mol Vis, 10, 1011–1018. [PubMed] [Google Scholar]
- 29. Smith T.S., Spitzbarth B., Li J., Dugger D.R., Stern-Schneider G., Sehn E., Bolch S.N., McDowell J.H., Tipton J., Wolfrum U.. et al. (2013) Light-dependent phosphorylation of Bardet-Biedl syndrome 5 in photoreceptor cells modulates its interaction with arrestin1. Cell Mol Life Sci, 70, 4603–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abd-El-Barr M.M., Sykoudis K., Andrabi S., Eichers E.R., Pennesi M.E., Tan P.L., Wilson J.H., Katsanis N., Lupski J.R., Wu S.M. (2007) Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet-Biedl syndrome. Vision Res, 47, 3394–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nishimura D.Y., Fath M., Mullins R.F., Searby C., Andrews M., Davis R., Andorf J.L., Mykytyn K., Swiderski R.E., Yang B.. et al. (2004) Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A, 101, 16588–16593. 10.1073/pnas.0405496101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scheidecker S., Hull S., Perdomo Y., Studer F., Pelletier V., Muller J., Stoetzel C., Schaefer E., Defoort-Dhellemmes S., Drumare I.. et al. (2015) Predominantly Cone-System Dysfunction as Rare Form of Retinal Degeneration in Patients With Molecularly Confirmed Bardet-Biedl Syndrome. Am J Ophthalmol, 160, 364–372 e361. [DOI] [PubMed] [Google Scholar]
- 33. Nager A.R., Goldstein J.S., Herranz-Perez V., Portran D., Ye F., Garcia-Verdugo J.M., Nachury M.V. (2017) An Actin Network Dispatches Ciliary GPCRs into Extracellular Vesicles to Modulate Signaling. Cell, 168, 252–263 e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pazour G.J., Baker S.A., Deane J.A., Cole D.G., Dickert B.L., Rosenbaum J.L., Witman G.B., Besharse J.C. (2002) The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol, 157, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salinas R.Y., Pearring J.N., Ding J.D., Spencer W.J., Hao Y., Arshavsky V.Y. (2017) Photoreceptor discs form through peripherin-dependent suppression of ciliary ectosome release. J Cell Biol, 216, 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Portran D., Schaedel L., Xu Z., Thery M., Nachury M.V. (2017) Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat Cell Biol, 19, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu Z., Schaedel L., Portran D., Aguilar A., Gaillard J., Marinkovich M.P., Thery M., Nachury M.V. (2017) Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science, 356, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shida T., Cueva J.G., Xu Z., Goodman M.B., Nachury M.V. (2010) The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci U S A, 107, 21517–21522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blanks J.C., Mullen R.J., LaVail M.M. (1982) Retinal degeneration in the pcd cerebellar mutant mouse. II. Electron microscopic analysis. J Comp Neurol, 212, 231–246. 10.1002/cne.902120303 [DOI] [PubMed] [Google Scholar]
- 40. Bosch Grau M., Masson C., Gadadhar S., Rocha C., Tort O., Marques Sousa P., Vacher S., Bieche I., Janke C. (2017) Alterations in the balance of tubulin glycylation and glutamylation in photoreceptors leads to retinal degeneration. J Cell Sci, 130, 938–949. [DOI] [PubMed] [Google Scholar]
- 41. LaVail M.M., Blanks J.C., Mullen R.J. (1982) Retinal degeneration in the pcd cerebellar mutant mouse. I. Light microscopic and autoradiographic analysis. J Comp Neurol, 212, 217–230. 10.1002/cne.902120302 [DOI] [PubMed] [Google Scholar]
- 42. Mattapallil M.J., Wawrousek E.F., Chan C.C., Zhao H., Roychoudhury J., Ferguson T.A., Caspi R.R. (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci, 53, 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Furuta Y., Lagutin O., Hogan B.L., Oliver G.C. (2000) Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis, 26, 130–132. [PubMed] [Google Scholar]
- 44. Kirschman L.T., Kolandaivelu S., Frederick J.M., Dang L., Goldberg A.F., Baehr W., Ramamurthy V. (2010) The Leber congenital amaurosis protein, AIPL1, is needed for the viability and functioning of cone photoreceptor cells. Hum Mol Genet, 19, 1076–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ku C.A., Chiodo V.A., Boye S.L., Hayes A., Goldberg A.F., Hauswirth W.W., Ramamurthy V. (2015) Viral-mediated vision rescue of a novel AIPL1 cone-rod dystrophy model. Hum Mol Genet, 24, 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wright Z.C., Singh R.K., Alpino R., Goldberg A.F., Sokolov M., Ramamurthy V. (2016) ARL3 regulates trafficking of prenylated phototransduction proteins to the rod outer segment. Hum Mol Genet, 25, 2031–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.